

Structures of the kinase domain of human tau-tubulin kinase 1 both without and with a bound inhibitor are reported.
Keywords: TTBK1, kinase domain, inhibitors
Abstract
Tau-tubulin kinase 1 (TTBK1) is a dual-specificity (serine/threonine and tyrosine) kinase belonging to the casein kinase 1 superfamily. TTBK1 is a neuron-specific kinase that regulates tau phosphorylation. Hyperphosphorylation of tau is implicated in the pathogenesis of Alzheimer’s disease. Two kinase-domain constructs of TTBK1 were expressed in a baculovirus-infected insect-cell system and purified. The purified TTBK1 kinase-domain proteins were crystallized using the hanging-drop vapor-diffusion method. X-ray diffraction data were collected and the structure of TTBK1 was determined by molecular replacement both as an apo structure and in complex with a kinase inhibitor.
1. Introduction
Regulation of microtubule dynamics is dependent, in part, upon tau, a multidomain protein that binds to and stabilizes microtubule assemblies. Tau is regulated by site-specific amino-acid phosphorylation by cellular protein kinases (Drechsel et al., 1992 ▶). Tau protein that has been hyperphosphorylated, truncated and misfolded has been implicated in the pathogenesis of several tauopathies, which are a family of progressive neurodegenerative diseases that are classified based on brain deposition of insoluble tau deposits (Lee & Leugers, 2012 ▶). The most prevalent tauopathy, Alzheimer’s disease (AD), spreads in a neuroanatomically specific manner, with progressive deposition of insoluble amyloid-containing plaques and insoluble neurofibrillary tangles (NFTs) that contain hyperphosphorylated tau (Braak et al., 1993 ▶; Holtzman et al., 2012 ▶). Therapeutics in the majority of clinical trials for AD have targeted the prevention of amyloid deposition and have shown very little to no efficacy in modulating disease progression (Holtzman et al., 2012 ▶), but the role of hyperphosphorylated tau in AD has been relatively untested in the clinic.
Tau-tubulin kinase 1 (TTBK1) is a dual-specificity (serine/threonine and tyrosine), neuronally expressed kinase that phosphorylates tau (Sato et al., 2006 ▶). TTBK1 contains 1321 amino acids, which include an N-terminal kinase domain and a unique polyglutamate domain (amino acids 733–771), the function of which remains under investigation. TTBK1 also contains several PEST (proline–glutamate–serine–threonine) sequences which have been hypothesized to be involved in the lysosomal processing of TTBK1 into a 100 kDa kinase domain-containing fragment (Sato et al., 2006 ▶). Several lines of evidence have implicated TTBK1 in AD progression. When overexpressed, TTBK1 promotes insoluble, hyperphosphorylated tau aggregation both in vitro and in vivo (Tomizawa et al., 2001 ▶; Sato et al., 2006 ▶, 2008 ▶; Xu et al., 2010 ▶). TTBK1 overexpression impairs spatial learning and decreases motor function in vivo (Sato et al., 2008 ▶; Xu et al., 2010 ▶). Also, when compared with age-matched normal brain, TTBK1 protein expression is increased in AD, where it has been shown to be associated with NFTs immunohistochemically (Sato et al., 2002 ▶, 2006 ▶, 2008 ▶). TTBK1 phosphorylates tau at Ser422, one of the earliest tau phosphorylation sites upregulated in AD pathology, and has been shown to co-localize with pS422-positive pre-tangles in AD brain (Sato et al., 2006 ▶; Mondragón-Rodríguez et al., 2008 ▶; Lund et al., 2013 ▶). Moreover, growing evidence suggests that modulating TTBK1 phosphorylation of tau could have a therapeutic benefit. Two distinct genetic populations have been identified that possess a single-nucleotide polymorphism (SNP) for a minor allelic variant of TTBK1, and this SNP has been associated with a decreased incidence of AD (Vázquez-Higuera et al., 2011 ▶; Yu et al., 2011 ▶). Additionally, active immunization against the pS422 site in tau-overexpressing THY-22 mice decreases tau phosphorylation in insoluble fractions and improves spatial learning (Troquier et al., 2012 ▶).
Despite this rapidly growing body of biological data postulating a role for TTBK1 in AD progression and pathology, very little is known about the structure of TTBK1 or of its close homologue TTBK2. Importantly, mutations in TTBK2 promoting protein truncation have been implicated in the development of spinocerebellar ataxia type 11 (Houlden et al., 2007 ▶; Edener et al., 2009 ▶), suggesting the need for selectivity against inhibiting TTBK2. Thus, with the hypothesis that designed selective TTBK1 inhibitors would serve as useful AD therapeutics, we cloned, expressed and purified TTBK1 kinase domains to enable the structure-based drug design of TTBK1 inhibitors. Homology modelling with TTBK2 shows a high degree of conservation amongst the cognate ligand-interacting sites within the TTBK1 and TTBK2 kinase domains, suggesting that identification of TTBK1-selective, ATP-competitive inhibitors would be challenging and would require structure-based drug design.
Here, we present crystal structures of apo and ligand-bound TTBK1 kinase domain. Just prior to the submission of this paper, another report of TTBK1 structures appeared (Xue et al., 2013 ▶). A report of the crystallization of TTBK2 (Kitano-Takahashi et al., 2007 ▶) also mentions that data had been collected from a TTBK2 crystal to 2.8 Å resolution, but as of December 2013 no paper had described the TTBK2 structure nor had the coordinates been deposited in the PDB.
2. Materials and methods
2.1. Construct design and cloning
Protein sequence numbering was defined by the NCBI RefSeq entries for TTBK1 (accession No. NP_115927.1) and TTBK2 (accession No. NP_775771.3). Based on protein sequence alignment with a crystallized TTBK2 kinase domain that had been reported previously (Kitano-Takahashi et al., 2007 ▶), TTBK1(14–343) was selected and expressed as an N-terminally His-tagged protein. This construct encompassed the kinase domain of TTBK1 (34–297) and included residues both N-terminal and C-terminal to the kinase domain. The coding region of human TTBK1(14–343) flanked by an NdeI site at the 5′ end and a TGA stop codon and an XhoI site at the 3′ end was gene-synthesized at GenScript USA Inc. (Piscataway, New Jersey, USA) and subcloned into a modified pFastBac1 vector (Invitrogen, Carlsbad, California, USA) with an N-terminal His-TVMV (Tobacco vein mottling virus protease) tag to generate His-TVMV-TTBK1(14–343)-pFB. The fidelity of the synthetic fragment was confirmed by DNA sequencing. An additional shorter TTBK1 construct, His-TVMV-TTBK1(14–320)-pFB, was generated by PCR from the longer construct and its sequence was verified. The domain structure of TTBK1 and the amino-acid sequences of the two TTBK1 constructs are shown in Fig. 1 ▶.
Figure 1.

(a) Schematic diagram of human TTBK1 (as derived from Sato et al., 2006 ▶). TTBK1 includes a dual kinase domain (KD), a conserved domain A (DA), a polyglutamate domain (PG) and a conserved domain B (DB). (b) The amino-acid sequence of the TTBK1 constructs that were designed for crystallization studies showing the longer construct (residues 14–343) and the shorter construct (residues 14–320). The N-terminal His6 affinity tag and TVMV protease cleavage site are underlined. Non-native residues following TVMV cleavage are denoted as red letters. Residues encompassing the TTBK1 kinase domain are denoted as cyan letters.
2.2. Expression
Baculovirus was generated for all constructs using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer’s protocol. Briefly, recombinant bacmid was isolated from transformed DH10Bac Escherichia coli competent cells (Invitrogen) and was used to transfect Spodoptera frugiperda (Sf9) insect cells (Invitrogen). The baculovirus was harvested 72 h post-transfection and a virus stock was prepared by infecting fresh Sf9 cells at a 1:1000 virus:cell ratio for 66 h.
For large-scale protein production, Sf9 cells (Expression System, Davis, California, USA) grown in ESF921 insect medium (Expression System) at 2 × 106 cells ml−1 were infected with virus stock at a 1:100 virus:cell ratio for 66 h. The production was carried out either on a 10 l scale in a 22 l cellbag (GE Healthcare Bioscience, Pittsburgh, Pennsylvania, USA) or on a 20 l scale in a 50 l cellbag using a WAVE-Bioreactor System 20/50 (GE Healthcare Bioscience). The infected cells were harvested by centrifugation at 2000 rev min−1 for 20 min at 277 K in a Sorvall RC12BP centrifuge. The cell pellets were stored at ∼200 K before the protein was purified.
2.3. Purification of TTBK1(14–343)
All steps were performed at 277 K unless noted otherwise. Frozen Sf9 cells harvested from 11 l culture were suspended in 350 ml buffer A (25 mM Na HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 20 mM imidazole, 2 mM dithiothreitol) plus six tablets of Complete EDTA-free protease-inhibitor cocktail (Roche Applied Science) and 20 U ml−1 Benzonase nuclease (EMD Millipore). The cells were lysed by nitrogen cavitation using a cell-disruption vessel (Parr Instrument Co.) pressurized at ∼1.7 MPa for 30 min. The lysate was clarified by sedimentation at 100 000g for 40 min (Beckman 45Ti rotor) and the supernatant was loaded onto a Nickel-Sepharose 6 Fast Flow (GE Healthcare) column (2.5 × 2.1 cm) equilibrated with buffer A. The column was washed with 20 volumes of buffer A and the protein was eluted with six volumes of buffer A plus 350 mM imidazole pH 7.5. The protein was concentrated to a volume of 8.5 ml using a Millipore Ultra 15 ml concentration unit with a 10 000 Da cutoff and applied onto a HiLoad 26/60 Superdex 200 column (GE Healthcare) equilibrated with buffer B (25 mM Na HEPES pH 7.5, 0.3 M NaCl, 10% glycerol, 0.2 mM EDTA, 2 mM DTT). The purified protein was pooled from a peak centred at 223 ml and the His tag was cleaved with His-TVMV protease (1:10 ratio of TVMV:TTBK1) overnight. The TVMV protease was removed by applying the reaction mixture onto a Nickel-Sepharose 6 Fast Flow (GE Healthcare) column (1.5 × 0.6 cm) equilibrated with buffer B plus 15 mM imidazole. The column-flowthrough fraction was concentrated to 2 ml and buffer-exchanged by applying it onto a PD10 desalting column (GE Healthcare) equilibrated in buffer C (20 mM Na HEPES pH 7.5, 250 mM NaCl, 10% glycerol, 0.5 mM EDTA, 2 mM DTT). The protein purity was ≥85% as determined by SDS–PAGE analysis and Coomassie Brilliant Blue staining (Fig. 2 ▶ a). Electrospray ionization mass spectrometry was used to confirm the identity of the protein, and the purified TTBK1 was determined to be predominantly nonphosphorylated with a minor amount of monophosphorylation. Protein concentrations for both the 14–343 and 14–320 constructs (§2.4) were determined by the method of Bradford (1976 ▶) using bovine serum albumin as a standard (Bio-Rad).
Figure 2.

SDS–PAGE analysis of (a) TTBK1(14–343) and (b) TTBK1(14–320) purified from baculovirus. The samples were electrophoresed on a 4–12% Bis-Tris NuPAGE gel (Invitrogen) and stained with SimplyBlue Coomassie stain (Invitrogen). Lane 1, molecular-mass markers (kDa); lane 2, 1 µg protein; lane 3, 3 µg protein; lane 4, 6 µg protein. The arrow to the right of lane 4 in (a) indicates the proteolytic fragment.
2.4. Purification of TTBK1(14–320)
TTBK1(14–320) was purified using the same methods as described for TTBK1(14–343). The TTBK1(14–320) protein purity was ≥85% as determined by SDS–PAGE analysis and Coomassie Brilliant Blue staining (Fig. 2 ▶ b). Electrospray ionization mass spectrometry confirmed the identity of the protein and the absence of any significant phosphorylation in this protein construct.
2.5. Synthesis of compound 3
The 3-({5-[(4-amino-4-methylpiperidin-1-yl)methyl]pyrrolo[2,1-f][1,2,4]triazin-4-yl}amino)-5-bromophenol (3) that was used for the cocrystallization experiments was synthesized from N-{(4-chloropyrrolo[1,2-f][1,2,4]triazin-5-yl)methyl}-N,N-diethylethanaminium bromide (1; Fink et al., 2011 ▶) as outlined in Fig. 3 ▶. A mixture of 1 (105 mg, 0.302 mmol) and 3-bromo-5-methoxyaniline (61 mg, 0.302 mmol) in dry acetonitrile (2 ml) was heated at 348 K for 1 h. After cooling to ∼293 K, 4-methylpiperidin-4-amine (207 mg, 0.604 mmol) and N,N-diisopropylethylamine (0.527 ml, 3.02 mmol) were added and the reaction was heated in a microwave reactor at 338 K for 1 h. Preparative HPLC afforded 5-[(4-amino-4-methylpiperidin-1-yl)methyl]-N-(3-bromo-5-methoxyphenyl)pyrrolo[1,2-f][1,2,4]triazin-4-amine (2; 52 mg, 39% yield) as a solid. MS (ESI): m/z = 447.20 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 12.02 (br s, 1H), 7.92 (s, 1H), 7.48–7.43 (m, 2H), 7.42–7.36 (m, 1H), 6.78 (d, J = 1.8 Hz, 1H), 6.48 (d, J = 2.4 Hz, 1H), 3.80 (s, 3H), 3.76 (s, 2 H), 3.03–2.27 (m, 4H), 1.76–1.62 (m, 2H), 1.53 (d, J = 12.2 Hz, 2H), 1.41 (br s, 2H), 1.16 (s, 3H). A solution of 2 (43 mg, 0.097 mmol) in dry dichloromethane (8 ml) under nitrogen was cooled to 196 K. Boron tribromide (1.0 ml, 1.0 M in dichloromethane, ten equivalents) was added dropwise and the reaction was allowed to warm to ∼293 K. After 18 h, water (2 ml) was added and the organic solvent was removed. Preparative HPLC afforded 3 (18 mg, 41% yield) as a white solid. MS (ESI): m/z = 433.18 (M + H)+. 1H NMR (500 MHz, CD3OD): δ 7.86 (s, 1H), 7.53 (d, J = 2.4 Hz, 1H), 7.41 (br s, 1H), 7.20 (br s, 1H), 6.75 (d, J = 1.8 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 3.86 (s, 2H), 2.65–2.69 (m, 4H), 1.72 (d, J = 4.3 Hz, 4H), 1.24 (s, 3H).
Figure 3.

Conversion of 3-bromo-5-methoxyaniline to compound 3. a, 3-Bromo-5-methoxyaniline, acetonitrile, heat and then 4-methylpiperidin-4-amine, DIPEA, microwave reaction; b, BBr3, dichloromethane.
2.6. Crystallization
2.6.1. TTBK1(14–343) + staurosporine
TTBK1(14–343; 46 µM) was incubated in buffer C with 140 µM staurosporine (Sigma; a threefold molar excess) overnight at 277 K. The mixture was concentrated to 16 mg ml−1 (423 µM) using a Millipore 15 ml concentration unit with a 10 000 Da cutoff. All crystallization experiments were carried out at room temperature (∼293 K). Given the limited amount of protein available, crystallization conditions were derived from a screen based on previous experience with kinase domains that consisted of six buffers (bis-tris pH 6.0, cacodylate pH 6.5, MES pH 6.5, ammonium acetate pH 7.0, HEPES pH 7.5 and Tris–HCl pH 8.5), 30%(w/v) PEG 5000 methyl ether and various unbuffered salts (ammonium sulfate, lithium sulfate, ammonium acetate, sodium acetate etc.), which were added to obtain a 100 mM solution, and organic additives. Clusters of very thin crystals were observed overnight. Crystallization conditions were optimized by adjusting the PEG 5000 methyl ether concentration and the ratio of protein solution to reservoir solution. Crystals were grown using the hanging-drop vapour-diffusion method in a drop consisting of 1.5 µl protein solution and 2 µl reservoir solution consisting of 100 mM ammonium acetate pH 7.0 buffer, 19%(w/v) PEG 5000 methyl ether and an additional unbuffered 100 mM ammonium acetate in Qiagen EasyXtal plates (Qiagen). The crystals appeared overnight and continued to grow for one week. Macroseeding was performed to obtain larger crystals for diffraction experiments by breaking the clusters with a metal probe and transferring fragments to a new drop on the tip of the probe (Fig. 4 ▶ a). Crystals were flash-cooled in liquid nitrogen for data collection using 25% glycerol and 75% reservoir solution as a cryoprotectant. Crystallization of TTBK1(14–320) in the absence of staurosporine yielded crystals with the same morphology under the same crystallization conditions.
Figure 4.

Crystals of TTBK1 obtained in the presence of (a) staurosporine and (b) compound 3.
2.6.2. TTBK1(14-320) + compound 3
TTBK1(14–320) was mixed with compound 3 by incubating TTBK1 (29.4 µM) in buffer C with 90 µM compound 3 (a threefold molar excess) overnight at 277 K. The protein was concentrated to 15 mg ml−1 (424 µM) using a Millipore 15 ml concentration unit with a 10 000 Da cutoff. Crystals were grown in a similar manner as for TTBK1(14–343) in the presence or absence of staurosporine, but the TTBK1(14–320) complex was more stable than the TTBK1(14–343) complex. The protein solution was mixed in a 1.5:2(v:v) ratio with a well solution consisting of 100 mM bis-tris propane pH 6.0, 24%(w/v) PEG 5000 methyl ether, 200 mM ammonium sulfate. Diffraction-quality crystals (Fig. 4 ▶ b) were formed by multiple rounds of seeding. Crystals were cryoprotected by transfer to 25% glycerol and 75% reservoir solution and were flash-cooled using liquid nitrogen.
2.7. Data collection and processing
2.7.1. Data collection and processing for crystals grown in the presence of staurosporine
Data were initially collected on beamline 17-ID at the Advanced Photon Source (APS; Argonne, Illinois, USA) using radiation of wavelength 1.0 Å at ∼100 K. A Pilatus 6M detector was used to collect 900 0.2° images. The data were integrated with XDS (Kabsch, 2010a ▶,b ▶) and scaled with SCALA (Winn et al., 2011 ▶) through the use of autoPROC (Global Phasing Ltd, Cambridge, England). The statistics are shown in Table 1 ▶.
Table 1. Data-collection results.
Values in parentheses are for the highest resolution shell.
| Compound | Apo | Apo | 3 (co-crystal) |
|---|---|---|---|
| Source | Advanced Photon Source, beamline 17-ID | Canadian Light Source, beamline 08ID-1 | Advanced Photon Source, beamline 17-ID |
| Wavelength | 1.0 | 0.9797 | 1.0 |
| Detector | Pilatus 6M | Rayonix MX-300 | Pilatus 6M |
| Crystal-to-detector distance (mm) | 450 | 280 | 180 |
| Rotation range/image () | 0.2 | 0.75 | 0.2 |
| Total rotation range () | 180 | 180 | 180 |
| Exposure time per image (s) | 0.2 | 0.8 | 0.2 |
| Space group | C2 | C2 | C2 |
| Unit-cell parameters (, ) | a = 171.7, b = 40.0, c = 49.4, = 103.8 | a = 170.3, b = 39.9, c = 50.4, = 104.3 | a = 169.5, b = 39.9, c = 50.5, = 105.3 |
| Mosaicity range () | 0.51.2 | 0.51.4 | 0.30.5 |
| Resolution range () | 47.962.54 (2.682.54) | 502.13 (2.212.13) | 38.771.42 (1.501.42) |
| Total No. of measured reflections | 34058 (5024) | 68682 (18769) | 198458 (60739) |
| Unique reflections | 10775 (1565) | 6839 (1868) | 28611 (8785) |
| Multiplicity | 3.2 (3.2) | 3.7 (3.7) | 3.3 (3.3) |
| Completeness (%) | 98.2 (99.4) | 99.4 (100.0) | 98.5 (98.2) |
| R merge † (%) | 8.4 (43.6) | 7.0 (49.0) | 4.1 (37.5) |
| Mean I/(I) | 8.4 (2.4) | 11.6 (4.0) | 13.0 (2.3) |
| Overall B from Wilson plot (2) | 69 | 48 | 19 |
R
merge = 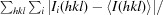
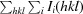 , where I
i(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations i of reflection hkl.
Data from a larger crystal were collected on the 08ID-1 beamline at the Canadian Light Source, Saskatoon, Saskatchewan, Canada (Grochulski et al., 2011 ▶) at ∼100 K. These data were integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). The statistics are shown in Table 1 ▶.
2.7.2. Data collection and processing for the crystal grown in the presence of compound 3
Data for TTBK1 complexed with compound 3 were collected on beamline 17-ID at APS, which yielded a 1.42 Å resolution data set. The wavelength used was 1.0 Å and the temperature was ∼100 K. The detector was a Pilatus 6M and 900 0.2° images were collected. The data were integrated with XDS (Kabsch, 2010a ▶,b) and scaled with SCALA (Winn et al., 2011 ▶) through the use of autoPROC (Global Phasing Ltd, Cambridge, England). The statistics are shown in Table 1 ▶.
2.8. Structure determination and refinement
The structure of TTBK1 was determined by molecular replacement using Phaser (McCoy et al., 2007 ▶) with the initial lower resolution data set and a model derived from casein kinase 1 (CK1; PDB entry 1cki; Longenecker et al., 1996 ▶), which at the time of structure determination was the most similar structure in the PDB at ∼37% sequence identity for the kinase domains. A model of CK1 was prepared by dividing it into N-terminal (residues 8–100) and C-terminal (residues 103–171, 178–218 and 231–295) domains. Non-identical residues were typically truncated to Ala, but similar residues were maintained or truncated minimally, e.g. if the sequence was Tyr in CK1 and Phe in TTBK1 then Phe would be retained. The statistics for the Phaser solution are shown in Table 2 ▶. Successive rounds of refinement and rebuilding used Coot (Emsley et al., 2010 ▶) and autoBUSTER (Blanc et al., 2004 ▶). However, some regions of the model remained suboptimal, so these data were abandoned when the higher resolution data became available. Refinement of the higher resolution structure also used Coot and autoBUSTER and the statistics are shown in Table 3 ▶. Residues 22–314 were interpretable without any breaks. Molecular superpositions were carried out with ALIGN (Satow et al., 1986 ▶).
Table 2. Molecular-replacement results.
RFZ, rotation-function Z-score; TFZ, translation-function Z-score; PAK, number of C packing clashes; LLG, log-likelihood gain.
| Domain | RFZ | TFZ | PAK | LLG | LLG |
|---|---|---|---|---|---|
| C-terminal | 9.4 | 8.9 | 0 | 161 | |
| N-terminal | 3.8 | 7.1 | 3 | 287 | 126 |
Table 3. Refinement results.
The values in parentheses for the resolution, R work and R free rows are for the highest resolution shell.
| Apo | 3 (co-crystal) | |
|---|---|---|
| Resolution () | 47.482.12 (2.252.12) | 32.21.42 (1.461.42) |
| R work | 0.204 (0.252) | 0.185 (0.226) |
| R free | 0.241 (0.322) | 0.209 (0.260) |
| Average B factor (2) | ||
| Overall | 54 | 23 |
| Protein (residues 22314) | 53 | 23 |
| Waters (No. of waters) | 56 (80) | 30 (202) |
| Non-water solvent | 82 (1 glycerol) | 23 (3 sulfates) |
| Ligand | 16 | |
| R.m.s.d., bond distances () | 0.010 | 0.010 |
| R.m.s.d., bond angles () | 1.1 | 1.0 |
| Ramachandran† (%) | ||
| Favoured and allowed | 98.5 | 99.6 |
| Disallowed | 0 | 0 |
| PDB code | 4nfm | 4nfn |
As defined by Laskowski et al. (1993 ▶).
The structure of TTBK1 grown in the presence of compound 3 was determined using the rigid-body fitting part of the CCP4 version of AMoRe (Winn et al., 2011 ▶; Navaza, 1994 ▶) followed by refinement in autoBUSTER. grade (Global Phasing Ltd, Cambridge, England) was used to generate a CIF restraint dictionary for compound 3 and rhofit (Global Phasing Ltd, Cambridge, England) was used to place the ligand. Refinement was completed using Coot and autoBUSTER and the statistics are shown in Table 3 ▶. Residues 22–314 were interpretable without any breaks. Display graphics were produced with PyMOL v.1.4 (Schrödinger).
3. Results and discussion
3.1. Protein expression and purification
Expression of TTBK1(14–343) in Sf9 insect cells infected with recombinant baculovirus resulted in a modestly overexpressed protein that was soluble and could be purified with a yield of 0.6 mg per litre of cell culture. An intact band migrating at the expected molecular size corresponding to a 37.8 kDa protein was observed on SDS–PAGE analysis (Fig. 2 ▶ a). SDS–PAGE analysis of protein stored at 277 K for 4 d revealed an additional lower band running at approximately 35 kDa (data not shown). A small amount of this fragment was also observed running just ahead of the main TTBK1(14–343) band when freshly purified preparations were analyzed by SDS–PAGE (Fig. 2 ▶ a, arrow). The suspicion that this band corresponded to a proteolytic fragment of TTBK1(14–343) was confirmed by analysis of the sample using electrospray ionization mass spectrometry. The mass of this fragment was consistent with the site of proteolysis occurring at residue 320 in the TTBK1(14–343) sequence. This degradation product persisted for more than one week of storage at 277 K, and analysis of the preparation by SDS–PAGE and mass spectrometry did not reveal further degradation (data not shown). Hence, the cleavage site at residue 320 represented a more stable fragment of TTBK1 and led to the design, expression and purification of TTBK1(14–320) for crystallization. Additionally, both purified TTBK1(14–343) and TTBK1(14–320) demonstrated high tau-dependent kinase activity in a biochemical assay (Gill et al., in preparation).
3.2. TTBK1 apo structure
The structure of TTBK1 crystallized in the presence of staurosporine showed no evidence of staurosporine binding. Subsequent analysis in an activity assay showed that staurosporine was not an inhibitor of TTBK1 and a fluorescent dye-binding thermal stabilization assay showed that the presence of staurosporine provided no stabilization against thermal unfolding of TTBK1(14–343), suggesting that staurosporine binding to the TTBK1 kinase domain was very weak or non-existent (data not shown).
The structure of TTBK1 resembles other protein kinases, which are characterized by a largely β-strand-containing N-terminal domain and a mostly α-helical C-terminal domain connected by an extended ‘hinge’ region (residues 108–111). The N-terminal domain contains the P-loop (residues 40–49) and the αC helix (residues 72–84). The C-terminal domain contains the DFG motif (residues 176–178) and the activation loop (residues 178–202), which although rather mobile, i.e. with high B factors, was traceable in its entirety (Fig. 5 ▶ a). The hinge region at the interface between the N- and C-terminal domains constitutes the adenine nucleotide-binding site. As described in §2, the structure of casein kinase 1 (PDB entry 1cki) was the source of the model used in molecular replacement to determine the structure of the TTBK1 kinase domain. When the C-terminal domain of 1cki was superimposed on the C-terminal domain of TTBK1, an additional 9° rotation was required to obtain the best superposition of the N-terminal domain of 1cki on TTBK1. This additional rotation showed the wisdom of searching for the C-terminal and N-terminal domains separately during molecular replacement (Fig. 5 ▶ b). The other large differences between the structure of CK1 and TTBK1 are the position of the P-loop, which is positioned away from the active site in TTBK1 compared with CK1, and the activation loop, which is differently arranged (Fig. 5 ▶ c).
Figure 5.

Structure of apo TTBK1. (a) The final 2F o − F c electron-density map is shown with the final model of the activation loop (178–202), showing that the electron density is reasonably good throughout the region. (b) Cartoon diagram of the superposition of CK1 (PDB entry 1cki) on TTBK1, with TTBK1 shown in cyan and 1cki shown in red. The C-terminal domains of CK1 and TTBK1 were superimposed and the transformation matrix was then applied to the entire 1ck1 structure, showing that the N-terminal domain of TTBK1 has rotated relative to its position in the 1cki structure. (c) After the superposition described in (b) the N-terminal domain of 1cki was superimposed on the N-terminal domain of TTBK1. This required an additional 9° rotation about the axis shown in both (b) and (c). This shows that the N-terminal domains superimpose reasonably well except for some local variation, for example around residues 45 (the P-loop), 69 and 100 in TTBK1, which correspond to residues 22, 47 and 78, respectively, in 1ck1. (d) The active site is shown with water molecules 501, 502, 503 and 504 labelled. Hydrogen bonds are shown as a series of black prolate ellipsoids.
Four water molecules are visible in the ATP-binding pocket (Fig. 5 ▶ d). Two water molecules (HOH502 and HOH501) form hydrogen bonds to the hinge region (Gln110 N and Gly111 O, respectively) and to each other. A third water molecule, HOH503, forms a hydrogen bond to HOH502 and is nearly close enough to Gln108 O to form a hydrogen bond. Together, they occupy the hydrophilic donor–acceptor–donor motif site of the adenine pocket. Another water molecule (HOH504) forms hydrogen bonds to the Glu77 side chain and Phe177 N.
The sugar and phosphate pockets are delineated by Ala161, Leu175, Asp176 and Asn159 and part of the relatively long activation loop (residues Gly178–Thr202) that forms a short β-strand (residues Arg181–Thr184) similar to those observed in CK1 and other kinase families (e.g. SRC).
The side chain of Lys63 was not visible in the apo structure. In other kinases, the equivalent conserved lysine residue often forms a salt bridge to the aspartate of the DFG motif (e.g. Lys33 and Asp145 in the CDK2 structure; PDB entry 4ek3; Y. N. Kang & J. A. Stuckey, unpublished work) or to an acidic residue from the αC helix (e.g. Lys38 and Glu52 in the CK1∊ structure; PDB entry 4hok; Long et al., 2012 ▶).
Comparison of the TTBK1 structure with a CK1∊ apo structure (PDB entry 4hok) also reveals structural similarities: the DFG motif adopts the active DFG-in conformation and the gatekeeper residue is a Met (Met107 in TTBK1) in both cases. Behind the gatekeeper residue is a small partly hydrophobic back pocket bounded by Met107, Val105, Lys63 and Leu81.
CK1∊ has been reported to adopt a DFG-out conformation stabilized by Phe55 (from the αC helix), which rotates and fills the phenylalanine pocket when in the inactive inhibitor-bound conformation (Long et al., 2012 ▶). This stabilization has been proposed to explain the observed selectivity between δ and ∊ isoforms for certain CK1 inhibitors despite the two sharing a highly conserved ATP pocket (Long et al., 2012 ▶). Given the sequence and structural similarities to CK1, it is possible that TTBK1 may also adopt such a DFG-out conformation. However, the equivalent residue to Phe55 in TTBK1 is a valine, which is not likely to provide stabilization of the DFG-out form. Nevertheless, such a potentially inactive conformation might still be induced in TTBK1 by its binding to certain inhibitors.
3.3. TTBK1–compound 3 structure
Compound 3 inhibits TTBK1 with an IC50 of 120 nM and TTBK2 with an IC50 of 170 nM (Gill et al., in preparation). The most remarkable feature of the complex of TTBK1 with compound 3 (Figs. 3 ▶ and 6 ▶ a) compared with the apo molecule is how little the overall structure needed to change to bind compound 3 (Figs. 6 ▶ c and 6 ▶ d). Placement of compound 3 was straightforward, although rhofit was not able to place the bromophenol moiety because the side chain of the gatekeeper residue Met107 rearranged to accommodate the ligand (Fig. 6 ▶ b). The terminal side-chain C atom of the Met107 gatekeeper residue is rotated by approximately 90°, allowing the bromo group of compound 3 to fit into the back pocket (Fig. 6 ▶ d). The side chain of Glu77 also rearranged to allow interaction with the phenol moiety. The Br atom of compound 3 was modelled as partially radiolyzed, i.e. at 2/3 occupancy rather than full occupancy. The hinge region forms a single hydrogen bond to compound 3 between Gly110 N and the pyrrolotriazinyl N atom (N7) (Figs. 6 ▶ b and 6 ▶ d). Compound 3, however, does form other hydrogen bonds to TTBK1. The phenolic hydroxyl moiety (O24) forms hydrogen bonds to Glu77 OE1, Asp176 OD1 and Asp176 OD2, and the piperidinyl amino N atom (N26) forms hydrogen bonds to Asn159 OD1 and Asp176 OD2 (Fig. 6 ▶ d). Binding of the ligand also displaces the four water molecules visible in the pocket in the apo structure (Fig. 5 ▶ d). The side chain of Lys63 is visible in this structure and forms a salt bridge with Glu77 from the αC helix (Fig. 6 ▶ d). The submicromolar potency of compound 3 can be explained from the structure by the following: (i) three good hydrogen bonds are formed with TTBK1, (ii) the bromophenyl and imidazopyrimidine moieties are involved in favourable hydrophobic interactions and (iii) the overall shape is complementary with the ATP pocket.
Figure 6.

Binding of compound 3 to TTBK1. (a) Two-dimensional representation of compound 3 oriented similarly to that in (b). An IUPAC name for compound 3 is 3-({5-[(4-amino-4-methylpiperidin-1-yl)methyl]pyrrolo[2,1-f][1,2,4]triazin-4-yl}amino)-5-bromophenol. (b) Initial F o − F c electron density contoured at 3 r.m.s.d. (slate) and 10 r.m.s.d. (orange) with the final model of the TTBK1–compound 3 complex. The hinge region (residues 108–111) is at the top of the figure and the gatekeeper residue (Met107) is on the right. C atoms are shown in cyan for TTBK1 and in orange for compound 3, N atoms are shown in blue, O atoms are shown in red, S atoms are shown in yellow, the Br atom is shown in violet and waters are represented by red spheres. (c) Cartoon representation of the placement of compound 3 in the context of the entire TTBK1 kinase domain, with a superposition of the apo structure on the complex structure. The complex structure is shown in cyan, the apo structure is shown in magenta and compound 3 is shown with C atoms in orange, N atoms in blue, the O atom in red and the Br atom in violet. (d) Superposition of the apo structure at the binding site on the structure of the complex with compound 3. The hinge region (residues 108–111) is at the top of the figure. C atoms are shown in cyan for the complex structure and magenta for the apo structure, N atoms are shown in blue, O atoms are shown in red, S atoms are shown in yellow, the Br atom is shown in violet and water molecules are shown as red spheres. Hydrogen bonds are shown as a series of black prolate ellipsoids. The figures show that only the side chains of Glu77, Met107 (gatekeeper residue) and to a lesser extent Asp176 move significantly upon binding compound 3.
In the recent TTBK1 structure (PDB entry 4btk; Xue et al., 2013 ▶) the gatekeeper methionine (107 in our structure; 131 in 4btk) is essentially unperturbed relative to the apo structure, and the phenol moiety of the 4btk ligand (DTQ) is perpendicular to the bromophenol moiety of compound 3. Potential reasons for this difference include (i) the hydrogen bond between the phenolic hydroxyl moiety and Glu77 and Asp176 favours this orientation, (ii) the same phenolic hydroxyl moiety is likely to clash with the stem of Lys63 if rotated into a similar orientation as the ligand in the 4btk structure, (iii) the bicyclic cores of the two ligands are not completely aligned, resulting in a slightly different vector connecting the core with the aniline N atoms of the respective substituents, and (iv) if the aniline ring of compound 3 is rotated to match the ligand structure in 4btk, the bromo moiety would potentially clash with HOH587 and HOH589, and may disrupt the hydrogen-bond network formed between these waters, HOH525 and the protein.
The piperidinyl amino N atom of compound 3 occupies a similar location as the Mg2+ ion in ATP-bound protein kinase structures (e.g. the CDK2 structure with PDB code 1hck and the ERK2 structure with PDB code 4gt3). The hydrogen bonds that it forms to Asn159 and Asp176 are similar in geometry to those that coordinate the ion in the ATP-bound structures. Note, however, that the recent TTBK1 structure with ADP bound (PDB entry 4btj; Xue et al., 2013 ▶) does not show a Mg2+ ion, but it would be expected to bind in a similar location coordinated by Asp176 and Asn159. As in the apo structure, the DFG motif adopts the DFG-in activated conformation and compound 3 may be classified as a type I inhibitor (Zuccotto et al., 2010 ▶). However, the residue composition and structure near the DFG phenylalanine pocket are similar to the imatinib-bound allosteric site of Abl kinase. This suggests that TTBK1 may also adopt a DFG-out conformation depending on the ligand bound. Indeed, given that the related CK1∊ structure was also shown to adopt the DFG-out conformation (Long et al., 2012 ▶), it may be feasible to design type II inhibitors for TTBK1. While TTBK1/2 selectivity may still be difficult to achieve owing to the high degree of sequence similarity, e.g. a sequence alignment between TTBK1 and TTBK2 shows 100% sequence conservation for the binding site defined by the 32 residues within 6 Å of the bound compound 3, the TTBK1 kinase-domain structure presented here provides insights that may help to guide the design of drugs for therapeutic use in Alzheimer’s disease.
Supplementary Material
PDB reference: tau-tubulin kinase 1, 4nfm
PDB reference: complex with inhibitor, 4nfn
Acknowledgments
We thank John Newitt for extensive comments on a draft of the manuscript. Data were collected by Rick Walter and Gina Ranieri of Shamrock Structures LLC using beamlines at the Advanced Photon Source and the Canadian Light Source. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman–Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of beamline 08ID-1 at the Canadian Light Source was supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada and the University of Saskatchewan.
References
- Blanc, E., Roversi, P., Vonrhein, C., Flensburg, C., Lea, S. M. & Bricogne, G. (2004). Acta Cryst. D60, 2210–2221. [DOI] [PubMed]
- Braak, H., Braak, E. & Bohl, J. (1993). Eur. Neurol. 33, 403–408. [DOI] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem. 72, 248–254. [DOI] [PubMed]
- Drechsel, D. N., Hyman, A. A., Cobb, M. H. & Kirschner, M. W. (1992). Mol. Biol. Cell, 3, 1141–1154. [DOI] [PMC free article] [PubMed]
- Edener, U., Kurth, I., Meiner, A., Hoffmann, F., Hübner, C. A., Bernard, V., Gillessen-Kaesbach, G. & Zühlke, C. (2009). J. Neurol. 256, 1856–1859. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fink, B. E., Norris, D., Mastalerz, H., Chen, P., Goyal, B., Zhao, Y., Kim, S.-H., Vite, G. D., Lee, F. Y., Zhang, H., Oppenheimer, S., Tokarski, J. S., Wong, T. W. & Gavai, A. V. (2011). Bioorg. Med. Chem. Lett. 21, 781–785. [DOI] [PubMed]
- Grochulski, P., Fodje, M. N., Gorin, J., Labiuk, S. L. & Berg, R. (2011). J. Synchrotron Rad. 18, 681–684. [DOI] [PubMed]
- Holtzman, D. M., Mandelkow, E. & Selkoe, D. J. (2012). Cold Spring Harb. Perspect. Med. 2, a011585. [DOI] [PMC free article] [PubMed]
- Houlden, H., Johnson, J., Gardner-Thorpe, C., Lashley, T., Hernandez, D., Worth, P., Singleton, A. B., Hilton, D. A., Holton, J., Revesz, T., Davis, M. B., Giunti, P. & Wood, N. W. (2007). Nature Genet. 39, 1434–1436. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kitano-Takahashi, M., Morita, H., Kondo, S., Tomizawa, K., Kato, R., Tanio, M., Shirota, Y., Takahashi, H., Sugio, S. & Kohno, T. (2007). Acta Cryst. F63, 602–604. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lee, G. & Leugers, C. J. (2012). Prog. Mol. Biol. Transl. Sci. 107, 263–293. [DOI] [PMC free article] [PubMed]
- Long, A. M., Zhao, H. & Huang, X. (2012). J. Med. Chem. 55, 10307–10311. [DOI] [PubMed]
- Longenecker, K. L., Roach, P. J. & Hurley, T. D. (1996). J. Mol. Biol. 257, 618–631. [DOI] [PubMed]
- Lund, H., Cowburn, R. F., Gustafsson, E., Strömberg, K., Svensson, A., Dahllund, L., Malinowsky, D. & Sunnemark, D. (2013). Brain Pathol. 23, 378–389. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mondragón-Rodríguez, S., Basurto-Islas, G., Santa-Maria, I., Mena, R., Binder, L. I., Avila, J., Smith, M. A., Perry, G. & García-Sierra, F. (2008). Int. J. Exp. Pathol. 89, 81–90. [DOI] [PMC free article] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Sato, S., Cerny, R. L., Buescher, J. L. & Ikezu, T. (2006). J. Neurochem. 98, 1573–1584. [DOI] [PubMed]
- Sato, S., Tatebayashi, Y., Akagi, T., Chui, D.-H., Murayama, M., Miyasaka, T., Planel, E., Tanemura, K., Sun, X., Hashikawa, T., Yoshioka, K., Ishiguro, K. & Takashima, A. (2002). J. Biol. Chem. 277, 42060–42065. [DOI] [PubMed]
- Sato, S., Xu, J., Okuyama, S., Martinez, L. B., Walsh, S. M., Jacobsen, M. T., Swan, R. J., Schlautman, J. D., Ciborowski, P. & Ikezu, T. (2008). J. Neurosci. 28, 14511–14521. [DOI] [PMC free article] [PubMed]
- Satow, Y., Cohen, G. H., Padlan, E. A. & Davies, D. R. (1986). J. Mol. Biol. 190, 593–604. [DOI] [PubMed]
- Tomizawa, K., Omori, A., Ohtake, A., Sato, K. & Takahashi, M. (2001). FEBS Lett. 492, 221–227. [DOI] [PubMed]
- Troquier, L., Caillierez, R., Burnouf, S., Fernandez-Gomez, F. J., Grosjean, M.-E., Zommer, N., Sergeant, N., Schraen-Maschke, S., Blum, D. & Buee, L. (2012). Curr. Alzheimer Res. 9, 397–405. [DOI] [PMC free article] [PubMed]
- Vázquez-Higuera, J. L., Mateo, I., Sánchez-Juan, P., Rodríguez-Rodríguez, E., Pozueta, A., Calero, M., Dobato, J. L., Frank-García, A., Valdivieso, F., Berciano, J., Bullido, M. J. & Combarros, O. (2011). J. Alzheimers Dis. 27, 291–297. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xu, J., Sato, S., Okuyama, S., Swan, R. J., Jacobsen, M. T., Strunk, E. & Ikezu, T. (2010). FASEB J. 24, 2904–2915. [DOI] [PubMed]
- Xue, Y., Wan, P. T., Hillertz, P., Schweikart, F., Zhao, Y., Wissler, L. & Dekker, N. (2013). ChemMedChem, 8, 1846–1854. [DOI] [PubMed]
- Yu, N.-N., Yu, J.-T., Xiao, J.-T., Zhang, H.-W., Lu, R.-C., Jiang, H., Xing, Z.-H. & Tan, L. (2011). Neurosci. Lett. 491, 83–86. [DOI] [PubMed]
- Zuccotto, F., Ardini, E., Casale, E. & Angiolini, M. (2010). J. Med. Chem. 53, 2681–2694. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: tau-tubulin kinase 1, 4nfm
PDB reference: complex with inhibitor, 4nfn