

Polyphenol oxidase 4 (PPO4) from the natural source A. bisporus was crystallized in its latent precursor form (pro-tyrosinase; Ser2–Thr565) using the 6-tungstotellurate(VI) salt Na6[TeW6O24]·22H2O as a crystallization additive.
Keywords: tyrosinases, zymogens, polyphenol oxidase 4, Agaricus bisporus, polyoxometalates
Abstract
Tyrosinase exhibits catalytic activity for the ortho-hydroxylation of monophenols to diphenols as well as their subsequent oxidation to quinones. Owing to polymerization of these quinones, brown-coloured high-molecular-weight compounds called melanins are generated. The latent precursor form of polyphenol oxidase 4, one of the six tyrosinase isoforms from Agaricus bisporus, was purified to homogeneity and crystallized. The obtained crystals belonged to space group C121 (two molecules per asymmetric unit) and diffracted to 2.78 Å resolution. The protein only formed crystals under low-salt conditions using the 6-tungstotellurate(VI) salt Na6[TeW6O24]·22H2O as a co-crystallization agent.
1. Introduction
Tyrosinases (EC 1.14.18.1 and 1.10.3.1) are type 3 copper enzymes that are widely distributed in nature and catalyze the two reactions inducing the formation of brown-coloured compounds called melanins (Sánchez-Ferrer et al., 1995 ▶). These reactions, the ortho-hydroxylation of monophenols and their subsequent oxidation to quinones, are of interest since the impairment and ageing-related browning of agricultural products represent a concern in the food industry. Additionally, the impact of the generated pigments as stress-resistance and immune-defence factors is a current area of research (Bell & Wheeler, 1986 ▶; Jacobson, 2000 ▶). Similar to other polyphenol oxidases (PPOs), tyrosinases are known to be expressed as inactive zymogens which are activated by the proteolytic removal of the C-terminal active-site-shielding domain of the enzyme (Yamaguchi et al., 1970 ▶). This maturation process as well as the supplementary functions of the C-terminal domain, e.g. copper incorporation, are currently an area of tyrosinase research (Faccio et al., 2013 ▶). Recently, the published crystal structure of a recombinantly expressed pro-tyrosinase from Aspergillus oryzae (aoTYR; Fujieda et al., 2013 ▶) demonstrated that the C-terminal domain plays a crucial role in the incorporation of copper ions into the active site.
In the white edible mushroom (Agaricus bisporus), six gene sequences (abPPO1–6) encoding tyrosinases are present (Wichers et al., 2003 ▶; Wu et al., 2010 ▶; Weijn et al., 2013 ▶; http://genome.jgi-psf.org/pages/search-for-genes.jsf?organism=Agabi_varbisH97_2). Although expression studies have shown that different isoforms are expressed in differing quantities depending on the growth stage and the tissue type (fruit-body compartment, mycelia etc.), no significant assignment with respect to functionality or responsibility could be established for any of these six isoforms (Weijn et al., 2013 ▶). However, the two by far most abundantly expressed isoforms are abPPO3 (UniProt C7FF04) and abPPO4 (UniProt C7FF05). The crystal structure of abPPO3 showed that the active form of the enzyme exhibits a hetero-tetrameric protein conformation (H2L2) involving a small subunit to which no functionality has yet been assigned (Ismaya et al., 2011 ▶). Recently, a biochemical study (Mauracher et al., 2014 ▶) comprehensively characterized abPPO4 in its zymogen (latent) form (Fig. 1 ▶). The protein was purified in its latent state, lacking only a small transmembrane anchor-containing C-terminal domain (46 amino acids). Several post-translational modifications as well as multiple strain-related mutations were additionally detected. abPPO4 has the highest sequence identity to abPPO3 among structurally known proteins (sequence identity 52%, solely active form); however, only structural knowledge of the C-terminal domain of aoTYR, which has very low sequence identity (11%), is presently available.
Figure 1.

Schematic illustration of the polypeptide chain of PPO4 mushroom tyrosinase. The polypeptide chain of active tyrosinase (core region) is coloured red. The C-terminal domain is coloured orange. The missing C-terminal tail is coloured purple. Aa, amino acids; A-TYR, active tyrosinase; L-TYR, latent tyrosinase.
Polyoxometalates (POMs) are discrete metal–oxo cluster anions of early transition metals in high oxidation states, exhibiting a unique structural and compositional variety (Pope, 1983 ▶; Pope & Kortz, 2012 ▶). POMs are potentially useful in many different areas owing to their thermal, redox, magnetic, optical and bioactive properties (Müller et al., 2001 ▶; Schnack et al., 2006 ▶; Kortz et al., 2009 ▶; Iqbal et al., 2013 ▶; Jahier et al., 2013 ▶). The shape, size and negative charge of POMs allow their binding to positively charged regions of proteins (Zhang et al., 2007 ▶). The use of POMs in protein crystallography has mostly been limited to protein crystal soaking, trying to utilize the POMS for phasing either by isomorphous replacement or anomalous scattering (Corey et al., 1962 ▶; Ladenstein et al., 1987 ▶; O’Halloran et al., 1987 ▶; Thygesen et al., 1996 ▶; Rudenko et al., 2003 ▶; Zebisch et al., 2012 ▶; Dahms et al., 2013 ▶) as well as using POMs as ‘contrast agents’ to display buried channels within protein structures (Dauter, 2005 ▶). Noteworthy is the famous crystal structure of ribosome D50S (Deinococcus radiodurans), where crystals were soaked with the Keggin-type POM salt K5H[PW12O40]·12H2O (Schluenzen et al., 2000 ▶; Harms et al., 2001 ▶; Pioletti et al., 2001 ▶).
In this work, the crystallization of abPPO4, purified by the method of Mauracher et al. (2014 ▶), with the 6-tungstotellurate(VI) salt Na6[TeW6O24]·22H2O as a co-crystallization agent is presented.
2. Materials and methods
2.1. Sample preparation
The protein was isolated and purified by the method described by Mauracher et al. (2014 ▶). Extraction of the enzyme from the natural source (mushrooms) with prevention of protein-interfering reactions (amino-acid oxidation, protein aggregation, protein cross-linking etc.) was established using a distinctly developed method that relies on detergent and soluble polymer (PEG) phase separations. The latent enzyme was kept in its precursor state by the extended usage of protease-inhibition agents and was purified to homogeneity by fast protein liquid chromatography (FPLC) using several ion-exchange columns. Purity was checked by SDS–PAGE and nanoESI–qTOF. For crystallization experiments, the protein was kept at a concentration of 10 mg ml−1 in 10 mM HEPES buffer pH 7.5.
2.2. Synthesis of Na6[TeW6O24]·22H2O
The hydrated sodium salt of hexatungstotellurate(VI) was synthesized according to a modified procedure (Roy & Mishra, 1978 ▶; Schmidt et al., 1986 ▶). Na2WO4·2H2O (5.0 g, 15.2 mmol) and Te(OH)6 (0.6 g, 2.6 mmol) were first dissolved in 100 ml water. The pH of the mixture was then adjusted to 5.0 using aqueous HCl solution (1 M), which was followed by heating at 383 K until three-quarters of the volume remained. The mixture was then cooled to room temperature and filtered. Slow evaporation of the filtrate at room temperature led to the formation of colourless crystals within one week. The crystals were collected and air-dried and their identity was confirmed by infrared spectroscopy in the solid state.
2.3. Protein crystallization
Initial crystallization screens were performed by the sitting-drop vapour-diffusion technique (96-well Crystal Quick plates, Greiner Bio-One) using a nanodispenser robot (Gryphon, Art Robbins). Screening over a broad variety of crystallization conditions (Crystallization Basic Kit for Membrane Proteins, Crystallization Low Ionic Strength Kit for Proteins and Crystallization Basic Kit for Proteins from Sigma–Aldrich; Pi-PEG Screen HTS, JBScreen Classic 1–10 and JBScreen Membrane 1–3 from Jena Bioscience) gave the first hits (microcrystalline precipitant) for crystallization.
The first microcrystals were obtained by the hanging-drop vapour-diffusion technique (15-well EasyXtal 10 × 15 plates, Qiagen) using 10% PEG 4000, 15 mM MgCl2, 25 mM Tris–HCl pH 7.5 at 291 K as the crystallization condition (Fig. 2 ▶ a). The final crystallization condition for the growth of single crystals suitable for diffraction measurements was 10% PEG 4000, 1 mM Na6[TeW6O24]·22H2O, 25 mM Tris–HCl pH 7.5 at 291 K using 1 µl protein solution (10 mg ml−1) and 0.5 µl reservoir solution in the hanging drop and 500 µl solution in the reservoir (Fig. 2 ▶ b). The first crystals appeared after 1–2 d and crystal growth came to a stop after approximately 5 d.
Figure 2.

Crystal images of PPO4 mushroom tyrosinase (left, total drop image; right, enlarged image). (a) Wispy microcrystals (sea urchins) obtained using MgCl2 as a crystallization additive (10% PEG 4000, 15 mM MgCl2, 25 mM Tris–HCl pH 7.5). (b) Flat rod-shaped crystals obtained using the POM as a crystallization additive [10% PEG 4000, 1 mM Na6[TeW6O24]·22H2O, 25 mM Tris–HCl pH 7.5].
2.4. Data collection and processing
Single crystals were harvested by transferring them with a cryoloop (10 µm, 0.1–0.2 mm; Hampton Research) into a 1 µl drop of cryoprotectant solution [20% PEG 4000, 25% PEG 400, 1 mM Na6[TeW6O24]·22H2O, 25 mM Tris–HCl pH 7.5] and were subsequently flash-cooled in liquid nitrogen. The diffraction of about 15 crystals of suitable size was measured at Diamond Light Source, Oxfordshire, England on the monochromatic (0.9173 Å) MX beamline I04-1 equipped with a PILATUS 2M detector. Data sets were collected at 100 K with an oscillation range of 0.5° and an exposure time of 0.5 s. The best crystal diffracted to 2.78 Å resolution using a crystal-to-detector distance of 300.9 mm. The obtained diffraction data sets were processed using the XDS program package (version March 30, 2013; Kabsch, 2010 ▶). The space group was determined using the program POINTLESS from the CCP4 program suite (v.6.3.0; Winn et al., 2011 ▶).
3. Results and discussion
By applying the method described by Mauracher et al. (2014 ▶), the obtained protein was purified to homogeneity; hence, any other non-target proteins or different isoforms (e.g. PPO3) could be removed entirely (SDS–PAGE, nanoESI–qTOF). However, the preparation showed a somewhat proteolytically ragged C-terminus such that five differing species possessing alternate C-terminal amino acids (polypeptide backbone Ser2–Gly563/Thr565/Gly568/Ala569/Thr570) occur. The species ending with Thr565 was the most abundant, at a ratio of about 6:1 (as determined by nanoESI–qTOF; Mauracher et al., 2014 ▶).
Initial attempts to crystallize the protein covering a wide range of crystallization conditions proved to be unsuccessful with regard to obtaining single crystals; however, a microcrystalline precipitate was obtained using 10% PEG 4000 in the pH range 7–8.5. The best results could be achieved by using magnesium chloride as an additive at rather low concentrations (5–20 mM). Low salt concentrations in a basic pH range appeared to be crucial in order to obtain wispy sea-urchin-like microcrystals (Fig. 2 ▶ a). However, alteration of any of the crystallization parameters (pH, precipitation agent, temperature or additives), either qualitatively and/or quantitatively, did not help to obtain single crystals. By substituting the magnesium chloride with the 6-tungstotellurate(VI) salt Na6[TeW6O24]·22H2O, crystals suitable for X-ray diffraction experiments were obtained (in 1–5 d). The functional concentration range of this POM under these conditions was 0.5–3 mM, with a clear optimum at 1 mM. In Fig. 2 ▶, the difference between the use of MgCl2 and POM as an additive under otherwise identical conditions is presented. The occurrence of multiple, rod-shaped, closely clustered and intergrown crystals was still not preventable, but some single crystals became detached. Such crystals of reasonable size (300 × 30 × 10 µm) were used for X-ray diffraction analysis (Fig. 2 ▶ b).
Statistics for the X-ray diffraction measurements are given in Table 1 ▶. The crystals belonged to space group C121, with unit-cell parameters a = 213.57, b = 83.73, c = 66.95 Å, β = 102.522°, and diffracted to a maximum resolution of 2.78 Å. Considering the precisely known molecular weight of 64.24 kDa per monomer and assuming the presence of two monomers (as proposed by phenix.xtriage from the PHENIX program suite; Adams et al., 2010 ▶) per asymmetric unit gives a Matthews coefficient (Matthews, 1968 ▶) of ∼2.29 Å3 Da−1 and a solvent content of 46%.
Table 1. Data-collection and processing statistics for A. bisporus tyrosinase (PPO4) crystals.
Values in parentheses are for the outermost resolution shell.
| Space group | C121 |
| Wavelength (Å) | 0.9173 |
| No. of images | 400 |
| Oscillation (°) | 0.5 |
| Resolution range (Å) | 48.14–2.784 (2.884–2.784) |
| Completeness (%) | 95.89 (96.04) |
| R merge † | 0.1657 (0.8243) |
| 〈I/σ(I)〉 | 8.70 (1.87) |
| Multiplicity | 4.0 (4.0) |
| Unit-cell parameters (Å, °) | a = 213.57, b = 83.73, c = 66.95, β = 102.522 |
| R p.i.m. ‡ | 0.093 (0.466) |
| CC1/2 | 0.987 (0.62) |
| No. of reflections collected | 110549 (11068) |
| No. of unique reflections | 27846 (2762) |
R
merge = 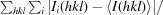
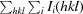 .
.
R
p.i.m. =  , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
We are currently attempting to solve the crystal structure by using molecular replacement and single-wavelength anomalous dispersion (MR–SAD; using AutoSol from the PHENIX program suite) provided by the POM (tungsten signal), respectively. Several search models for MR are available: A. bisporus PPO3 (sequence identity 52%, solely active form; Ismaya et al., 2011 ▶), Aspergillus oryzae tyrosinase; sequence identity 21%, full-length pro-enzyme; Fujieda et al., 2013 ▶) and Octopus dofleini haemocyanin (sequence identity 16% for copper-binding domain; Cuff et al., 1998 ▶).
Acknowledgments
The authors are grateful to the University of Vienna for financial support of the graduate training program entitled ‘Functional Molecules’ (grant No. IK I041-N) as well as for financial support by the ‘Fonds zur Förderung der wissenschaftlichen Forschung’ (FWF) under P25217-N28. The cost action CM1203 PoCheMoN is acknowledged. We would also like to thank Dr Alice Douangamath, beamline (I04-1) scientist at Diamond Light Source (Oxfordshire, England), for very kind support during data collection (proposal No. MX8476).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bell, A. A. & Wheeler, M. H. (1986). Annu. Rev. Phytopathol. 24, 411–451.
- Corey, R. B., Stanford, R. H. Jr, Marsh, R. E., Leung, Y. C. & Kay, L. M. (1962). Acta Cryst. 15, 1157–1163.
- Cuff, M. E., Miller, K. I., van Holde, K. E. & Hendrickson, W. A. (1998). J. Mol. Biol. 278, 855–870. [DOI] [PubMed]
- Dahms, S. O., Kuester, M., Streb, C., Roth, C., Sträter, N. & Than, M. E. (2013). Acta Cryst. D69, 284–297. [DOI] [PMC free article] [PubMed]
- Dauter, Z. (2005). C. R. Chim. 8, 1808–1814.
- Faccio, G., Arvas, M., Thöny-Meyer, L. & Saloheimo, M. (2013). J. Inorg. Biochem. 121, 37–45. [DOI] [PubMed]
- Fujieda, N., Yabuta, S., Ikeda, T., Oyama, T., Muraki, N., Kurisu, G. & Itoh, S. (2013). J. Biol. Chem. 288, 22128–22140. [DOI] [PMC free article] [PubMed]
- Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F. & Yonath, A. (2001). Cell, 107, 679–688. [DOI] [PubMed]
- Iqbal, J., Barsukova-Stuckart, M., Ibrahim, M., Ali, S. U., Khan, A. A. & Kortz, U. (2013). Med. Chem. Res. 22, 1224–1228.
- Ismaya, W. T., Rozeboom, H. J., Weijn, A., Mes, J. J., Fusetti, F., Wichers, H. J. & Dijkstra, B. W. (2011). Biochemistry, 50, 5477–5486. [DOI] [PubMed]
- Jacobson, E. S. (2000). Clin. Microbiol. Rev. 13, 708–717. [DOI] [PMC free article] [PubMed]
- Jahier, C., Mal, S. S., Al-Oweini, R., Kortz, U. & Nlate, S. (2013). Polyhedron, 57, 57–63.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kortz, U., Müller, A., van Slageren, J., Schnack, J., Dalal, N. S. & Dressel, M. (2009). Coord. Chem. Rev. 253, 2315–2327.
- Ladenstein, R., Bacher, A. & Huber, R. (1987). J. Mol. Biol. 195, 751–753. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mauracher, S. G., Molitor, C., Michael, C., Kragl, M., Rizzi, A. & Rompel, A. (2014). Phytochemistry, 10.1016/j.phytochem.2013.12.016. [DOI] [PMC free article] [PubMed]
- Müller, A., Luban, M., Schröder, C., Modler, R., Kögerler, P., Axenovich, M., Schnack, J., Canfield, P., Bud’ko, S. & Harrison, N. (2001). ChemPhysChem, 2, 517–521. [DOI] [PubMed]
- O’Halloran, T. V., Lippard, S. J., Richmond, T. J. & Klug, A. (1987). J. Mol. Biol. 194, 705–712. [DOI] [PubMed]
- Pioletti, M., Schlünzen, F., Harms, J., Zarivach, R., Glühmann, M., Avila, H., Bashan, A., Bartels, H., Auerbach, T., Jacobi, C., Hartsch, T., Yonath, A. & Franceschi, F. (2001). EMBO J. 20, 1829–1839. [DOI] [PMC free article] [PubMed]
- Pope, M. T. (1983). Heteropoly and Isopoly Oxometalates. New York: Springer.
- Pope, M. T. & Kortz, U. (2012). Encyclopedia of Inorganic and Bioinorganic Chemistry. Chichester John Wiley & Sons. 10.1002/9781119951438.eibc0185.pub2.
- Roy, S. K. & Mishra, H. C. (1978). J. Indian Chem. Soc. 55, 188–195.
- Rudenko, G., Henry, L., Vonrhein, C., Bricogne, G. & Deisenhofer, J. (2003). Acta Cryst. D59, 1978–1986. [DOI] [PubMed]
- Sánchez-Ferrer, Á., Neptuno Rodríguez-López, J., García-Cánovas, F. & García-Carmona, F. (1995). Biochim. Biophys. Acta, 1247, 1–11. [DOI] [PubMed]
- Schluenzen, F., Tocilj, A., Zarivach, R., Harms, J., Gluehmann, M., Janell, D., Bashan, A., Bartels, H., Agmon, I., Franceschi, F. & Yonath, A. (2000). Cell, 102, 615–623. [DOI] [PubMed]
- Schmidt, K. J., Schrobilgen, G. J. & Sawyer, J. F. (1986). Acta Cryst. C42, 1115–1118.
- Schnack, J., Brüger, M., Luban, M., Kögerler, P., Morosan, E., Fuchs, R., Modler, R., Nojiri, H., Rai, R. C., Cao, J., Musfeldt, J. L. & Wei, X. (2006). Phys. Rev. B, 73, 094401.
- Thygesen, J., Weinstein, S., Franceschi, F. & Yonath, A. (1996). Structure, 4, 513–518. [DOI] [PubMed]
- Weijn, A., Bastiaan-Net, S., Wichers, H. J. & Mes, J. J. (2013). Fungal Genet. Biol. 55, 42–53. [DOI] [PubMed]
- Wichers, H. J., Recourt, K., Hendriks, M., Ebbelaar, C. E., Biancone, G., Hoeberichts, F. A., Mooibroek, H. & Soler-Rivas, C. (2003). Appl. Microbiol. Biotechnol. 61, 336–341. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wu, J., Chen, H., Gao, J., Liu, X., Cheng, W. & Ma, X. (2010). Biotechnol. Lett. 32, 1439–1447. [DOI] [PubMed]
- Yamaguchi, M., Hwang, P. M. & Campbell, J. D. (1970). Can. J. Biochem. 48, 198–202. [DOI] [PubMed]
- Zebisch, M., Krauss, M., Schäfer, P. & Sträter, N. (2012). J. Mol. Biol. 415, 288–306. [DOI] [PubMed]
- Zhang, G., Keita, B., Craescu, C. T., Miron, S., de Oliveira, P. & Nadjo, L. (2007). J. Phys. Chem. B, 111, 11253–11259. [DOI] [PubMed]