

Abstract
Irradiation of the copper(II)-superoxide synthetic complexes [(TMG3tren)CuII(O2)]+ (1) and [(PV-TMPA)CuII(O2)]+ (2) with visible light resulted in direct photo-generation of O2 gas at low temperature (from −40 °C to −70°C for 1 and from −125 °C to −135 °C for 2) in 2-methyltetrahydrofuran (MeTHF) solvent. The yield of O2 release was wavelength dependent: λexc = 436 nm, ϕ = 0.29 (for 1), ϕ = 0.11 (for 2), and λexc = 683 nm, ϕ = 0.035 (for 1), ϕ = 0.078 (for 2), which was followed by fast O2-recombination with [(TMG3tren)CuI]+ (3) and [(PV-TMPA)CuI]+ (4). Enthalpic barriers for O2 re-binding to the copper(I) center (~ 10 kJ mol−1) and for O2 dissociation from the superoxide compound 1 (45 kJ mol−1) were determined. TD-DFT studies, carried out for 1, support the experimental results confirming the dissociative character of the excited states formed upon blue or red light laser excitation.
Copper-containing proteins play a major role in O2 transport and activation in biology. Thus, CuI/O2 reactions and subsequent transformations are critical in this setting as well as in practical systems.1 Initial O2 adducts of copper(I) must form in all cases, including in O2- carriers, oxygenases (oxygen transfer to the substrate) and oxidases (substrate oxidized by O2), but these first formed species often further react with other electron/proton sources (which may be the substrate) to give Cun-peroxo, CuII-hydroperoxo2,3 or perhaps Cun-oxyl1b, 4 active species or intermediates. In peptidylglycine α-hydroxylating monooxygenase5 and dopamine β-monooxygenase,6 such O2 activation occurs at a single copper center. An X-ray structure of a pre-catalytic complex along with chemical7 and computational studies,4a, 8 suggested an end-on bound CuII-superoxide species serves as the enzyme reactive intermediate effecting substrate hydrogen abstraction, further implicating the (bio)chemical importance of initially formed CuI/O2 1:1 adducts, i.e., CuII-superoxide species.
Here, for the first time, we show that O2 can be photo-ejected directly from the 1:1 mononuclear copper/O2 compounds [(TMG3tren)CuII(O2)]+ (1) and [(PV-TMPA)CuII(O2)]+ (2) using either 436 nm or 683 nm pulsed laser light (Scheme 1). Interestingly, a different yield for O2 release was observed with these two excitation wavelengths which is different if compared to the O2 photo-release found in heme systems, like myoglobin.9 Temperature-dependent kinetic and thermodynamic studies have been carried out to elucidate the nature of the barriers and the stability of the species involved in the O2 binding and dissociation processes. Data are corroborated by DFT calculations that help to a) explain why O2 photo-release is observed and b) interpret the experimentally observed excitation wavelength dependent quantum yield for the O2 photo-release through new insights into the evolution of the excited states along the copper-oxygen reaction coordinate. To the best of our knowledge, this is the first time that a direct O2 photo-ejection from 1:1 copper-superoxide adducts has been shown to occur.
Scheme 1.

Oxygenation of 3 at low temperature in MeTHF was accompanied by a drastic color change of the solution, from colorless to green, forming the previously well characterized compound 110 and leading to the red spectrum shown in Figure 1A. Oxygenation of 4 at low temperature also yielded the previously characterized mononuclear copper/O2 species 2 (see Supporting Information for UV-visible spectra).11 Cleavage of the copper-oxygen bond was, then, induced upon laser excitation of 1 and 2 (λexc = 436 or 683 nm) as shown by the transient absorption spectral data collected after laser excitation, for 1. These spectra were in complete agreement with that expected for O2 photo-release from 1 to yield 3 (Figure 1B) and from 2 to yield 4 (see Supporting Information). The products of the reaction (3, O2 and 4, O2, respectively) were excitation wavelength independent, although the quantum yields differed markedly: ϕ = 0.29 for 1 and ϕ = 0.11 for 2 (λexc = 436 nm), ϕ = 0.035 for 1 and ϕ = 0.078 for 2 (λexc = 683 nm). The appearance of the products, 3 and 4, occurred within the instrument response time indicating an O2 time release of less than 10 ns.
Figure 1.
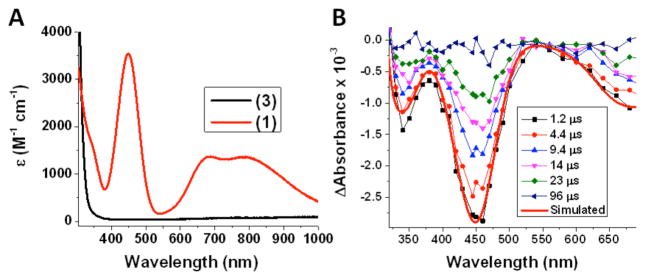
(A) Absorption spectrum of 1 (red line) obtained from oxygenation of 3 (black line) at 218 K in MeTHF. (B) Transient absorption difference spectra collected at the indicated delay times after 436 nm laser excitation (15 mJ/pulse, 8–10 ns fwhm) of 1 in MeTHF at 218 K. Overlaid in red on the experimental data is a simulated spectrum (Abs(3) - Abs(1) ).
The follow-up thermal reaction of [(TMG3tren)CuI]+ (3) with O2 led to the formation of the initial compound 1 as is shown in Figure 1B. Kinetic parameters for O2 coordination to 3 were quantified based on microsecond time scale data. Thus, a plot of the observed rate constants versus the O2 concentration under pseudo-first-order conditions (excess of O2) revealed a linear correlation that allowed the determination of the second-order rate constants for O2 coordinating to 3, i.e., kO2 = 2.1 × 106 M−1s−1 at −80 °C. For the same temperature, this compares to kO2 = 6.6 × 105 M−1s−1 for 4 (Table 1).
Table 1.
Comparison of Kinetic and Thermodynamic Parameters for O2 binding and dissociation for [(L)Cu]+ adducts.
| TMG3trenc | |||
|---|---|---|---|
| kO2 | k−O2 | KO2 | |
| ΔH‡ or ΔH0 a | 10 ± 6 | 45 ± 7 | −40 ± 2 |
| ΔS‡ or ΔS0 b | −70 ± 26 | 42 ± 34 | −134 ± 11 |
| k or K 25°C | (2.7 ± 1.2) · 107 | (1.5 ± 0.8) · 107 | ≈ 1 |
| k or K −80°C | (2.1 ± 1.0) · 106 | (5.2 ± 2.0) · 102 | (6.3 ± 1.9) · 103 |
| PV-TMPAc | |||
|---|---|---|---|
| kO2 | k−O2 | KO2 | |
| ΔH‡ or ΔH0 a | 9 ± 1 | - | - |
| ΔS‡ or ΔS0 b | −97 ± 7 | - | - |
| k or K 25°C | (4.8 ± 2.8) · 107 | - | - |
| k or K −80°C | (6.6 ± 3.5) · 105 | - | - |
| TMPAd | |||
|---|---|---|---|
| kO2 | k−O2 | KO2 | |
| ΔH‡ or ΔH0 a | 7.62 | 58.0 | −48.5 |
| ΔS‡ or ΔS0 b | −45.1 | 105 | −140 |
| k or K 25°C | 1.3 · 109 | 1.3 · 108 | 15.4 |
| k or K −80°C | 1.4–1.6 · 108 | 240 | 6.5 · 105 |
ΔH, kJ mol−1
ΔS, J K−1 mol−1
In MeTHF, this work
In THF, determined through flash-and-trap method.13 Values for [(TMPA)-Cu] in MeTHF have been found to be the same as in THF within experimental errors.
The linear plots of kobs vs [O2] had a positive intercept that was indicative of the presence of an equilibrium between the reacting species, O2 and 3 (see Supporting Information). Such a positive intercept was not observed for the coordination of O2 to 4, instead, indicating a quantitative formation of 2 from 4 and O2. Consequently, rate constants for O2 dissociation from 2 and equilibrium constants for the reaction between 4 and O2 to give 2 could not be determined here. However, we were able to determine the equilibrium constant at several temperatures in MeTHF solvent through benchtop titration experiments for the binding of O2 to 3, to give 1 (Table 1 and Supporting Information). Equilibrium constant values were also determined from laser experiments as follows. In pseudo-first-order conditions, the rate law for O2 binding to 3 is expressed by the equation kobs = kO2 [O2] + k−O2 where kobs is the observed rate constant, kO2 is the second-order rate constant for the binding between 3 and O2, and k−O2 is the first-order rate constant for the dissociation reaction of O2 from 1 (see section 6 of Supporting Information). The values of k−O2 and kO2 were determined from laser experiments, as a function of temperature through which the equilibrium constants were determined from the ratios kO2 / k−O2. Van’t Hoff analysis (see Section 5 of the SI) of the equilibrium constants determined with the two different methods (titration experiments and laser experiments) led to the same thermodynamic parameters within the experimental errors and are consistent with values found in a previous report by Roth and co-workers (Table S1 and Figures S2 and S5). Furthermore, equilibrium constants found in this work follow a trend with solvent dielectric constant (ε ) that was previously established.12 The equilibrium constant should favor the superoxide adduct as ε increases because of the stabilization of the charge separation present in 1. In fact, the equilibrium constant for the formation of 1 (KO2) determined here at −60 °C fits well into a linear correlation together with the previously determined KO2 values in DMF (3030 and 4340)12 and in chlorobenzene (216)13 at −60 °C as a function of ε (see Supporting Information).
A comparison of activation and thermodynamic parameters determined in this study with those previously reported for the [(TMPA)CuII(O2)]+ adduct in MeTHF using [(TMPA)CuI(CO)]+ and the “flash-and-trap” method are also given in Table 1 (see Chart 1 for structure of ligands). This complex has been very well studied and it is the ‘parent’ ligand of PV-TMPA.1a,13,14 The “flash-and-trap” experiments, previously employed for [(L)CuI(CO)]+ (L = ligand) compounds, allowed characterization of O2 binding to copper(I) after CO photo-release through competitive coordination of CO and O2.13,14 The kinetic data obtained through the direct O2 photo-ejection method described here are more straightforward to analyze compared to that of the “flash-and-trap” method where the competitive binding of CO needs to be taken into account. Furthermore, in fast time scale studies of heme-copper oxidases, it has been shown that the presence of CO and starting with a metal-CO adduct may interfere or alter the mechanism or rate of O2 binding.15 The activation parameters found for the compounds studied here are quite similar to those previously determined by the flash-and-trap method, providing strong evidence for the reliability of the new method we have employed here to study the reactivity of mononuclear copper compounds with O2.
Chart 1.
TMG3tren, PV-TMPA, and TMPA offer an analogous coordination sphere to the copper ion, all being tetradentate chelating ligands to study under the same experimental conditions (solvent, temperature, etc). The activation enthalpy found for the binding of O2 to 3 and 4 falls within the same range (~ 10 kJ mol−1). On the basis of the crystal structure of the starting compound [(PV-TMPA)CuI]+,11 the coordination within 4 mostly likely also includes an interaction between the copper(I) ion and the O-atom of the pivilamido group. As a consequence, one would expect a higher activation enthalpy for the reaction between O2 and 4 compared with that between O2 and 3 as the Cu(I)-O interaction needs to be “disrupted” by O2 coordination to 4 but not for 3. Since the ΔH‡ values for the binding of O2 to 3 and 4 fall, instead, into the same range, this suggests a quite weak interaction for the CuI-O(carbonyl) coordination in 4. The activation entropy estimated for the reaction involving O2 coordination to 3 and to 4 is, instead, smaller for the latter. This suggests a mechanism where O2 coordination to 4 leads to a “highly ordered” transition state where both O2 and the pivalamido O-atom are interacting with the copper center; for O2 reacting with 3, there is of course no pivalamido group present.
The activation enthalpy and entropy for O2 coordination to [(TMPA)CuI]+ previously determined (Table 1) are smaller and less negative, respectively, compared with those found for 3 and 4. This can be interpreted on the basis of a stronger Cu-O2 interaction in the transition state for [(TMPA)CuI]+ compared to that for 3 and 4 due to an “easier” spatial approach of O2 to the copper(I) in [(TMPA)CuI]+. In fact, the presence of guanidino groups which extend out away from the copper and its ligands in 3, and of the CuI-O(carbonyl) coordination, in 4, would support this hypothesis. The less negative activation entropy found for the coordination of O2 to [(TMPA)CuI]+ could reflect a smaller molecular reorganization occurring upon O2 binding to [(TMPA)CuI]+ due to the absence of guanidino groups or specific Cu(I)-O interactions in [(TMPA)CuI]+ compared with 3 and 4. Similar arguments can be used to interpret the difference between the activation enthalpy found for the O2 dissociation from [(TMPA)CuII(O2)]+ with the “flash-and-trap” method and those found, here, for 1 and 2, although the large activation entropy found for O2 dissociation from [(TMPA)CuII(O2)]+ seems unclear.
TD-DFT calculations are in line with the previously assigned electronic ground state for 1.10,12,16. In this rather peculiar electronic structure, the central copper ion is in a d9 configuration and coordinated to a superoxide ligand. The singly occupied MOs are of copper 3dz2 and O2•−-π*v character. The orthogonality of these two orbitals leads to a S = 1 ground state multiplicity in which the spin in both SOMOs are aligned parallel (see Supporting Information). In a spin-unrestricted description, the highest occupied spin-down orbital has mainly oxygen π*σ-character and it is bonding with respect to the CuII-superoxide Cu-O bond. The lowest unoccupied orbitals in the spin-down manifold are the empty partner orbitals of the two SOMOs. Importantly, the unoccupied 3dz2 orbital is strongly σ-antibonding with respect to the Cu-O bond. Excitation from the bonding π*σ·-based orbital to the antibonding dz2 orbital corresponds to a ligand-to-metal charge transfer (LMCT) excitation that formally leads to a Cu(I)-3O2 electronic configuration. Importantly, this excitation leads to a dramatic weakening of the Cu-O bond to the point that the excited state becomes dissociative (Figure 2).
Figure 2.

TD-DFT calculated excited state potential energy surfaces (PESs) as a function of copper-oxygen bond distance.
As it is evident from Figure 3, there is an avoided crossing of the dz2 and π* orbitals upon elongation of the Cu-O bond, resulting in a change from a triplet Cu(II) superoxide ground state [d8] dz21π*3 to a triplet Cu(I)-3O2 (d10π*2) state at the dissociation limit. The triplet ground state potential energy surface of 1 (Figure 2, green line) shows a minimum at a Cu-O distance of about 1.9 Å. The calculated excited state energy at the same Cu-O bond distance was 18,843 cm−1 (530 nm) for the d-d and 21,635 cm−1 (462 nm) for the LMCT transition, consistent with the experimentally observed electronic transitions for these states. Moreover, the character of both excited states at a Cu-O bond distance of 1.9 Å is dissociative (Figure 2). The LMCT excited state (blue line) crosses the ground state at a Cu-O distance of about 4.5 Å. As the dissociative LMCT state crosses the d-d excited states (shown in red) there is an opportunity for the system to cross from one of the d-d excited surfaces to the dissociative LMCT surface. Hence, there can also be O2 dissociation following d-d excitation, provided that these states live long enough to reach the crossing regime. The exact crossing probability will depend on many details the discussion of which is outside the scope of this work. Given the smaller oscillator strengths for the d-d absorptions and the finite probability for surface hopping, much lower quantum yields are theoretically predicted for d-d excitations. This is in agreement with the observations for O2 photo-release observed experimentally following excitation of 1 with either red (ϕ683 = 0.035) or blue light (ϕ436 = 0.29). The theoretical results are also consistent with the activation enthalpy for the O2 dissociation from 1 observed experimentally (ΔH‡experim = 45 kJ mol−1 vs ΔH‡comput = 67 kJ mol−1).
Figure 3.

TD-DFT calculated energy and shape of the beta HOMO and beta LUMO orbitals as a function of copper-oxygen bond distance.
Finally, the crossing between the ground state (green) and LMCT (blue) surfaces explains the fact that an association barrier is observed for O2-rebinding. The calculated barrier from TD-DFT (~ 24 kJ mol−1) is in the right ballpark but slightly overestimates the experimentally measured barrier (~ 10 kcal mol−1).
Summarizing, we report here the first example of a photodissociation of molecular oxygen from cupric-superoxide complexes, thus also representing a new approach to study the kinetics and the thermodynamics of formation of 1:1 ligand-copper(I)/O2 compounds. Copper-oxygen bond breaking is induced in [based orbital to the antibonding TMG3tren based orbital to the antibonding CuII based orbital to the antibonding O2 based orbital to the antibonding]+ and [based orbital to the antibonding PV-TMPA based orbital to the antibonding CuII based orbital to the antibonding O2 based orbital to the antibonding]+ through laser excitation either into the LMCT band, using 436 nm light, or into the d-d electronic transition, using 683 nm light. Interestingly, the quantum yield for O2 release was wavelength dependent. TD-DFT studies elucidated the O2 photo-release event occurring upon irradiation with red light on the basis of a) population of a molecular orbital (3dz2) that has strong σ-antibonding character along the Cu-O bond and b) energy surface crossing between the d-d and the LMCT excited states to lead to O2 release. Such findings add new insights into the observed wavelength dependent Cu-O2 photochemistry which differs markedly from that observed with hemes, where for example, the O2 adduct of myoglobin releases O2 with a quantum yield of 0.3 following Soret (λexc = 488 nm) or Q (λexc = 580 nm) band excitation.9 Formation and decay of [(TMG3tren)CuI]+ and [(PV-TMPA)CuI]+ formed in situ have been observed and both activation and thermodynamic parameters for the Cu/O2 reactions have been determined. Additional experimental studies are on their way to further characterize the excited states involved in the copper-oxygen bond breaking process using ultra-fast laser spectroscopy.
Supplementary Material
Acknowledgments
K.D.K. acknowledges financial support of this research from the National Institutes of Health, R01 GM28962. G.J.M. is grateful for research support from the National Science Foundation CHE-1213357.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, spectra, explanations, DFT calculations and supporting diagrams. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fukuzumi S, Karlin KD. Coord Chem Rev. 2013;257:187. doi: 10.1016/j.ccr.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Solomon EI, Ginsbach JW, Heppner DE, Kieber-Emmons MT, Kjaergaard CH, Smeets PJ, Tian L, Woertink JS. Faraday Discuss. 2011;148:11. doi: 10.1039/c005500j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Allen SE, Walvoord RR, Padilla-Salinas R, Kozlowski MC. Chem, Rev. 2013;113:6234. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maiti D, Lee DH, Gaoutchenova K, Wurtele C, Holthausen MC, Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Angew Chem, Int Ed. 2008;47:82. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 3.Maiti D, Narducci Sarjeant AA, Karlin KD. Inorg Chem. 2008;47:8736. doi: 10.1021/ic800617m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Comba P, Knoppe S, Martin B, Rajaraman G, Rolli C, Shapiro B, Stork T. Chemistry. 2008;14:344. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]; (b) Decker A, Solomon EI. Curr Opin Chem Biol. 2005;9:152. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]; (c) Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]; (d) Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J Am Chem Soc. 2006;128:12817. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]; (e) Huber SM, Ertem MZ, Aquilante F, Gagliardi L, Tolman WB, Cramer CJ. Chemistry. 2009;15:4886. doi: 10.1002/chem.200802338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Blackburn NJ, Rhames FC, Ralle M, Jaron S. J Biol Inorg Chem. 2000;5:341. doi: 10.1007/pl00010663. [DOI] [PubMed] [Google Scholar]; (b) Prigge ST, Mains RE, Eipper BA, Amzel LM. Cell Mol Life Sci. 2000;57:1236. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart LC, Klinman JP. Annu Rev Biochem. 1988;57:551. doi: 10.1146/annurev.bi.57.070188.003003. [DOI] [PubMed] [Google Scholar]
- 7.(a) Evans JP, Ahn K, Klinman JP. J Biol Chem. 2003;278:49691. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]; (b) Klinman JP. Chem Rev. 1996;96:2541. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; (c) Klinman JP. J Biol Chem. 2006;281:3013. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]; (d) Bauman AT, Yukl ET, Alkevich K, McCormack AL, Blackburn NJ. J Biol Chem. 2006;281:4190. doi: 10.1074/jbc.M511199200. [DOI] [PubMed] [Google Scholar]
- 8.(a) Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]; (b) Cramer CJ, Tolman WB. Acc Chem Res. 2007;40:601. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen P, Bell J, Eipper BA, Solomon EI. Biochemistry. 2004;43:5735. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 9.Ye X, Demidov A, Champion PM. J Am Chem Soc. 2002;124:5914. doi: 10.1021/ja017359n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Schatz M, Raab V, Foxon SP, Brehm G, Schneider S, Reiher M, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem Int Ed. 2004;43:4360–4363. doi: 10.1002/anie.200454125. [DOI] [PubMed] [Google Scholar]; (b) Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]; (c) Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Wurtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg Chem. 2010;49:9450. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J Am Chem Soc. 2011;133:1702. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. J Am Chem Soc. 2007;129:14697. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 13.Fry HC, Scaltrito DV, Karlin KD, Meyer GJ. J Am Chem Soc. 2003;125:11866. doi: 10.1021/ja034911v. [DOI] [PubMed] [Google Scholar]
- 14.Lucas HR, Meyer GJ, Karlin KD. J Am Chem Soc. 2010;132:12927. doi: 10.1021/ja104107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Elinarsdóttir O, Funatogawa C, Soulimane T, Szundi I. Biochim Biophys Acta. 2012;1817:672. doi: 10.1016/j.bbabio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Szundi I, Funatogawa C, Fee JA, Soulimane T, Elinarsdóttir Ó. Proc Nat Acad Sci. 2010;107:21010. doi: 10.1073/pnas.1008603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Poater A, Cavallo L. Inorg Chem. 2009;48:4062. doi: 10.1021/ic802269v. [DOI] [PubMed] [Google Scholar]; (b) Zapata-Rivera J, Caballol R, Calzado CJ. J Comput Chem. 2011;32:1144. doi: 10.1002/jcc.21697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.