

Abstract
Significance: Exposure to ionizing radiation (IR) as the result of nuclear accidents or terrorist attacks is a significant threat and a major medical concern. Hematopoietic stem cell (HSC) injury is the primary cause of death after accidental or intentional exposure to a moderate or high dose of IR. Protecting HSCs from IR should be a primary goal in the development of novel medical countermeasures against radiation. Recent Advances: Significant progress has been made in our understanding of the mechanisms by which IR causes HSC damage. The mechanisms include (i) induction of HSC apoptosis via the p53-Puma pathway; (ii) promotion of HSC differentiation via the activation of the G-CSF/Stat3/BATF-dependent differentiation checkpoint; (iii) induction of HSC senescence via the ROS-p38 pathway; and (iv) damage to the HSC niche. Critical Issues: Induction of apoptosis in HSCs and hematopoietic progenitor cells is primarily responsible for IR-induced acute bone marrow (BM) injury. Long-term BM suppression caused by IR is mainly attributable to the induction of HSC senescence. However, the promotion of HSC differentiation and damage to the HSC niche can contribute to both the acute and long-term effects of IR on the hematopoietic system. Future Directions: In this review, we have summarized a number of recent findings that provide new insights into the mechanisms whereby IR damages HSCs. These findings will provide new opportunities for developing a mechanism-based strategy to prevent and/or mitigate IR-induced BM suppression. Antioxid. Redox Signal. 20, 1447–1462.
Introduction
After the discovery of X-rays by Wilhelm Röntgen in 1895, Warren and Whipple (161) and Shouse et al. (143) first reported that dogs exposed to a high dose of X-rays developed fatal hematopoietic toxicity. The devastating effects of ionizing radiation (IR) on human health were discovered in the wake of the first atomic bomb explosions in 1945 when thousands of Hiroshima and Nagasaki atomic bomb victims died of IR. They showed that IR-induced hematopoietic failure was the primary cause of death after exposure to a moderate or high dose of total body irradiation (TBI). The pioneering studies by Jacobson and his colleagues in 1940s demonstrated that lead shielding of the spleen or one entire hind leg or transplantation of splenocytes protected mice from the lethal effect of IR (71, 72). Lorenz et al. soon described a similar finding in which they showed that intravenous infusions of bone marrow (BM) cell suspensions protected mice against IR (95). The radioprotective effects of the spleen and BM cell suspensions were initially ascribed to a “humoral factor” (72) but then attributed to the transplanted cells (43, 100, 121, 150). The identity of those cells that were capable of protecting animals from IR-induced lethal hematopoietic damage remained elusive until early 1960s when Till and McCulloch discovered hematopoietic stem cells (HSCs) (15, 106, 148). They showed that HSCs are sensitive to radiation and can self-renew and give rise to multiple lineages of progeny after transplantation into lethally irradiated animals. Till and McCulloch's landmark discovery laid the foundation for modern stem cell and radiation biology research (15, 106, 148). Since then, significant progress has been made in our understanding of the mechanisms by which IR causes hematopoietic damage. Below is a brief summary of some of these recent findings uncovering the mechanisms of action of IR on HSCs. We plan to focus our discussion on the mechanisms whereby IR induces HSC injury and the implication of HSC injury to IR-induced BM suppression in mouse because IR-induced damage to human HSCs has not been well studied. In addition, IR-induced hematopoietic genomic instability and malignancies will not be discussed here either because they have been extensively reviewed by others recently (96, 115).
The Hierarchy of the Murine Hematopoietic System and HSC Niche
As demonstrated by Till and McCulloch in their pioneering works, the cells that were originally believed to be HSCs identified in their colony-forming units-spleen (CFU-S) assay were heterogeneous because they had variable capacity for self-renewal (15, 106, 148). This finding provoked a series of investigations aimed at identification, purification, and characterization of HSCs and their progeny. Through decades of research, HSCs and their progeny, including multipotent progenitors (MPPs) and hematopoietic progenitor cells (HPCs), can now be prospectively isolated in high purity using multiparameter flow cytometry and a large array of monoclonal antibodies against various cell surface molecules (Fig. 1). Murine HSCs and MPPs do not express mature hematopoietic cell lineage markers (e.g., Lin−), such as B220, CD4, CD8, Gr-1, Mac-1, and Ter-119, but express c-Kit and Sca-1 (82). They are collectively called LSK (Lin−sca1+c-kit+) cells, whereas HPCs are LS−K+ (Lin−sca1−c-kit+) cells (82). HSCs and MPPs can be separated according to their expression of CD150 and CD48 (78). Specifically, HSCs are CD150+CD48−LSK cells and MPPs are CD150+/−CD48+LSK cells. Alternative strategies using other cell surface markers and dye effluxing have also been used to identify and isolate HSCs. These include the identification of HSCs as CD34−LSK cells (124), Thy1loFlk-2−LSK cells (26), and the Hoechst-effluxing side population cells (50). More recently, according to the expression of CD34, CD150, and CD48, HSCs can be further differentiated into long-term or dormant HSCs (CD34−CD150+CD48−LSK cells) and short-term HSCs (CD34+CD150+CD48−LSK cells) (166).
FIG. 1.
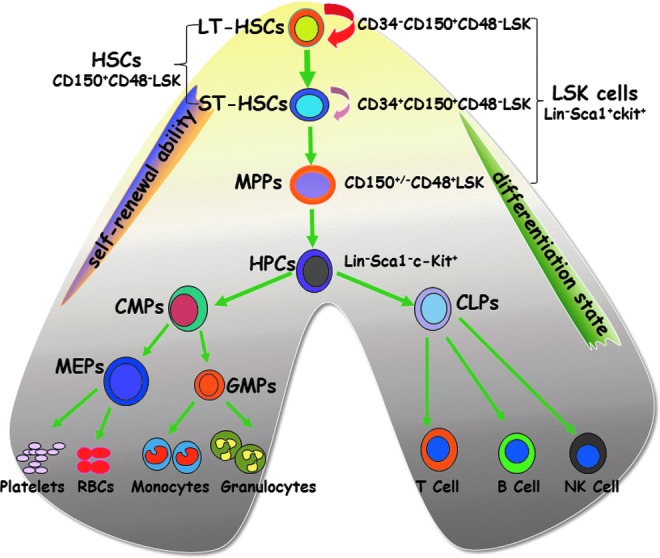
A hierarchical model of the murine hematopoietic system. Long-term hematopoietic stem cell (LT-HSCs, CD34−CD150+CD48−LSK cells) reside at the top of the hierarchy and have the ability to self-renew, proliferate, and differentiate into short-term HSCs (ST-HSCs, CD34+CD150+CD48−LSK cells), multipotent progenitors (MPPs, CD150+/−CD48+LSK cells), and hematopoietic progenitor cells (HPCs, Lin−sca1−c-kit+ or LSK− cells). HPCs can proliferate and differentiate into different lineages of mature blood cells via common lymphoid progenitors (CLPs) as well as common myeloid progenitors (CMPs) and their progeny, such as megakaryocyte/erythroid progenitors (MEPs) and granulocyte/monocyte progenitors (GMPs).
The hematopoietic system is organized in a hierarchical manner. The rare HSCs reside at the top of the hierarchy and have the ability to self-renew, proliferate, and differentiate into different lineages of peripheral blood cells through MPPs and HPCs (131, 163) (Fig. 1). HSCs are quiescent under steady-state conditions and serve as a reserve that protects the hematopoietic system from exhaustion under various stress conditions (165). In contrast, MPPs and HPCs are proliferating cells with limited and no self-renewal ability, respectively. The proliferation and differentiation of MPPs and HPCs satisfy the needs of normal hematopoiesis and also allow the hematopoietic system to react swiftly and effectively to meet demands for increased production of mature cells during hematopoietic crises, such as loss of blood, hemolysis, or infection. If MPPs and HPCs are depleted by an exogenous stressor, such as chemotherapy and/or IR, acute myelosuppression occurs. Under such circumstances, HSCs can undergo self-renewing proliferation and differentiation to repopulate MPPs and HPCs and restore homeostasis. However, if HSCs are injured or their self-renewing ability is impaired, long-term or permanent damage to the hematopoietic system occurs and BM failure and death of the organism may occur (159).
Normally, the quiescent HSCs reside in the osteoblastic niche adjacent to the endosteal bone surface (7, 91, 165) (Fig. 2). These cells represent the long-term repopulating and the most primitive HSCs. The osteoblastic niche provides HSCs with a special environment that supports their self-renewal. This is likely achieved in part by extensive interactions between HSCs and the niche via a variety of soluble factors, such as Wnt (145), bone morphogenetic proteins (49), thrombopoietin (171), interleukin-3, and interleukin-6 (13); various adhesion molecules, including CXCL12-CXCR4 and N-cadherin (58); and different signaling pathways, for example, stem cell factor/c-Kit, Jagged/Notch, and angiopoietin-1/Tie2 (Ang-1/Tie2) (8, 18, 110). These intricate interactions promote HSC self-renewal not only by increasing HSC survival but also by keeping them quiescent to prevent HSCs from exhaustion. In addition, sinusoidal endothelial cells (SECs) in BM have been revealed to function as an alternative HSC niche called the vascular niche (9) (Fig. 2). The vascular niche plays an important role in hematopoietic development during embryonic and fetal development. It is also involved in the regulation of HSC/HPC mobilization, proliferation, and differentiation in response to hematopoietic stress (57). Therefore, HSCs may use either osteoblasts or endothelial cells as their niche under different circumstances to maintain a fine balance between quiescence and proliferation or self-renewal and differentiation, as well as to respond to stress. Furthermore, recent studies have identified additional types of cells that contribute to the compositions of the HSC niche, which include perivascular cells, CXCL12-abundant reticular cells, mesenchymal stem cells, macrophages, regulatory T cells, and Schwann cells along with sympathetic nerve fibers (Fig. 2) (25, 44, 107, 123, 170). These cells can also regulate various HSC activities directly and/or indirectly via affecting the function of the osteoblastic and vascular niche.
FIG. 2.
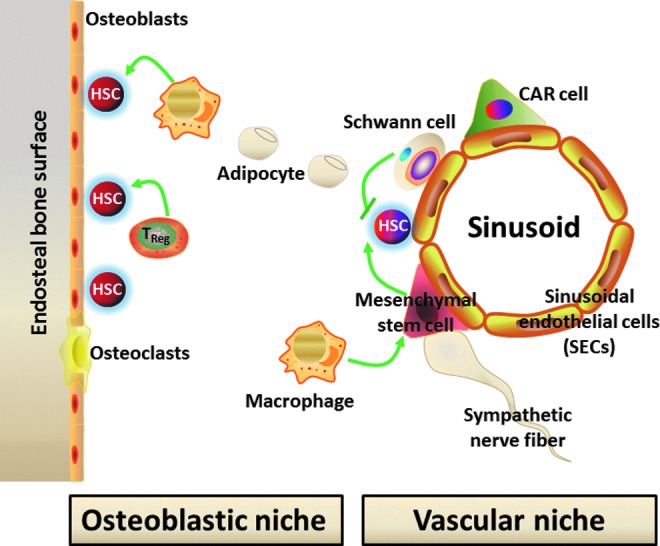
The HSC niche. The HSC niche includes the endosteal osteoblastic niche and vascular niche made up by osteoblasts and sinusoidal endothelial cells (SECs), respectively. In addition, additional types of cells contribute to the compositions of the HSC niche, which include perivascular cells, mesenchymal stem cells, adipocytes, CXCL12-abundant reticular (CAR) cells, macrophages, regulatory T cells (TReg), and Schwann cells along with sympathetic nerve fibers.
Mechanisms of IR-Induced Cell Injury
Cell and tissue injury as a result of exposure to IR occurs by the direct ionization of cellular macromolecules or by the reaction of macromolecules with free radicals generated by the radiolysis of water. Since water constitutes about 75% of the mass of cells and tissues, the majority of energy of a low linear energy transfer radiation such as X-rays and γ-irradiation is mainly deposited in water to produce free radicals that, in turn, significantly contribute to IR-induced injury. Moreover, free radical-mediated cell injury can be enhanced in the presence of oxygen due to the formation of various reactive oxygen species (ROS). Ionization and/or reaction with free radicals/ROS disrupt the structure and function of DNA, lipids, and proteins, which lead to metabolic and functional alterations and ultimately to a cell injury or death. Among various macromolecules, DNA is considered to be the most critical molecular target for IR-induced cell injury and death (152).
IR induces several different types of damage to DNA, which include base damages and changes, cross linking, single-strand breaks, and double-strand breaks (DSBs). Among them, DSBs are the most detrimental DNA damage to a cell because they can ultimately lead to chromosome breaks and translocations that are associated with many human diseases, including cancer if they are left unrepaired or misrepaired. Fortunately, DSBs can effectively trigger a series of cellular reactions termed DNA damage response (DDR) to ensure the rapid detection and repair of DSBs or to remove the damaged cells via the induction of apoptosis and senescence to maintain genome integrity. Specifically, DSBs are recognized by ataxia telangiectasia mutated (ATM). Once activated, ATM phosphorylates various downstream substrates to initiate a signaling cascade that regulates DNA repair, cell cycle checkpoints, and cell survival. For example, phosphorylation of checkpoint kinase 2 at Thr68 and/or that of p53 at Ser15 by ATM activate the G1–S cell cycle checkpoint. Phosphorylation of p53 by ATM can upregulate the expression of p21, proapoptotic proteins such as Puma, and p16 to induce cell cycle arrest, apoptosis, and senescence (Fig. 3), respectively, in a cell type- and a cell context-dependent manner, because many intrinsic and extrinsic factors can affect cellular responses to IR-induced DNA damage (120). In addition, IR-induced oxidative stress can also activate the mitogen-activated protein kinase (MAPK) p38, which in turn causes cellular senescence by the induction of p16 expression and/or interacting with p53 (70) (Fig. 3).
FIG. 3.
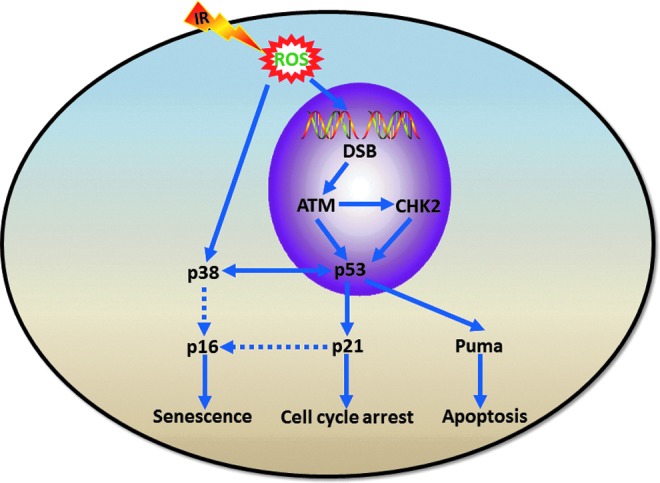
Ionizing radiation (IR)-induced DNA damage response (DDR). DNA double-strand breaks (DSBs) induced by IR activate ataxia telangiectasia mutated (ATM), which in turn phosphorylates checkpoint kinase 2 (CHK2) and p53. Phosphorylation of p53 by ATM upregulates the expression of p21, proapoptotic proteins such as Puma and p16 to induce cell cycle arrest, apoptosis, and senescence, respectively, in a cell type- and a cell context-dependent manner. In addition, IR-induced reactive oxygen species (ROS) can also activate p38, which in turn causes cellular senescence by induction of p16 expression and/or interacting with p53.
Mechanisms of IR-Induced HSC Injury
Within a few hours or days after exposure to a significant dose of TBI (Table 1), a series of characteristic clinical complications termed the acute radiation syndrome (ARS) appear (116, 162). The hematopoietic syndrome occurs at TBI doses in the range of 2–7.5 Gy in humans (3–10 Gy in rodents) and is caused by severe depletion of blood elements due to BM suppression; the gastrointestinal syndrome occurs after doses >5.5 Gy of TBI; and the neurovascular syndrome occurs following large doses of TBI (>20 Gy), indicating that the hematopoietic system is the most radiosensitive tissue of the body. The severity and duration of BM suppression is dose dependent at TBI >1 Gy. Acute and transient myelosuppression typically results from exposure to a moderate dose (<3.5 Gy) of TBI, which primarily damages HPCs and MPPs that are rapidly proliferating and thus highly sensitive to IR (30, 104). A persistent myelosuppression or BM failure occurs as a result of severe injury to HSCs after exposure to a high dose (>3.5 Gy) of TBI (158, 160).
Table 1.
Ionizing Radiation Dose and Acute Radiation Syndrome
| ARS | ||||
|---|---|---|---|---|
| IR dose (Gy) | Hematologic | Gastrointestinal | Neurologic | Prognosis |
| <1.0 | + | − | − | ∼100% survival |
| 1.0–2.0 | + | − | − | >90% survival |
| 2.0–3.5 | ++ | − | − | 90%–50% survival |
| 3.5–5.5 | +++ | + | − | 50% death within 3.5–6 weeks |
| 5.5–7.5 | +++ | ++ | − | death probable in 2–3 weeks |
| 7.5–10 | +++ | +++ | − | death probable in 1–2.5 weeks |
| 10–20 | +++ | +++ | +++ | death certain in 5–12 days |
| >20 | +++ | +++ | +++a | death certain in 2–5 days |
plus cardiovascular syndrome.
−, no effects;+, mild;++, moderate;+++, severe.
Modification from Waselenko et al. (162).
In addition, exposure to a moderate- or high-dose TBI also induces residual (or long-term) BM injury manifested by a decrease in HSC reserves and fitness and an impairment in HSC self-renewal (42, 104, 147, 153). Unlike acute BM injury, residual BM damage is latent. Patients and animals with residual BM injury usually have normal blood cell counts under normal homeostatic conditions despite a decrease in HSC reserves (104, 147). Because of this latency, the clinical implications of residual BM injury have been largely overlooked. Moreover, the importance of long-term BM damage is further obscured by the seemingly complete recovery of peripheral blood cell counts, BM cellularity, and the number of CFU, especially after the treatment with hematopoietic growth factors. In fact, the use of hematopoietic growth factors may worsen IR- and chemotherapy-induced residual BM damage by promoting HSC and HPC proliferation and differentiation at the expense of HSC self-renewal (59, 105, 108). This could lead to an accelerated exhaustion of HSCs and further compromise the long-term recovery of BM hematopoietic function. Although residual BM damage is latent, it is long lasting, shows little tendency for recovery, and can lead to the development of hypoplastic anemia or a meylodysplastic syndrome at a later time or after additional hematopoietic stress, such as subsequent cycles of consolidation cancer treatment or BM transplantation (104).
Several mechanisms have been proposed to explain how IR as well as some chemotherapeutic agents induces HSC injury (Fig. 4), including (i) quantitative reduction in HSCs due to induction of cell death or apoptosis and differentiation; (ii) qualitative changes in HSC replicative function resulting from induction of HSC senescence; and/or (iii) damage to BM stromal cells or the HSC niche (37, 108, 109). Because BM failure resulting from severe damage to HSCs is the primary life-threatening injury after exposure to a moderate or high dose of IR, understanding the mechanisms by which IR causes HSC injury can lead to the development of new medical countermeasures against IR. Therefore, we will discuss each of these mechanisms in more details below.
FIG. 4.
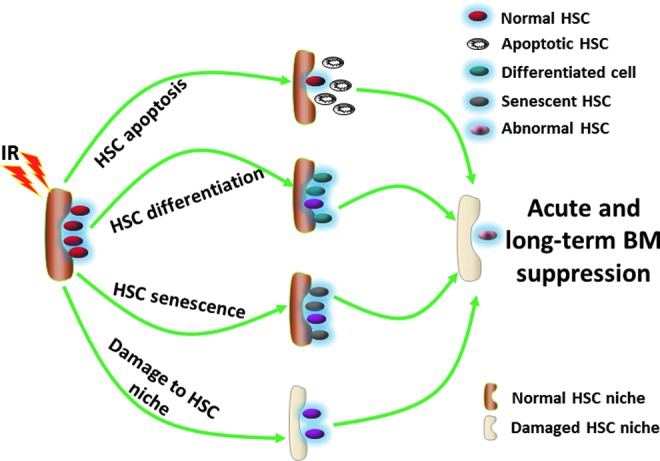
Mechanisms of IR-induced HSC damage and bone marrow (BM) injury. The mechanisms include IR-induced: (i) HSC apoptosis; (ii) HSC differentiation; (iii) HSC senescence; and (iv) damage to the HSC niche.
IR-Induced Apoptosis in HSCs
Apoptosis is an orderly and regulated form of cell death via a genetically controlled process (76, 101). The characteristics of an apoptotic cell include externalization of phosphatidylserine on the outer leaflet of the plasma membrane, cell shrinkage, condensation of the nuclear chromatin, fragmentation of the nucleus and DNA, and cellular membrane blebbing (76, 101). In coordination with cell proliferation and differentiation, apoptosis contributes to the maintenance of hematopoietic homeostasis by regulating the size of hematopoietic lineages (164). Dysregulation of apoptosis in hematopoietic cells can result in many pathological conditions (164). It has been suggested that the induction of apoptosis in BM cells, including HSCs, may be primarily responsible for the induction of ARS in the hematopoietic system after TBI (34, 104). The evidence that IR can damage HSCs by induction of apoptosis is increasing. First, IR is a potent inducer of apoptosis in many different cell types (55). Second, overexpression of an antiapoptotic protein, bcl-2, throughout the hematopoietic compartment protects mice against IR-induced hematopoietic failure and death (34). HSCs isolated from bcl-2 transgenic mice are more resistant to IR-induced damage in vitro (34). In contrast, bcl-2 deficiency sensitizes murine HSCs to IR (65). Third, HSCs from p53-deficient mice are less sensitive to IR than are those from wild-type mice (29, 63, 86) and treatment with a p53 inhibitor protected mice from IR-induced lethal damage by suppression of p53-dependent apoptosis (81). In addition, using flow cytometric analysis of Annexin V and/or 7-AAD staining in LSK cell population enriched with HSCs (56, 122), we demonstrated that IR-induced cell death in LSK cells mainly by apoptosis but not by necrosis (108, 109).
More recently, several groups of investigators reported that Puma, a downstream target of p53 and a proapoptotic BH3-only protein, plays a critical role in mediating IR-induced HSC apoptosis and ARS (140, 172). They showed that Puma was selectively induced by IR in LSK cells and LSK cells from Puma knockout mice were insensitive to IR-induced apoptosis. As such, Puma deficiency in mice confers resistance to high-dose radiation in a hematopoietic cell-autonomous manner. In contrast, other p53 targets, such as Bim and Noxa, play a minor or moderate role in IR-induced apoptosis in hematopoietic cells (41, 84). It has been well established that Puma can block the interactions of antiapoptotic proteins with the proapoptotic effectors, Bax and Bak. This leads to the interruption of mitochondrial integrity and mitochondrial release of caspase-activating factors, such as cytochrome c and Apaf-1. In turn, cytochrome c and Apaf-1 activate the initiator caspase-9 and then the effector caspases (e.g., caspases-3, -6, and -7) whose activation leads to the final stage of cell-self destruction (98) (Fig. 5). We assumed that this caspase cascade may also play an important role in mediating IR-induced HSC apoptosis downstream of Puma. This assumption is supported by our observation that inhibition of caspase activities with z-VAD abrogated IR-induced LSK cell apoptosis. In addition, z-VAD treatment also prevented the IR-induced decrease in LSK cell numbers and significantly attenuated the IR-induced reduction in day-28 and -35 cobblestone area-forming cells that correspond to the primitive HSCs with long-term repopulating ability (108). Together, these findings suggest that induction of HSC apoptosis not only is one of the primary causes of IR-induced HSC depletion but also significantly contributes to IR-induced ARS in the hematopoietic system. All these findings indicate that targeting the p53-Puma pathway and inhibition of caspases may represent a novel strategy to protecting HSCs from IR injury, particularly considering that transient inhibition of p53 activity with an inhibitor did not increase IR-induced carcinogenesis while Puma knockout actually reduced IR-induced tumorigenesis in mice (27, 84, 111).
FIG. 5.
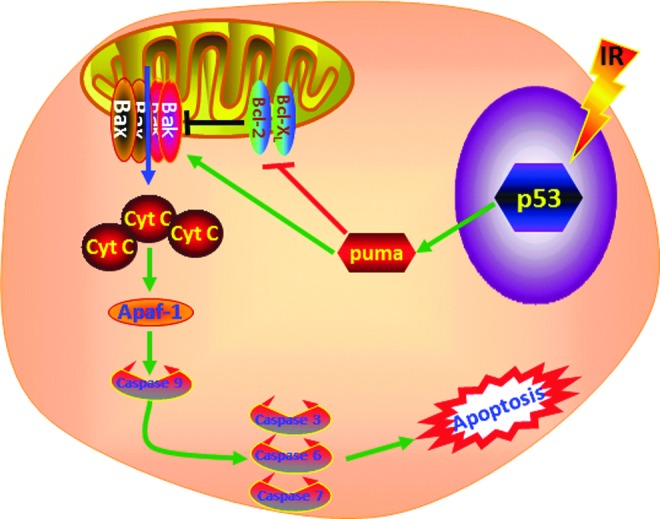
Mechanisms of IR-induced HSC apoptosis. As shown in Figure 3, IR-induced DDR includes the activation of p53 and induction of the Puma, a downstream target of p53. Puma can block the interactions of antiapoptotic proteins such as Bcl-2 and Bcl-xl with the proapoptotic effectors Bax and Bak. This leads to the interruption of mitochondrial integrity and mitochondrial release of caspase-activating factors, such as cytochrome c and Apaf-1. In turn, cytochrome c and Apaf-1 activate the initiator caspase-9 and then the effector caspases (e.g., caspases-3, -6 and -7) whose activation leads to the final stage of cell-self destruction of HSCs by apoptosis.
IR-Induced HSC Differentiation
HSCs have the ability to not only self-renewal but also differentiate to different lineages of blood cells. These two functions have to be tightly regulated because inhibition of HSC differentiation can increase HSC self-renewal that may result in abnormal HSC expansion and leukemia, whereas promotion of HSC differentiation can decrease HSC self-renewal that may lead to HSC premature exhaustion and BM failure. Furthermore, HSC differentiation to different lineages of blood cells has to be balanced to prevent lineage skewing. An increasing body of evidence demonstrates that DDR plays an important role in the regulation of stem cell differentiation. For example, Lin et al. (92) reported that the induction of DSBs by doxorubicin promoted mouse embryonic stem cell (ESC) differentiation in a p53-dependent manner (Fig. 6). This is because the activation of p53 downregulates the expression of Nanog that is required for ESC self-renewal. Inomata et al. (66) showed that exposure of melanocyte stem cells (MSCs) to IR abrogated MSC self-renewal by the induction of MSC differentiation in the hair follicle bugle without affecting their apoptosis or senescence, resulting in MSC depletion and hair graying. IR-induced MSC differentiation can be enhanced by ATM deficiency, suggesting that DDR initiated by ATM plays an important role in protecting MSCs from premature differentiation in response to genotoxic stress. In addition, the results from several studies also demonstrate that neuronal stem cell (NSC) differentiation can be regulated by IR. Ozeki et al. reported that exposure to IR accelerated astrocyte differentiation of surviving NSCs (126). Similar finding was also observed in subventricular zone NSCs in which p53 activity increased after brain irradiation, and loss of p53 resulted in increased NSC proliferation and self-renewal but impaired neuronal differentiation (47). However, the opposite effect of IR on NSC differentiation was also reported in several studies in which it was shown that IR can inhibit NSC differentiation in vitro (75) and in vivo (114). The contradictory effects of IR on NSC differentiation may be attributable to the doses of IR and cellular origins of NSCs used in those different studies.
FIG. 6.
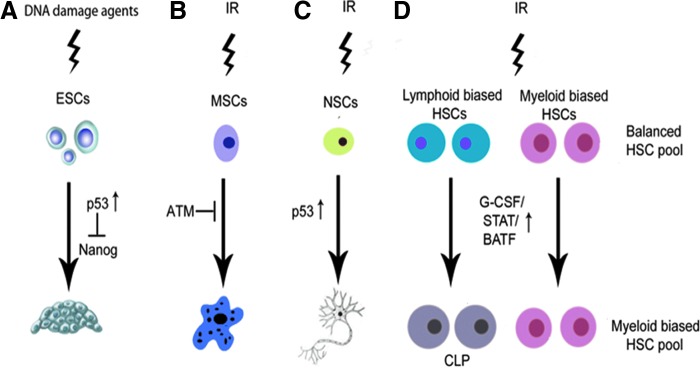
Mechanisms of DNA damage and IR-induced stem cell differentiation. (A) DNA damage in embryonic stem cells (ESCs) activates p53 to promote ESC differentiation by downregulating the expression of Nanog. (B) IR promotes melanocyte stem cell (MSC) differentiation, which can be repressed by ATM. (C) IR increases neuronal stem cell (NSC) differentiation in a p53-dependent manner. (D) IR promotes HSC differentiation to CLPs by activating the G-CSF/STAT/BATF-dependent differentiation checkpoint, resulting in a significant reduction in HSCs, particularly in the lymphoid biased pool of HSC, and myeloid skewing.
However, whether IR affects HSC differentiation was not known until a recent study reported by Wang et al. (155). In this study, they showed that DNA damage in HSCs resulting from telomere dysfunction and after exposure to IR depleted HSCs by promoting HSC differentiation to lymphoid lineage. The enhanced lymphoid differentiation was attributed to the activation of the granulocyte colony-stimulating factor (G-CSF)/Stat3/BATF-dependent differentiation checkpoint in HSCs. G-CSF knockout, Stat3 knockdown, or BATF deletion improved HSC self-renewal and function in response to IR or telomere shortening. More importantly, it was found that lymphoid-biased HSCs were more sensitive to the induction of differentiation after the activation of the G-CSF/Stat3/BATF-dependent differentiation checkpoint by IR than myeloid-biased HSCs, resulting in myeloid skewing in irradiated mice. These findings demonstrate that the induction of HSC differentiation via the G-CSF/Stat3/BATF pathway not only plays an important role in mediating IR-induced HSC injury but also is a causal factor for IR-induced myeloid lineage skewing. Although targeting the G-CSF/Stat3/BATF-pathway can inhibit IR-induced HSC injury via the inhibition of IR-induced HSC differentiation, it also results in the accumulation of DNA damage in HSCs (155), which may increase the risk of HSC transformation and leukemia development after IR. Therefore, inhibitors of this pathway may have limited utility as radiation protectants.
IR-Induced Senescence in HSCs
Cellular senescence
In the early 1960s, Hayflick and Moorhead demonstrated that normal human diploid fibroblasts have finite growth potential (73). The intrinsic replicative lifespan of a cell appears to be determined by telomere length (20). Without the expression of telomerase, telomeric sequences shorten each time DNA replicates. When the telomeres reach a critically short length (∼4 kb) after a certain number of cell doublings, known as the Hayflick limit, cells stop dividing and are irreversibly arrested at the G1 phase, entering replicative senescence (20, 102). The occurrence of replicative senescence has been demonstrated for most cell types. Exceptions include ESCs and the majority of tumor-derived cell lines (20, 102). This is because these cells express high levels of telomerase. A moderate level of telomerase activity is detectable in HSCs (3, 52, 54, 134). This activity is needed to maintain the normal function of HSCs, as a deficiency in telomerase activity can lead to telomere shortening and reduction in HSC transplantation ability, as seen in late generations of telomerase RNA component (TERC) null mice (54). In addition, the development of aplastic anemia or marrow failure has been observed in patients with telomerase deficiency, due to mutations in telomerase reverse transcriptase (TERT) or TERC (168). However, overexpression of TERT in HSCs maintains the length of HSC telomeres but fails to extend HSC lifespan in a serial BM transplantation setting (4). It is therefore the subject of intense debate whether HSCs are characterized by finite cell replicative ability and undergo replicative senescence after extensive proliferation (2, 39).
In addition, many types of human and animal cells undergo senescence after exposure to oxidative and genotoxic stress (including IR) in a species-independent manner (138). This also occurs when cells are subjected to an oncogenic stress and/or aberrant activation of the p38 pathway (138). The senescence induced by oxidative, genotoxic, and oncogenic stress is also referred to as premature senescence to differentiate it from replicative senescence (102, 138). This is because cells undergoing stress-induced premature senescence have a shortened intrinsic replicative lifespan without significant erosion in telomeres. However, cells undergoing premature senescence are morphologically indistinguishable from replicatively senescent cells and exhibit many of the characteristics ascribed to replicatively senescent cells (20, 102, 138). These changes include an enlarged and flattened appearance, increased senescence-associated β-galactosidase (SA-β-gal) activity, and elevated expression of p16 (20, 102, 138). Moreover, premature and replicative senescence share common induction pathways (20, 102, 138). Therefore, senescence has been frequently used as a general term to describe cells undergoing either premature or replicative senescence.
HSC senescence
The first evidence that HSCs can undergo senescence was observed in Bmi1−/− mice. It was found that mice lacking the Bmi1 gene developed progressive BM hypoplasia and died early (<2 months) after birth (31, 87). Although Bmi1−/− mice had a normal pool of fetal liver HSCs, transplantation of their fetal liver HSCs to a lethally irradiated recipient resulted only in a transient reconstitution of the hematopoietic system (31, 87). This suggests that the mutant fetal liver HSCs have the ability to proliferate and differentiate into HPCs enabling transient reconstitution of the BM but cannot self-renew and generate HSCs to ensure long-term hematopoietic engraftment. Deficiency in self-renewal was also found in neural and leukemia stem cells lacking Bmi1, indicating that Bmi1 is a general regulator of stem cell self-renewal (31, 87, 113). Bmi1 is a member of the Polycomb group of transcriptional repressors. Its downstream targets include the gene products of the Ink4a/Arf locus, for example, p16 and Arf. HSCs from Bmi1−/− mice express increased levels of p16 and Arf (31, 87, 113). Enforced expression of p16 and Arf in HSCs induces cell cycle arrest and apoptosis, respectively, whereas p16 knockout partially restores the ability of Bmi1−/− stem cells to self-renew (31, 87, 113).
Similarly, it has been hypothesized that IR and chemotherapy cause residual BM injury primarily by the induction of HSC senescence, which impairs HSC replication and self-renewal leading to the reduction in HSC reserves (59, 105, 119). Impairment in HSC self-renewal has been well documented in patients and animals after exposure to TBI or treatment with various chemotherapeutic agents that can cause residual BM injury (59, 105, 147, 151). For example, BM HSCs from mice after exposure to IR or receiving chemotherapy generated fewer CFU-S and repopulating units in lethally irradiated recipients after BM transplantation (45, 105, 119, 151). Similar impairments of HSC self-renewal capacity and long-term repopulating ability were observed in patients undergoing autologous transplantation after TBI and/or dose-intensified chemotherapy (22, 35, 36). However, direct evidence to demonstrate that HSCs undergo senescence after exposure to IR or a chemotherapeutic agent was lacking until our recent studies. In these studies, we found that exposure to IR or busulfan treatment induced HSC senescence in vitro and in vivo (108, 109). The senescent HSCs induced by IR and busulfan had diminished clonogenic activity and expressed increased levels of SA-β-gal, p16, and Arf. Interestingly, a shortening of the intrinsic replicative capacity of HSCs or loss of HSC self-renewal after exposure to IR does not affect HSC differentiation to generate various HPCs and more mature progeny before their final exhaustion. Moreover, HPCs from irradiated mice showed neither abnormalities nor did they exhibit the signs of senescence. These findings indicate that IR can selectively induce HSC senescence.
Reactive oxygen species
The mechanism by which IR induces HSC senescence remains to be elucidated but may be attributable to increased production of ROS because a large body of evidence suggests that ROS plays an important role in mediating the induction of HSC senescence under various pathological conditions (69, 112, 127, 149, 167). For example, it was found that ATM−/− mice exhibit progressive failure of hematopoietic function with aging (69). The failure is attributed primarily to HSC premature exhaustion or senescence resulting from an increased production of ROS, as treatment of ATM−/− mice with N-acetyl-cysteine (NAC) can restore the function of HSCs and prevent the development of BM failure. It was subsequently reported that triple-FoxO (FoxO1, FoxO3, and FoxO4) knockout mice also developed hematopoietic abnormalities (149). The number of HSCs and their long-term repopulating activity were markedly reduced after the deletion of FoxOs. These defects were associated with increased production of ROS in HSCs. Treatment of FoxOs knockout mice with NAC reversed the defects of HSCs and hematopoietic abnormalities. It appears that FoxO3 is the primary regulator of HSCs because the deletion of FoxO3 alone in mice can recapitulate most of the phenotypes observed in the triple-FoxO knockout mice (112, 167). The molecular basis for FoxOs to regulate ROS production in HSCs is mainly attributed to their transcriptional regulation of the expression of superoxide dismutase and catalase (149). In addition, there is a mechanistic link between ATM and FoxO3 in the regulation of ROS production in HSCs as FoxO3 is essential for ATM expression (167). Increased production of ROS in association with HSC defect has been observed in several other pathological conditions, such as deletion of Bmi1 (127, 136), MDM2 (1) and TSC1 (23), Fanconi anemia mutation (38), and aging (68). Similarly, we showed that exposure of mice to a sublethal dose of TBI induced a persistent increase in ROS production in HSCs (157). The induction of chronic oxidative stress was associated with sustained increases in oxidative DNA damage in HSCs, inhibition of HSC clonogenic function, and induction of HSC senescence (32, 90, 157). Treatment of the irradiated mice with NAC after TBI significantly attenuated IR-induced inhibition of HSC clonogenic function and reduction of HSC long-term engraftment after transplantation (157). These findings provide the foremost direct evidence demonstrating that TBI induces long-term BM suppression and HSC senescence, at least in part via induction of oxidative stress in HSCs. In addition, it suggests that antioxidants such as NAC may be used as an effective strategy to mitigate IR-induced residual BM injury.
Production of ROS is one of the by-products of mitochondrial respiration, and mitochondria have frequently been considered to be the main source of cellular-derived ROS (12). It has been shown that cells, including HSCs, from Bmi1−/− mice exhibit abnormal mitochondrial function resulting in increased production of ROS (94). In addition, increased production of ROS in HSCs from TSC1−/− mice has been attributed to the elevation of mitochondrial biogenesis and oxidative activities (23). However, compared to their progeny, HSCs are dormant and have fewer mitochondria (129, 130). It has also been shown that HSCs primarily utilize glycolysis rather than mitochondrial oxidative phosphorylation for adenosine triphosphate production (5). Thus, it has yet to be determined whether mitochondria play a major role in contributing to the increased production of ROS in HSCs under various pathological conditions. Recently, an increasing body of evidence demonstrates that cells can also actively produce ROS through a family of tightly regulated NADPH oxidases (NOXs) that are homologues of the phagocyte oxidase (Phox or NOX2) (16, 85). ROS produced by NOXs participate in the regulation of many cell functions and have been implicated in the pathogenesis of different diseases. Five different NOXs are expressed in different tissues or cells with distinctive functions and mechanisms of regulation in a tissue- or cell-specific manner (16, 85). The expression of NOX1, 2, and 4 and various regulatory subunits of NOXs has been detected in human HSCs (129, 130). It was estimated that NOX-mediated extramitochondrial oxygen consumption accounts for about half of the endogenous cell respiration in human HSCs (130). Interestingly, our recent studies showed that NOX1, 2, and 4 are also expressed in mouse BM HSC-enriched Lin−Sca1+c-kit+ cells, whereas HPCs, Lin− cells, and mononuclear cells from mouse BM express NOX1 and 2, but not NOX4, suggesting that the expression of NOX4 is downregulated upon HSC differentiation and that NOX4 may play an important role in the regulation of HSC function (157). More importantly, it was found that the expression of NOX4 was upregulated, whereas the expression NOX1 and 2 was unchanged in HSCs after TBI. Because NOX4 is a constitutively active NOX and ROS production by NOX4 is regulated at the transcriptional level (16, 85), the finding that IR upregulates NOX4 in HSCs implies that NOX4 may primarily mediate the IR-induced increase in ROS production in HSCs (Fig. 7). This suggestion is supported by the finding that diphenyleneiodonium, but not apocynin, inhibits IR-induced elevation of ROS production in HSCs because NOX4 is not sensitive to apocynin inhibition, whereas other NOXs are (16, 85, 137, 157). In addition, our recent study showed that resveratrol, a potent antioxidant and a putative activator of Sirtuin 1, can ameliorate TBI-induced long-term BM injury by inhibiting radiation-induced chronic oxidative stress and senescence in HSCs in part by the downregulation of NOX4 expression (173). However, additional experiments will be required to confirm the role of NOX4 in IR-induced chronic oxidative stress and senescence in HSCs by utilizing a more specific approach, such as genetically knocking out NOX4 in HSCs.
FIG. 7.
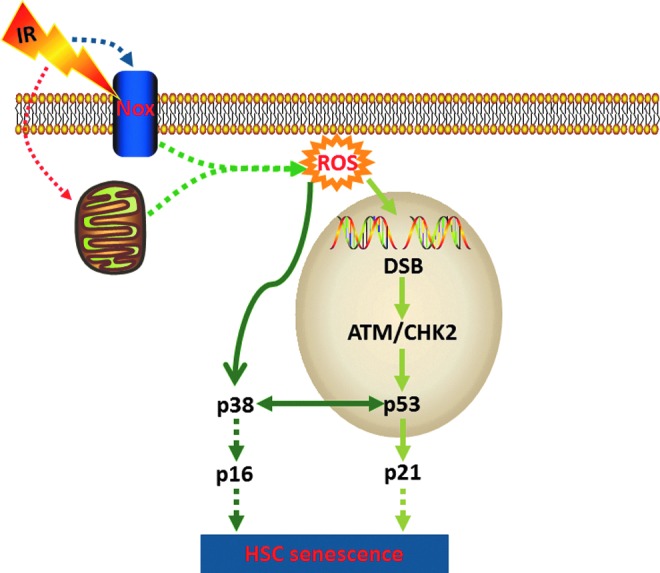
Hypothetic mechanisms of IR-induced HSC senescence. IR may increase the production of ROS by mitochondria and/or NADPH oxidases (NOXs) in HSCs. ROS can induce HSC senescence by stimulating the p38 pathway and/or activating the ATM-CHK2-p53-p21 pathway via induction of DSBs. Both pathways could converge at p16 for the induction of HSC senescence.
The specific species of ROS induced by IR in HSCs have yet to be determined but may include superoxide and hydrogen peroxide, because both of them can be produced by IR-induced dysfunctional mitochondria and NOX4. In addition, it has been shown that IR can increase the production of nitric oxide (NO) in BM stromal cells via NO synthases 1 and 2 (40, 51). However, whether NO also plays a role in mediating IR-induced damage to HSCs remains to be investigated. The mechanisms by which IR increases mitochondrial production of ROS have been extensively studied (11, 48), which may include increases in mitochondrial electron transport chain complex II activity due to mutations in succinate dehydrogenase subunit C (10) and cell cycle arrest (169) and Drp1-dependent mitochondrial fission (80). In contrast, little is known about how IR upregulates NOX4 expression at the present. Furthermore, an increasing body of literatures has shown that many of the DDR elements are also involved in sensing oxidative stress and regulating ROS production. For example, it has been shown that ATM can be directly activated by oxidation, which in turn can modulate oxidative stress by regulating mitochondrial function and metabolism via p53, AMP-activated protein kinase, mammalian target of rapamycin, and hypoxia-inducible factor 1 (33, 93). Therefore, it will be of a great interest to determine whether the activation of DDR by IR also contributes to the persistent increase in ROS production in HSCs via perturbation of mitochondrial function and/or upregulation of NOX4.
p38
An increasing body of evidence suggests that ROS cause damage to HSCs not by a nonspecific cytotoxic effect as previously hypothesized, but at least in part via the activation of p38 (139). p38 is a member of the MAPK family of signal transduction kinases, which can be activated in a sequential order (mitogen-activated or extracellular signal-regulated kinase kinase [MEKK]-MAPK kinase 3/6 [MKK3/6]-p38) after exposure to stress (83). In addition, oxidative stress can also activate p38 via the activation of apoptosis signal-regulating kinase 1 (ASK1) (103) and/or inactivation of protein tyrosine phosphatases (PTPs), such as MAPK phosphatases (77, 128). Normally, ASK-1 forms an inactive complex with the repressor protein thioredoxin in a cell. The formation of this complex is dependent on the presence of a reduced form of an intramolecular disulfide bridge between two cysteine residues of thioredoxin. Oxidation of thioredoxin by ROS causes the dissociation of ASK-1 from thioredoxin, resulting in the activation of ASK1 by oligomerization, interaction with TNF receptor-associated factor-2/6, and threonine autophosphorylation (103). It has been shown that ROS production from Nox4 can activate p38 via the activation of ASK-1 (24). In addition, oxidation of the catalytic cysteine of PTPs by ROS can reversibly inactivate PTPs (125), which in turn can increase p38 activity. It remains to be determined whether ROS can activate p38 in HSCs through which of these mechanisms.
Activation of p38 regulates a variety of cellular processes such as inflammation, cell cycle arrest, and apoptosis in a cell type-specific manner. There is an increasing body of evidence demonstrating that p38 plays a critical role in the induction of senescence in response to a variety of stimuli via upregulating p16 (6, 46). For example, it was shown that a high level of Ras or Raf activation in human normal fibroblasts induced senescence by stimulating a sustained activation of p38, which in turn upregulated the expression of p16 (6). Activation of the p38 pathway also contributes to the induction of p16 and cellular senescence after DNA damage resulting from exposure to genotoxic and oxidative stress and telomere shortening due to extensive replication (79, 99, 154) (Fig. 7). Furthermore, activation of p38 by ectopic transfection of MKK3 and/or MKK6 increases p16 expression and induces senescence. In contrast, inhibition of p38 activity or downregulation of p38 expression attenuates the induction of p16 and cellular senescence by oncogenic stress, DNA damage, and telomere shortening (70, 79, 99, 154).
In addition, activation of p38 has been implicated in BM suppression in various pathological conditions, including aplastic anemia and myelodysplastic syndromes (118, 175). Furthermore, recently, it was shown that mutation of the ATM gene and knockout of the FoxO3 gene induced premature senescence/exhaustion of HSCs (69, 112, 149). The induction of HSC senescence/exhaustion was associated with an elevated production of ROS, a selective activation of p38, and an upregulation of p16 in HSCs. Pharmacological inhibition of p38 activity rescued the defects of HSCs from ATM mutants and FoxO3 knockout mice (69, 112, 149). These findings indicate that p38 plays an important role in the regulation of HSC self-renewal and its activation by oxidative stress can mediate the induction of HSC senescence via regulation of p16 (68). Therefore, we recently examined whether IR causes hematopoietic suppression in part by inducing hematopoietic cell senescence through the activation of the p38 pathway and whether pharmacological inhibition of p38 can attenuate IR-induced residual BM injury (89, 156). In this study, we found that p38 was selectively activated in irradiated hematopoietic cells and this activation sustained up to 5 weeks after IR in a long-term BM cell culture assay. Inhibition of p38 activity with a specific inhibitor, for example, SB203580, attenuated IR-induced suppression of BM hematopoietic cell function in association with a significant reduction in p16 expression and SA-β-gal activity. Moreover, our in vivo data show that inhibition of p38 attenuated IR-induced residual BM suppression. These results suggest that p38 activation plays a role in mediating IR-induced hematopoietic cell senescence and BM suppression and that pharmacological inhibition of the p38 pathway with a specific inhibitor can be further exploited for amelioration of IR-induced residual BM injury (89, 156).
Ink4a and Arf
The Ink4a-Arf locus encodes two tumor suppressors, p16 and Arf (97, 141). The transcripts for these proteins have different first exons (α for p16 and β for Arf ) but share exons 2 and 3. However, there is no amino acid sequence similarity between these two proteins due to the use of alternative reading frames for their translation. p16 is a potent cyclin-dependent kinase (CDK) 4/6 inhibitor. By inhibiting CDK4/6 activity, p16 causes Rb hypophosphorylation and suppresses the expression of E2F-dependent genes, resulting in the restriction of G1/S cell cycle progression and the formation of senescence-associated heterochromatic foci (SAHF) (97, 117, 141). Once SAHF are formed after the engagement of the p16-Rb pathway, the cells become permanently growth arrested and senescent. It has therefore been suggested that diverse stimuli can induce cellular senescence via various upstream signal transduction cascades, including the p38 and p53-p21 pathways, that converge on the p16-Rb pathway, whose activation provides an inescapable barrier preventing senescent cells from re-entering the cell cycle. This suggestion is supported by the finding that the activation of p53 and induction of p21 in cells undergoing senescence are transient events that occur during the onset of senescence and then subside when the expression of p16 starts rising (19, 132, 146). Inactivation of p53 before upregulation of p16 can prevent senescence induction. However, once p16 is highly expressed, cell cycle arrest becomes irreversible by the downregulation of p53, indicating that the activation of the p53-p21 pathway plays an important role in the initiation of senescence, but induction of p16 is required for the maintenance of senescence (14, 19). In agreement with this suggestion, we found that IR induced p53 activation and p21 expression in HSCs before the induction of p16 (109, 160). Whereas p53 activation and p21 upregulation gradually declined within a few weeks after IR, p16 expression in irradiated HSCs remained elevated and the cells subsequently became senescent, exhibiting positive SA-β-gal staining (Fig. 7). In contrast, the biological action of Arf relies on the p53 pathway. This is because Arf can directly bind to MDM2 and cause the accumulation of p53 by segregating MDM2 from p53 and by inhibiting MDM2's E3 ubiquitin protein ligase activity for p53 (97, 141, 142). Therefore, the activation of p53 by Arf can induce not only cellular senescence but also apoptosis, depending on which gene down-stream of p53 is induced following its activation.
Upregulation of p16 and Arf has been implicated in mediating the induction of cellular senescence in a variety of cells including HSCs. For example, increased expression of p16 and Arf was found in HSCs from Bmi1−/− mice (127). However, it appears that p16 but not Arf plays an important role in mediating the induction of Bmi1−/− HSC senescence (127). In addition, it has been found that knockout of both the p16 and Arf genes in mice significantly increases the clonal expansion of HSCs in vitro but modestly promotes HSC self-renewal in vivo (87, 113). However, knockout of the Arf gene alone does not provide any advantage for HSC/HPC expansion and self-renewal (113). In contrast, knockout p16 increases the lifespan of HSCs by promoting HSC self-renewal (74, 144). Furthermore, mutation of the ATM gene also results in upregulation of p16 and Arf in HSCs (69, 113). Inactivation of the p16-Rb pathway by retroviral transfection of HPV E7 proteins restores the reproductive function of ATM−/− HSCs, whereas inhibition of the Arf-p53 pathway by E6 transfection has no such effect (67). These findings suggest that p16 plays a more significant role than Arf in regulation of HSC self-renewal and induction of HSC senescence, even though both proteins are overexpressed in senescent HSCs. Increased expression of p16 and Arf has been found in IR-induced senescent LSK cells (160). However, their roles in mediating IR-induced HSC senescence and long-term BM suppression remain to be investigated.
IR-Induced Damage to the HSC Niche
As discussed earlier in the review, a complex of cellular and molecular components constitutes the HSC niche in BM stroma and the HSC niche is crucial for the maintenance of HSC self-renewal capacity and reserves (7, 91, 165). IR-induced damage to various components of the HSC niche not only contributes to HSC injury but also affects the recovery of HSCs after IR. There is a large body of literature demonstrating that IR induces BM stroma injury in a dose- and time-dependent manner, which was extensively reviewed in previous publications (17, 53, 60, 104). IR-induced BM stromal cell damage is at least in part attributable to the induction of senescence because Carbonneau et al. (21) reported recently that exposure of mice to IR induced senescence in BM stromal cells in a p16/Arf-dependent manner. The induction of BM stromal cell senescence contributes to IR-induced residual damage to BM environment that can influence hematopoiesis.
However, IR damage to specific components of the HSC niche and the impact of the damage on HSCs were not known until recently. Hooper et al. (64) reported that BM SECs are highly sensitive to IR. They showed that exposure of mice to a sublethal dose of TBI resulted in regression of SECs while exposure to a lethal dose of TBI induced severe damage to SECs and required BM transplantation to regenerate. SEC damage induced by IR may be mediated in part by the activation of acid sphingomyelinase (aSMase), which induces endothelial cell apoptosis via increased production of ceramide (135). Inhibition of SECs regeneration via blocking vascular endothelial growth factor receptor 2 signaling in irradiated mice prevented hematopoietic reconstitution (Fig. 8). In contrast, transplantation of endothelial cells into lethally irradiated mice improved their survival by promoting HSC regeneration and hematopoietic recovery (28, 88). Similarly, infusion of endothelial progenitor cells into irradiated mice not only accelerated the recovery of the vascular niche but also promoted HSC reconstitution (133). The mechanisms by which SECs promote HSC reconstitution after IR remain to be elucidated but may be partially attributable to their expression of angiopoietin-like protein 3 (28, 174) and pleiotrophin (61, 62). Compared to BM SECs, endosteal osteoblasts, a major component of the osteoblastic niche, are relatively radioresistant. After BM radioablation, endosteal osteoblasts underwent rapid expansion in response to megakaryocyte-derived mesenchymal growth factors such as platelet-derived growth factor-β and basic fibroblast growth factor to promote HSC engraftment and hematopoietic reconstitution after BM transplantation by restoring the damaged HSC niche (37). However, it is not known whether the stimulation of osteoblast proliferation by IR can result in induction of replicative senescence as reported by Carbonneau et al. (21). In addition, whether IR can also cause damage to other cells in the HSC niche has yet to be determined.
FIG. 8.
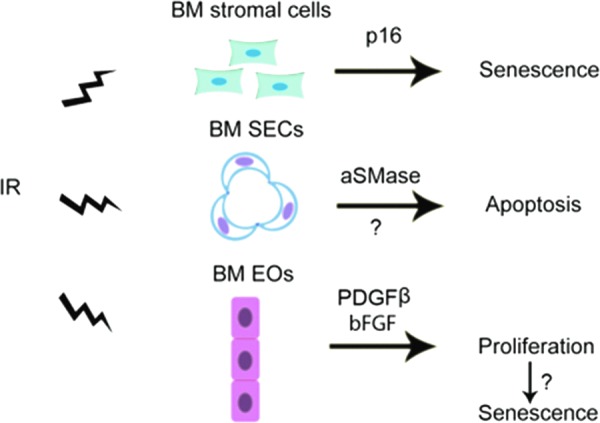
Mechanisms of IR-induced damage to the HSC niche. IR can damage the HSC niche by: (i) induction of BM stromal cell senescence in a p16/Arf-dependent manner; (ii) induction of SEC apoptosis probably via activation of acidic sphingomyelinase (aSMase); and (iii) stimulation of endosteal osteoblast (EO) proliferation through megakaryocyte-derived mesenchymal growth factors, such as platelet-derived growth factor-β (PDGFβ) and basic fibroblast growth factor (bFGF), which may eventually lead to osteoblast senescence.
Conclusion
Exposure to IR as the result of nuclear accidents or terrorist attacks is a significant threat and a major medical concern. When such an event occurs, many victims may receive a moderate or high dose of TBI that can cause significant civilian casualties due to IR-induced normal tissue damage. Among various tissues and organs, BM is the most radiosensitive tissue in the body. Acute and transient BM suppression typically results from exposure to a moderate dose of TBI, which primarily damages HPCs and to a lesser degree HSCs. In this circumstance, HSCs can undergo self-renewing proliferation and differentiation to repopulate HPCs and restore hematopoietic homeostasis. However, if the dose of TBI is too high, IR also severely damages HSCs and impairs their ability to self-renew by induction of HSC apoptosis, differentiation, and senescence and damage to the HSC niche, which may eventually lead to BM failure and organism death. Because HSC injury is the primary cause of death after accidental or intentional exposure to a moderate or high dose of IR, protecting HSCs from IR should be a primary goal in the development of novel medical countermeasures against radiation. This development depends on a better understanding of the mechanisms by which IR causes HSC damage. Therefore, in this review, we have focused our discussions on a number of recent findings that provide new insights into the mechanisms whereby IR damages HSCs. These findings will provide new opportunities for developing a mechanism-based strategy to prevent and mitigate IR-induced BM suppression.
Abbreviations Used
- ARS
acute radiation syndrome
- ASK1
apoptosis signal-regulating kinase 1
- ATM
ataxia telangiectasia mutated
- BM
bone marrow
- CDK
cyclin-dependent kinase
- CFU-S
colony-forming unit-spleen
- CLPs
common lymphoid progenitors
- DDR
DNA damage response
- DSB
double-strand break
- ESC
embryonic stem cell
- G-CSF
granulocyte colony-stimulating factor
- HPC
hematopoietic progenitor cell
- HSC
hematopoietic stem cell
- IR
ionizing radiation
- LSK
Lin−sca1+c-kit+ cells
- MAPK
mitogen-activated protein kinase
- MPP
multipotent progenitor
- MSC
melanocyte stem cell
- NAC
N-acetyl-cysteine
- NO
nitric oxide
- NOX
NADPH oxidase
- NSC
neuronal stem cell
- PTP
protein tyrosine phosphatases
- ROS
reactive oxygen species
- SA-β-gal
senescence-associated β-galactosidase
- SAHF
senescence-associated heterochromatic foci
- SEC
sinusoidal endothelial cell
- TBI
total body irradiation
- TERC
telomerase RNA component
- TERT
telomerase reverse transcriptase
Acknowledgments
We apologize to authors whose contributions were not directly cited owing to space limitations. The authors thank the previous and current members of Dr. Zhou's Laboratory for their work and support. The research conducted in Dr. Zhou's Laboratory was supported in part by grants from the National Institutes of Health (R01-CA122023 and AI080421), a grant from the National Natural Science Foundation of China (NSFC 30828011), and a grant from the Edward P. Evan's Foundation, the Winthrop Rockefeller Endowment for Leukemia Research, and the Arkansas Research Alliance Scholarship from the Arkansas Science & Technology Authority.
References
- 1.Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, You MJ, Hirschi KK, and Lozano G. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7: 606–617, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allsopp RC. and Weissman IL. Replicative senescence of hematopoietic stem cells during serial transplantation: does telomere shortening play a role? Oncogene 21: 3270–3273, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Allsopp RC, Morin GB, DePinho R, Harley CB, and Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 102: 517–520, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Allsopp RC, Morin GB, Horner JW, DePinho R, Harley CB, and Weissman IL. Effect of TERT over-expression on the long-term transplantation capacity of hematopoietic stem cells. Nat Med 9: 369–371, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Andersen CA. Noninvasive assessment of lower extremity hemodynamics in individuals with diabetes mellitus. J Vasc Surg 52: 76S–80S, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Antonchuk J, Sauvageau G, and Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell 109: 39–45, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Arai F. and Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci 1106: 41–53, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, and Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118: 149–161, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, Jin DK, Dias S, Zhang F, Hartman TE, Hackett NR, Crystal RG, Witte L, Hicklin DJ, Bohlen P, Eaton D, Lyden D, de Sauvage F, and Rafii S. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10: 64–71, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Aykin-Burns N, Slane BG, Liu AT, Owens KM, O'Malley MS, Smith BJ, Domann FE, and Spitz DR. Sensitivity to low-dose/low-LET ionizing radiation in mammalian cells harboring mutations in succinate dehydrogenase subunit C is governed by mitochondria-derived reactive oxygen species. Radiat Res 175: 150–158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azzam EI, Jay-Gerin JP, and Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 327: 48–60, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balaban RS, Nemoto S, and Finkel T. Mitochondria, oxidants, and aging. Cell 120: 483–495, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Barria E, Mikels A, and Haas M. Maintenance and self-renewal of long-term reconstituting hematopoietic stem cells supported by amniotic fluid. Stem Cells Dev 13: 548–562, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, and Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J 22: 4212–4222, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becker AJ, Mcculloch EA, and Till JE. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 197: 452–454, 1963 [DOI] [PubMed] [Google Scholar]
- 16.Bedard K. and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Bierkens JG, Hendry JH, and Testa NG. The radiation response and recovery of bone marrow stroma with particular reference to long-term bone marrow cultures. Eur J Haematol 43: 95–107, 1989 [DOI] [PubMed] [Google Scholar]
- 18.Broudy VC. Stem cell factor and hematopoiesis. Blood 90: 1345–1364, 1997 [PubMed] [Google Scholar]
- 19.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120: 513–522, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Campisi J, Kim SH, Lim CS, and Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol 36: 1619–1637, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Carbonneau CL, Despars G, Rojas-Sutterlin S, Fortin A, Le O, Hoang T, and Beausejour CM. Ionizing radiation-induced expression of INK4a/ARF in murine bone marrow-derived stromal cell populations interferes with bone marrow homeostasis. Blood 119: 717–726, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Cartron G, Herault O, Benboubker L, Clement N, Bernard MC, Roingeard F, Desbois I, Colombat P, Binet C, and Domenech J. Quantitative and qualitative analysis of the human primitive progenitor cell compartment after autologous stem cell transplantation. J Hematother Stem Cell Res 11: 359–368, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, and Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205: 2397–2408, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, and David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J Immunol 176: 5720–5724, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, Tanaka M, Merad M, and Frenette PS. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208: 261–271, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen JL. and Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc Natl Acad Sci U S A 98: 14541–14546, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, and Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443: 214–217, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Chute JP, Muramoto GG, Salter AB, Meadows SK, Rickman DW, Chen B, Himburg HA, and Chao NJ. Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 109: 2365–2372, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui YF, Zhou PK, Woolford LB, Lord BI, Hendry JH, and Wang DW. Apoptosis in bone marrow cells of mice with different p53 genotypes after gamma-rays irradiation in vitro. J Environ Pathol Toxicol Oncol 14: 159–163, 1995 [PubMed] [Google Scholar]
- 30.Dainiak N. Hematologic consequences of exposure to ionizing radiation. Exp Hematol 30: 513–528, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Danet GH, Pan Y, Luongo JL, Bonnet DA, and Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest 112: 126–135, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diaz-Montero CM, Wang Y, Shao L, Feng W, Zidan AA, Pazoles CJ, Montero AJ, and Zhou D. The glutathione disulfide mimetic NOV-002 inhibits cyclophosphamide-induced hematopoietic and immune suppression by reducing oxidative stress. Free Radic Biol Med 52: 1560–1568, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ditch S. and Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 37: 15–22, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domen J, Gandy KL, and Weissman IL. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood 91: 2272–2282, 1998 [PubMed] [Google Scholar]
- 35.Domenech J, Cartron G, Clement N, Estienne MH, Herault O, Truglio D, Benboubker L, Roingeard F, Desbois I, Colombat P, and Binet C. Persistent decrease in proliferative potential of marrow CD34(+)cells exposed to early-acting growth factors after autologous bone marrow transplantation. Bone Marrow Transplant 29: 557–562, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Domenech J, Linassier C, Gihana E, Dayan A, Truglio D, Bout M, Petitdidier C, Delain M, Petit A, Bremond JL, and Et A. Prolonged impairment of hematopoiesis after high-dose therapy followed by autologous bone marrow transplantation. Blood 85: 3320–3327, 1995 [PubMed] [Google Scholar]
- 37.Dominici M, Rasini V, Bussolari R, Chen X, Hofmann TJ, Spano C, Bernabei D, Veronesi E, Bertoni F, Paolucci P, Conte P, and Horwitz EM. Restoration and reversible expansion of the osteoblastic hematopoietic stem cell niche after marrow radioablation. Blood 114: 2333–2343, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du W, Adam Z, Rani R, Zhang X, and Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal 10: 1909–1921, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Effros RB. and Globerson A. Hematopoietic cells and replicative senescence. Exp Gerontol 37: 191–196, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Epperly MW, Cao S, Zhang X, Franicola D, Shen H, Greenberger EE, Epperly LD, and Greenberger JS. Increased longevity of hematopoiesis in continuous bone marrow cultures derived from NOS1 (nNOS, mtNOS) homozygous recombinant negative mice correlates with radioresistance of hematopoietic and marrow stromal cells. Exp Hematol 35: 137–145, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, Adams JM, Strasser A, and Villunger A. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood 106: 4131–4138, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleenor CJ, Marusyk A, and DeGregori J. Ionizing radiation and hematopoietic malignancies: altering the adaptive landscape. Cell Cycle 9: 3005–3011, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ford CE, Hamerton JL, Barnes DW, and Loutit JF. Cytological identification of radiation-chimaeras. Nature 177: 452–454, 1956 [DOI] [PubMed] [Google Scholar]
- 44.Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo CC, Tsuyuzaki H, Sato T, Cote D, Sykes M, Strom TB, Scadden DT, and Lin CP. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474: 216–219, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner RV, Lerner C, Astle CM, and Harrison DE. Assessing permanent damage to primitive hematopoietic stem cells after chemotherapy using the competitive repopulation assay. Cancer Chemother Pharmacol 32: 450–454, 1993 [DOI] [PubMed] [Google Scholar]
- 46.Geest CR. and Coffer PJ. MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol 86: 237–250, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, and Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci 26: 1107–1116, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gius D. and Spitz DR. Redox signaling in cancer biology. Antioxid Redox Signal 8: 1249–1252, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Goldman DC, Bailey AS, Pfaffle DL, Al MA, Christian JL, and Fleming WH. BMP4 regulates the hematopoietic stem cell niche. Blood 114: 4393–4401, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodell MA, Brose K, Paradis G, Conner AS, and Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183: 1797–1806, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorbunov NV, Pogue-Geile KL, Epperly MW, Bigbee WL, Draviam R, Day BW, Wald N, Watkins SC, and Greenberger JS. Activation of the nitric oxide synthase 2 pathway in the response of bone marrow stromal cells to high doses of ionizing radiation. Radiat Res 154: 73–86, 2000 [DOI] [PubMed] [Google Scholar]
- 52.Goytisolo FA, Samper E, Martin-Caballero J, Finnon P, Herrera E, Flores JM, Bouffler SD, and Blasco MA. Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J Exp Med 192: 1625–1636, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenberger JS. Toxic effects on the hematopoietic microenvironment. Exp Hematol 19: 1101–1109, 1991 [PubMed] [Google Scholar]
- 54.Greenwood MJ. and Lansdorp PM. Telomeres, telomerase, and hematopoietic stem cell biology. Arch Med Res 34: 489–495, 2003 [DOI] [PubMed] [Google Scholar]
- 55.Harms-Ringdahl M, Nicotera P, and Radford IR. Radiation induced apoptosis. Mutat Res 366: 171–179, 1996 [DOI] [PubMed] [Google Scholar]
- 56.Hasper HJ, Weghorst RM, Richel DJ, Meerwaldt JH, Olthuis FM, and Schenkeveld CE. A new four-color flow cytometric assay to detect apoptosis in lymphocyte subsets of cultured peripheral blood cells. Cytometry 40: 167–171, 2000 [PubMed] [Google Scholar]
- 57.Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, Hicklin DJ, Zhu Z, Bohlen P, Witte L, Hendrikx J, Hackett NR, Crystal RG, Moore MA, Werb Z, Lyden D, and Rafii S. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med 8: 841–849, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haug JS, He XC, Grindley JC, Wunderlich JP, Gaudenz K, Ross JT, Paulson A, Wagner KP, Xie Y, Zhu R, Yin T, Perry JM, Hembree MJ, Redenbaugh EP, Radice GL, Seidel C, and Li L. N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell 2: 367–379, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Hellman S. and Botnick LE. Stem cell depletion: an explanation of the late effects of cytotoxins. Int J Radiat Oncol Biol Phys 2: 181–184, 1977 [DOI] [PubMed] [Google Scholar]
- 60.Hendry JH. The cellular basis of long-term marrow injury after irradiation. Radiother Oncol 3: 331–338, 1985 [DOI] [PubMed] [Google Scholar]
- 61.Himburg HA, Harris JR, Ito T, Daher P, Russell JL, Quarmyne M, Doan PL, Helms K, Nakamura M, Fixsen E, Herradon G, Reya T, Chao NJ, Harroch S, and Chute JP. Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep 2: 964–975, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, Chi JT, Salter AB, Lento WE, Reya T, Chao NJ, and Chute JP. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med 16: 475–482, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hirabayashi Y, Matsuda M, Matumura T, Mitsui H, Sasaki H, Tukada T, Aizawa S, Yoshida K, and Inoue T. The p53-deficient hemopoietic stem cells: their resistance to radiation-apoptosis, but lasted transiently. Leukemia 11Suppl 3: 489–492, 1997 [PubMed] [Google Scholar]
- 64.Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, Kopp HG, Shido K, Petit I, Yanger K, James D, Witte L, Zhu Z, Wu Y, Pytowski B, Rosenwaks Z, Mittal V, Sato TN, and Rafii S. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 4: 263–274, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoyes KP, Cai WB, Potten CS, and Hendry JH. Effect of bcl-2 deficiency on the radiation response of clonogenic cells in small and large intestine, bone marrow and testis. Int J Radiat Biol 76: 1435–1442, 2000 [DOI] [PubMed] [Google Scholar]
- 66.Inomata K, Aoto T, Binh NT, Okamoto N, Tanimura S, Wakayama T, Iseki S, Hara E, Masunaga T, Shimizu H, and Nishimura EK. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 137: 1088–1099, 2009 [DOI] [PubMed] [Google Scholar]
- 67.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, and Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431: 997–1002, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, and Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12: 446–451, 2006 [DOI] [PubMed] [Google Scholar]
- 69.Ito K, Takubo K, Arai F, Satoh H, Matsuoka S, Ohmura M, Naka K, Azuma M, Miyamoto K, Hosokawa K, Ikeda Y, Mak TW, Suda T, and Hirao A. Regulation of reactive oxygen species by Atm is essential for proper response to DNA double-strand breaks in lymphocytes. J Immunol 178: 103–110, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Iwasa H, Han J, and Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 8: 131–144, 2003 [DOI] [PubMed] [Google Scholar]
- 71.Jacobson LO. Evidence for a humoral factor (or factors) concerned in recovery from radiation injury: a review. Cancer Res 12: 315–325, 1952 [PubMed] [Google Scholar]
- 72.Jacobson LO. Hematopoietic responses to radiation injury. Annu Rev Med 7: 345–352, 1956 [DOI] [PubMed] [Google Scholar]
- 73.Hayflick L. and Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585–621, 1961 [DOI] [PubMed] [Google Scholar]
- 74.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, and Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443: 421–426, 2006 [DOI] [PubMed] [Google Scholar]
- 75.Kanzawa T, Iwado E, Aoki H, Iwamaru A, Hollingsworth EF, Sawaya R, Kondo S, and Kondo Y. Ionizing radiation induces apoptosis and inhibits neuronal differentiation in rat neural stem cells via the c-Jun NH2-terminal kinase (JNK) pathway. Oncogene 25: 3638–3648, 2006 [DOI] [PubMed] [Google Scholar]
- 76.Kerr JF, Wyllie AH, and Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev 27: 253–261, 2008 [DOI] [PubMed] [Google Scholar]
- 78.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121: 1109–1121, 2005 [DOI] [PubMed] [Google Scholar]
- 79.Kirito K, Fox N, and Kaushansky K. Thrombopoietin stimulates Hoxb4 expression: an explanation for the favorable effects of TPO on hematopoietic stem cells. Blood 102: 3172–3178, 2003 [DOI] [PubMed] [Google Scholar]
- 80.Kobashigawa S, Suzuki K, and Yamashita S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem Biophys Res Commun 414: 795–800, 2011 [DOI] [PubMed] [Google Scholar]
- 81.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, and Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285: 1733–1737, 1999 [DOI] [PubMed] [Google Scholar]
- 82.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, and Weissman IL. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol 21: 759–806, 2003 [DOI] [PubMed] [Google Scholar]
- 83.Kyriakis JM. and Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807–869, 2001 [DOI] [PubMed] [Google Scholar]
- 84.Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, and Villunger A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev 24: 1602–1607, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 43: 332–347, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JM. and Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A 90: 5742–5746, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lessard J. and Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255–260, 2003 [DOI] [PubMed] [Google Scholar]
- 88.Li B, Bailey AS, Jiang S, Liu B, Goldman DC, and Fleming WH. Endothelial cells mediate the regeneration of hematopoietic stem cells. Stem Cell Res 4: 17–24, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li D, Wang Y, Wu H, Lu L, Zhang H, Chang J, Zhai Z, Zhang J, Wang Y, Zhou D, and Meng A. Mitigation of ionizing radiation-induced bone marrow suppression by p38 inhibition and G-CSF administration. J Radiat Res 52: 712–716, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H, Wang Y, Pazhanisamy SK, Shao L, Batinic-Haberle I, Meng A, and Zhou D. Mn(III) meso-tetrakis-(N-ethylpyridinium-2-yl) porphyrin mitigates total body irradiation-induced long-term bone marrow suppression. Free Radic Biol Med 51: 30–37, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li L. and Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol 21: 605–631, 2005 [DOI] [PubMed] [Google Scholar]
- 92.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, and Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7: 165–171, 2005 [DOI] [PubMed] [Google Scholar]
- 93.Liu D. and Xu Y. p53, oxidative stress, and aging. Antioxid Redox Signal 15: 1669–1678, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, and Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459: 387–392, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lorenz E, Uphoff D, Reid TR, and Shelton E. Modification of irradiation injury in mice and guinea pigs by bone marrow injections. J Natl Cancer Inst 12: 197–201, 1951 [PubMed] [Google Scholar]
- 96.Lorimore SA, Coates PJ, and Wright EG. Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene 22: 7058–7069, 2003 [DOI] [PubMed] [Google Scholar]
- 97.Lowe SW. and Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 13: 77–83, 2003 [DOI] [PubMed] [Google Scholar]
- 98.Maas RA, Oei HL, Venema-Kemper S, Koch G, and Bongers J. Dose-response effects of inactivated Newcastle disease vaccines: influence of serologic assay, time after vaccination, and type of chickens. Avian Dis 43: 670–677, 1999 [PubMed] [Google Scholar]
- 99.Madlambayan GJ, Rogers I, Kirouac DC, Yamanaka N, Mazurier F, Doedens M, Casper RF, Dick JE, and Zandstra PW. Dynamic changes in cellular and microenvironmental composition can be controlled to elicit in vitro human hematopoietic stem cell expansion. Exp Hematol 33: 1229–1239, 2005 [DOI] [PubMed] [Google Scholar]
- 100.Main JM. and Prehn RT. Successful skin homografts after the administration of high dosage X radiation and homologous bone marrow. J Natl Cancer Inst 15: 1023–1029, 1955 [PubMed] [Google Scholar]
- 101.Majno G. and Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146: 3–15, 1995 [PMC free article] [PubMed] [Google Scholar]
- 102.Marcotte R. and Wang E. Replicative senescence revisited. J Gerontol A Biol Sci Med Sci 57: B257–B269, 2002 [DOI] [PubMed] [Google Scholar]
- 103.Matsuzawa A. and Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta 1780: 1325–1336, 2008 [DOI] [PubMed] [Google Scholar]
- 104.Mauch P, Constine L, Greenberger J, Knospe W, Sullivan J, Liesveld JL, and Deeg HJ. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol Biol Phys 31: 1319–1339, 1995 [DOI] [PubMed] [Google Scholar]
- 105.Mauch P, Rosenblatt M, and Hellman S. Permanent loss in stem cell self renewal capacity following stress to the marrow. Blood 72: 1193–1196, 1988 [PubMed] [Google Scholar]
- 106.Mcculloch EA. and Till JE. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat Res 13: 115–125, 1960 [PubMed] [Google Scholar]
- 107.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'Ayan A, Enikolopov GN, and Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466: 829–834, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meng A, Wang Y, Brown SA, Van Zant G, and Zhou D. Ionizing radiation and busulfan inhibit murine bone marrow cell hematopoietic function via apoptosis-dependent and -independent mechanisms. Exp Hematol 31: 1348–1356, 2003 [DOI] [PubMed] [Google Scholar]
- 109.Meng A, Wang Y, Van Zant G, and Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 63: 5414–5419, 2003 [PubMed] [Google Scholar]
- 110.Mercher T, Cornejo MG, Sears C, Kindler T, Moore SA, Maillard I, Pear WS, Aster JC, and Gilliland DG. Notch signaling specifies megakaryocyte development from hematopoietic stem cells. Cell Stem Cell 3: 314–326, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, and Strasser A. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev 24: 1608–1613, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, Chen C, Hosokawa K, Nakauchi H, Nakayama K, Nakayama KI, Harada M, Motoyama N, Suda T, and Hirao A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1: 101–112, 2007 [DOI] [PubMed] [Google Scholar]
- 113.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, and Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425: 962–967, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Monje ML, Mizumatsu S, Fike JR, and Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med 8: 955–962, 2002 [DOI] [PubMed] [Google Scholar]
- 115.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res 159: 581–596, 2003 [DOI] [PubMed] [Google Scholar]
- 116.Moroni M, Coolbaugh TV, Lombardini E, Mitchell JM, Moccia KD, Shelton LJ, Nagy V, and Whitnall MH. Hematopoietic radiation syndrome in the Gottingen minipig. Radiat Res 176: 89–101, 2011 [DOI] [PubMed] [Google Scholar]
- 117.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, and Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716, 2003 [DOI] [PubMed] [Google Scholar]
- 118.Navas TA, Mohindru M, Estes M, Ma JY, Sokol L, Pahanish P, Parmar S, Haghnazari E, Zhou L, Collins R, Kerr I, Nguyen AN, Xu Y, Platanias LC, List AA, Higgins LS, and Verma A. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood 108: 4170–4177, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neben S, Hellman S, Montgomery M, Ferrara J, and Mauch P. Hematopoietic stem cell deficit of transplanted bone marrow previously exposed to cytotoxic agents. Exp Hematol 21: 156–162, 1993 [PubMed] [Google Scholar]
- 120.Norbury CJ. and Hickson ID. Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol 41: 367–401, 2001 [DOI] [PubMed] [Google Scholar]
- 121.Nowell PC, Cole LJ, Habermeyer JG, and Roan PL. Growth and continued function of rat marrow cells in x-radiated mice. Cancer Res 16: 258–261, 1956 [PubMed] [Google Scholar]
- 122.Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, and Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood 80: 3044–3050, 1992 [PubMed] [Google Scholar]
- 123.Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, and Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33: 387–399, 2010 [DOI] [PubMed] [Google Scholar]
- 124.Osawa M, Hanada K, Hamada H, and Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273: 242–245, 1996 [DOI] [PubMed] [Google Scholar]
- 125.Ostman A, Frijhoff J, Sandin A, and Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem 150: 345–356, 2011 [DOI] [PubMed] [Google Scholar]
- 126.Ozeki A, Suzuki K, Suzuki M, Ozawa H, and Yamashita S. Acceleration of astrocytic differentiation in neural stem cells surviving X-irradiation. Neuroreport 23: 290–293, 2012 [DOI] [PubMed] [Google Scholar]
- 127.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, and Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423: 302–305, 2003 [DOI] [PubMed] [Google Scholar]
- 128.Patterson KI, Brummer T, O'Brien PM, and Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J 418: 475–489, 2009 [DOI] [PubMed] [Google Scholar]
- 129.Piccoli C, D'Aprile A, Ripoli M, Scrima R, Lecce L, Boffoli D, Tabilio A, and Capitanio N. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochem Biophys Res Commun 353: 965–972, 2007 [DOI] [PubMed] [Google Scholar]
- 130.Piccoli C, Ria R, Scrima R, Cela O, D'Aprile A, Boffoli D, Falzetti F, Tabilio A, and Capitanio N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J Biol Chem 280: 26467–26476, 2005 [DOI] [PubMed] [Google Scholar]
- 131.Reya T. Regulation of hematopoietic stem cell self-renewal. Recent Prog Horm Res 58: 283–295, 2003 [DOI] [PubMed] [Google Scholar]
- 132.Robles SJ. and Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 16: 1113–1123, 1998 [DOI] [PubMed] [Google Scholar]
- 133.Salter AB, Meadows SK, Muramoto GG, Himburg H, Doan P, Daher P, Russell L, Chen B, Chao NJ, and Chute JP. Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood 113: 2104–2107, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Samper E, Fernandez P, Eguia R, Martin-Rivera L, Bernad A, Blasco MA, and Aracil M. Long-term repopulating ability of telomerase-deficient murine hematopoietic stem cells. Blood 99: 2767–2775, 2002 [DOI] [PubMed] [Google Scholar]
- 135.Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, and Kolesnick R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86: 189–199, 1996 [DOI] [PubMed] [Google Scholar]
- 136.Schuringa JJ. and Vellenga E. Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr Opin Hematol 17: 294–299, 2010 [DOI] [PubMed] [Google Scholar]
- 137.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, and Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Serrano M. and Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 13: 748–753, 2001 [DOI] [PubMed] [Google Scholar]
- 139.Shao L, Li H, Pazhanisamy SK, Meng A, Wang Y, and Zhou D. Reactive oxygen species and hematopoietic stem cell senescence. Int J Hematol 94: 24–32, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shao L, Sun Y, Zhang Z, Feng W, Gao Y, Cai Z, Wang ZZ, Look AT, and Wu WS. Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells against high-dose radiation. Blood 115: 4707–4714, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sharpless NE. and DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev 9: 22–30, 1999 [DOI] [PubMed] [Google Scholar]
- 142.Sherr CJ. and Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev 10: 94–99, 2000 [DOI] [PubMed] [Google Scholar]
- 143.Shouse SS, Warren SL, and Whipple GH. II. Aplasia of marrow and fatal intoxication in dogs produced by roentgen radiation of all bones. J Exp Med 53: 421–435, 1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stepanova L. and Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood 106: 827–832, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, Haug JS, Peng L, Zhong XB, Suda T, and Li L. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell 150: 351–365, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Te PR, Okorokov AL, Jardine L, Cummings J, and Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62: 1876–1883, 2002 [PubMed] [Google Scholar]
- 147.Testa NG, Hendry JH, and Molineux G. Long-term bone marrow damage in experimental systems and in patients after radiation or chemotherapy. Anticancer Res 5: 101–110, 1985 [PubMed] [Google Scholar]
- 148.Till JE. and Mcculloch EA. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res 14: 213–222, 1961 [PubMed] [Google Scholar]
- 149.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, and Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128: 325–339, 2007 [DOI] [PubMed] [Google Scholar]
- 150.Trentin JJ. Mortality and skin transplantability in x-irradiated mice receiving isologous, homologous or heterologous bone marrow. Proc Soc Exp Biol Med 92: 688–693, 1956 [DOI] [PubMed] [Google Scholar]
- 151.van Os R, Robinson S, Sheridan T, Mislow JM, Dawes D, and Mauch PM. Granulocyte colony-stimulating factor enhances bone marrow stem cell damage caused by repeated administration of cytotoxic agents. Blood 92: 1950–1956, 1998 [PubMed] [Google Scholar]
- 152.Verheij M. and Bartelink H. Radiation-induced apoptosis. Cell Tissue Res 301: 133–142, 2000 [DOI] [PubMed] [Google Scholar]
- 153.von Wangenheim KH, Peterson HP, and Feinendegen LE. Residual radiation effect in the murine hematopoietic stem cell compartment. Radiat Environ Biophys 25: 93–106, 1986 [DOI] [PubMed] [Google Scholar]
- 154.Waegell WO, Higley HR, Kincade PW, and Dasch JR. Growth acceleration and stem cell expansion in Dexter-type cultures by neutralization of TGF-beta. Exp Hematol 22: 1051–1057, 1994 [PubMed] [Google Scholar]
- 155.Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, Schambach A, Wuestefeld T, Dauch D, Schrezenmeier H, Hofmann WK, Nakauchi H, Ju Z, Kestler HA, Zender L, and Rudolph KL. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 148: 1001–1014, 2012 [DOI] [PubMed] [Google Scholar]
- 156.Wang Y, Liu L, and Zhou D. Inhibition of p38 MAPK attenuates ionizing radiation-induced hematopoietic cell senescence and residual bone marrow injury. Radiat Res 176: 743–752, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang Y, Liu L, Pazhanisamy SK, Li H, Meng A, and Zhou D. Total body irradiation causes residual bone marrow injury by induction of persistent oxidative stress in murine hematopoietic stem cells. Free Radic Biol Med 48: 348–356, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang Y, Probin V, and Zhou D. Cancer therapy-induced residual bone marrow injury-mechanisms of induction and implication for therapy. Curr Cancer Ther Rev 2: 271–279, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang Y, Schulte BA, and Zhou D. Hematopoietic stem cell senescence and long-term bone marrow injury. Cell Cycle 5: 35–38, 2006 [DOI] [PubMed] [Google Scholar]
- 160.Wang Y, Schulte BA, LaRue AC, Ogawa M, and Zhou D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 107: 358–366, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Warren SL. and Whipple GH. Roentgen ray intoxication: I. unit dose over thorax negative-over abdomen lethal. Epithelium of small intestine sensitive to x-rays. J Exp Med 35: 187–202, 1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Waselenko JK, MacVittie TJ, Blakely WF, Pesik N, Wiley AL, Dickerson WE, Tsu H, Confer DL, Coleman CN, Seed T, Lowry P, Armitage JO, and Dainiak N. Medical management of the acute radiation syndrome: recommendations of the Strategic National Stockpile Radiation Working Group. Ann Intern Med 140: 1037–1051, 2004 [DOI] [PubMed] [Google Scholar]
- 163.Weissman IL, Anderson DJ, and Gage F. Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu Rev Cell Dev Biol 17: 387–403, 2001 [DOI] [PubMed] [Google Scholar]
- 164.Wickremasinghe RG. and Hoffbrand AV. Biochemical and genetic control of apoptosis: relevance to normal hematopoiesis and hematological malignancies. Blood 93: 3587–3600, 1999 [PubMed] [Google Scholar]
- 165.Wilson A, Laurenti E, and Trumpp A. Balancing dormant and self-renewing hematopoietic stem cells. Curr Opin Genet Dev 19: 461–468, 2009 [DOI] [PubMed] [Google Scholar]
- 166.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, and Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135: 1118–1129, 2008 [DOI] [PubMed] [Google Scholar]
- 167.Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R, and Ghaffari S. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem 283: 25692–25705, 2008 [DOI] [PubMed] [Google Scholar]
- 168.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, and Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 352: 1413–1424, 2005 [DOI] [PubMed] [Google Scholar]
- 169.Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, and Inanami O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med 53: 260–270, 2012 [DOI] [PubMed] [Google Scholar]
- 170.Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, and Nakauchi H. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147: 1146–1158, 2011 [DOI] [PubMed] [Google Scholar]
- 171.Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, Gomei Y, Iwasaki H, Matsuoka S, Miyamoto K, Miyazaki H, Takahashi T, and Suda T. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 1: 685–697, 2007 [DOI] [PubMed] [Google Scholar]
- 172.Yu H, Shen H, Yuan Y, XuFeng R, Hu X, Garrison SP, Zhang L, Yu J, Zambetti GP, and Cheng T. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose gamma-irradiation. Blood 115: 3472–3480, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang H, Zhai Z, Wang Y, Zhang J, Wu H, Wang Y, Li C, Li D, Lu L, Wang X, Chang J, Hou Q, Ju Z, Zhou D, and Meng A. Resveratrol ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice. Free Radic Biol Med 54: 40–50, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zheng J, Huynh H, Umikawa M, Silvany R, and Zhang CC. Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood 117: 470–479, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou L, Opalinska J, and Verma A. p38 MAP kinase regulates stem cell apoptosis in human hematopoietic failure. Cell Cycle 6: 534–537, 2007 [DOI] [PubMed] [Google Scholar]