

Abstract
Carbon nanotubes are a natural choice as gas sensor components given their high surface to volume ratio, electronic properties, and capability to mediate chemical reactions. However, a realistic assessment of the interaction of the tube wall and the adsorption processes during gas phase reactions has always been elusive. Making use of ultraclean single-walled carbon nanotubes, we have followed the adsorption kinetics of NO2 and found a physisorption mechanism. Additionally, the adsorption reaction directly depends on the metallic character of the samples. Franck–Condon satellites, hitherto undetected in nanotube–NOx systems, were resolved in the N 1s X-ray absorption signal, revealing a weak chemisorption, which is intrinsically related to NO dimer molecules. This has allowed us to identify that an additional signal observed in the higher binding energy region of the core level C 1s photoemission signal is due to the C=O species of ketene groups formed as reaction byproducts . This has been supported by density functional theory calculations. These results pave the way toward the optimization of nanotube-based sensors with tailored sensitivity and selectivity to different species at room temperature.
Keywords: carbon nanotube sensors, X-ray absorption, physisorption, chemisorption, photoemission
Among the extraordinary physical and chemical properties that carbon nanotubes (CNTs) have, their large surface area to volume ratio is particularly appealing for applications where the adsorption of other molecules plays an important role.1−8 Several studies are available, which use both multiwalled (MW) and single-walled (SW) CNTs aiming at understanding the mechanisms that affect their electronic properties upon exposure to different molecular species.9−14 The literature in this field is very rich, but to achieve a gas sensing device that overcomes the problems related to accuracy and long-term stability without compromising sensitivity and selectivity15−19 requires further investigation. Crucial matters such as gas uptake, recovery time and reversibility depend on the physicochemical characteristics of the nanotube-molecule system. Also, the behavior of MWCNTs in a sensor can exhibit great differences from the same device employing SWCNTs, and the performance is also different if the tubes are used in the form of films, as individual nanocomponents or as bulk product.20 Fundamental questions still need to be addressed, such as the nanotube mechanical deformation, the presence of synthesis-related defects, residuals from chemical treatments and functionalization. All of these can have a significant influence on the sensor’s performance.
One of the ways to use SWCNTs as sensing objects is by analyzing the changes in their electric response while interacting with gas molecules. In this context, most studies deal with the use of nanotube-material that is either highly defective or nonpurified, which obliges us to rethink the established sensing principles, or at least how to describe the sensing mechanism addressed to a specific nanotube-based sensor. It has often been proposed that the intrinsic defective sites and those created during the SWCNTs processing are responsible for the changes in their electrical response.21−23 Defects in SWCNTs are believed to promote the formation of highly active adsorption sites, which can also serve for the condensation of chemical species at high vapor concentrations. On the other hand, residual contaminants in purified bundles have also been associated with changes in the electrical response of SWCNTs, in particular regarding the exposure to oxygen.17,24 Therefore, despite the many studies related to the adsorption of oxygen on bulk and individual tube devices, there remain open questions regarding the real role of the nanotubes in sensing experiments vs the function of impurities. Conclusive answers can only be given if ultraclean SWCNT samples are used, because only in this manner is it possible to find the basic phenomena responsible for the changes of their electronic properties when used as sensors.25,26
Taking advantage of the great improvements in the purification and sorting of nanotubes in the past few years, we have made use of ultraclean and metallicity sorted nanotube material in order to understand the sensing mechanisms of NO and NO2, which are two examples of poisonous gases that offer an interesting comparative advantage. Related studies have reported that the electrical conductance of CNTs increases dramatically upon NO2 gas exposure.3,27 Additionally, NO2 has experimentally been identified as a charge acceptor, which consistently agrees with most theoretical predictions. While temperature programmed desorption experiments have estimated an adsorption energy of about 0.38 eV (37 kJ/mol) for NO2 on highly oriented pyrolitic graphite (HOPG),28 results in the literature based on density functional theory (DFT) calculations are quite scattered. For example, most calculated binding energies of NO2 are within the range 0.40–0.80 eV (39–77 kJ/mol),5,29−34 while others have predicted weaker or almost no binding.27,35−37 This disagreement between computational studies is partially explained by the different calculation methods employed and the nanotube adsorption sites considered. The mixed chirality of the samples, the inherent difficulties in describing long-range van der Waals interactions and the multireference character of NO2 (requiring multiple Slater determinants for a proper description) contribute to the mismatch between theory and experiment. However, shifts of core-level eigenvalues are expected to be rather insensitive to van der Waals interactions and the multireference character of the molecules.
We have studied the reaction pathway of NO2 in the presence of SWCNTs as a function of their metallic character. This has been done by employing X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS).38,39 Opposite to the reported chemisorption on metallicity mixed samples, we have encountered a predominant physisorption process when using metallicity sorted material. However, a slight chemisorption can still be identified, which is explained also by the separate metallic and semiconducting character of the samples. Purely metallic SWCNTs exhibit twice as high reactivity when compared to semiconducting ones, suggesting that conduction electrons play the major role in the adsorption process. Our combined experimental and theoretical study of the C 1s core-level shifts in photoemission due to adsorption of NO2 and its reaction byproducts contributes to answering the central questions of NOx adsorption. Specifically, whether this adsorption is covalent chemisorption or charge transfer mediated, and if it changes for metallic vs semiconducting SWCNTs.
This study paves the way toward controlled sensing using SWCNTs ruling out the effect of impurities. It reveals key aspects that can be applied to different reactive and poisonous species, and it provides an insight toward a real improvement in the design of sensors with tailored and heightened sensitivity and selectivity at room temperature.
Results and Discussion
When using SWCNTs in gas sensing, the cleanliness of the material is often not sufficiently taken into account. This is crucial to identify the inherent nanotube capability to interact with the potential molecular adsorbates, which otherwise could not be distinguished. The presence of oxygen is also often disregarded. Therefore, we have removed it by resistive heating and corroborated this with a broad survey scan before our experiments exposing the nanotubes to NO and NO2. This allows us to safely study the reaction pathways that the nitrogen oxides undergo exclusively in the presence of SWCNTs.
We have first inspected the C 1s responses in high resolution XPS (Figure 1) and XAS (not shown here) of the pristine samples according to the established footprints,38,39 which confirmed their cleanliness and metallic and semiconducting purity. The samples were then dosed at 10, 30, 70, 155, and 260 L to observe the spectral evolution of the C 1s line. The upper panels of Figure 1 show the changes in the C 1s line recorded in XPS for both types of samples upon exposure to a 70 L dose of NO (Figure 1a) and NO2 (Figure 1b). This case has been chosen because it has a midrange dosage, to better illustrate the results. Here, the exposure to NO produces an intensity attenuation of 12% and an increase of the full width at half-maximum (fwhm) to 0.41 eV in the C 1s of pristine semiconducting samples. In the case of metallic tubes exposed to NO, the intensity is reduced by 17% and the line exhibits an even larger broadening to 0.64 eV. The exposure to a comparable dosage of NO2 not only induces a larger shift (∼ 150 meV) but also a larger broadening. The shift of the C 1s line to lower binding energies in comparison to pristine samples for the metallic (from 284.48 to 284.37 eV) and semiconducting tubes (from 284.43 to 284.34 eV)39 is the first evidence of a different core-level shift from what has been previously reported for unsorted material.17,24 Additionally, at saturation doping, the intensity attenuation is around 25 and 70% for the semiconducting and metallic samples, respectively. This is another remarkably different result from previous studies on unsorted SWCNT-material, where the C 1s response of sorted SWCNTs upon exposure to NO2 was conversely four times more intense than for NO. On the basis of this, in the following, we will center our attention to the case of NO2 adsorption, where the changes are more evident, as seen in Figure 1b.
Figure 1.

C 1s photoemission spectra recorded on the semiconducting (top) and metallic (bottom) SWCNT-samples after exposure to 70 L of (a) NO and (b) NO2, compared to their pristine corresponding material. (c) Binding energy shift and (d) overall area and of the C 1s line and as a function of increasing NO2 dosage observed for semiconducting and metallic samples.
We have analyzed the spectral evolution of the C 1s line upon exposure to NO2 at the different dosages described above. The C 1s shift from the main peak position is shown in Figure 1c, whereas Figure 1d depicts the change of the area under the curve relative to the pristine material. This exponential dependence with increasing dosage in both the C 1s relative area and the peak downshift can have different explanations. These include the oxidation of the tubes by the NO2 molecules, the chemisorption of molecular species as products of a reaction or simply the presence of reactive defects. Whatever the case may be, these new observations strongly suggest that the reaction pathways of the NOx molecules are very different to what has been reported so far. Previous studies suggested that metal impurities were responsible for the tremendous increased sensitivity to NO2 absorption.19 However, changes in the C 1s and N 1s (discussed below) in photoemission strongly hint that working with purified and sorted samples provides additional information. This requires rethinking of the adsorption processes that have been proposed so far in similar studies with unpurified SWCNTs.
To investigate the adsorption processes at the highest possible coverage, only the responses after exposure to 260 L, close to the saturation dosage, are now considered. Figure 2a provides insight in the vicinity of the main peak of the C 1s in XPS, which is proportionally downshifted about 180 meV for both types of samples at this dosage. Additionally, upon exposure to NO2, a component independent from the π plasmon satellite and the shakeup features related to low energy π–π* interband transitions40 is observed. This feature is located at 286.4 and 286.2 eV for the metallic and the semiconducting samples, respectively. Its intensity appears to be directly related to the hybridization of the system and clearly depends on the metallicity of the samples, as it is about five times higher for metallic tubes. In order to understand the nature of this feature, we have performed DFT calculations on semiconducting (10,0) and metallic (6,6) SWCNTs.
Figure 2.

(a) C 1s XPS spectra recorded on the metallic (bottom) and semiconducting (top) SWCNT-samples after exposure to the 260 L saturation limit of NO2. An additional spectrum at 70 L for the metallic sample is depicted for comparison purposes (see text). (b) C 1s simulated spectra for defective semiconducting (10,0) (top) and metallic (6,6) (bottom) SWCNTs with adsorbed atomic oxygen extracting a carbon atom to form a ketene group at the monovacancy. The maximum of the simulated C 1s and the corresponding experimental spectrum were aligned. The inset shows a ball-and-stick model with carbon and oxygen atoms as gray and red spheres, respectively. An isosurface representation (0.11 e/Å3/2) of the ketene C=O C 1s orbital is also shown.
We have investigated various NOx adsorbed molecules, and introduced a monovacancy defect on the SWCNTs with various C–N and C–O adsorbed configurations to understand the origin of this signal. In general, our calculations could not reproduce this feature at the binding energy values observed experimentally using nitrogen-containing molecules. The calculated separation between the C 1s main peak and this feature was never more than 1.1 eV to higher binding energies, with a charge transfer of 0.33 and 0.38 electrons from the (10,0) and (6,6) SWCNTs to NO2, respectively. We found only a slight chemisorption of NO2 at high coverages but in general it was physisorbed. Therefore we focused on the possible adsorption of oxygen.
On a pristine nanotube, the adsorption of molecular oxygen is weak, with a small charge transfer, and in this case no significant shift of the C 1s is observed.32,41−44 Therefore, we introduced a monovacancy and observed the adsorption of atomic oxygen. The monovacancy resembles a bicyclo[7.3.0]dodecane, which consists of a nine-membered ring and a five-membered ring. In this monovacancy, there is one unsaturated hexagon carbon with divalent carbene character that sticks out of the SWCNT, as shown in Figure 2b. To this electrophile site, we attach a CO molecule leading to the formation of a ketene group. This describes the likely scenario of an atomic oxygen extracting a carbon atom and creating a defect in the lattice.45−47 For both SWCNTs the calculated C=C bond length is 1.31 Å and the C=O bond length is 1.17 Å, as is the case for the ethenone molecule (see Supporting Information). The calculated C 1s core-levels for the C=O species of the ketene group are 286.67 and 286.33 eV for the (6,6) and (10,0) SWCNTs, respectively, as shown in Figure 2b. This is in semiquantitative agreement with the experimentally observed features in the C 1s spectra at 286.4 and 286.2 eV for the metallic and semiconducting samples, respectively. The differences in the core-level shifts between the metallic and semiconducting samples is also reproduced, and may be related to the different charges of 0.64 and 0.62 e calculated for the C=O species in the (6,6) and (10,0) SWCNTs, respectively. For all other oxygen containing groups considered, such as carbonyls and epoxides, the C 1s core levels are at most 1.6 eV, as shown in Supporting Information. This clearly demonstrates that these experimentally observed features are due to the C=O species of ketene groups formed as reaction byproducts.
To provide further confirmation that the features observed at high binding energy in the C 1s photoemission signal are due to a C=O species in a ketene group, we performed separate calculations for isolated molecules containing a variety of C–N and C–O chemical environments. Our results clearly showed that the experimentally observed feature can only be attributed to a C=O species within a ketene group. Further details are provided as Supporting Information. As described below in more detail, this atomic oxygen is available via a new reaction pathway, which is generated by the photoinduced interaction with synchrotron radiation. This observation contrasts with previous studies on less purified metallicity mixed samples.17,19,24
Figure 3 shows the N 1s spectra recorded for metallic (a) and semiconducting (b) samples, compared to the previously reported N 1s signal from mixed SWCNTs19 exposed to a comparable dose of NO2 (c). The spectral deconvolution has been done with Voigtian functions, taking into account the experimental resolution. At the 260 L saturation dosage the N/C ratios are ∼15 and ∼8% for the metallic and the semiconducting tubes, respectively. This means that the metallic tubes are twice as prone to adsorb NO2 (and products of its reaction) than their semiconducting counterparts. The strongest component identified at 405.5 eV in Figure 3a,b corresponds to NO2, which is remarkably different from the case observed in the mixed samples. This suggests that the reaction pathway is different when only ultraclean nanotubes are used. The chemical reactions are different and take place at a different speed from what has been reported for unsorted material. NO2 is weakly hybridized with the SWCNTs, and the relative intensities of the reaction products as compared to the main NO2 contribution are much smaller than previously reported. The other components that can be identified are NO (∼401.2 eV), N2O2 (∼403 eV), N2O4 and NO3 (∼407.1 eV). Bearing this in mind, we can now analyze the type of reaction that occurs once the NO2 molecules approach the nanotube’s surface.48
Figure 3.
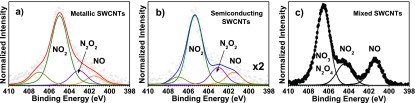
N 1s core level photoemission spectra recorded at 100 K of metallic (a) and semiconducting (b) SWCNTs after exposure to NO2 at the saturation limit of 260 L, compared to the N 1s spectrum reported in ref (19) (c), where the highest binding energy component corresponds to N2O4 and NO3. This component has a minor contribution in the right and middle spectra corresponding to the separated samples. Also note that the intensity of the response for the metallic sample is twice as high as that corresponding to the semiconducting one.
Previous studies on unsorted SWCNT–material17,24 proposed NO and NO3 (or N2O4) as major products of the chemical reaction as can be clearly observed in Figure 3c. However, these are hardly observed in our spectra in Figure 3a,b. This can be related to a preferential physisorption of NO2 on the metallicity sorted SWCNTs and its inability to dissociate rapidly due to the much smaller number of defects and almost nonexistent metallic impurities in the clean separated samples. From the weaker N 1s signal strength it can be inferred that semiconducting SWCNTs induce more dissociation of NO2 than the metallic ones. It is then clear that physisorption is the predominant adsorption mechanism so far, but it cannot be ruled out that a weak chemisorption can take place. Previous work on unsorted material49 reported the following chemical reactions based on the pathways for NO2 proposed by Huffman et al.:48 2(NO2) → NO3 + NO and 2(NO2 + NO)→2NO3 + N2. However, these equations are in contrast to our observations, where NO2 is by far the strongest component after it interacts with the nanotubes. One explanation for this difference could be the low NO2 decomposition rate in comparison to the gas injection rate. The differences from our new results on ultraclean metallicity-sorted SWCNTs compared to the data reported for unpurified material are compelling evidence that the effect of impurities is still not understood and may not be neglected.
As mentioned above, the core level N 1s XPS signal includes the response from NO and NO2 molecules. Experimental evidence of stoichiometric equilibrium of these monomers with their dimers (N2O2 and N2O4) at low temperatures was reported in ref (48). Such a qualitative analysis to identify the dimers using XPS alone is complicated. Therefore, to further understand our data we performed XAS on the N 1s edge. This technique is more sensitive and allows the analysis of samples with an ultralow amount of dopants/adsorbates.50Figure 4 shows the overall XAS response in the N 1s recorded for the metallic and semiconducting samples exposed to a dose of 70 L of NO (bottom) and NO2 (top). The response for NO onto metallic tubes is five times more intense than that for the semiconducting ones. In the case of exposure to NO2, it is about twice as intense. The clearly discernible features in all spectra correspond to the different π* resonances related to the N-containing species. However, the spectra corresponding to NO2 exposure are much richer in nature and clearly exhibit additional signals above 402 eV. In studies of adsorption of NO2 on Ni(100), the features between 400 and 402 eV have previously been assigned to transitions into unoccupied π* orbitals of NO and NO dimers.51 The additional intensity at around 401.3 eV in the two top spectra of Figure 4 can be assigned to the lowest unoccupied π orbital of NO2 as reported in refs (51−53). The component at 403.3 eV is also associated to the π* of NO2, whereas the one at 405.1 eV corresponds to N2O4.52,54,55 This confirms that the small component at high binding energies in the spectra in Figure 3a,b can be related to N2O4, and it is minimal. Therefore, it can be related to direct physisorption of a small amount of NO2 dimers.
Figure 4.

N 1s XAS spectra of sorted SWCNTs upon the exposure of a comparable dose of NO and NO2.
We will now focus on the N 1s close-up into the region between 400 and 402 eV. To the best of our knowledge, the nicely revealed fine structure in the N 1s edge has not been investigated in studies with carbon nanotubes. In molecular systems, this fine structure is assigned to vibrational shake ups, the so-called Franck–Condon satellites (FCS). Studies identifying FCS by XAS and electron energy loss spectroscopy (EELS) in gas phase experiments have been widely reported, mostly dealing with CO and N2.56,57 Also in the gas phase, studies on NO and its dimers have found a separation of the FCS of 235 meV.51,58 Much less is reported on the observation of FCS of gases adsorbed on substrates. Among these, investigations of CO adsorption on different metals (e.g., Ni, Rh or Cu) have shown that the line profile is dominated by the excitation of the stretch vibrations of the adsorbed molecules.59−61 In general, the shakeup intensity decreases with increasing chemisorption strength, which is related to the electronic relaxation of the adsorbate. Hence, in physisorbed and weakly chemisorbed systems, the adsorbate retains its molecular character with the molecular orbital closely resembling the free molecule. In strongly chemisorbed molecules, adsorbate–substrate hybrid orbitals are formed. Their influence on observations for gas absorption on different substrates is well understood.62 We have additionally observed that the absorption on SWCNTs leads to an upshift of the energy for NO and to a downshift for NO2.
Turning to a quantitative analysis of the N 1s XAS π* response, the first FCS at around 400.8 eV varies slightly in position when adsorption occurs onto metallic or semiconducting samples. This suggests that the strongest interaction is the one between the NO dimer and the nanotubes independently from the metallicity of the sample, for both NO and NO2 adsorption. Therefore, we assign the π* response of the FCS to the NO dimers. The relative percentage area of the π* response corresponding to NO and the (NO)2 FCS compared to the πNO2* is summarized in Table 1.
Table 1. Relative Percentage Area of the π* Response Corresponding to NO and the N2O2 FCS Compared to the πNO2*.
| dose (L) | π* (NO)2 | π* NO | π1* NO2 |
|---|---|---|---|
| Semiconducting SWCNTs: | |||
| 30 | 74.7 | 6.1 | 19.2 |
| 70 | 71.6 | 9.1 | 19.3 |
| 260 | 60.1 | 8.8 | 31.1 |
| Metallic SWCNTs: | |||
| 30 | 75.5 | 15.9 | 8.6 |
| 70 | 71.3 | 16.9 | 11.8 |
| 260 | 67.4 | 14.6 | 18.0 |
The region between 400 and 402 eV in the N 1s XAS can be analyzed regarding the first π1* resonances of NO2 and NO, and a set of correlated Voigtians revealing the FCS, which are separated by the phonon frequency and broadened by the lifetime. The top panels in Figure 5 show the corresponding line shape analysis of spectra recorded on the metallic (Figure 5a) and semiconducting (Figure 5b) samples after an exposure to 70 L of both NO2 and NO. The πNO resonance at ca. 400.8 eV represents the first unoccupied π orbital, which can either be related to adsorbed NO or to its dimers. The intensity variation of this component explains why the NO molecules are coadsorbed on the surface of the nanotubes at different rates depending on the nanotube’s metallicity. When the samples were exposed to NO2, the spectral response is surprisingly similar. Only an additional peak at 401.1 eV corresponding to the π* was observed, which is the transition into the lowest unoccupied πNO2* orbital of NO2.53 This is a further indication that many of the adsorbed NO2 molecules undergo a dissociation reaction with the SWCNT forming NO and NO dimers. A quantitative analysis of the vibrational frequencies and the relative strength of the FCS as compared to the π* signal of NO and NO2 provides information related to the relative hybridization strength of the adsorbed species and their nature. In the case of NO, the stretching frequency (ν) is around 236 meV, which corresponds to the C2ν asymmetric N–O stretching mode of the dimer (N2O2), which is close to NO. To gain further information, the changes in the XAS response were also studied as a function of NO2 dosage. These are depicted in Figure 5c,d, comparing the metallic and semiconducting samples after exposure to 70 and 260 L of NO2. The vibrational frequencies of the FCS become slightly harder for higher doses and for metallic samples.
Figure 5.

Close-up into Franck–Condon satellites region of the main peak of N 1s XAS response for (a) metallic and (b) semiconducting SWCNTs. These spectra were recorded upon the exposure to 70 L of NO and NO2. The bottom spectra show a comparison of the response obtained upon exposure to 70 and 260 L of NO2 in both the metallic (c) and semiconducting (d) samples. The shaded areas represent only the components corresponding to molecular NO product of the NO2 decomposition. For clarity, the spectral contribution of the Franck–Condon satellites in the lower pannels is not depicted, which of course has been included to obtain the total fits.
Table 2 summarizes the values of the FCS frequencies at 70 L exposure and at the saturation dosage of 260 L to NO2. The frequency hardening with dosage and metallicity can be explained by an increased hybridization with the SWCNT. This is consistent with previous experiments of FCS on metallic substrates.59−61 Concomitantly, the increased hybridization also reveals a change in the relative intensity of the π* response of NO2 as compared to NO and the NO dimer FCS. These results are shown in Table 1. The relative NO2 intensity increases with increasing NO2 dosage, and it is about three times stronger for the semiconducting nanotubes. This further confirms that part of the NO2 is transformed into NO dimers following the endothermic chemical reaction of 2NO2 → 2NO + 2O proposed in ref (48). The oxygen from the chemical reaction with the SWCNTs may be physisorbed as O2 or chemisorbed on pristine SWCNTs forming epoxide groups or at defects as carbonyl or ketene groups. However, it is the C=O species of ketene groups that gives rise to the extra peak in the C 1s XPS response, as explained above. The higher NO2 response at the saturation dosage is related to the higher coverage, which lowers the molecular interaction with the SWCNTs. The apparent sensitivity of the NO dimer to the electronic nature of the surfaces upon adsorption represents an ideal situation to understand the differences found between semiconducting and metallic SWCNTs while exposed to different gases.
Table 2. Frequencies of the FCSs in Figure 5c,d.
| semiconducting SWCNTs | metallic SWCNTs | |
|---|---|---|
| 70 L | 232 ± 2 meV | 229 ± 2 meV |
| 260 L | 235 ± 2 meV | 233 ± 2 meV |
The last part of our experiments involved heating up the SWCNTs covered with NO2 in order to estimate a lower bound for the desorption energy in metallicity-sorted material. After the saturation dose was reached and properly measured at low temperatures, thermal desorption was studied by resistive heating. Because of fast thermal desorption the whole desorption cycle could not be followed by in situ measurements as in previous studies on the adsorption and desorption of different gases and solvents on surfaces of mixed nanotubes.28,52 However, using the partial pressure of the analysis chamber and the heating time allowed us to estimate a lower bound for the desorption energy. For both metallicities desorption started after 2 min at 140 K, and it finished shortly after recovering the base pressure of the chamber. Here, a complete recovery of the C 1s line of the pristine SWCNTs and vanishing of the N 1s and O 1s responses was observed. We estimate a lower bound of the activation energy for desorption (Ea) of about 20 kJ/mol for the semiconducting tubes and 37 kJ/mol for the metallic ones. Previous results on HOPG28 reported an Ea of 37 kJ/mol comparable to our estimation for metallic tubes. However, the purely semiconducting tubes interestingly show a much lower estimated value. In any case, both types of metallicity sorted nanotubes exhibit much lower values than the calculated Ea of 114 kJ/mol reported for unsorted nanotubes.52 The lower desorption/adsorption energy for the semiconducting sample is also consistent with a weaker hybridization of the adsorbed nitric oxides due to the absence of conduction electrons. Concomitant with the strong interaction of nitroxides with different metallic systems, this study paves the way to tailor the interaction with SWCNTs by advanced filling reactions with metals and metal compounds.
Conclusions
We have investigated the physicochemical effects in the interaction of nitrogen oxides and the outer wall of SWCNTs, which are sorted according to metallic character. Using ultrapure separated SWCNTs has allowed us to identify that the chemical reactions are different and take place at different rates from what has been reported for unsorted material. The adsorption reaction is charge transfer mediated physisorption, which directly depends on the metallic character of the samples, as it is twice as high for the metallic tubes as for the semiconducting ones. Franck–Condon satellites were resolved in the N1s on this gas-nanotube system, revealing a weak chemisorption, which is related to NO dimer molecules. Complementary experiments on the desorption of the system show a very fast recovery and hence provide evidence of the reversibility of the process. This study settles the limit for the strength of the interaction of SWCNTs with nitroxides and paves the way to tailor the tube’s interaction with different compounds such as metals during filling reactions. This will allow one to control the selectivity of the interaction with different nitroxides toward heightened sensitivity and selectivity of nanotube sensors at room temperature.
Methods
Experimental Details
SWCNTs synthesized by the arc-discharge method, purified and separated into metallic and semiconducting tubes were deposited on sapphire substrates. XPS and XAS studies were conducted at the SuperESCA beamline at the ELETTRA synchrotron,63 where the emitted photoelectrons are collected by a 150 mm hemispherical analyzer with a time-delay detector mounted at 70° with respect to the incident beam. The samples were placed on a vertical Ta holder, mounted on a manipulator that allows cooling down to about 100 K and annealing up to 1800 K. The experimental chamber had a base pressure of 2.5 × 10–10 mbar. In order to remove any remaining impurities, the samples were outgassed in situ by a combined resistive and electron-beam heating system up to 900 K. The exceptional purity of the samples was confirmed by wide range high resolution (HR) photoemission spectroscopy (PES) survey scans.39,40 For HR fast PES and XAS measurements, the samples were cooled down with liquid N2. The different nanotube samples were exposed to pure O2, NO and NO2 gases, which were inserted through a needle valve. During the gas sensing experiments the temperature of the samples was kept constant at 100 K, and the gas pressure below 10–8 mbar. Different Langmuir (1L ≈ 1.33 × 10–6 mbar·s) gas doses were used up to the saturation range (∼260 L). Core level spectra were recorded for the C 1s, N 1s and O 1s signals using photon energies of 400, 525, and 650 eV, with corresponding overall energy resolution of 100, 150, and 200 meV. Measurements on Ta 4f and Ta 4d5/2 were used for calibration. XAS was carried out using the Auger yield of the respective core levels.
Computational Details
DFT calculations were performed using the real-space grid implementation of the projector augmented wave function (PAW) method in the GPAW code64−66 within the local density approximation (LDA).67 A grid spacing of h ≈ 0.2 Å, k-point sampling of (1 × 1 × 3), and electronic temperature of 0.1 eV was used, with all energies extrapolated to T → 0 K. All structures were relaxed until a maximum force below 0.05 eV/Å was reached. We included spin-polarization whenever necessary, e.g., for open-shell systems. All-electron calculations within the PAW method were used to model core-level shifts within the initial state method.68 XPS spectra were simulated using the density of states (DOS) by applying a Lorentzian broadening, assuming an inverse lifetime of 0.27 eV, and aligning the maximum of each DOS with their experimental counterparts.
Semiconducting and metallic nanotubes were modeled by pristine (10,0) and (6,6) SWNTs and SWNTs with a monovacancy. A vacuum layer of more than 10 Å was used to remove interactions between repeated images perpendicular to the tube axis. Unit cells of 8.526 and 9.845 Å along the tube axis were used for the pristine (10,0) and (6,6) SWNTs, respectively. The (10,0) and (6,6) SWNTs with a monovacancy were modeled with 17.052 and 17.229 Å long cells, respectively. To ensure an absolute energy reference for the C 1s core-levels between calculations, nonperiodic boundary conditions were employed perpendicular to the nanotube axis by fixing the electron density and Kohn–Sham wave functions to zero at the cell boundary.
Acknowledgments
This work was supported by the Austrian Science Fund through Project FWF P21333-N20 and by the EU Proposal No. 20105285 for ELETTRA. P.A. was supported by a Marie Curie Intra European Fellowship within the Seventh European Community Framework Programme. D.J.M. acknowledges funding through the Spanish “Juan de la Cierva” program (JCI-2010-08156). We acknowledge funding by the European Research Council Advanced Grant DYNamo (ERC-2010-AdG -Proposal No. 267374), Spanish Grants (FIS2010-21282-C02-01 and PIB2010US-00652), “Grupos Consolidados UPV/EHU del Gobierno Vasco” (IT-319-07 and IT-578-13), and the European Commission project CRONOS (280879-2 CRONOS CP-FP7) and POCAONTAS (FP7-PEOPLE-2012-ITN). A.G. thanks the COST ACTION EuNetAir (No. TD1105).
Supporting Information Available
Sample characterization; additional simulated C 1s spectra for molecular oxygen, epoxide and carbonyl groups on SWCNTs and molecular species; absolute energies and atomic coordinates for all optimized geometries. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Collins P. G.; Bradley K.; Ishigami M.; Zettl A. Extreme Oxygen Sensitivity of Electronic Properties of Carbon Nanotubes. Science 2000, 287, 1801. [DOI] [PubMed] [Google Scholar]
- Sumanasekera G. U.; Adu C. K. W.; Fang S.; Eklund P. C. Effects of Gas Adsorption and Collisions on Electrical Transport In Single-Walled Carbon Nanotubes. Phys. Rev. Lett. 2000, 85, 1096. [DOI] [PubMed] [Google Scholar]
- Kong J.; Franklin N. R.; Zhou C. W.; Chapline M. G.; Peng S.; Cho K. J.; Dai H. J. Nanotube Molecular Wires as Chemical Sensors. Science 2000, 287, 622. [DOI] [PubMed] [Google Scholar]
- Kong J.; Chapline M. G.; Dai H. Functionalized Carbon Nanotubes for Molecular Hydrogen Sensors. Adv. Mater. 2001, 13, 1384. [Google Scholar]
- Chang H.; Lee J. D.; Lee S. M.; Lee Y. H. Adsorption of NH3 and NO2 Molecules on Carbon Nanotubes. Appl. Phys. Lett. 2001, 79, 3863. [Google Scholar]
- Modi A.; Koratkar N.; Lass E.; Wei B.; Ajayan P. M. Miniaturized Gas Ionization Sensors using Carbon Nanotubes. Nature 2003, 424, 171. [DOI] [PubMed] [Google Scholar]
- Chopra S.; McGuire K.; Gothard N.; Rao A. M.; Pham A. Selective Gas Detection Using a Carbon Nanotube Sensor. Appl. Phys. Lett. 2003, 83, 2280. [Google Scholar]
- Mallouk T. E.; Yang P. Chemistry at the Nano Bio Interface. J. Am. Chem. Soc. 2009, 131, 7937–7939. [DOI] [PubMed] [Google Scholar]; PMID: 19507897.
- Zhao J. J.; Buldum A.; Han J.; Lu J. P. Gas Molecule Adsorption in Carbon Nanotubes and Nanotube Bundles. Nanotechnology 2002, 13, 195. [Google Scholar]
- Cao Q.; Rogers J. A. Ultrathin Films of Single-Walled Carbon Nanotubes for Electronics and Sensors: A Review of Fundamental and Applied Aspects. Adv. Mater. 2009, 21, 29. [Google Scholar]
- Kauffman D. R.; Star A. Carbon Nanotube Gas and Vapor Sensors. Angew. Chem., Int. Ed. Engl. 2008, 47, 6550. [DOI] [PubMed] [Google Scholar]
- Goldoni A.; Petaccia L.; Lizzit S.; Larciprete R. Sensing Gases with Carbon Nanotubes: a Review of the Actual Situation. J. Phys.: Condens. Matter 2010, 22, 01300. [DOI] [PubMed] [Google Scholar]
- Mowbray D. J.; Morgan C.; Thygesen K. S. Influence of O2 and N2 on the Conductivity of Carbon Nanotube Networks. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 79, 195431. [Google Scholar]
- Garca-Lastra J. M.; Mowbray D. J.; Thygesen K. S.; Rubio A.; Jacobsen K. W. Modeling Nanoscale Gas Sensors under Realistic Conditions: Computational Screening of Metal-Doped Carbon Nanotubes. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 81, 245429. [Google Scholar]
- Bekyarova E.; Kalinina I.; Itkis M. E.; Beer L.; Cabrera N.; Haddon R. C. Mechanism of Ammonia Detection by Chemically Functionalized Single-Walled Carbon Nanotubes: In Situ Electrical and Optical Study of Gas Analyte Detection. J. Am. Chem. Soc. 2007, 129, 10700–10706. [DOI] [PubMed] [Google Scholar]; PMID: 17696430.
- Kordas K.; Mustonen T.; Tóth G.; Jantunen H.; Lajunen M.; Soldano C.; Talapatra S.; Kar S.; Vajtai R.; Ajayan P. M. Inkjet Printing of Electrically Conductive Patterns of Carbon Nanotubes. Small 2006, 2, 1021. [DOI] [PubMed] [Google Scholar]
- Goldoni A.; Larciprete R.; Petaccia L.; Lizzit S. Single-Wall Carbon Nanotube Interaction with Gases: Sample Contaminants And Environmental Monitoring. J. Am. Chem. Soc. 2003, 125, 11329. [DOI] [PubMed] [Google Scholar]
- Valentini L.; Armentano I.; Kenny J. M.; Cantalini C.; Lozzi L.; Santucci S. Sensors for Sub-Ppm NO2 Gas Detection Based on Carbon Nanotube Thin Films. Appl. Phys. Lett. 2003, 82, 961. [Google Scholar]
- Larciprete R.; Petaccia L.; Lizzit S.; Goldoni A. The Role of Metal Contact in the Sensitivity of Single-Walled Carbon Nanotubes to NO2. J. Phys. Chem. C 2007, 111, 12169. [Google Scholar]
- Sayago I.; Santos H.; Horrillo M.; Aleixandre M.; Fernández M.; Terrado E.; Tacchini I.; Aroz R.; Maser W.; Benito A.; et al. Carbon Nanotube Networks as Gas Sensors for NO2 Detection. Talanta 2008, 77, 758–764. [Google Scholar]
- Gruner G. Carbon Nanotube Transistors for Biosensing Applications. Anal. Bioanal. Chem. 2006, 384, 322. [DOI] [PubMed] [Google Scholar]
- Huang J.; Ng A. L.; Piao Y.; Chen C.-F.; Green A. A.; Sun C.-F.; Hersam M. C.; Lee C. S.; Wang Y. Covalently Functionalized Double-Walled Carbon Nanotubes Combine High Sensitivity and Selectivity in the Electrical Detection of Small Molecules. J. Am. Chem. Soc. 2013, 135, 2306–2312. [DOI] [PubMed] [Google Scholar]
- Boussaad S.; Diner B. A.; Fan J. Influence of Redox Molecules on the Electronic Conductance of Single-Walled Carbon Nanotube Field-Effect Transistors: Application to Chemical and Biological Sensing. J. Am. Chem. Soc. 2008, 130, 3780–3787. [DOI] [PubMed] [Google Scholar]
- Goldoni A.; Petaccia L.; Gregoratti L.; Kaulich B.; Barinov A.; Lizzit S.; Laurita A.; Sangaletti L.; Larciprete R. Spectroscopic Characterization of Contaminants and Interaction with Gases in Single-Walled Carbon Nanotubes. Carbon 2004, 42, 2099. [Google Scholar]
- Yanagi K.; Udoguchi H.; Sagitani S.; Oshima Y.; Takenobu T.; Kataura H.; Ishida T.; Matsuda K.; Maniwa Y. Transport Mechanisms in Metallic and Semiconducting Single-Wall Carbon Nanotube Networks. ACS Nano 2010, 4, 4027. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Chen Z.; Du X.; Logan J.; Sippel J.; Nikolou M.; Kamaras K.; Reynolds J.; Tanner D.; Hebard A.; et al. Transparent, Conductive Carbon Nanotube Films. Science 2004, 305, 1273. [DOI] [PubMed] [Google Scholar]
- Santucci S.; Picozzi S.; Di Gregorio F.; Lozzi L.; Cantalini C.; Valentini L.; Kenny J. M.; Delley B.; NO2; Gas C. O. Adsorption on Carbon Nanotubes: Experiment and Theory. J. Chem. Phys. 2003, 119, 10904. [Google Scholar]
- Ulbricht H.; Zacharia R.; Cindir N.; Hertel T. Thermal Desorption of Gases and Solvents from Graphite and Carbon Nanotubes Surfaces. Carbon 2006, 44, 2931. [Google Scholar]
- Sövall P.; So S.; Kasemo B.; Franchy R.; Ho W. NO2 Adsorption on Graphite at 90 K. Chem. Phys. Lett. 1990, 172, 125–130. [Google Scholar]
- Peng S.; Cho K. J. Chemical Control of Nanotube Electronics. Nanotechnology 2000, 11, 57. [Google Scholar]
- Zhang Y.-H.; Chen Y.-B.; Zhou K.-G.; Liu C.-H.; Zeng J.; Zhang H.-L.; Peng Y. Improving Gas Sensing Properties of Graphene by Introducing Dopants and Defects: a First-Principles Study. Nanotechnology 2009, 20, 185504. [DOI] [PubMed] [Google Scholar]
- Jijun Z.; Alper B.; Jie H.; Jian P. L. Gas Molecule Adsorption in Carbon Nanotubes and Nanotube Bundles. Nanotechnology 2002, 13, 195. [Google Scholar]
- Yim W.-L.; Gong X. G.; Liu Z.-F. Chemisorption of NO2 on Carbon Nanotubes. J. Phys. Chem. B 2003, 107, 9363–9369. [Google Scholar]
- Seo K.; Park K. A.; Kim C.; Han S.; Kim B.; Lee Y. H. Chirality- and Diameter-Dependent Reactivity of NO2 on Carbon Nanotube Walls. J. Am. Chem. Soc. 2005, 127, 15724–15729. [DOI] [PubMed] [Google Scholar]
- Tang S.; Cao Z. Adsorption of Nitrogen Oxides on Graphene and Graphene Oxides: Insights from Density Functional Calculations. J. Chem. Phys. 2011, 134, 044710. [DOI] [PubMed] [Google Scholar]
- Leenaerts O.; Partoens B.; Peeters F. M. Adsorption of H2O, NH2, CO, NO2 and NO on Graphite: a First-Principles Study. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 77, 125416. [Google Scholar]
- Dai J.; Giannozzi P.; Yuan J. Adsorption of Pairs of NO2 Molecules on Single-Walled Carbon Nanotubes and Formation of NO + NO2 from NO2. Surf. Sci. 2009, 603, 3234–3238. [Google Scholar]
- Ayala P.; Shiozawa H.; De Blauwe K.; Miyata Y.; Follath R.; Kataura H.; Pichler T. An X-Ray Absorption Approach to Mixed and Metallicity-Sorted Single-Walled Carbon Nanotubes. J. Mater. Sci. 2010, 45, 5318. [Google Scholar]
- Ayala P.; Miyata Y.; De Blauwe K.; Shiozawa H.; Feng Y.; Yanagi K.; Kramberger C.; Silva S.; Follath R.; Kataura H.; et al. Disentanglement of the Electronic Properties of Metallicity-Selected Single-Walled Carbon Nanotubes. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 205427. [Google Scholar]
- Kramberger C.; Rauf H.; Shiozawa H.; Knupfer M.; Buchner B.; Pichler T.; Batchelor D.; Kataura H. Unraveling Van Hove Singularities in X-Ray Absorption Response of Single-Wall Carbon Nanotubes. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 75, 235437. [Google Scholar]
- Grujicic M.; Cao G.; Rao A.; Tritt T.; Nayak S. UV-Light Enhanced Oxidation of Carbon Nanotubes. Appl. Surf. Sci. 2003, 214, 289. [Google Scholar]
- Giannozzi P.; Car R.; Scoles G. Oxygen Adsorption on Graphite and Nanotubes. J. Chem. Phys. 2003, 118, 1003. [Google Scholar]
- Dag S.; Goulseren O.; Yildirim T.; Ciraci S. Oxygenation of Carbon Nanotubes: Atomic Structure, Energetics and Electronic Structure. Phys. Rev. B: Condens. Matter Mater. Phys. 2003, 67, 165424. [Google Scholar]
- Jhi S. H.; Louie S. G.; Cohen M. L. Electronic Properties of Oxidized Carbon Nanotubes. Phys. Rev. Lett. 2000, 85, 1710. [DOI] [PubMed] [Google Scholar]
- Xiao B.; Zhao J.-x.; Ding Y.-h.; Sun C.-c. Theoretical Investigation of the Interaction between Carbon Monoxide and Carbon Nanotubes with Single-Vacancy Defects. ChemPhysChem 2010, 11, 3505–3510. [DOI] [PubMed] [Google Scholar]
- Nongnual T.; Limtrakul J. Healing of a Vacancy Defect in a Single-Walled Carbon Nanotube by Carbon Monoxide Disproportionation. J. Phys. Chem. C 2011, 115, 4649–4655. [Google Scholar]
- Pei S.; Cheng H.-M. The Reduction of Graphene Oxide. Carbon 2012, 50, 3210–3228. [Google Scholar]
- Huffman R. E.; Davidson N. Shock Waves in Chemical Kinetics—the Thermal Decomposition of NO2. J. Am. Chem. Soc. 1959, 10, 2311. [Google Scholar]
- Larciprete R.; Fabris S.; Sun T.; Lacovig P.; Baraldi A.; Lizzit S. Dual Path Mechanism in the Thermal Reduction of Graphene Oxide. J. Am. Chem. Soc. 2011, 133, 17315. [DOI] [PubMed] [Google Scholar]
- Ayala P.; Arenal R.; Loiseau A.; Rubio A.; Pichler T. The Physical and Chemical Properties of Heteronanotubes. Rev. Mod. Phys. 2010, 82, 1843. [Google Scholar]
- Geisler H.; Odörfer G.; Illing G.; Jaeger R.; Freund H. J.; Watson G.; Plummer E. W.; Neuber M.; Neumann M. NO2 Adsorption on Ni(100): a Comparison of NO2 with CO2 Adsorption. Surf. Sci. 1990, 234, 237. [Google Scholar]
- Ellison M. D.; Crotty M. J.; Koh D.; Spray R. L.; Tate K. E. Adsorption of NH3 and NO2 on Single-Walled Carbon Nanotubes. J. Phys. Chem. B 2004, 108, 7938. [Google Scholar]
- Pedio M.; Casero E.; Nannarone S.; Giglia A.; Mahne N.; Hayakawa K.; Benfatto M.; Hatada K.; Felici R.; Cerda J.; et al. NEXAFS Study of Nitric Oxide Layers Adsorbed from a Nitrite Solution onto a Pt (111) Surface. J. Phys. Chem. C 2008, 112, 10161. [Google Scholar]
- Holland R.; Maier W. Infrared Absorption Spectra of Nitrogen Oxides in Liquid Xe. Isomerization of N2O3. J. Chem. Phys. 1983, 78, 2928. [Google Scholar]
- Mélen F.; Pokorni F.; Herman M. Vibrational Band Analysis of N2O4. Chem. Phys. Lett. 1992, 194, 181. [Google Scholar]
- Chen C. T.; Ma Y.; Sette F. K-Shell Photoabsorption of the N2 Molecule. Phys. Rev. A 1989, 40, 6737. [DOI] [PubMed] [Google Scholar]
- Medhurst L.; Heimann P.; Siggel M.; Shirley D.; Chen C. T.; Ma Y.; Modesti F.; Sette Vibrationally S. Resolved Threshold Photoemission of N2 And CO at the N and C K-Edges. Chem. Phys. Lett. 1992, 193, 493. [Google Scholar]
- Saito T.; Esaka F.; Furuya K.; Kikuchi T.; Imamura M.; Matsubayashi N.; Shimada H. XAS and XPS Studies on Molecular and Dissociative Adsorption of Nitric Oxide on Rh. J. Electron Spectrosc. Relat. Phenom. 1998, 88, 763. [Google Scholar]
- Kivimäki A.; Kempgens B.; Maier K.; Köppe H. M.; Piancastelli M. N.; Neeb M.; Bradshaw A. M. Vibrationally Resolved O 1s Photoelectron Spectrum of CO2: Vibronic Coupling and Dynamic Core-Hole Localization. Phys. Rev. Lett. 1997, 79, 998. [Google Scholar]
- Foehlisch A.; Hasselström J.; Karis O.; Väterlein P.; Mårtensson N.; Nilsson A.; Heske C.; Stichler M.; Keller C.; Wurth W.; et al. Franck-Condon Breakdown in Core-Level Photoelectron Spectroscopy of Chemisorbed CO. Chem. Phys. Lett. 1999, 315, 194. [Google Scholar]
- Ehara M.; Nakatsuji H.; Matsumoto M.; Hatamoto T.; Liu X. J.; Lischke T.; Prümper G.; Tanaka T.; Makochekanwa C.; Hoshino M.; et al. Symmetry-Dependent Vibrational Excitation in N1s Photoionization of NO2: Experiment and Theory. J. Chem. Phys. 2006, 124, 124311. [DOI] [PubMed] [Google Scholar]
- Brown W. A.; King D. A. NO Chemisorption and Reactions on Metal Surfaces: a New Perspective. J. Phys. Chem. B 2000, 104, 2578. [Google Scholar]
- Abrami A.; Barnaba M.; Battistello L.; Bianco A.; Brena B.; Cautero G.; Chen Q. H.; Cocco D.; Comelli G.; Contrino S.; et al. Super Esca—First Beamline Operating at Elettra. Rev. Sci. Instrum. 1995, 66, 1618. [Google Scholar]
- Mortensen J. J.; Hansen L. B.; Jacobsen K. W. Real-Space Grid Implementation of the Projector Augmented Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 035109. [Google Scholar]
- Enkovaara J.; Rostgaard C.; Mortensen J. J.; Chen J.; Dulak M.; Ferrighi L.; Gavnholt J.; Glinsvad C.; Haikola V.; Hansen H.; et al. Electronic Structure Calculations with GPAW: a Real-Space Implementation of the Projector Augmented-Wave Method. J. Phys.: Condens. Matter 2010, 22, 253202. [DOI] [PubMed] [Google Scholar]
- Bahn S. R.; Jacobsen K. W. An Object-Oriented Scripting Interface to a Legacy Electronic Structure Code. Comput. Sci. Eng. 2002, 4, 56. [Google Scholar]
- Perdew J. P.; Zunger A. Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems. Phys. Rev. B: Condens. Matter Mater. Phys. 1981, 23, 5048. [Google Scholar]
- Cabellos J. L.; Mowbray D. J.; Goiri E.; El-Sayed A.; Floreano L.; de Oteyza D. G.; Rogero C.; Ortega J. E.; Rubio A. Understanding Charge Transfer in Donor-Acceptor/Metal Systems: A Combined Theoretical and Experimental Study. J. Phys. Chem. C 2012, 116, 17991–18001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.