

The crystallization of the recombinant catalytic domain of the human membrane type 1 matrix metalloproteinase is described. The crystals diffracted to 2.24 Å resolution.
Keywords: membrane type 1 matrix metalloproteinase
Abstract
Membrane type 1 matrix metalloproteinase (MT1-MMP) belongs to the large family of zinc-dependent endopeptidases termed MMPs that are located in the extracellular matrix. MT1-MMP was crystallized at 277 K using the vapour-diffusion method with PEG as a precipitating agent. Data sets for MT1-MMP were collected to 2.24 Å resolution at 100 K. The crystals belonged to space group P43212, with unit-cell parameters a = 62.99, c = 122.60 Å. The crystal contained one molecule per asymmetric unit, with a Matthews coefficient (V M) of 2.90 Å3 Da−1; the solvent content is estimated to be 57.6%.
1. Introduction
Matrix metalloproteinases (MMPs) comprise a family of zinc-dependent endopeptidases found in the extracellular matrix (Tallant et al., 2010 ▶; Murphy & Nagase, 2008 ▶). They play a major role in the degradation of extracellular matrix substrates such as collagens, gelatin, fibronectin and others during embryogenesis, wound healing, bone resorption etc. (Vargová et al., 2012 ▶). More than 20 members of the MMP family are expressed in the human organism, many of which have been associated with common diseases such as atherosclerosis, arthritis, tumourigenesis, cancer and neuronal pathologies (Tallant et al., 2010 ▶; Murphy & Nagase, 2008 ▶). Therefore, researchers worldwide have been trying to find specific peptide-based inhibitors or other low-molecular-weight inhibitors for pharmaceutical application (Gupta & Patil, 2012 ▶; Ndinguri et al., 2012 ▶). However, the degree of specificity required for pharmaceutical use has not yet been obtained. It is now believed that specificity is not primarily obtained via the recognition of target peptides by the active-site cleft but through multiple interfacial interactions between a target substrate protein and the MMP (Murphy & Nagase, 2008 ▶; Udi et al., 2013 ▶). This hypothesis is supported by the fact that native MMP inhibitors, for example tissue inhibitors of metalloproteinases (TIMP), are also proteins, which allow interaction specificity to be gained through multiple surface–surface interactions (Murphy & Nagase, 2008 ▶; Grossman et al., 2010 ▶).
Upon expression, the MMPs contain an N-terminal pro-domain motif, which inhibits protein activity through binding of a pro-domain Cys to the active-site Zn2+ (Tallant et al., 2010 ▶; Nagase & Woessner, 1999 ▶; Vargová et al., 2012 ▶). Zymogenic activation of the enzyme allows the substrate peptides to enter the active site for metal-catalyzed hydrolysis. Besides the active-site Zn2+, MMPs contain a second Zn2+-binding site and two Ca2+-binding sites of unknown function. The catalytic domain downstream of the pro-domain is linked to a haemopexin-like domain via the so-called hinge region in most MMPs (Nagase & Woessner, 1999 ▶). The purpose of the haemopexin-like domains is not clear, but they may be important for substrate recognition and enzyme regulation. A group of MMPs, including MMP-14 to MMP-17, MMP-24 and MMP-25, are membrane-anchored proteins, with MMP-14 to MMP-16 and MMP-24 having an integral membrane-spanning domain located C-terminal to the haemopexin-like domain. These proteins are termed MT-MMPs, where MMP-14 is specifically named MT1-MMP.
Of the 168 X-ray structures of MMP catalytic domains currently available from the PDB, only MMP-1 (PDB entry 3shi; Bertini et al., 2012 ▶), MMP-2 (gelatinase A; PDB entry 1qib; Dhanaraj et al., 1999 ▶), MMP-3 (stromelysin; PDB entries 1qia and 1qic; Pavlovsky et al., 1999 ▶), MMP-8 (PDB entry 2oy4; Bertini et al., 2006 ▶) and MMP-12 (PDB entry 2oxu; Bertini et al., 2006 ▶) were crystallized in the uninhibited form, with only the crystals of MMP-1, MMP-8 and MMP-12 diffracting to a resolution of <2.5 Å. Detailed structural knowledge of the catalytic domains in all stages of the enzymatic activity is indispensable for understanding the catalytic process and thus the controlled manipulation of the enzymatic activity. The structure of the catalytic domain of none of the membrane-associated MMPs has been solved to date except for MT1-MMP in complex with TIMP-2 (PDB entries 1buv and 1bqq; Fernandez-Catalan et al., 1998 ▶) and in complex with a mutant of TIMP-1 (PDB entry 3ma2; Grossman et al., 2010 ▶). Here, we report the crystallization and diffraction of the uninhibited catalytic domain of human MT1-MMP.
2. Materials and methods
2.1. Purification
The plasmid containing the catalytic domain of human MT1-MMP together with the hinge linker to the haemopexin-like domain (residues 112–292) as described in Grossman et al. (2011 ▶) was transformed into Escherichia coli BL21 (DE3) (Novagen). Expression was carried out at 310 K in LB medium for 5 h after induction with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at OD600 nm = 0.8. Upon harvesting, the cells were sonicated on ice for five cycles of 1 min (Branson Sonifier, 70% output power, 1 s pulse duration). Inclusion bodies were harvested by centrifugation (6000g, 277 K, 15 min) and then subjected to extensive washing using a Potter–Elverheim homogenizer. The inclusion bodies were washed two times with 30 mM Tris–HCl pH 7.4, 30 mM NaCl, 1 mM EDTA, 2%(v/v) Tween 20, 5 mM β-mercaptoethanol, two times with 30 mM Tris–HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol and finally two times with 30 mM Tris–HCl pH 7.4, 30 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol. The inclusion bodies were then homogenized in 6 M urea, 50 mM Tris–HCl pH 8.5 and kept at 293 K for 2 h before ultracentrifugation (100 000g, 277 K, 30 min). Folding was carried out through dialysis (12–14 kDa molecular-weight cutoff) against a tenfold volume of 50 mM Tris–HCl pH 8.5, 100 mM NaCl, 10 mM CaCl2, 10 µM zinc acetate, 2 M urea (overnight at 293 K). The solution was then changed to 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM CaCl2, 10 µM zinc acetate, 1 M urea (24 h at 277 K). Finally, the protein was dialyzed two times against 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM CaCl2, 1 µM zinc acetate (24 h at 277 K). The solution was concentrated to 10 mg ml−1 at 277 K using Amicon Ultra-0.5 centrifugal filter devices (10 kDa molecular-weight cutoff; Millipore) and the solution was then exchanged for zinc-free buffer. The quality of the samples was determined by SDS–PAGE.
2.2. Crystallization
Screening of crystallization conditions was performed by the sitting-drop vapour-diffusion method with a Phoenix dispensing robot (Art Robbins Instruments, California, USA). The crystallization experiments consisted of 0.3 µl protein solution mixed with 0.3 µl reservoir solution and were equilibrated against 80 µl reservoir solution at 277 K. After 4–6 weeks, small crystals of MT1-MMP were obtained under several conditions from the PEG/Ion and PEG/Ion2 screening kits (Hampton Research, California, USA): (i) 0.1 M sodium malonate pH 4.0, 20%(w/v) PEG 3350, (ii) 4%(v/v) Tacsimate pH 6.0, 12%(w/v) PEG 3350 and (iii) 0.2 M ammonium nitrate, 20%(w/v) PEG 3350 pH 6.2. To obtain crystals suitable for X-ray diffraction, the crystals from condition (iii) were soaked in buffer consisting of 0.3 M ammonium nitrate, 25%(w/v) PEG 3350 pH 6.2 and 20%(v/v) glycerol as a cryoprotectant.
2.3. Data collection
Diffraction data sets were collected on beamline BL14.2 at BESSY II (Hermholtz-Zentrum Berlin, Germany). The detector was a Rayonix MX-225 (Rayonix, Illinois, USA). A complete native data set was collected to 2.24 Å resolution at an X-ray wavelength of 0.92 Å. Data collection for the Zn and Ca atoms was performed at X-ray wavelengths of 1.24 Å (zinc peak), 1.34 Å (zinc remote) and 1.95 Å (calcium). Diffraction images were indexed and integrated using the program XDS (Kabsch, 2010 ▶). Scaling was carried out using XSCALE (Kabsch, 2010 ▶). Molecular replacement and initial refinement were carried out using MOLREP (Vagin & Teplyakov, 2010 ▶) and REFMAC5 (Murshudov et al., 2011 ▶), respectively, from the CCP4 program package (Winn et al., 2011 ▶).
3. Results and discussion
MT1-MMP was expressed as inclusion bodies and subsequently folded. Characterization by SDS–PAGE showed that after dialysis the protein consists of a mixture of the pure catalytic domain (apparent molecular mass 21 kDa) and a form containing the hinge region (apparent molecular mass 24 kDa) (Fig. 1 ▶). The mature protein was generated by successive autoproteolysis after 1 d at 293 K as demonstrated by SDS–PAGE (Fig. 1 ▶). The catalytic activity of the mature MT1-MMP was demonstrated by a fluorogenic assay monitoring the peptide cleavage of Mca-Lys-Pro-Leu-Gly-Leu-Lys(Dnp)-Ala-Arg-NH2, where Mca is (7-methoxycoumarin-4-yl)acetyl and Dnp is N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl (Neumann et al., 2004 ▶).
Figure 1.
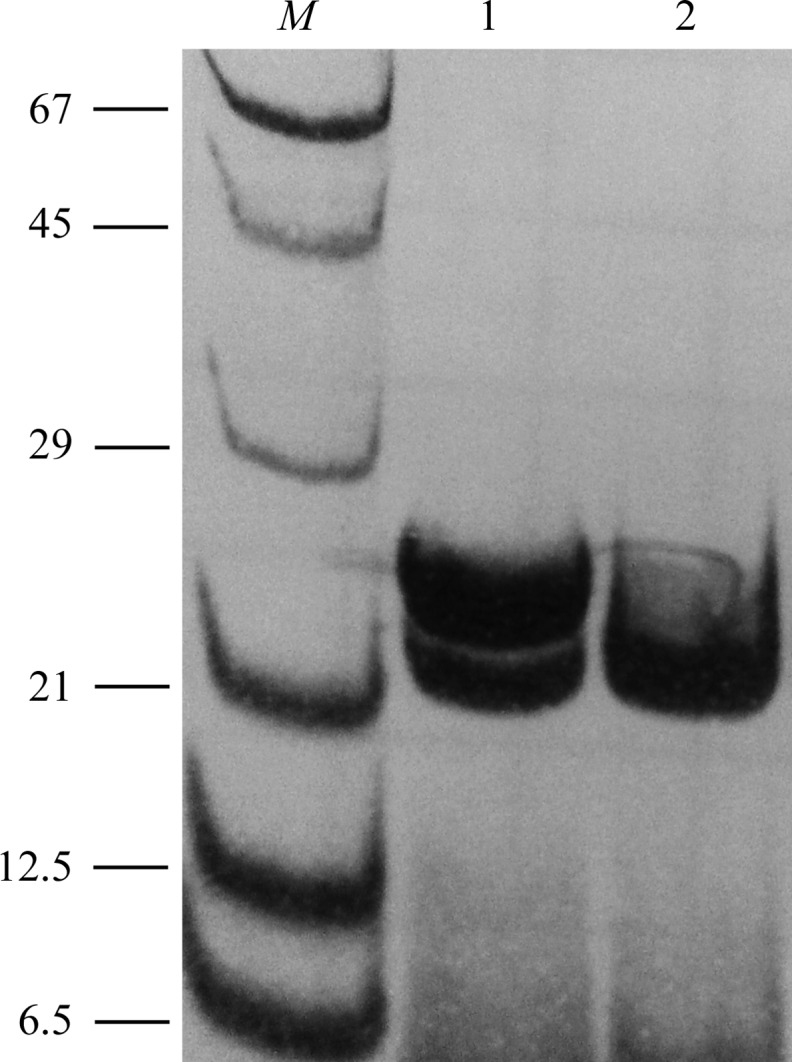
Autoproteolytic activity of MT1-MMP documented by SDS–PAGE (12%). Lane 1, recombinantly expressed human MT1-MMP catalytic domain together with the hinge region after the last dialysis step performed in 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM CaCl2, 1 µM zinc acetate; lane 2, sample as in lane 1 but kept for 24 h at 293 K prior to analysis. 20 µl of the samples were incubated with 10 µl 25 mM Tris–HCl pH 6.8, 1 mM Na2EDTA, 10%(v/v) glycerol, 5%(v/v) β-mercaptoethanol, 0.025%(w/v) bromophenol blue for 4 min at 343 K. Protein bands were stained with 0.1%(w/v) Coomassie Brilliant Blue G-250 in 10%(v/v) acetic acid and 40%(v/v) methanol. Lane M contains molecular-mass marker (labelled in kDa).
MT1-MMP was crystallized at 277 K using the vapour-diffusion method with PEG as the precipitating agent. The crystal dimensions were typically 0.1 × 0.1 × 0.05 mm (Fig. 2 ▶). The crystal of MT1-MMP diffracted to 2.24 Å resolution (Fig. 3 ▶). The data-collection conditions are summarized in Table 1 ▶. The crystal belonged to space group P43212 or P41212. The unit-cell parameters were a = 62.99, c = 122.60 Å. The calculated Matthews coefficient (V M; Matthews, 1968 ▶) of 2.90 Å3 Da−1, with a solvent content of 57.6%, indicates the presence of one molecule per asymmetric unit.
Figure 2.

Crystals of the catalytic domain of human MT1-MMP. They were obtained after four weeks from 0.3 µl protein solution mixed with 0.3 µl reservoir solution [0.2 M ammonium nitrate, 20%(w/v) PEG 3350 pH 6.2] at 277 K.
Figure 3.

Diffraction pattern of MT1-MMP (native data). The crystal-to-detector distance was maintained at 250 mm. 100 images of 1.0° oscillation angle and 3.0 s exposure time each were collected.
Table 1. X-ray data-collection statistics for the catalytic domain of MT1-MMP.
Values in parentheses are for the outermost resolution shell.
| Native | Zn peak | Zn remote | Ca peak | |
|---|---|---|---|---|
| Wavelength (Å) | 0.92 | 1.24 | 1.34 | 1.95 |
| Space group | P43212 | P43212 | P43212 | P43212 |
| Unit-cell parameters | ||||
| a (Å) | 62.99 | 63.02 | 63.05 | 63.08 |
| c (Å) | 122.60 | 122.64 | 122.68 | 122.79 |
| Resolution (Å) | 44.54–2.24 (2.37–2.24) | 44.56–2.46 (2.61–2.46) | 44.59–2.62 (2.78–2.62) | 44.61–2.88 (3.05–2.88) |
| No. of observed reflections | 98084 | 201819 | 161826 | 151387 |
| No. of unique reflections | 12491 | 16520 | 13552 | 10689 |
| R merge † | 0.105 (0.998) | 0.080 (0.705) | 0.065 (0.519) | 0.163 (1.145) |
| Completeness | 0.998 (0.993) | 0.962 (0.806) | 0.952 (0.777) | 0.998 (0.989) |
| 〈I/σ(I)〉 | 16.8 (2.3) | 27.3 (2.3) | 31.2 (2.7) | 20.4 (1.9) |
| CC1/2 | 99.8 (70.1) | 99.9 (68.4) | 99.9 (83.0) | 99.7 (68.4) |
R
merge = 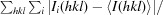
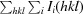 .
.
The molecular-replacement method was applied using the program MOLREP. The coordinates of the MT1-MMP–TIMP-2 complex (PDB entry 1bqq; Fernandez-Catalan et al., 1998 ▶) were used as a search model. A solution of the rotation and translation parameters for one MT1-MMP molecule in space group P43212 was obtained (the weighted R factor and score were 0.518 and 0.505, respectively). In space group P41212, the weighted R factor and score were 0.574 and 0.383, respectively. Calculation of the electron-density map by molecular replacement confirmed the space group to be P43212 with one monomer in the asymmetric unit. Zn-MAD data for calculation of the anomalous difference map were obtained from a crystal of MT1-MMP at X-ray wavelengths of 1.24 and 1.34 Å. The anomalous difference map revealed peaks that correspond to the zinc ions. Furthermore, Ca-SAD data were obtained at an X-ray wavelength of 1.95 Å, revealing peaks corresponding to calcium ions in the anomalous difference map. Model building and refinement are now in progress.
Acknowledgments
We are grateful for the technical assistance of Inge Heise, Norbert Dickmann and Yvonne Brandenburger (MPI for Chemical Energy Conversion, Mülheim, Germany). We thank the staff of beamline BL14.2 at BESSY II (Helmholtz-Zentrum Berlin, Germany) for their assistance during data collection. This work was supported by the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft. Financial support was also obtained from the Max Planck Society.
References
- Bertini, I., Calderone, V., Cerofolini, L., Fragai, M., Geraldes, C. F. G. C., Hermann, P., Luchinat, C., Parigi, G. & Teixeira, J. M. C. (2012). FEBS Lett. 586, 557–567. [DOI] [PubMed]
- Bertini, I., Calderone, V., Fragai, M., Luchinat, C., Maletta, M. & Yeo, K. J. (2006). Angew. Chem. Int. Ed. 45, 7952–7955. [DOI] [PubMed]
- Dhanaraj, V., Williams, M. G., Ye, Q.-Z., Molina, F., Johnson, L. L., Ortwine, D. F., Pavlovsky, A., Rubin, J. R., Skeean, R. W., White, A. D., Humblet, C., Hupe, D. J. & Blundell, T.L. (1999). Croat. Chem. Acta, 72, 575–591.
- Fernandez-Catalan, C., Bode, W., Huber, R., Turk, D., Calvete, J. J., Lichte, A., Tschesche, H. & Maskos, K. (1998). EMBO J. 17, 5238–5248. [DOI] [PMC free article] [PubMed]
- Grossman, M., Born, B., Heyden, M., Tworowski, D., Fields, G. B., Sagi, I. & Havenith, M. (2011). Nature Struct. Mol. Biol. 18, 1102–1108. [DOI] [PMC free article] [PubMed]
- Grossman, M., Tworowski, D., Dym, O., Lee, M. H., Levy, Y., Murphy, G. & Sagi, I. (2010). Biochemistry, 49, 6184–6192. [DOI] [PubMed]
- Gupta, S. P. & Patil, V. M. (2012). Matrix Metalloproteinase Inhibitors, edited by S. P. Gupta, pp. 35–56. Basel: Springer.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Murphy, G. & Nagase, H. (2008). Mol. Aspects Med. 29, 290–308. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nagase, H. & Woessner, J. F. Jr (1999). J. Biol. Chem. 274, 21491–21494. [DOI] [PubMed]
- Ndinguri, M. W., Bhowmick, M., Tokmina-Roszyk, D., Robichaud, T. K. & Fields, G. B. (2012). Molecules, 17, 14230–14248. [DOI] [PMC free article] [PubMed]
- Neumann, U., Kubota, H., Frei, K., Ganu, V. & Leppert, D. (2004). Anal. Biochem. 328, 166–173. [DOI] [PubMed]
- Pavlovsky, A. G., Williams, M. G., Ye, Q.-Z., Ortwine, D. F., Purchase, C. F. II, White, A. D., Dhanaraj, V., Roth, B. D., Johnson, L. L., Hupe, D. J., Humblet, C. & Blundell, T. L. (1999). Protein Sci. 8, 1455–1462. [DOI] [PMC free article] [PubMed]
- Tallant, C., Marrero, A. & Gomis-Rüth, F. X. (2010). Biochim. Biophys. Acta, 1803, 20–28. [DOI] [PubMed]
- Udi, Y., Fragai, M., Grossman, M., Mitternacht, S., Arad-Yellin, R., Calderone, V., Melikian, M., Toccafondi, M., Berezovsky, I. N., Luchinat, C. & Sagi, I. (2013). J. Mol. Biol. 425, 2330–2346. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vargová, V., Pytliak, M. & Mechírová, V. (2012). Matrix Metalloproteinase Inhibitors, edited by S. P. Gupta, pp. 1–33. Basel: Springer.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.