

Abstract
Radial glia serve as the resident neural stem cells in the embryonic vertebrate nervous system, and their proliferation must be tightly regulated to generate the correct number of neuronal and glial cell progeny in the neural tube. During a forward genetic screen, we recently identified a zebrafish mutant in the kif11 loci that displayed a significant increase in radial glial cell bodies at the ventricular zone of the spinal cord. Kif11, also known as Eg5, is a kinesin-related, plus-end directed motor protein responsible for stabilizing and separating the bipolar mitotic spindle. We show here that Gfap+ radial glial cells express kif11 in the ventricular zone and floor plate. Loss of Kif11 by mutation or pharmacological inhibition with S-trityl-L-cysteine (STLC) results in monoastral spindle formation in radial glial cells, which is characteristic of mitotic arrest. We show that M-phase radial glia accumulate over time at the ventricular zone in kif11 mutants and STLC treated embryos. Mathematical modeling of the radial glial accumulation in kif11 mutants not only confirmed an ~226x delay in mitotic exit (likely a mitotic arrest), but also predicted two modes of increased cell death. These modeling predictions were supported by an increase in the apoptosis marker, anti-activated Caspase-3, which was also found to be inversely proportional to a decrease in cell proliferation. In addition, treatment with STLC at different stages of neural development uncovered two critical periods that most significantly require Kif11 function for stem cell progression through mitosis. We also show that loss of Kif11 function causes specific reductions in oligodendroglia and secondary interneurons and motorneurons, suggesting these later born populations require proper radial glia division. Despite these alterations to cell cycle dynamics, survival, and neurogenesis, we document unchanged cell densities within the neural tube in kif11 mutants, suggesting that a mechanism of compensatory regulation may exist to maintain overall proportions in the neural tube. We propose a model in which Kif11 normally functions during mitotic spindle formation to facilitate the progression of radial glia through mitosis, which leads to the maturation of progeny into specific secondary neuronal and glial lineages in the developing neural tube.
Keywords: Kif11, Eg5, kinesin, radial glia, neural stem cell, mitosis, zebrafish, interneuron, motorneuron, oligodendrocyte, mathematical modeling
INTRODUCTION
Neurogenesis relies on precise cell cycle control of stem cell and progenitor cell proliferation mediated through an array of extrinsic and intrinsic molecular mechanisms (Demir et al., 2009; Sommer and Rao, 2002; Takahashi et al., 1995). During neurulation in mammals, or cavitation of the neural rod in teleosts, proliferative stem cell populations are established within the ventricular zone positioned about the midline of the developing neural tube (Ciruna et al., 2006; Copp et al., 2003; Watters, 2006). Pseudostratified neuroepithelial cells serve as the first neural stem cell population undergoing several rounds of symmetrical cell divisions to equally amplify cell populations on the two halves of the central nervous system (CNS) (Hollyday, 2001; Kriegstein and Alvarez-Buylla, 2009; Tawk et al., 2007). Later, neuroepithelial cells undergo asymmetric divisions that contribute to the first stages of neurogenesis, as well as transform into radial glial cells that serve as the neural stem cell population throughout life in zebrafish, but only until postnatal development in mammals (Grandel and Brand, 2013; Kriegstein and Alvarez-Buylla, 2009; Suter et al., 2009).
Radial glia retain the bipolar morphology of their neuroepithelial predecessors; however, unlike neuroepithelial cells, radial glia express Glial fibrillary acidic protein (Gfap), which is an intermediate filament protein and the most common and comprehensive marker for astroglial cells (Bignami and Dahl, 1977; Zhang, 2001). As neuroepithelial and radial glial cells progress through the cell cycle they demonstrate interkinetic nuclear migration, in which the nucleus translocates to the luminal surface where mitosis is performed (Alexandre et al., 2010; Gotz and Huttner, 2005; Leung et al., 2011). Importantly, radial glial cells function as self-renewing stem cells that possess both neurogenic and gliogenic potential ((Malatesta et al., 2000; Noctor et al., 2002) and reviewed in (Ihrie and Alvarez-Buylla, 2008; Pinto and Gotz, 2007)). In zebrafish, it has been suggested that radial glia give rise to motorneurons and oligodendrocytes through the creation of a common progenitor cell population called oligodendrocyte progenitor cells (OPCs) (Kim et al., 2008). Under the influence of external signaling systems such as the Notch and Hedgehog pathways, OPCs contribute to the creation of oligodendrocytes and both primary and secondary motorneurons (Huang et al., 2012; Kim et al., 2008; Park et al., 2002; Park et al., 2004; Shin et al., 2007). Much still remains to be discovered about the mechanisms controlling radial glial proliferation and the neuronal and glial cell lineages radial glia create.
The zebrafish, Danio rerio, has emerged as a powerful genetic model system for the study of cell cycle control during vertebrate neurogenesis. Mutant loss of function analyses have uncovered essential cell cycle genes that range from regulating cell division throughout the embryo (futile cycle, (Dekens et al., 2003); crash&burn, (Shepard et al., 2005); cassiopeia, (Pfaff et al., 2007)) to more neural restricted control (curly fry, (Song et al., 2004); cug2, (Kim et al., 2011)). Recently, we took part in a screen of zebrafish mutants that were originally isolated during a forward genetic insertional mutagenesis screen (Amsterdam et al., 2004), and identified a class of genes necessary for the proper number and patterning of radial glial cells in the spinal cord (Barresi et al., 2010). An allele of the kinesin family member 11 gene (kif11hi3112a) displayed a dramatic increase in the number of radial glial somas throughout the spinal cord (Barresi et al., 2010). Whether this increase was due to changes in radial glial proliferation rates, defects in the mechanisms of cell division, or impairments in proper interkinetic nuclear migration remained to be investigated.
Kif11, also known as Eg5 (Bim-C class of kinesins), is a plus-end directed heterotetrameric motor protein capable of simultaneously moving along two microtubules (Kapitein et al., 2005; Krzysiak et al., 2008; Sawin and Mitchison, 1995; Valentine et al., 2006a; Valentine et al., 2006b). Eg5 has been shown to directly associate with the anti-parallel, interpole microtubules of the mitotic spindle and generate the necessary bipolar forces to push centrosomes away from one another (Uzbekov et al., 1999; Valentine et al., 2006a). In support of this proposed function for Kif11/Eg5, pharmacological inhibition of Eg5 in Xenopus egg cultures causes mitotic arrest by preventing chromosome segregation through the reduction of the bipolar spindle into a monopolar or monoaster spindle (Cochran et al., 2005; Gartner et al., 2005; Gruber et al., 2005; Kapoor et al., 2000; Mayer et al., 1999; Miyamoto et al., 2004; Muller et al., 2007; Sarli and Giannis, 2006). eg5 is expressed in the mouse blastula and eg5 knock-out mice die prior to gastrulation, which demonstrates that Eg5 is required for early cleavage events in the mouse (Castillo and Justice, 2007; Chauviere et al., 2008; Ferhat et al., 1998). Unfortunately, the early lethality of eg5 knock-out mice makes it impossible to investigate the in vivo role of kif11/eg5 during the later developmental events of embryogenesis and beyond.
In this study, we characterized the in vivo role of the kinesin motor protein Kif11, and defined a specific role for Kif11 in early neural stem cell division and neurogenesis in the zebrafish spinal cord. Loss of Kif11 caused the progressive accumulation of mitotically arrested radial glial somas at the ventricular zone of the spinal cord. We experimentally supported the predictions made by mathematical modeling that severely delayed mitotic exit, reduced cell cycle entry, and increased programmed cell death are all critical factors that influence Kif11-dependent radial glial proliferation. Using loss of Kif11 as a method for indirect lineage analysis, we showed specific reductions in secondary neuronal cell types and maturing oligodendroglial cells. We propose that kif11 plays a critical role in facilitating the separation of the mitotic spindle in radial glia, which has both direct and indirect implications for proper neurogenesis in the developing zebrafish neural tube.
METHODS
Zebrafish
Zebrafish were maintained according to Smith College Institutional Animal Care and Use Committee (IACUC) and Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) regulations. Adult fish experienced a 12h light-dark cycle, maintained at ~28°C, and fed a daily diet of live brine shrimp and Gemma Micro (GM) 300 dry mix (Skretting/Bio-Oregon). Embryos were maintained at a constant temperature of 28.5°C in E3 embryo medium according to highly accepted procedures for zebrafish care ((Westerfield, 2007); Zebrafish International Resource Center, ZIRC). Lines utilized in this study were kif11hi3112a (provided by N. Hopkins, MIT), AB (wild type) (provided by C. Lawrence, Harvard University), Tg(gfap:EGFP) (provided by P. Raymond, University of Michigan), Tg(olig2:EGFP) (provided by B. Appel, University of Colorado), Tg(gata2:EGFP) (provided by S. Lin, UCLA), and Tg(mnx1:EGFP) (obtained from ZIRC).
To identify kif11 mutants, head tissue from labeled kif11hi3112a embryos was digested overnight in Proteinase K in TE and genotyped using the Multiplex PCR Kit (Qiagen). The following primers were used: forward 5′-GCA GCC ACT CAC TTT TAA AGT ATG AC-3′, reverse 5′-GTG CAG TCC TAA CTA TTG AGT-3′, and viral reverse 5′-TCA GTT CGC TTC TCG CTT C-3′.
For RT-PCR analysis, kif11hi3112a embryos were staged to 48hpf and anesthetized in 4.2% Tricaine methanesulfonate (Argent Chemical Laboratories, MS-222) in E3 (Westerfield, 2007). Tail tissue was preserved in RNAlater (Qiagen, cat. no: 1017980) at −20°C, while remaining tissue was digested overnight in 10mg/mL Proteinase K in TE and processed for embryonic genotyping. Corresponding tail tissue was then processed for RNA isolation and RNA cleanup as previously described (Peterson and Freeman, 2009). RNA cleanup and cDNA synthesis were performed using the Qiagen RNEasy Protect Mini Kit (cat. no. 74124) and the Qiagen QuantiTect Reverse Transcription Kit (cat. no. 205311) respectively per manufacturer instruction and as described (Peterson and Freeman, 2009). PCR on cDNA was performed using the following kif11 primers: forward 5′-GGT CTA CTC TTA AGC AAG ATC GGC-3′ and reverse 5′-CTT CAA TTT GTT TGG CAG AAG GGC-3′. β-actin was used as a control: forward 5′-TGG TAT TGT GAT GGA CTC TGG-3′ and reverse 5′-AGC ACT GTG TTG GCA TAC AGG-3′.
Pharmacological inhibition of Kif11
S-trityl-L-cysteine (MP Biomedicals), Dimethylenastron (Alexis Biochemicals), and Monastrol (Tocris Bioscience) were each dissolved to 100mM in Dimethyl sulfoxide (DMSO) (Fisher Scientific) and further diluted to 10μM, 100μM, 0.5mM, 0.625mM, 0.75mM, 0.875mM, and 1.0mM in embryo medium (E3). Experimental Kif11 inhibitor and vehicle control (DMSO) embryos were treated at 5hpf and incubated at 28.5°C until desired age.
In situ hybridization and Immunohistochemistry
Whole mount and fluorescent in situ hybridizations were conducted on 27hpf wild type AB, Tg(gfap:EGFP), and Tg(olig2:EGFP) embryos using the cb1 probe conjugated to kif11 (eg5) mRNA (ZIRC) using published protocols (Jowett, 1997; Thisse and Thisse, 2008).
Whole mount immunohistochemistry was conducted as previously described (Barresi et al., 2010) with some modifications. To study neuronal populations (anti-GABA and anti-Islet-1), embryos were fixed in 4% formaldehyde, 0.05% glutaraldehyde, 5mM EGTA, 5mM MgSO4, 0.1% Triton-X in Phosphate buffer (PB) for 1 hour (Dekens et al., 2003). All other antibody labeling was conducted in embryos fixed in 4% paraformaldehyde (Ted Pella) in PB for 2 hours at room temperature or overnight at 4C. The following primary antibodies were used: rabbit anti-goldfish GFAP (1:400, generously donated by Dr. Samuel Nona), mouse anti-acetylated Tubulin (1:800, Sigma), mouse anti-Zrf1 (1:4, ZIRC), mouse anti-phosphohistone H3 (1:1000, Cell Signaling), mouse anti-Islet-1 (39.4D5, 1:200, DSHB), rabbit anti-GABA (1:1000, Sigma), mouse anti-α-Tubulin (1:500, Sigma), mouse anti-BrdU (G3G4, 5ug/mL, DSHB), and rabbit anti-active Caspase-3 (1:500, BD Pharmingen). Tissue sections were obtained at 14μm thickness with a Leica cryostat and processed for labeling per (Devoto et al., 1996). DNA was visualized in sectioned tissue with Hoescht stain (1:30,000, Invitrogen). Imaging was conducted using structural illumination with the AxioImager Z1 equipped with ApoTome (Zeiss). Z-stacks were collected at an optical slice thickness of 0.53μm at 400X magnification and 0.31μm at 630X magnification for all whole mounts and all sections respectively.
S-phase labeling with BrdU
Embryos were treated with 5-bromo-2′-deoxyuridine (BrdU) as previously described (Kim et al., 2011; Shepard et al., 2004) with slight modifications. Manually dechorionated kif11hi3112a embryos were incubated in E3 medium on ice for 5 minutes and then incubated in 10mM BrdU (Sigma, catalog no: B9285) in 15% DMSO/E3 medium for 20 minutes on ice. Embryos were then fixed using 4% paraformaldehyde for 2 hours at room temperature. BrdU detection was performed using anti-G3G4 (anti-BrdU, 5μg/mL, DSHB) as described (Shepard et al., 2004).
Mathematical Modeling
To further investigate the difference in the cell division process occurring in wild-type neural tubes as compared with the kif11 mutants, we built and analyzed ordinary differential equation models consisting of three dynamic variables reflecting the number of radial glia cells at different stages of the cell cycle:
G(t), the number of quiescent (non-dividing) radial glial cells at time t,
S(t), the number of radial glia in S-phase at time t, and
M(t), the population of radial glial cells undergoing mitosis at time t.
The starting model, built to reflect the dynamics for the wild-type population, is schematically illustrated in the Figure 4A with the exclusion of the cell death variable (D(t)) used to evaluate kif11 mutant cell dynamics. Resulting parameter values are presented in Table 1.
Figure 4.
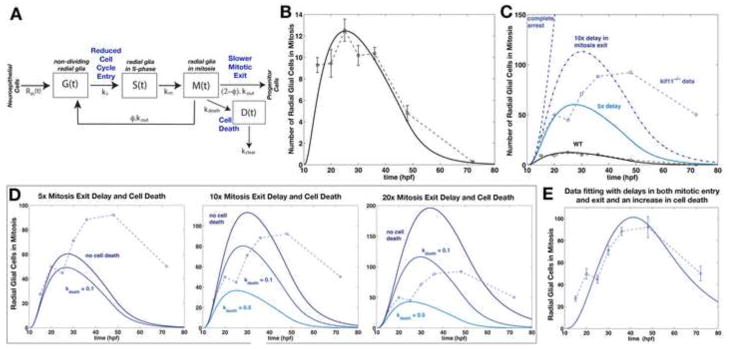
Mathematical Model Variant 1 predicts arrested mitosis, cell death, and reduced entry into the cell cycle in kif11−/− mutants. (A) Schematic of radial glial cell division through mitosis under Model Variant 1. Assumed changes in kif11−/− are shown in blue. Parameter values and their definition are listed in Table 1 (for wild-type) and Table 2 (for kif11−/−). (B) Best-fit simulation result from the mathematical model showing the dynamics of M-phase radial glia in wild type. (C) Mathematical model testing whether simple accumulation of the number of wild type PH3 positive radial glia can account for the number of mitotic radial glia in kif11−/− mutants. Simulation results fail to capture kif11−/− data with complete mitotic arrest or a 5x or 10x delay in mitosis exit. (D) Mathematical model testing the combined effects of a 5x, 10x, or 20x delay in mitosis exit with 0%, 10% or 50% cell death occurring in M-phase cells. Mitosis exit delay and cell death alone also fail to capture the full dynamics of kif11−/− data. (E) Mathematical modeling of mitosis exit delay with cell death and reduced entry into S-phase achieves a good fit to the kif11−/− M-phase radial glial data. (B–E) Raw data is displayed as dashed lines while data fitting curves are represented as solid lines.
Table 1.
Parameter values characterizing radial glia division in wild-type.
| Parameter | Definition | Values | 95% Interval* |
|---|---|---|---|
| Rin | Maximum numbers of radial glial generated by the neuroephithelial | 49.93 cells/hr | 47.86–52.01 |
| φ | Average number of daughter cells that remains as glial cells | 0.182 | 0.140–0.223 |
| tend | Time at which neuroepithelial are fully differentiated | 46.15 hpf | 44.55–47.75 |
| ks | Rate of entry into S-phase | 0.667/hr | |
| km | Rate of glia entry into M-phase | 0.171/hr | |
| kout | Rate of exit from mitosis | 2.4/hr |
95% confident intervals are given for parameter values obtained from data-fitting.
The following underlying assumptions were used in constructing the model:
We assume that, starting from tinit = 10 hpf to an ending time tend hpf, neuroepithelial cells generate radial glial cells at a linearly decreasing rate of the form Rin (1−t/tend) during that period. At tinit, the rate of generation of glial cells is at its highest value, Rin (1−tinit/tend), and then begins to decrease, reaching zero at tend. We assume prior to tinit = 10 hpf, the neuroepithelial population builds up and reaches its peak at 10 hpf. The neuroepithelial population then began to decrease by differentiating after 10 hpf, which marks the first signs of any gfap expression (Bernardos and Raymond, 2006). In our model after tend, we assume most (if not all) neuroepithelial cells have differentiated and do not continue to generate radial glia.
- In radial glial cells, the cell division process occurs with fixed constant rates. That is, each radial glial cell completes each stage of the cell cycle in a fixed period of time. The following rates used were based on previous measurements of neural stem cell cycle progression in the zebrafish hindbrain (Leung et al., 2011):
Following a cell division, the two resulting daughter cells can turn into progenitor cells or radial glial cells. We assume that asymmetric division occurs with probability p, and symmetric differentiation (producing two progenitor cells) occurs with probability q. The remainder of the time, symmetric renewal can also occur (with two radial glial cells produced) with probability 1-p-q.
Based on these assumptions, the dynamics of the cell populations follow the differential equations:
where φ = [2 − (p + 2q)], the average number of daughter cells (from a radial glial division) that either continue the generation of radial glial cells or become progenitor cells. The value of φ is restricted between 0 to 2. When 0 ≤ φ < 1, most daughter cells turn into progenitors. Full asymmetric division corresponds to φ = 1, and if φ > 1 most daughter cells remains as radial glial cells.
Unknown parameter values for this model are obtained by data fitting. The best-fit parameter set, , is obtained in the least-square sense. That is, given a data set consisting of the number of mitotic radial glial cells at different time points, (ti, mi), the following objective function is minimized:
where is obtained by solving the differential equations above at time ti using the parameter values . The least-square problem is numerically solved using a standard constrained optimization package (specifically fmincon in MATLAB). The parameter values are constrained to be positive with further restriction of 0<φ<2, and tend > tinit (as discussed above). We also performed bootstrapping to obtain the 95% confidence interval for each parameter values. 100 data sets were generated by sampling from the Gfap and PH3 double positive raw data presented in Figure 2I. For each data set, constrained least-square optimization was performed; each final parameter value was obtained by taking the mean and the 95% confidence interval was computed.
Figure 2.

Loss of kif11 causes an accumulation of radial glia in mitosis. (A–H) Lateral views of kif11−/− sibling neural tubes fixed at timepoints throughout neurogenesis and labeled for radial glia (Gfap), Phosphohistone H3 (PH3) and acetylated Tubulin (AT). Within all siblings, most Gfap+ cells labeled with our antibody were PH3+, indicating these cells were actively proliferating at the time of fixation (A,E insets). At all time points assayed, there were significantly more Gfap+/PH3+ cells in kif11−/− embryos compared with wild type siblings (E–H, quantified in I, asterisks), as well as more PH3+ nuclei (J, shaded and black bars). No change in the number of Gfap+ cells was seen between 20–25hpf and 36–48hpf within kif11−/− (I, brackets). Of all proliferating cells (J), it was noted that an unidentified population (PH3+, Gfap−) is also kif11 dependent in both wild type and kif11−/− neural tubes. Error bars delineate the standard error of the mean. Asterisks indicate a statistical significance (t-test, two tailed, assuming equal variances, p<0.05).
We performed the same procedure to analyze the kif11−/− data. We first assumed that the mutation causes a delay in mitotic exit, which was done by decreasing the value of the mitosis exit rate kout. All other parameter values are assumed to remain the same during the acquisition of these models. In addition, we further tested the possibility of changes in the rate of cell death (both Model Variant 1 and 2) and entry into the cell cycle (only Model Variant 1) as important parameters influencing the number of cells seen in kif11 mutants. To account for potential increases in apoptosis of cells following mitotic arrest only (Fig. 4A), we added an additional term to the equation for M(t):
and a variable D(t), representing the turnover of the population of dying/dead cells
Here we assumed that a single dead cell is cleared within 3 hours (kclear = 1/3 hr = 0.33/hr), which is based on live cell recordings of apoptotic induced cell death in radial glial cells within the ~48hpf spinal cord (Bashiruddin and Barresi, unpublished observations).
In Model Variant 1, we also evaluated potential reductions in the proliferation rate for radial glia. To do this we allowed the possibility that the G1 phase might be lengthened (reducing the value of ks). Data-fitting was performed to find the best-fit value for all parameters (Table 2).
Table 2.
Best-fit parameter values for kif11 data under Model Variant 1: Mitotic exit delay leading to cell death and reduced cell cycle entry.
| Parameter | WT Values | kif11−/− Values | Parameter | WT Values | kif11−/− Values | kif11−/− 95% Interval |
|---|---|---|---|---|---|---|
| Rin | 49.93 cells/hr | 49.93 cells/hr | ks | 0.667/hr | 0.0731/hr | 0.0667–0.0794 |
| φ | 0.182 | 0.182 | kout | 2.4/hr | 0.1562/hr | 0.1400–0.1724 |
| tend | 46.15 hpf | 46.15 hpf | kdeath | 0 | 0.0261/hr | 0.0163–0.0359 |
| km | 0.171/hr | 0.171/hr | kclear | - | 0.33/hr | - |
Kif11 Model Variant 2 was designed to analyze whether the kif11−/− data could be explained by delayed mitotic exit with increased cell death occurring at all stages of the cell cycle (Fig. 6A), however this model lacks any slowing of cell cycle entry. To account for additional cell death occurring at G-phase, S-phase, and M-phase, the following terms were added to this model:
Figure 6.

Mathematical Model Variant 2 predicts arrested mitosis and two modes of cell death drive the number of M-phase radial glia in kif11−/− mutants. Assumed changes impacting kif11−/− are shown in blue. (A) Schematic of radial glial cell division through mitosis under Model Variant 2. (B) Mathematical modeling of significant mitosis exit delay with cell death occurring at all stages of the cell cycle achieves a good fit to the kif11−/− M-phase radial glial data. (C) Application of the radial glial cell division mathematical Model Variant 2 for data fitting of total cell counts for anti-Activated Caspase 3 positive cells in kif11−/− mutants. (D, E) Application of the radial glial cell division mathematical Model Variant 2 for data fitting of total cell counts for BrdU incorporation in wild type (D) and kif11−/− (E). (B–E) Raw data is displayed as dashed lines while data fitting curves are represented as solid lines.
Here kdeath2 corresponds to the death rate due to mitotic arrests while kdeath1 represents the death rate due to other processes (e.g. cell cycle check points or toxicity-induced-death from neighboring apoptotic cells). Data-fitting was again performed to find the best-fit value for all parameters under Model Variant 2 (Table 3).
Table 3.
Best-fit parameter values for kif11 data under Model Variant 2: Mitotic exit delay leading to two modes of cell death and no change in cell cycle entry.
| Parameter | WT Values | kif11−/− Values | Parameter | WT Values | kif11−/− Values | 95% Interval |
|---|---|---|---|---|---|---|
| Rin | 49.93 cells/hr | 49.93 cells/hr | kout | 2.4/hr | 0.0106/hr | 0.0086–0.0127 |
| φ | 0.182 | 0.182 | kdeath1 | 0 | 0.4372/hr | 0.4244–0.4499 |
| tend | 46.15 hpf | 46.15 hpf | kdeath2 | 0 | 0.009/hr | 0.0071–0.0108 |
| ks | 0.667/hr | 0.667/hr | kclear | - | 0.33/hr | |
| km | 0.171/hr | 0.171/hr |
Imaging, Quantification and Statistics
All neural tube imaging was performed between somite regions 9 through 13. Images for glial proliferation were focused on the ventricular zone, which was captured by an average of 55 optical sections (0.53μm per slice) centered on the midline acquired from a lateral view of the neural tube. Data for neuronal populations, BrdU+ nuclei, and activated Caspase-3+ cells were derived from a lateral view of the full Z-stack of the neural tube. Blind cell counts of 3-dimensional Z-stacks and/or maximum intensity projections were conducted manually using the Adobe Photoshop counting tool. Gfap+ somas were identified by their characteristic spherical shape, and secondarily confirmed with colocalization with PH3 and/or Hoechst labeling depending on the experiment. Specific neurons in anti-GABA+3 and anti-ISL+ populations were identified by location along the D/V axis, soma size, and axon trajectory (Appel and Chitnis, 2002; Appel and Eisen, 1998; Bernhardt et al., 1992; Lewis and Eisen, 2003; Song et al., 2004). Oligodendrocyte progenitor cells visualized with Tg(olig2:EGFP) transgenic embryos were counted at different stages of their development; immature OPCs in the ventral column of the spinal cord showed extensive overlapping GFP expression, therefore they were only counted as either double labeled for PH3 or triple labeled with PH3 and Gfap. However, once a Tg(olig2:EGFP) GFP+ cell had begun to emerge from the ventral column and become positioned more dorsally we counted these as “migratory OPCs”. All values obtained were conveyed as the mean ± standard error of the mean. Significance of the results was reported using two-tailed t-tests assuming equal variance at an alpha level of 0.05.
RESULTS
kif11 is expressed by radial glial cells and is required for the correct number of luminal localized radial glial somas
As part of a large screen of insertional mutants, we identified kif11hi3112a to be in a class of genes required for proper radial glial development in the zebrafish spinal cord (Barresi et al., 2010). Expanding on our previous characterization, homozygous kif11hi3112a mutants exhibited an eight-fold increase in the number of Gfap+ radial glial somas that were all concentrated at the luminal surface of the ventricular zone (Fig.1A–E; wt avg. = 10.8±0.54, n=40; kif11−/− avg. = 81.1±4.34, n=24, p<0.0001). Quantification of the total number of nuclei in neural tube cross sections at three different axial positions showed no statistically significant difference between kif11hi3112a mutants and wild type siblings (Fig. 1E; wt avg. = 58.4±4.89, n=11; kif11−/− avg. = 58.2±6.85, n=19, p>0.88), suggesting that the increase in Gfap+ somas in kif11hi3112a mutants is not the result of an increase in the number of cells in the neural tube.
Figure 1.

Characterization of kif11 expression in radial glia and its effect on radial glial soma localization. (A–D) Kif11 siblings were grown to 30hpf and labeled for radial glia (Gfap) and acetylated Tubulin (AT) to identify glial and axon defects. In wild type siblings a small population of Gfap+ radial glial somas can be visualized at the luminal surface in a maximum intensity projection of optical sections along the parasagittal plane that encompass the ventricular zone (A) as well as in cross section of the neural tube (B). An increase in the number of radial glial somas can be seen the neural tube of kif11−/− mutants (C,D). (E) Quantification of Gfap+ somas counted from maximum intensity projections of parasagittal optical slices within the neural tube and the total number of nuclei in transverse cross sections of the neural tube showed an 8-fold increase in the number of Gfap+ cell bodies in kif11−/− mutants, but no change in the number of total nuclei between siblings. (F) RT-PCR analysis of kif11 expression between pooled wild type/heterozygote siblings and kif11−/− at 48hpf. β-actin was used as a loading control. (G–M) Wild type kif11 mRNA was present throughout the forebrain (G, lateral; H, dorsal) and trunk (I, J–M) at 27hpf, concentrated at the luminal surface of the VZ (J,K) and into the floor plate (J,K, insets). (K–M) Fluorescent kif11 detection (K) within a Tg(gfap:EGFP) embryo (L) shows that kif11 mRNA is present down the length of the ventricular zone and colocalizes with cells expressing GFP associated with the lumen of the VZ (M).
The two recessive alleles of the kif11 mutant, kif11hi486 and kif11hi3112a, contain viral insertions within the second intron, and the kif11hi486 loci has been shown result in a 90% reduction in kif11 gene expression (Amsterdam et al., 2002). Similarly, reverse transcriptase (RT) PCR analysis of 48hpf kif11hi3112a homozygous mutants displayed a severe reduction in kif11 transcripts as compared with their wild type siblings (Fig. 1F). This suggests that while the kif11 alleles do not cause null mutations, they do represent a significant knock-down of kif11 transcript that results in embryonic lethality by 5dpf ((Amsterdam et al., 2004), our own observations). All analyses of the kif11 mutation throughout this study were performed on the kif11hi3112a allele.
We hypothesized that if any residual Kif11 activity remained in the kif11 mutant, then additional methods to knock down its function would result in more severe phenotypes. We tested the efficacy of three known pharmacological inhibitors of Eg5 on zebrafish: Monastrol, Dimethylenastron (DME), and S-trityl-L-cysteine (STLC) (DeBonis et al., 2004; Gartner et al., 2005; Kapoor et al., 2000; Kozielski et al., 2008; Mayer et al., 1999; Muller et al., 2007). Comprehensive dose response analysis in wild type (AB) embryos revealed that a concentration of 0.875mM STLC yielded maximal effects that phenocopied the mutant and did not cause any additional gross morphological malformations to the neural tube (Sup. Fig. 1A–K). Using STLC as a working pharmacological inhibitor in zebrafish, we next tested if the remaining kif11 transcript in kif11 mutants (Fig. 1F) contributes to radial glial development. Homozygous kif11+/+ wild type, heterozygous kif11+/− siblings, and homozygous kif11−/− mutants treated with 0.875mM STLC from 5hpf to 30hpf showed the same increase in the number of Gfap+ somas as kif11−/− mutants treated with the DMSO vehicle control, and importantly, no statistically significant difference was seen between any of the treated groups (Sup. Fig.1L). This result confirmed that homozygous kif11−/− mutants represent a phenotypically null mutation and that heterozygous genotypes possess no phenotypic consequence. Lastly, while we did determine through RT-PCR that kif11 is maternally expressed from the one-cell-stage and retained throughout embryogenesis (data not shown), lack of phenotypic change following STLC treatment in kif11 mutants also demonstrates that there is little to no compensation from these maternal contributions on the radial glial population.
We further investigated the role Kif11 plays in radial glial development by examining the pattern of kif11 transcript expression throughout the embryonic CNS. Whole-mount in situ hybridization on wild type (AB) tissue revealed concentrated kif11 expression throughout the ventricular zone of the brain and spinal cord extending from the roofplate to the floorplate at 27hpf (Fig. 1G–J, arrows). kif11 expression was also detected outside the nervous system in the eye and ventral portions of the trunk (Fig. 1H–J). Fluorescent in situ hybridization of kif11 expression showed higher levels of expression in the dorsal two-thirds of the neural tube, but was consistently detected throughout the ventricular zone and floorplate (Fig. 1J, inset; K, inset). kif11 transcript was detected in radial glial cells as determined by overlapping expression with Green Fluorescent Protein (GFP) positive cells in Tg(gfap:EGFP) transgenic embryos (Fig. 1K–M).
Loss of kif11 causes an accumulation of M-phase arrested radial glia over time
In kif11 mutants, the increase in the number of Gfap+ somas at the luminal surface was seen without a change in overall number of nuclei at 30hpf, which suggested that the proportion of radial glia attempting division may be altered in kif11 mutants. We labeled kif11 siblings for the M-phase marker anti-Phosphohistone H3 (PH3) and anti-Gfap to determine if Gfap+ radial glial somas were actively undergoing mitosis at any time throughout embryonic neurogenesis (Pfaff et al., 2007; Shepard et al., 2005). Between the ages of 15 and 72hpf, a majority of Gfap+ somas in wild type siblings and nearly all in kif11 mutants were positive for PH3 (wt mean, 83.5%; kif11−/− mean, 98.9%; Fig. 2A,E insets). These results suggest that with our labeling method, most Gfap+ somas assayed were undergoing mitosis in both wild type and kif11 mutants. It is important to note that while the anti-Gfap antibodies we are using label all astroglial processes, we only observed subcellular expression within the cell body during the process of division (Fig. 2, 3). Such a change in Gfap protein localization within a dividing cell has been described for cells in culture, in which the phosphorylation state of Gfap can influence its differential positioning at the cleavage furrow ((Inagaki et al., 1994; Yasui et al., 1998); reviewed in (Middeldorp and Hol, 2011)).
Figure 3.

Loss of Kif11 causes monoaster spindle formation in radial glial cells during division. Kif11 siblings and wild type embryos treated with either STLC or DMSO control from 5–30hpf were labeled for radial glia (Gfap), microtubules (α-Tubulin) (microtubules), and histones (Hoechst stain) to identify mitotic spindle phenotypes. (A) Kif11 wild type siblings and (I) vehicle control (DMSO) treated embryos exhibit normal spindles in M-phase (enlarged in B–D and J–L, respectively) while (E) kif11−/− mutants and (M) STLC treated embryos display monoastral spindles characteristic of mitotic arrest (enlarged in F–H and N–P, respectively). White dots designate presumptive locations of centrosomes. For greater clarity Hoechst is displayed as a greyscale image in the enlargements (D, H, L, P).
Quantitative analysis of the mitotic index in kif11 wild type and mutant siblings showed different patterns of M-phase cells over time. The number of M-phase radial glia recorded in wild type siblings remained constant at approximately 10 cells between 15hpf and 36hpf, after which mitotically active cells declined gradually to near zero by 72hpf (Fig. 2A–D, I, hatched black line, J, white and grey bars). In contrast, kif11 mutants showed significantly more Gfap+ cells in M-phase at 15hpf (wt avg. = 11.16±0.72, n=13; kif11−/− avg. = 27.83±3.23, n=23, p<0.0001) that increased steadily at an average rate of 16 cells per hour (Fig. 2E–H, I, red line, J, black and hatched bars) until 48hpf, which was followed by a dramatic drop in the number of PH3+ radial glia and total PH3+ cells (Fig. 2I,J). Additionally, there were a significant percentage of PH3+ nuclei that did not co-express Gfap, which were consistently present in both wild type and kif11 mutants at all ages examined (wt avg. = 55.5%; kif11−/− avg. = 29.2%).
Since Kif11/Eg5 has a known role in mitotic spindle function, we next sought to determine if the accumulation of M-phase radial glia in kif11 mutants was due to a mechanistic failure of mitotic spindle separation. We labeled kif11 siblings for anti-alpha-Tubulin and anti-Gfap to visualize spindle morphology in radial glial cells (Fig. 3). Wild type spindle morphologies were unperturbed, showing microtubule arrangements consistent with metaphase, anaphase or telophase (Fig. 3A–D; not all spindle morphologies shown). In contrast, kif11 mutants showed Gfap+ cell somas exhibiting a single monoastral spindle phenotype (Fig. 3E–H). Previous cell culture studies using Eg5 specific pharmacological inhibitors have shown similar monoaster spindle formation that results in mitotic arrest (DeBonis et al., 2004; Kapoor et al., 2000; Muller et al., 2007; Valensin et al., 2009). To confirm that our previous pharmacological inhibition assay also results in monoaster spindle formation in radial glia, vehicle control and STLC treated wild type (AB) embryos were also assayed for anti-alpha-Tubulin and anti-Gfap. Similar to kif11 mutants, STLC treated wild type embryos displayed a single monoastral spindle phenotype within Gfap+ cells as compared to the unaffected spindles of DMSO control treated embryos (Fig. 3I–P). These data indicate that the loss of Kif11 causes defective mitotic spindle formation, which potentially leads to mitotic arrest and the accumulation of radial glial somas at the ventricular zone.
Mathematical modeling reveals kif11-dependent changes in cell cycle rate and cell survival
We next employed mathematical modeling to theoretically test whether accumulation of mitotically arrested cells alone can fully account for the number of radial glial somas we have quantified, or predict whether additional parameters of cell division are also impacted by the loss of kif11. We generated a differential equations model for radial glial cell division that considered several influencing parameters: the radial glial generating potential of neuroepithelial cells over time Rin(t), non-dividing radial glial cells G(t), and radial glial cells in S-phase S(t) or in mitosis M(t) (Fig. 4A). Performing data-fitting on this model yielded a close fit to the wild type raw data with robust parameter estimation as indicated by the 95% confidence interval (Fig. 4B; Table 1). According to our model, the resulting best-fit parameter values indicated that neuroepithelial proliferation ends after ~48 hpf. In addition, radial glial cell division is dominated by differentiation, such that only 9% (~0.18/2) of daughter cells remain as radial glial cells while the rest become progenitors.
We next applied this mathematical model towards testing whether simple mitotic arrest alone could account for the apparent accumulation of M-phase radial glia in kif11 mutants (Fig. 2). When manipulating kout to simulate a full mitotic arrest or a 5x or 10x delay in the rate of mitotic exit (decrease in kout), accumulation alone does not fit the quantification seen in kif11 mutants (Fig. 4C). Rather our model predicted that a greater number of radial glial cells should be present at each time point for accumulation alone to be valid (Fig. 4C), suggesting that additional parameters are impacting the dynamics (number and/or rate) of radial glia in mitosis.
It is known that cells arrested in mitosis for a prolonged period of time can trigger apoptosis (Kozielski et al., 2008; Marcus et al., 2005); therefore, we next evaluated a constant rate of cell death for cells that are in the mitotic phase of the cell cycle (kdeathM). The population of apoptotic cells, D(t), is assumed to be cleared within a time-scale of 3 hours (kclear) in the embryonic spinal cord (Bashiruddin and Barresi, unpublished observations). Taking these two processes into account, we then varied the values of kout to achieve a 5x, 10x, or 20x delay in mitosis exit and kdeath to reflect cells dying at a rate of either 10% or 50% per hour (Fig. 4D). Simulating an increase in cell death caused the number of cells to decrease, however this resulted in a poor fit to the kif11 mutant data at later time points. Further investigation by varying these two parameters (kout and kdeath) never produced an accurate fit to the kif11 mutant data (Fig. 4D). Increasing the delay of mitotic exit by 10x (decreasing kout) did increase the number of cells that produced a better fit to the kif11 mutant data at earlier time points as opposed to only a 5x delay, but this simulation still poorly fit the later time points. An even longer delay in mitotic exit (20x) paired with either a 10% or 20% cell death of radial glia improved the fit to the kif11 mutant M-phase radial glial data at the later time points yet worsened the fit for the earlier data points. These results allow us to conclude that delays in mitosis exit and subsequent cell death cannot fully explain the experimental data.
We next considered whether reducing the proliferation rate of radial glia in addition to mitotic exit delay and cell death would best fit the M-phase radial population captured in kif11 mutants. To theoretically test this we simulated a lengthening of the G1-phase of the cell cycle (reducing the value of ks) and performed data-fitting to find the best-fit value for the parameters kout, kdeath, and km (Table 2). This model, called Model Variant 1, predicted that the radial glial population has an approximate 9x lengthening of the G1-phase of the cell cycle to obtain a well aligned fit of M(t) to the kif11 mutant data. This slower rate of cell cycle entry was obtained as a best fit together with an average ~15x longer stay in M-phase and ~2% of those cells in mitosis undergoing cell death per hour (Fig. 4E).
Our mathematical Model Variant 1 suggested that the accumulated M-phase radial glia in the kif11 mutant was the combined result of cell death, delayed cell cycle entry, and significant mitotic exit delay. To biologically verify these predictions, we first quantified the number of cells undergoing apoptosis in the neural tube through anti-activated Caspase-3 labeling. Increased cell death was detected at 15hpf in the neural tube of kif11 mutants as compared with wild type siblings (wt avg. = 2.15±0.9, n=20; kif11−/− avg. = 15.4±5.9, n=16, p<0.05) and the number of apoptotic cells significantly increased by 20hpf to a level that remained relatively constant throughout the time course (1.33±0.54, n=48; kif11−/− avg. = 54.69±6.75, n=29, p<0.001; Fig. 5A,B,E). To determine if loss of kif11 causes a delay in cell cycle entry, we next assayed the number of cells entering S-phase through BrdU pulse-fix labeling. Kif11 siblings were briefly exposed to BrdU at different developmental stages and fixed immediately after treatment to capture all cells in the neural tube undergoing DNA replication at that stage. Quantification of BrdU incorporation at 15hpf, 20hpf, 25hpf, 30hpf and 36hpf revealed that cells in the neural tube of kif11 mutants did not show any change at 15hpf (wt avg. = 186.3 cells±7.6, n=15; kif11−/−, avg. 181.5 cells±6.0. n=15; p=0.62), but did show an approximately 24% reduction at each later time point as compared with wild type siblings (Pooled averages for 20hpf to 36hpf: wt avg. = 214.6 cells, n=57; kif11−/− avg. = 163.8 cells, n=58; p<0.0001; Fig. 5C–E). As predicted by our mathematical model, these results confirm that cell cycle entry and apoptosis are parameters also affected by the loss of kif11.
Figure 5. kif11 mutants have inversely proportional changes to the number of cells dying and proliferating.

(A,B) Lateral view of the 25hpf neural tube labeled for anti-activated Caspase-3 (green) and anti-Gfap (red) antibodies in wild type (A) and kif11−/− mutants (B). (C,D) Lateral view of the 25hpf neural tube labeled for anti-BrdU (green) and anti-Gfap (red) antibodies in wild type (C) and kif11−/− (D). (E) Quantification of cell death (dashed lines) and BrdU incorporation (solid lines). Anti-activated Caspase-3 labeling was increased in kif11−/− mutants from 15hpf to 72hpf (E, dashed blue line), whereas the number of cells in S-phase was reduced from 20hpf to 36hpf in kif11−/− mutants (E, solid black line) as compared with wild type embryos (black lines). (F) Graphical representation of the difference between the number of cells dying (dashed line) or BrdU positive (solid line) between kif11 siblings.
Mitotic arrest and progressive cell death determine cell number in kif11 neural tubes
If our mathematical model is accurately capturing all the parameters of neural stem cell division, then the quantitative measures of BrdU incorporation and apoptosis should be comparable to the representative trends of S(t) and D(t) respectively in this model. Comparing to the wild type BrdU data, S(t) matches the temporal dynamics of the data, but is approximately 20% lower in magnitude (Sup. Fig. 2A). A lower value is to be expected, since the collected data represents the total number of cells in S-phase at a given time post-fertilization, whereas our model is only considering radial glial cells in division. Performing the same comparison for the kif11 mutant yielded outputs from Model Variant 1 that failed to capture many aspects of the new experimental measurements of BrdU incorporation and cell death. First, the dynamics of S(t) were too slow as compared with the BrdU data collected in kif11 mutants; such that, a peak was obtained approximately 20 hours later than that observed in the raw BrdU data set (Sup. Fig. 2B). Second, the level of cell death D(t) obtained was also far too low in comparison to the counted number of apoptotic cells in the kif11 mutant over time (Sup. Fig. 2C). Although we successfully performed data-fitting for M(t) using Model Variant 1, we dispensed this model and sought to incorporate other critical factors to be incorporated into the mathematical model.
By examining the difference in the number of BrdU positive cells and apoptotic cells between wild type and kif11 mutants, we observed that the number of cells in the mutant positive for activated Caspase-3 is similar to the number of cells that should be experiencing S-phase according to the wild type data (Fig. 5E; means = 45.7 act. Caspase-3+ cells in kif11−/− - wt and 41.6 BrdU+ cells in wt - kif11−/−). Moreover, the trend of these difference curves suggested an inverse relationship between cell death and proliferation; such that, a peak in cell death is followed by a subsequent drop in BrdU incorporation and later observed drops in cell death were similarly followed by relative increases in BrdU incorporation (Fig. 5F). This data analysis suggests that the presumed mitotic arrest-induced cell death may progressively establish an unhealthy environment in the neural tube, which results in more extensive cell death at all stages of the cell cycle that could in turn influence the rate or number of cells actively proliferating.
Previously, we found using Model Variant 1 that the level of cell death is far too low in comparison to the observed experimental data (only 2% of mitotic radial glia undergo apoptosis per hour in Model Variant 1). To theoretically test whether cell death occurring at all stages of the cell cycle could better represent the dynamics of radial glial cell division in kif11 mutants we generated Model Variant 2 for best fit analysis (Fig. 6A). In Model Variant 2, we divided cell death parameters into kdeath1 and kdeath2, which represent the cell death due to a potentially toxic environment and due to mitotic arrest respectively (Fig. 6A). kdeath1 occurs during the G1 and S-phases of the cell cycle while kdeath2 is during M-phase. To focus on the impact of cell death as a driving force, no delays in cell cycle entry were factored into this Model Variant 2. As before, we performed data-fitting on M(t) to find the best-fit value for the following parameters: kout, kdeath, and kdeath2, as listed in Table 3. Data fitting for M(t) using Model Variant 2 obtained a close fit to the kif11 mutant mitotic radial glial data (Fig. 6B). Importantly this model yielded outputs that were more closely aligned to the BrdU and activated Caspase-3 data for both wild type and kif11 mutants (Fig. 6C–E) as compared with the simulations obtained using Model Variant 1. Specifically, Model Variant 2 showed improved S(t) temporal correspondence with the BrdU data, however it was still significantly lower in magnitude (Fig. 6E). Similarly, D(t) obtained from Model Variant 2 was now well aligned with the cell death data in both magnitude and temporal correspondence from 15 to 36hpf; however, the resulting D(t) curve failed to capture an observed secondary peak in cell death at 48hpf (Fig. 6C). While Model Variant 2 does not completely capture all aspects seen in all the raw data collected, simulations obtained using best-fit parameter values suggest that a near mitotic arrest of radial glial cells paired with two rates of cell death during the different phases of the cell cycle are sufficient to explain most of the dynamics witnessed in the kif11 mutant.
Cell density is maintained in kif11 mutants
Due to the cell death and changes in cell cycle dynamics seen in kif11 mutants, we revisited our earlier observation that the total number of cells in the neural tube remains unchanged between kif11 siblings at 30 hpf (Fig. 1E). We extended this data by quantifying the total cell density of the neural tube over time in wild type and kif11 mutants. Similar to our previous results, the average number of total nuclei was not statistically different between wild type and kif11 mutants with the exception of slight decreases at 25hpf and 48hpf in kif11 mutants (Sup. Fig. 3A,B). At no time was the area of the neural tube significantly different (Sup. Fig. 3A,C). Importantly, the calculated cell densities (cells/μm2) within wild type and kif11 mutant neural tubes showed remarkably little variation from 15 hpf to 72hpf (Sup. Fig. 3D). Although, due to the significant differences seen in the total number of nuclei at 25hpf and 48hpf, their corresponding cell densities recorded as statistically different. However, from a biological perspective, the unchanged nature of nuclei number, area, and resulting density in kif11 mutants paired with the similar proportions and behavioral trends of BrdU incorporation and cell death over time (Fig. 5F) implies there is a tight level of compensatory regulation between these processes to maintain overall proportions in the neural tube.
Kif11 has two temporal phases of mitotic influence
A steady increase in the number of M-phase radial glia in kif11 mutants suggests Kif11 may exhibit a temporally consistent influence over neurogenesis. To test if there were temporal changes in the requirement of Kif11 for the radial glial population, we treated wild type (AB) embryos with 0.875mM STLC or DMSO starting at different ages (5, 10, 15, 20, and 25hpf; Fig. 7A). Embryos remained in their treatment until 30hpf and were immunolabeled for anti-Gfap and anti-PH3 to assay for mitotic radial glia. No change in the number of M-phase radial glia was seen across vehicle control treated embryos (Fig. 7B–D, H, white and grey bars). In contrast, all ages treated with STLC prior to 25hpf displayed dramatic increases in the number of M-phase radial glia with respect to their controls (Fig. 7E–G, H, hatched and black bars; p<0.0001). Comparison of experimental groups indicated they were also statistically significant to each other with the exception of 10 and 15hpf, where no significant change in the number of PH3+ cells alone (p=0.88) or double labeled with Gfap in the soma (p=0.5) was seen (Fig. 7H, bracket). The difference in mitotic index between 5 and 10hpf indicated that an average of 46.7 PH3+ cells had accumulated (cells per laterally imaged neural tube between somites 9 and 13), 65.2% of which were co-expressing Gfap (7H). A similar increase in the mitotic index of on average 43.7 PH3+ cells was additionally recorded between 15 and 20hpf, 85.8% of which were co-labeled for Gfap. The number of mitotic cells continued to increase between 20 and 25hpf, yet at a reduced amount compared with earlier time periods (13.81 PH3+; 9.31 Gfap+). Given the assumption in these calculations that no PH3+ cells are progressing though mitosis, these data suggest that Kif11 is not required between 10 and 15hpf but does play a significant role in cell division of neural stem cells during an early phase (5–10hpf) and a later phase (15–25hpf).
Figure 7.
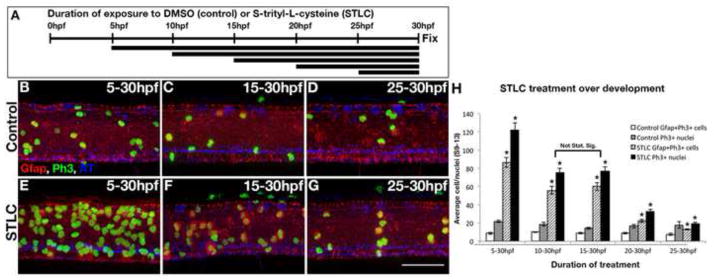
Kif11 function is most required during two distinct periods for proper cell division. (A) Schematic illustrating time of exposure to S-trityl-L-cysteine (STLC) or vehicle control (dimethyl sulfoxide) and duration of treatment between groups. (B–G) Lateral views of post-treatment embryos labeled for radial glia (Gfap), Ph3 and AT. (H) In all test groups, there were significantly more Gfap+/Ph3+ and Gfap−/Ph3+ cells in STLC treated embryos (E–G) compared with their controls (B–D) at 5–30hpf, 10–30hpf, 15–30hpf, 20–30hpf, and 25–30hpf. Error bars delineate the standard error of the mean. Asterisks indicate a statistical significance (t-test, two tailed, assuming equal variances, p<0.05).
Loss of Kif11 causes reductions in secondary interneurons and motorneurons
Arresting key progenitor populations in mitosis could have severe consequences for the proper generation of neuronal and glial cell fates during neurogenesis. We hypothesized that Kif11 deficient embryos could be used as a novel tool to determine which neuronal and glial cell types may rely specifically on Kif11-dependent radial glia as their stem cell population. To determine whether any neuronal populations in the spinal cord were affected following loss of Kif11, we labeled for interneurons, sensory and motor neurons using anti-GABA and anti-Islet-1 (Isl) antibodies (Appel and Eisen, 1998; Bernhardt et al., 1992; Myers et al., 1986; Park et al., 2004). GABA labeling within vehicle control and wild type kif11 siblings identified four classes of interneurons (dorsal longitudinal ascending (DoLA), commissural secondary ascending (CoSA), ventral longitudinal descending (VeLD), and Kolmer–Agduhr (KA)) with distinct dorsoventral domains throughout the neural tube (Fig. 8A,E (Appel and Eisen, 1998; Bernhardt et al., 1992)); however, certain interneuron populations showed unique phenotypes following loss of Kif11 function at 30hpf (Fig. 8B,F). Specifically, both kif11 mutants and STLC treated embryos displayed a disorganized CoSA interneuron domain, along with VeLD and KA interneurons that frequently exhibited aberrant, dorsally growing axonal pathfinding errors (Fig. 8B,F, arrowheads). Quantitative analysis of the total number of GABA+ interneurons showed a 22% decrease in kif11 mutants (wt avg. = 53.02±0.95, n=43; kif11−/− avg. = 41.3±1.04, n=37; p<0.0001) and a 28% decrease in STLC treated embryos (DMSO avg. = 55.81±0.86; n=43; STLC avg. = 40.02±1.21, n=41; p<0.0001; Fig. 8I). Quantification of specific GABA+ interneuronal cell types revealed a clear and specific reduction of CoSA interneurons in kif11 mutants (kif11−/− avg. = 13.41±0.68; p<0.0001) and STLC treated embryos (STLC avg. = 12.27±0.88; p<0.0001) as compared with their respective controls (wt avg. = 23.19±0.58; DMSO avg. = 25.84±0.57) (Fig. 8I). These results suggest that the significant loss of CoSA interneurons nearly accounts for the total number of GABAergic interneurons lost in kif11 mutants or STLC treated embryos. Importantly, analysis of GABA+ immunolabeling over time from 24 hpf to 36 hpf in kif11 mutants and STLC treated embryos revealed a consistent deficit in the CoSA population of interneurons relative to wild type and control embryos (Fig. 8J), while DoLA, VeLD, and KA showed no significant reduction in number over this same time span (Sup. Fig. 4). The lack of a phenotype or change in number over time in these other interneuronal populations provides confidence that the CoSA cell reductions (and other phenotypes described here) are specific and not due to any general developmental delay.
Figure 8.

Kif11 is required for motorneurons and CoSA interneurons. Wild type/vehicle control (A,C,E,G) and kif11−/−/STLC treated embryos (B,D,F,H) were labeled for anti-GABA (A,B,E,F) to label distinct DoLA, CoSA, VeLD, and KA interneurons (denoted D,C,V, and K, respectively), as well as anti-ISL (C,D,G,H) to label Rohon Beard sensory neurons and motor neurons (denoted R and M, respectively). Decreases in GABA+ cell populations as well as axon pathfinding errors were found in Kif11 deficient embryos as compared with controls (asterisks in B and F label cell body, arrowheads mark axon trajectory). (I) Quantification of neuronal populations identified decreases specifically in CoSA and MN populations within Kif11 deficient embryos (asterisks, p<0.05) as compared with controls. (J) Time course analysis of GABA+ cell types at 24hpf, 30hpf and 36hpf shows a consistent number of GABA+ cells both lost and retained over time in kif11 mutants and STLC treated embryos.
Islet-1 labeling at 30hpf in wild type sibling or DMSO control treated embryos clearly distinguishes dorsally positioned Rohon Beard sensory neurons (wt avg. = 24.9±0.69, n=32; DMSO avg. = 22.49±0.56, n=35) from the more ventrally positioned motor neuron (MN) populations (wt avg. = 67.47±6.45; DMSO avg. = 78.91±2.56) (Fig. 8C,G; Song et al., 2004). These neuronal populations were both present in 30hpf kif11 mutants and STLC treated embryos (Fig. 8D, H); however, while the number of Rohon Beard sensory neurons was largely unaffected (kif11−/− avg. = 26.1±1.16, n=25, p>0.29; STLC avg. = 20.5±0.96, n=26, p>0.06), Isl+ motorneurons were significantly decreased by 23% in kif11 mutants (kif11−/− avg. = 52.16±4.13, p<0.01) and 45% in STLC treated embryos (STLC avg. = 43.65±2.32, p<0.0001) (Fig. 8I). Because the Islet-1 antibody cannot differentiate between primary and secondary motorneurons (Appel et al., 1995; Hutchinson and Eisen, 2006), therefore we treated Tg(mnx1:GFP) and Tg(gata2:EGFP) embryos with STLC starting at 5hpf (Fig. 9). Examination of Tg(mnx1:GFP) transgenic embryos at early somitogenesis stages can show the formation of primary motorneurons over time (Flanagan-Steet et al., 2005, Seredick et al., 2012). Treatment of Tg(mnx1:GFP) transgenic embryos with STLC from 5 hpf to 18, 24 or 30 hpf did not reveal any qualitative loss in primary motorneurons (Fig. 9A–F). In contrast, gata2 drives expression of GFP in secondary motorneurons and a small population of interneurons at 48hpf (Meng et al., 1997). A severe reduction in gata2 driven expression of GFP positive cells (58% loss) was detected in the spinal cords of 5–48hpf STLC treated embryos relative to DMSO treated control embryos (DMSO avg. = 186.62±4.05, n=34; STLC avg. = 78.52±5.16, n=25; p<0.0001; Fig. 9G–I). These results suggest that secondary motorneurons, as opposed to primary motorneurons, are most affected following loss of Kif11-dependent radial glial division.
Figure 9.
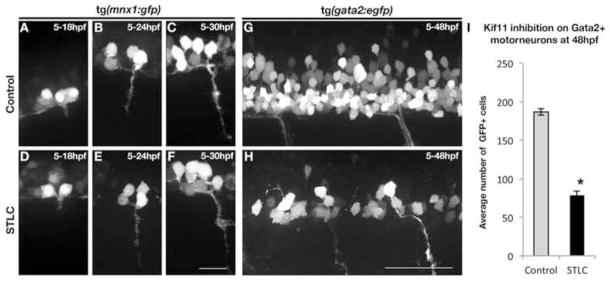
Kif11 is required for secondary motorneurons but not primary motorneurons. (A–F) Visualization of primary motorneuron development in somite 11 over time using the Tg(mnx1:GFP) transgenic line. No difference in the formation of primary motorneurons was observed in STLC treated Tg(mnx1:GFP) transgenic embryos (D–F) as compared to control DMSO treated embryos (A–C). (G,H) Lateral view of the neural tube of Tg(gata2:EGFP) transgenic embryos at 48hpf to assess secondary motorneurons in control DMSO treated (G) or STLC treated transgenic embryos from 5–48hpf (H). (I) A significant decrease in GFP+ neurons was seen in STLC treated Tg(gata2:EGFP) embryos. Error bars delineate the standard error of the mean. Asterisks indicate a statistical significance (t-test, two tailed, assuming equal variances, p<0.05).
Kif11 is necessary for the development of radial glial derived oligodendrocytes
To determine whether Kif11 mediated radial glial division is necessary for different stages of oligodendrocyte development, Tg(olig2:EGFP) embryos were treated with 0.875mM STLC at 5hpf and remained in treatment until fixation at 24hpf (initiation of olig2 expression), 36hpf (specification of OPCs), or 72hpf (oligodendrocyte maturation and axon myelination) (Kirby et al., 2006; Shin et al., 2003) (Fig. 10). At 24hpf there was no change to the pattern or number of GFP expressing cells in the ventral OPC layer between treatment groups (p>0.33; Fig. 10A,E,I); however, the number of dorsally migrating OPCs in the 5–36hpf treatment group was reduced compared with controls (DMSO avg. = 18.93±1.24, n=33; STLC avg. = 7.17±0.88, n=23; p<0.0001; Fig. 10B,F,I). This trend was consistent in the 5–72hpf treatment group (DMSO avg. = 14.93±1.32, n=23; STLC avg. = 8.54±1.15, n=19; p<0.0001; Fig. 10C,G,I) with no statistically significant difference seen in the reduction of dorsal migrating OPCs between the 5–36hpf and 5–72hpf treatment groups (Fig. 10I; p>0.98). Importantly, some GFP+ cells exhibited morphologies characteristic of cell death (Fig. 10G, arrowhead), and the presence of these irregular GFP+ cells increased with each older treatment group. We confirmed that these irregular GFP+ cells co-express anti-activated Caspase-3 in Tg(olig2:EGFP) embryos treated with STLC from 5 to 72hpf, indicating these cells are undergoing apoptosis (Fig. 10G, inset). These irregularly shaped cells were not included in the above quantification of migrating OPCs. Lastly, Tg(olig2:EGFP) embryos were also treated with STLC from 24–72hpf to discern whether changes in the Olig2+ populations were the result of a requirement for Kif11 function after olig2 expression and OPC specification (Kirby et al., 2006; Park et al., 2007; Shin et al., 2003). No difference in the number of migrating OPCs was seen following a 24 to 72hpf STLC treatment (Fig. 8D,H,I). These data suggest that the loss of early Kif11 function causes significant reductions in migrating OPCs and oligodendrocytes.
Figure 10.
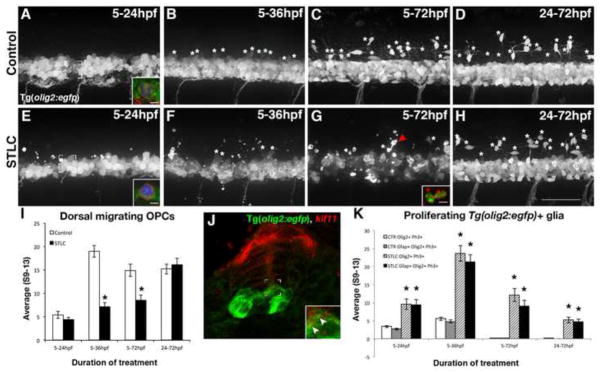
Kif11 is required for oligodendrocyte development. Lateral views of Tg(olig2:EGFP) embryos treated with either vehicle control (A–D) or STLC (E–H) at 5hpf and fixed at time points critical for oligodendrocyte development (5–24hpf, 5–36hpf, 5–72hpf, and 24–72hpf). Dorsal migrating OPCs (white asterisks) were present in control embryos older than 24hpf (B–D), which were significantly reduced in STLC treated groups (F–H, quantified in I, black asterisks p>0.05), except when treated from 24–72hpf (no statistical significance from the vehicle control, p>0.51). Irregular GFP+ fragments (G arrowhead) co-labeled with anti-Activated Caspase-3 (G, inset, red fluorescence) indicative of cell death. (J) Kif11 mRNA detection within 27hpf Tg(olig2:EGFP) embryos indicated that a small portion of olig2 cells associated with the ventricular zone colocalize with kif11 expression (J inset, arrowheads). Triple-labeled (GFP+, Ph3+, Gfap+) cells were identified in most control and treatment groups (A,E brackets; insets are maximum intensity projection of extracted Z-stack series in A,E; quantified in K), and were significantly increased in all STLC treated groups (K, asterisks). Error bars delineate the standard error of the mean. Asterisks indicate a statistical significance (t-test, two tailed, assuming equal variances, p<0.05).
To determine how Kif11 may be influencing oligodendroglial development, we performed whole mount in situ hybridization for kif11 in 30hpf Tg(olig2:EGFP) embryos. We detected a subset of GFP+ cells expressing kif11 transcript that were restricted to the luminal surface (Fig. 10J, inset), which is consistent with the location of proliferating progenitor populations (Kirby et al., 2006; Park et al., 2007; Shin et al., 2003). Assays for anti-Gfap and anti-PH3 antibody expression in STLC treated Tg(olig2:EGFP) embryos revealed that nearly all mitotic olig2:EGFP+ cells were also Gfap+ (Fig. 10A,E inset; full qualitative data not shown; K, grey and black bars). Furthermore, while no difference in the OPC migratory pattern was seen at 24hpf, there was a significant increase in the number of mitotic OPCs in this STLC treatment group (DMSO avg. = 3.41±0.27, n=27; STLC avg. = 9.55±0.26, n=18; p<0.0001; Fig. 10K). The number of M-phase OPCs increased greater than 2-fold in the 5–36hpf treatment group compared with the 5–24hpf treatment group (DMSO avg. = 5.58±0.45; STLC avg. = 23.65±0.4; p<0.0001; Fig. 10K). While the longest treatment period of 5–72hpf still showed significant increases in the number of M-phase OPCs as compared with DMSO treated controls (DMSO avg. = 0.125±0.13; STLC avg. = 12.18±0.13; p<0.0001), this increase was not statistically significant from the 5–24hpf treatment group (p>0.2). Lastly, while no defects were detected in oligodendrocyte differentiation in the 24–72hpf STLC treatment group (Fig. 7D,H,K), there was still a substantial increase in the number of M-phase OPCs (DMSO avg. = 0.09±0.05, n=40; STLC avg. = 5.22±0.01, n=37; p<0.0001; Fig. 10K). This temporal manipulation of Kif11 lends support to a radial glial origin for OPCs, and suggests that OPCs require Kif11 for proper maturation into oligodendrocytes.
DISCUSSION
In this study, we explored the role of the kinesin motor protein Kif11 in mediating radial glial cell division and the impact its loss may have on neurogenesis during the development of the zebrafish spinal cord. We present a model whereby Kif11 functions to mediate spindle pole separation in radial glial cells of the developing neural tube. Loss of Kif11 function causes mitotic arrest in radial glial cells leading to increases in cell death and subsequent reductions in cell proliferation (Fig. 11A). In addition, there are indirect lineage consequences resulting from mitotically arrested radial glia that support a specific dependence on radial glial origin by later-born secondary motorneurons, CoSA interneurons, and oligodendrocytes (Fig. 11B). Lastly, despite all the changes in cell cycle dynamics, cell numbers and cell survival, the appropriate cell density within the neural tube is maintained over the course of neurogenesis, which suggests the presence of a currently unidentified mechanism of compensatory regulation. This investigation demonstrates a clear cell biological role for Kif11 in the division of stem cell populations in vivo during neural development, and it has revealed the presence of a remarkably robust mechanism to maintain size and cell densities in the neural tube.
Figure 11.

Model of the role Kif11 plays in neural stem cell division and neurogenesis. (A) Illustration of one half of the neural tube depicting the consequences of a loss of kif11 to neural stem cell division. Neuroepithelial cells (orange) transition into radial glial cells (orange/red) that undergo division at the ventricular surface to generate progenitor cells (yellow) and/or additional radial glia (red). Upon loss of kif11, radial glia cells arrest in mitosis (lower red cell with monoaster spindle), which leads to increases in cell death (red apoptotic cell exhibiting cell blebbing). We propose that cell death is progressively initiated in cells at all stages of the cell cycle as opposed to cells just arrested in mitosis (three arrows). Moreover we propose that the consequential loss of cells by apoptosis will show reductions in the number of cells entering S-phase; however, we cannot rule out that negative feedback on neighboring parental stem cell populations signals a slow down in cell cycle entry (dashed inhibitory lines). (B) Examination of neuronal and glial cell fates in kif11−/− mutants provides an indirect method of lineage tracing from radial glial cells. Along the left side of this illustrated spinal cord shows the cell types that require Kif11 function, and therefore likely possess radial glial origins. Cell types that were patterned normally in the absence of functional Kif11 are illustrated along the right side of this spinal cord, and thus likely are not derived from radial glia.
Mathematical Modeling of Neurogenesis
We employed mathematical modeling to help identify and better understand the cellular and developmental processes potentially impacted by the loss of kif11. We constructed a mathematical model that only tracks changes in the population of radial glia at different stages of the cell cycle. The models were built to test out the most probable, biologically relevant effects of kif11 mutations, namely changes in proliferation and mitotic arrest that may then lead to cell death. However, due to a lack of existing and completely applicable quantitative measures related to neurogenesis in zebrafish, our models are currently constructed in the most simplistic form. Limitations of the current model include a coarse representation of the temporal changes in the neuroepithelial population (represented as a linear decrease from 10 hpf). Additionally, stem and progenitor cell renewal and differentiation may variably change over time, and these temporal changes to the division types and differentiation of radial glia and their progeny over time is not represented in our model. Despite these and likely other potential limitations, comparison between the output of the mathematical model in its current simplified form and the experimental data has already revealed that mitotic arrest, toxicity-induced cell death, and reduced cell cycle entry are possible additional effects of a loss of Kif11 function. Incorporating a more realistic neuroepithelial and radial glia maturation as well as testing out different feedback mechanisms for the regulation of cell proliferation and differentiation form a promising future direction.
Kif11 is required for radial glial cell progression through mitosis
Our analysis of Kif11 function in the developing zebrafish nervous system provides in vivo confirmation of past cell culture studies to dissect Eg5 function in cell division and it also reveals broader implications of the role this kinesin plays in neurogenesis. Plus-end directed, tetrameric kinesins exert forces that slide interpole microtubules toward the centrosomes, which contribute to spindle pole and chromosome separation (Sawin and Mitchison, 1990; Sawin et al., 1992; Sawin and Mitchison, 1991). Loss of Eg5 function in cultured Xenopus oocytes and human cancer cells causes monoaster spindle formation leading to mitotic arrest in these cells (Kapoor et al., 2000; Miyamoto et al., 2004; Sawin and Mitchison, 1995). We show that in fact radial glial cells display similar monoaster spindles at the ventricular zone in kif11 mutants (Fig. 3). Therefore, we propose that as a radial glial cell enters the cell cycle its nucleus migrates to the ventricular zone where the mitotic spindle collapses inward forming a monoaster microtubule morphology during M-phase. This disruption of radial glial mitotic spindles causes significant delays in exiting mitosis, which likely represents a full mitotic arrest in these cells leading to the progressive accumulation of radial glial somas at the ventricular zone over time.
The kif11 zebrafish mutant offers a unique tool to assay radial glial development throughout embryonic neurogenesis. Eg5 knock-out is lethal in mice prior to gastrulation (Castillo and Justice, 2007; Chauviere et al., 2008); in contrast, kif11 zebrafish mutants progress through the early stages of gastrulation and demonstrate distinct later phases of Kif11 dependence. We did verify that maternal contributions of kif11 are present in wild type, and, because kif11hi3112 is a recessive mutation, similar maternal contributions of Kif11 may partially support early embryonic development in kif11 mutants. However, earlier inhibition of Kif11 by STLC treatment in kif11 siblings did not alter radial glial number (Sup. Fig. 1L), indicating that any maternally expressed kif11 does not play a significant functional role in early CNS development. Proteins redundant with Kif11 mitotic function, such as the Kif15/Hklp2 motor protein in HeLa cells (Tanenbaum et al., 2009) and the Ncd motor protein in Drosophila embryos, have been documented previously (Sharp et al., 1999; Wilson et al., 2004); however, whether compensation by another molecular motor for Kif11 exists in zebrafish remains to be elucidated. Based on these and our data we propose that cell division in the early zebrafish embryo either operates in a kif11-independent manner or uses redundant motor proteins that functionally compensate for deficiencies in Kif11, whereas later cell divisions during neurogenesis are dependent on Kif11 function.
Loss of Kif11 has consequences to overall cell cycle regulation in the neural tube
Mathematical modeling predicted that delays in mitotic exit alone could not account for the number of accumulating mitotic radial glial somas in kif11 mutants. Our initial mathematical Model Variant 1 predicted that increases in the rate of cell death and decreases in the rate of cell cycle entry both paired with near mitotic arrest appropriately captured the number of M-phase radial glia in kif11 mutants (Fig. 4). While we did experimentally confirm the presence of increased apoptosis and decreased S-phase cells in kif11 mutants (Fig. 5), Model Variant 1 failed to capture S-phase and cell death data (Sup. Fig. 2). Mathematical Model Variant 2 was able to reproduce not only the mitotic radial glial data but also partially fit the collected BrdU and activated Caspase-3 data (Fig. 6). Model Variant 2 predicted that a mitotic arrest characterized by a 226x (94 hour) delay in completing mitosis with two forms of cell death (1% cell death due to mitotic arrest and 44% death due to worsening environmental conditions) together were the primary influencing factors for the number of M-phase radial glia during neurogenesis in kif11 mutants (Fig. 6B). A logical next step analysis would be to mathematically model the multiple cell death modes but paired with a slowing of cell cycle entry. We completed this analysis, and while it improved slightly the data fitting to S(t), it worsened the data fitting to M(t) and D(t) as compared with either Model Variant 1 or 2 (data not shown). This lends greater support to our conclusion that mitotic arrest paired with cell death present across all the stages of the cell cycle governs the number of M-phase radial glia in kif11 mutants.
Increased apoptosis is a logical predication of our model, as mitotic arrest due to loss of Eg5 function has been shown to lead to cell death (Kozielski et al., 2008; Marcus et al., 2005). We hypothesize that as mitosis arrested cells undergo cell death a progressively toxic environment will be generated over time, which could reduce the survivability of cells at all stages of the cell cycle. We propose that the reductions in the number of cells positive for BrdU incorporation are represented by the loss of cells due to death. Radial glial mitotic arrest and subsequent cell death could reduce the number of proliferating daughter cells (radial glial self-renewal or progenitor cell), which would show less BrdU uptake over time. This speculation is supported by the similar proportion and inverse relationship of cells undergoing death and simultaneously failing to enter S-phase over time (Fig. 5E,F).
As supported by our mathematical Model Variant 2, we favor the interpretation that decreases in the number of cells in S-phase in kif11 mutants is the result of less cells due to increased cell death; however, we cannot rule out the possibility that cell cycle entry is being directly altered by changes in cell signaling. Examination of Notch signaling in the outer subventricular zone of the mouse cortex provides some support for a mechanism of daughter-derived mitogenic signaling. Continued proliferation of outer radial glial (oRG) cells through activation of Notch depends on Delta mediated signaling from their daughter cells called intermediate progenitors (Kawaguchi et al., 2008a; Kawaguchi et al., 2008b; Shitamukai et al., 2011; Yoon et al., 2008; Zhang et al., 2007). Notch signaling has been demonstrated in the zebrafish neural tube to regulate neuronal and glial cell fate decisions, and asymmetric expression of the Notch signaling regulator Numb is seen in the developing neural progenitor during radial glial division in the mouse cortex (Alexandre et al., 2010; Kim et al., 2008). Therefore it is possible that changes in the number of radial glial derived progenitor cells could alter important daughter-derived signals that serve to maintain the rate of division in the parent stem cell population in the zebrafish spinal cord.
Maintaining size and cell density in the neural tube
If increases in cell death and reductions in proliferation were the only parameters involved, then there should be a reduction in the total number of cells in the neural tube. Interestingly, we showed that the total number of cells, the cross-sectional area of, and resulting cell density within the neural tube were largely unchanged in kif11 mutants over time as compared with wild type embryos (Fig. 1E; Sup. Fig. 3). These conflicting results suggest that some compensatory mechanism is actively maintaining neural tube size and cell density. One possible contribution for compensation is the potential for an M-phase cell to actually exit mitosis and undergo cytokinesis. The number of cells in M-phase in kif11 mutants begins to plateau at 36hpf and then drops after 48hpf (Fig. 2I). While constant cell death represents a major factor in interpreting cell number in the neural tube of kif11 mutants, it is possible that some small portion of neural stem cells delayed in M-phase are still successfully completing mitosis and contribute to neurogenesis. However, it is unlikely that a small contribution of successful mitoses can completely account for the maintenance of correct cell densities particularly over the entire time course.
An active mechanism of feedback regulation may exist to maintain the overall size and proportion of cells that make up the neural tube. Mitotic delays and cell death might logically employ a strategy to increase proliferation in kif11 mutants, perhaps through a similar mechanism of Notch mediated signaling as described above. However, we documented a decrease in the number of cells in S-phase (Fig. 5C,D). Alternatively, compensation might be achieved in kif11 mutants by extending the period of time in which the parental neuroepithelial cell type is maintained for sustained self-renewing symmetrical divisions. We did mathematically model a specific increase in the proliferation of the neuroepithelial population (Rin) to theoretically test whether specific alterations to the population of early stem cells might better fit the data; while increases to Rin did yield a much closer fit to the magnitude of BrdU positive cell counts, it failed to fit the raw data for mitotic radial glia and cell death (data not shown). An additional, and not necessarily mutually exclusive, mechanism of feedback regulation might involve alterations to cell movement in response to biomechanical changes at the cellular and tissue level. For instance, as cells die in kif11 mutants, it may change the cell-to-cell forces inside the neural tube in a manner that could reduce the migration of progenitor cells fated to leave the neural tube, such as the dorsally migrating neural crest cells or ventrally exiting perineurula glia (Kucenas et al., 2008; Kuo and Erickson, 2010; Raible et al., 1992). Quantification and live cell imaging of such migratory cell types in and out of the neural tube would be required to test this hypothesized model of altered cell movement due to changes in the physical forces of the developing neural tube.
The timing of Kif11 function during neurogenesis
Although we present evidence that kif11 is necessary for the mitotic progression of radial glia throughout neurogenesis, it was unclear whether the requirement of Kif11 function varies over this period of development. Using STLC to pharmacologically inhibit Kif11 during different periods of development, we discovered a critical requirement for Kif11 in M-phase radial glial cells between 5–10hpf and again between 15–20hpf (Fig. 7). As defined in previous studies, gfap expressing radial glia are not present prior to 10hpf in zebrafish (Bernardos and Raymond, 2006; Marcus and Easter, 1995), yet inhibition of Kif11 from 5–30hpf with STLC does cause significantly more mitotic arrest in radial glial cells as opposed to the 15–30 hpf treatment. These results suggest that at least some aspect of neuroepithelial cell development requires Kif11 for proper cell division. Therefore it is plausible that our time course of Kif11 inhibition captured a pause in observable proliferation that may occur during the natural transition from neuroepithelial cell to radial glial cell development, as opposed to specific temporal changes in Kif11 dependence. Thus, we interpret our temporal analysis of Kif11 function to suggest that Kif11 is consistently required over the course of neural development. To support this idea, a closer examination of cell type specific proliferation rates during the transition of neuroepithelial to radial glial cell development would be required.
Loss of kif11 provides indirect analysis of the radial glial lineage
Because loss of kif11 results in arrested radial glial cells that ultimately undergo apoptosis, we hypothesized that we could use these mutants and STLC treated embryos as a system to indirectly determine the neuronal and glial cell types that depend on radial glia for their generation. Our math model predicts that radial glial cell division is normally dominated by differentiation, such that only ~9% of daughter cells remain as proliferating radial glia while the rest become committed progenitor cells (Table 1, 0.182 cells/2 possible outcomes). If a cell type is derived from radial glia, then it should be reduced in number following Kif11 loss. We found that three specific cell populations were reduced in number: secondary motorneurons, commissural secondary ascending (CoSA) interneurons, and oligodendrocyte lineage cells (Figs. 8, 9, 10). Therefore, we propose that at least a portion of these three cell populations originate from radial glial cells.
In zebrafish, the developing neural tube is comprised of both primary neurons and later developing secondary neurons that are temporally regulated and have specific functions (Lewis and Eisen, 2003). Primary neurons are large, born early (9–10hpf), and consist of Rohon Beard sensory neurons, interneurons, and primary motorneurons, whereas secondary neurons are smaller, born later (13–14hpf), and are predominantly comprised of interneurons and secondary motorneurons (Beattie et al., 1997; Bernhardt et al., 1990; Kimmel et al., 1994; Lewis and Eisen, 2003; Myers et al., 1986). Labeling for Gfap has shown that radial glia are not specified in the neural tube until 10–12hpf (Bernardos and Raymond, 2006; Marcus and Easter, 1995), suggesting that radial glia in the zebrafish may not significantly contribute to primary neuron populations. Loss of Kif11 supports this later cell fate dependence on radial glia, as early-born neuronal populations assayed at 30hpf (Rohon Beard cells, DoLA, VeLD and KA interneurons, and Tg(mnx:GFP)+ motorneurons) were unaffected, whereas the secondary neuronal cell types assayed (CoSA, and Tg(gata2:GFP)+ secondary motorneurons) and later-born glial populations (Tg(olig2:EGFP)+ dorsal migrating OPCs and oligodendrocytes) were all significantly reduced (Figs. 8,9,10). Importantly, the significant loss of CoSA interneurons nearly matches the total number of GABA+ interneurons lost in kif11 mutants or STLC treated embryos, suggesting that CoSA interneurons are the only GABA+ interneuron cell type that may be derived from radial glial precursors at these stages. This interpretation is strengthened not only by the consistent proportion of CoSA interneurons being lost over time, but also by the consistent number of DoLA, VeLD and KA interneurons completely unaffected over time (Fig. 8; Sup. Fig. 4). Interestingly, while VeLD and KA interneurons were present in correct numbers, we did observe some axonal pathfinding errors by these neurons (Fig. 8B,F). Increasing evidence is lending support to a distinct role for kif11/eg5 in axonal growth (Myers and Baas, 2007; Nader, et. al. 2008; Barresi, 2010; Nader et. al., 2012). However, whether the disruption in proper KA and VeLD axonal anatomy is indirectly related to an unhealthy guidance environment or due more specifically to a required role of Kif11 in cytoskeletal dynamics during axonal growth remains to be determined.
OPCs represent an important progenitor cell population for both motorneurons and oligodendrocytes. While we show that kif11 is co-expressed in a subset of OPCs present at the lumen, we suggest they were originally derived from radial glia. In our assay, nearly all Tg(olig2:EGFP) positive cells undergoing mitosis were also simultaneously expressing Gfap (Fig. 10); therefore, we hypothesize that the reduction in dorsal migrating OPCs was most likely due to arrested radial glia that serve to generate OPCs destined for oligodendroglia. These results further support previous studies that suggest a significant portion of oligodendroglial cells have a radial glial origin (Kim et al., 2008; Kirby et al., 2006; Kriegstein and Alvarez-Buylla, 2009; Park et al., 2004). Furthermore, STLC drug treatments initiated at 24hpf and sustained until 72hpf showed that Kif11-dependent progenitors dividing later in development did not significantly contribute to dorsal migrating OPCs (Fig. 10D,H,I,K). This suggests that the portion of the Tg(olig2:EGFP) positive cells reduced following our earlier STLC treatment assays originated from radial glial-derived OPCs prior to 24hpf. Based on the cell type markers we have examined, it is possible that proper radial glial development is ultimately required for all later born, secondary neuronal and glial lineages; a speculation that can be tested as additional cell type specific makers become available.
Interestingly, approximately 30–60% of proliferating cells in the neural tube of both wild type and Kif11-deficient embryos did not express Gfap (Fig. 2G). These non-Gfap-expressing proliferative cells likely represent neuroepithelial or progenitor cells caught in our quantitative analyses. The persistence of these mitotic, non-Gfap-expressing cells following later stage inhibition of Kif11 suggests that potentially some progenitor or transient amplifying cells still require kif11 for proper progression through mitosis. More detailed marker and lineage analyses are needed to verify and characterize the origin of these kif11 dependent, Gfap-negative cells.
Our findings identify Kif11/Eg5 as essential for neural stem cells to progress through mitosis during neural tube development in zebrafish. Characterizing mitotic arrest of radial glial cells has revealed critical consequences to cell survival, cell cycle rates, the maintenance of cell density in the neural tube, and the development of secondary neuronal and glial lineages. In addition, the approach of this study demonstrates the predictive powers mathematical modeling can bare on our understanding of neural development. Further refinement of this model with greater quantitative data about the parameters influencing neural stem cell development will serve to strengthen the accuracy of its predictions and potentially broaden its applicability to other stem cell niches. Lastly, our characterization of the neuronal and glial cell types affected by the loss of Kif11 and subsequent mitotic arrest of radial glia has contributed to our understanding of radial glial derived lineages. It will be important to further this lineage analysis with more direct approaches as well as directly testing the requirement of radial glial cells to the development of certain cell types. Such continued studies of the relatively simple zebrafish neural tube can move the field closer to establishing a comprehensive model of vertebrate neurogenesis.
Supplementary Material
Highlights.
Kif11 is required for radial glial cell division during neural tube development.
We built a mathematical model of neural stem cell division.
Loss of Kif11 causes mitotic arrest in radial glial cells leading to apoptosis.
Indirect lineage analyses suggest radial glia generate later born neurons and glia.
Acknowledgments
We thank Nancy Hopkins for the kif11hi3112a line, Pamela Raymond for the Tg(gfap:EGFP) line, Bruce Appel for the Tg(olig2:EGFP) line, and Shuo Lin and Richard Dorsky for the Tg(gata2:EGFP) line. Special thanks to Samuel Nona for providing us with the anti-Gfap antibody. We thank all the Biomath students of the 2012 Frontiers course, in particular Julie Richardson, Ria Deshpande, and Erin Fernandes whose modeling was influential in our thinking about this work. We thank Rebecca Bernardos for protocol support during some of this work, and all the members of the Barresi lab for valuable feedback on this research. We truly appreciate all the efforts of the Smith College Animal Care staff for fish care and husbandry. This work was funded by Smith College, NIH HD060023, NSF 1054168, and NSF 0615594 to MJB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandre P, Reugels AM, Barker D, Blanc E, Clarke JD. Neurons derive from the more apical daughter in asymmetric divisions in the zebrafish neural tube. Nat Neurosci. 2010;13:673–679. doi: 10.1038/nn.2547. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel B, Chitnis A. Neurogenesis and specification of neuronal identity. Results Probl Cell Differ. 2002;40:237–251. doi: 10.1007/978-3-540-46041-1_12. [DOI] [PubMed] [Google Scholar]
- Appel B, Eisen JS. Regulation of neuronal specification in the zebrafish spinal cord by Delta function. Development. 1998;125:371–380. doi: 10.1242/dev.125.3.371. [DOI] [PubMed] [Google Scholar]
- Appel B, Korzh V, Glasgow E, Thor S, Edlund T, Dawid IB, Eisen JS. Motoneuron fate specification revealed by patterned LIM homeobox gene expression in embryonic zebrafish. Development. 1995;121:4117–4125. doi: 10.1242/dev.121.12.4117. [DOI] [PubMed] [Google Scholar]
- Barresi MJ, Burton S, Dipietrantonio K, Amsterdam A, Hopkins N, Karlstrom RO. Essential genes for astroglial development and axon pathfinding during zebrafish embryogenesis. Developmental dynamics : an official publication of the American Association of Anatomists. 2010;239:2603–2618. doi: 10.1002/dvdy.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie CE, Hatta K, Halpern ME, Liu H, Eisen JS, Kimmel CB. Temporal separation in the specification of primary and secondary motoneurons in zebrafish. Dev Biol (N Y 1985) 1997;187:171–182. doi: 10.1006/dbio.1997.8604. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Raymond PA. GFAP transgenic zebrafish. Gene Expr Patterns. 2006;6:1007–1013. doi: 10.1016/j.modgep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Bernhardt RR, Chitnis AB, Lindamer L, Kuwada JY. Identification of spinal neurons in the embryonic and larval zebrafish. J Comp Neurol. 1990;302:603–616. doi: 10.1002/cne.903020315. [DOI] [PubMed] [Google Scholar]
- Bernhardt RR, Patel CK, Wilson SW, Kuwada JY. Axonal trajectories and distribution of GABAergic spinal neurons in wildtype and mutant zebrafish lacking floor plate cells. J Comp Neurol. 1992;326:263–272. doi: 10.1002/cne.903260208. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. Specificity of the glial fibrillary acidic protein for astroglia. J Histochem Cytochem. 1977;25:466–469. doi: 10.1177/25.6.69656. [DOI] [PubMed] [Google Scholar]
- Castillo A, Justice MJ. The kinesin related motor protein, Eg5, is essential for maintenance of pre-implantation embryogenesis. Biochemical and biophysical research communications. 2007;357:694–699. doi: 10.1016/j.bbrc.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauviere M, Kress C, Kress M. Disruption of the mitotic kinesin Eg5 gene (Knsl1) results in early embryonic lethality. Biochemical and biophysical research communications. 2008;372:513–519. doi: 10.1016/j.bbrc.2008.04.177. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Jenny A, Lee D, Mlodzik M, Schier AF. Planar cell polarity signalling couples cell division and morphogenesis during neurulation. Nature. 2006;439:220–224. doi: 10.1038/nature04375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JC, Gatial JE, 3rd, Kapoor TM, Gilbert SP. Monastrol inhibition of the mitotic kinesin Eg5. The Journal of biological chemistry. 2005;280:12658–12667. doi: 10.1074/jbc.M413140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–793. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- DeBonis S, Skoufias DA, Lebeau L, Lopez R, Robin G, Margolis RL, Wade RH, Kozielski F. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol Cancer Ther. 2004;3:1079–1090. [PubMed] [Google Scholar]
- Dekens MP, Pelegri FJ, Maischein HM, Nusslein-Volhard C. The maternal-effect gene futile cycle is essential for pronuclear congression and mitotic spindle assembly in the zebrafish zygote. Development. 2003;130:3907–3916. doi: 10.1242/dev.00606. [DOI] [PubMed] [Google Scholar]
- Demir O, Singh S, Klimaschewski L, Kurnaz IA. From birth till death: neurogenesis, cell cycle, and neurodegeneration. Anatomical record. 2009;292:1953–1961. doi: 10.1002/ar.20980. [DOI] [PubMed] [Google Scholar]
- Devoto SH, Melancon E, Eisen JS, Westerfield M. Identification of separate slow and fast muscle precursor cells in vivo, prior to somite formation. Development. 1996;122:3371–3380. doi: 10.1242/dev.122.11.3371. [DOI] [PubMed] [Google Scholar]
- Ferhat L, Cook C, Chauviere M, Harper M, Kress M, Lyons GE, Baas PW. Expression of the mitotic motor protein Eg5 in postmitotic neurons: implications for neuronal development. J Neurosci. 1998;18:7822–7835. doi: 10.1523/JNEUROSCI.18-19-07822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner M, Sunder-Plassmann N, Seiler J, Utz M, Vernos I, Surrey T, Giannis A. Development and biological evaluation of potent and specific inhibitors of mitotic Kinesin Eg5. Chembiochem. 2005;6:1173–1177. doi: 10.1002/cbic.200500005. [DOI] [PubMed] [Google Scholar]
- Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Grandel H, Brand M. Comparative aspects of adult neural stem cell activity in vertebrates. Development genes and evolution. 2013;223:131–147. doi: 10.1007/s00427-012-0425-5. [DOI] [PubMed] [Google Scholar]
- Gruber J, Manninga H, Tuschl T, Osborn M, Weber K. Specific RNAi mediated gene knockdown in zebrafish cell lines. RNA Biol. 2005;2:101–105. doi: 10.4161/rna.2.3.2060. [DOI] [PubMed] [Google Scholar]
- Hollyday M. Neurogenesis in the vertebrate neural tube. Int J Dev Neurosci. 2001;19:161–173. doi: 10.1016/s0736-5748(00)00093-9. [DOI] [PubMed] [Google Scholar]
- Huang P, Xiong F, Megason SG, Schier AF. Attenuation of Notch and Hedgehog signaling is required for fate specification in the spinal cord. PLoS genetics. 2012;8:e1002762. doi: 10.1371/journal.pgen.1002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson SA, Eisen JS. Islet1 and Islet2 have equivalent abilities to promote motoneuron formation and to specify motoneuron subtype identity. Development. 2006;133:2137–2147. doi: 10.1242/dev.02355. [DOI] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez-Buylla A. Cells in the astroglial lineage are neural stem cells. Cell Tissue Res. 2008;331:179–191. doi: 10.1007/s00441-007-0461-z. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Nakamura Y, Takeda M, Nishimura T, Inagaki N. Glial fibrillary acidic protein: dynamic property and regulation by phosphorylation. Brain Pathol. 1994;4:239–243. doi: 10.1111/j.1750-3639.1994.tb00839.x. [DOI] [PubMed] [Google Scholar]
- Jowett T. Tissue in situ hybridization: methods in animal development. John Wiley & Sons, Inc; New York: 1997. [Google Scholar]
- Kapitein LC, Peterman EJ, Kwok BH, Kim JH, Kapoor TM, Schmidt CF. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005;435:114–118. doi: 10.1038/nature03503. [DOI] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. The Journal of cell biology. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi A, Ikawa T, Kasukawa T, Ueda HR, Kurimoto K, Saitou M, Matsuzaki F. Single-cell gene profiling defines differential progenitor subclasses in mammalian neurogenesis. Development. 2008a;135:3113–3124. doi: 10.1242/dev.022616. [DOI] [PubMed] [Google Scholar]
- Kawaguchi D, Yoshimatsu T, Hozumi K, Gotoh Y. Selection of differentiating cells by different levels of delta-like 1 among neural precursor cells in the developing mouse telencephalon. Development. 2008b;135:3849–3858. doi: 10.1242/dev.024570. [DOI] [PubMed] [Google Scholar]
- Kim H, Shin J, Kim S, Poling J, Park HC, Appel B. Notch-regulated oligodendrocyte specification from radial glia in the spinal cord of zebrafish embryos. Dev Dyn. 2008;237:2081–2089. doi: 10.1002/dvdy.21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HT, So JH, Jung SH, Ahn DG, Koh W, Kim NS, Kim SH, Lee S, Kim CH. Cug2 is essential for normal mitotic control and CNS development in zebrafish. BMC developmental biology. 2011;11:49. doi: 10.1186/1471-213X-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Warga RM, Kane DA. Cell cycles and clonal strings during formation of the zebrafish central nervous system. Development. 1994;120:265–276. doi: 10.1242/dev.120.2.265. [DOI] [PubMed] [Google Scholar]
- Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, Kelsh RN, Appel B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat Neurosci. 2006;9:1506–1511. doi: 10.1038/nn1803. [DOI] [PubMed] [Google Scholar]
- Kozielski F, Skoufias DA, Indorato RL, Saoudi Y, Jungblut PR, Hustoft HK, Strozynski M, Thiede B. Proteome analysis of apoptosis signaling by S-trityl-L-cysteine, a potent reversible inhibitor of human mitotic kinesin Eg5. Proteomics. 2008;8:289–300. doi: 10.1002/pmic.200700534. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzysiak TC, Grabe M, Gilbert SP. Getting in sync with dimeric Eg5. Initiation and regulation of the processive run. The Journal of biological chemistry. 2008;283:2078–2087. doi: 10.1074/jbc.M708354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucenas S, Takada N, Park HC, Woodruff E, Broadie K, Appel B. CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat Neurosci. 2008;11:143–151. doi: 10.1038/nn2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo BR, Erickson CA. Regional differences in neural crest morphogenesis. Cell Adh Migr. 2010;4:567–585. doi: 10.4161/cam.4.4.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L, Klopper AV, Grill SW, Harris WA, Norden C. Apical migration of nuclei during G2 is a prerequisite for all nuclear motion in zebrafish neuroepithelia. Development. 2011;138:5003–5013. doi: 10.1242/dev.071522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KE, Eisen JS. From cells to circuits: development of the zebrafish spinal cord. Prog Neurobiol. 2003;69:419–449. doi: 10.1016/s0301-0082(03)00052-2. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127:5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- Marcus AI, Peters U, Thomas SL, Garrett S, Zelnak A, Kapoor TM, Giannakakou P. Mitotic kinesin inhibitors induce mitotic arrest and cell death in Taxol-resistant and -sensitive cancer cells. The Journal of biological chemistry. 2005;280:11569–11577. doi: 10.1074/jbc.M413471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus RC, Easter SS., Jr Expression of glial fibrillary acidic protein and its relation to tract formation in embryonic zebrafish (Danio rerio) J Comp Neurol. 1995;359:365–381. doi: 10.1002/cne.903590302. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Meng A, Tang H, Ong BA, Farrell MJ, Lin S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc Natl Acad Sci U S A. 1997;94:6267–6272. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93:421–443. doi: 10.1016/j.pneurobio.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Miyamoto DT, Perlman ZE, Burbank KS, Groen AC, Mitchison TJ. The kinesin Eg5 drives poleward microtubule flux in Xenopus laevis egg extract spindles. J Cell Biol. 2004;167:813–818. doi: 10.1083/jcb.200407126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C, Gross D, Sarli V, Gartner M, Giannis A, Bernhardt G, Buschauer A. Inhibitors of kinesin Eg5: antiproliferative activity of monastrol analogues against human glioblastoma cells. Cancer Chemother Pharmacol. 2007;59:157–164. doi: 10.1007/s00280-006-0254-1. [DOI] [PubMed] [Google Scholar]
- Myers PZ, Eisen JS, Westerfield M. Development and axonal outgrowth of identified motoneurons in the zebrafish. J Neurosci. 1986;6:2278–2289. doi: 10.1523/JNEUROSCI.06-08-02278.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci. 2002;22:3161–3173. doi: 10.1523/JNEUROSCI.22-08-03161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HC, Mehta A, Richardson JS, Appel B. olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev Biol. 2002;248:356–368. doi: 10.1006/dbio.2002.0738. [DOI] [PubMed] [Google Scholar]
- Park HC, Shin J, Appel B. Spatial and temporal regulation of ventral spinal cord precursor specification by Hedgehog signaling. Development. 2004;131:5959–5969. doi: 10.1242/dev.01456. [DOI] [PubMed] [Google Scholar]
- Park HC, Shin J, Roberts RK, Appel B. An olig2 reporter gene marks oligodendrocyte precursors in the postembryonic spinal cord of zebrafish. Dev Dyn. 2007;236:3402–3407. doi: 10.1002/dvdy.21365. [DOI] [PubMed] [Google Scholar]
- Peters NT, Kropf DL. Kinesin-5 motors are required for organization of spindle microtubules in Silvetia compressa zygotes. BMC Plant Biol. 2006;6:19. doi: 10.1186/1471-2229-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SM, Freeman JL. RNA isolation from embryonic zebrafish and cDNA synthesis for gene expression analysis. Journal of visualized experiments : JoVE. 2009 doi: 10.3791/1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff KL, Straub CT, Chiang K, Bear DM, Zhou Y, Zon LI. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Molecular and cellular biology. 2007;27:5887–5897. doi: 10.1128/MCB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Gotz M. Radial glial cell heterogeneity--the source of diverse progeny in the CNS. Prog Neurobiol. 2007;83:2–23. doi: 10.1016/j.pneurobio.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Raible DW, Wood A, Hodsdon W, Henion PD, Weston JA, Eisen JS. Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev Dyn. 1992;195:29–42. doi: 10.1002/aja.1001950104. [DOI] [PubMed] [Google Scholar]
- Sarli V, Giannis A. Inhibitors of mitotic kinesins: next-generation antimitotics. Chem Med Chem. 2006;1:293–298. doi: 10.1002/cmdc.200500045. [DOI] [PubMed] [Google Scholar]
- Sawin K, Mitchison T. Cell biology. Motoring in the spindle. Nature. 1990;345:22–23. doi: 10.1038/345022a0. [DOI] [PubMed] [Google Scholar]
- Sawin KE, LeGuellec K, Philippe M, Mitchison TJ. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature. 1992;359:540–543. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ. Poleward microtubule flux mitotic spindles assembled in vitro. J Cell Biol. 1991;112:941–954. doi: 10.1083/jcb.112.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci U S A. 1995;92:4289–4293. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DJ, Yu KR, Sisson JC, Sullivan W, Scholey JM. Antagonistic microtubule-sliding motors position mitotic centrosomes in Drosophila early embryos. Nat Cell Biol. 1999;1:51–54. doi: 10.1038/9025. [DOI] [PubMed] [Google Scholar]
- Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J, Finley KR, Pfaff KL, Hersey C, Zhou Y, Barut B, Freedman M, Lee C, Spitsbergen J, Neuberg D, Weber G, Golub TR, Glickman JN, Kutok JL, Aster JC, Zon LI. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13194–13199. doi: 10.1073/pnas.0506583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard JL, Stern HM, Pfaff KL, Amatruda JF. Analysis of the cell cycle in zebrafish embryos. Methods in cell biology. 2004;76:109–125. doi: 10.1016/s0091-679x(04)76007-0. [DOI] [PubMed] [Google Scholar]
- Shin J, Park HC, Topczewska JM, Mawdsley DJ, Appel B. Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 2003;25:7–14. doi: 10.1023/B:MICS.0000006847.09037.3a. [DOI] [PubMed] [Google Scholar]
- Shin J, Poling J, Park HC, Appel B. Notch signaling regulates neural precursor allocation and binary neuronal fate decisions in zebrafish. Development. 2007;134:1911–1920. doi: 10.1242/dev.001602. [DOI] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:3683–3695. doi: 10.1523/JNEUROSCI.4773-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer L, Rao M. Neural stem cells and regulation of cell number. Progress in neurobiology. 2002;66:1–18. doi: 10.1016/s0301-0082(01)00022-3. [DOI] [PubMed] [Google Scholar]
- Song MH, Brown NL, Kuwada JY. The cfy mutation disrupts cell divisions in a stage-dependent manner in zebrafish embryos. Dev Biol (N Y 1985) 2004;276:194–206. doi: 10.1016/j.ydbio.2004.08.041. [DOI] [PubMed] [Google Scholar]
- Suter DM, Tirefort D, Julien S, Krause KH. A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells. 2009;27:49–58. doi: 10.1634/stemcells.2008-0319. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Macurek L, Janssen A, Geers EF, Alvarez-Fernandez M, Medema RH. Kif15 cooperates with eg5 to promote bipolar spindle assembly. Current biology : CB. 2009;19:1703–1711. doi: 10.1016/j.cub.2009.08.027. [DOI] [PubMed] [Google Scholar]
- Tawk M, Araya C, Lyons DA, Reugels AM, Girdler GC, Bayley PR, Hyde DR, Tada M, Clarke JD. A mirror-symmetric cell division that orchestrates neuroepithelial morphogenesis. Nature. 2007;446:797–800. doi: 10.1038/nature05722. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Uzbekov R, Prigent C, Arlot-Bonnemains Y. Cell cycle analysis and synchronization of the Xenopus laevis XL2 cell line: study of the kinesin related protein XlEg5. Microsc Res Tech. 1999;45:31–42. doi: 10.1002/(SICI)1097-0029(19990401)45:1<31::AID-JEMT3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Valensin S, Ghiron C, Lamanna C, Kremer A, Rossi M, Ferruzzi P, Nievo M, Bakker A. KIF11 inhibition for glioblastoma treatment: reason to hope or a struggle with the brain? BMC Cancer. 2009;9:196. doi: 10.1186/1471-2407-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine MT, Fordyce PM, Block SM. Eg5 steps it up! Cell Div. 2006a;1:31. doi: 10.1186/1747-1028-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine MT, Fordyce PM, Krzysiak TC, Gilbert SP, Block SM. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nat Cell Biol. 2006b;8:470–476. doi: 10.1038/ncb1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters C. Video views and reviews: neurulation and the fashioning of the vertebrate central nervous system. CBE Life Sci Educ. 2006;5:99–103. doi: 10.1187/cbe.06-02-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book, A guide for the laboratory use of zebrafish (Danio rerio) 5. University of Oregon Press; Eugene: 2007. [Google Scholar]
- Wilson PG, Simmons R, Saighal S. Novel nuclear defects in KLP61F-deficient mutants in Drosophila are partially suppressed by loss of Ncd function. Journal of cell science. 2004;117:4921–4933. doi: 10.1242/jcs.01334. [DOI] [PubMed] [Google Scholar]
- Yasui Y, Amano M, Nagata K, Inagaki N, Nakamura H, Saya H, Kaibuchi K, Inagaki M. Roles of Rho-associated kinase in cytokinesis; mutations in Rho-associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol. 1998;143:1249–1258. doi: 10.1083/jcb.143.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Koo BK, Im SK, Jeong HW, Ghim J, Kwon MC, Moon JS, Miyata T, Kong YY. Mind bomb 1-expressing intermediate progenitors generate notch signaling to maintain radial glial cells. Neuron. 2008;58:519–531. doi: 10.1016/j.neuron.2008.03.018. [DOI] [PubMed] [Google Scholar]
- Zhang C, Li Q, Jiang YJ. Zebrafish Mib and Mib2 are mutual E3 ubiquitin ligases with common and specific delta substrates. J Mol Biol. 2007;366:1115–1128. doi: 10.1016/j.jmb.2006.11.096. [DOI] [PubMed] [Google Scholar]
- Zhang SC. Defining glial cells during CNS development. Nat Rev Neurosci. 2001;2:840–843. doi: 10.1038/35097593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.