

Abstract
Background and Aims
MADS-box transcriptional regulators play important roles during plant development. Based on phylogenetic reconstruction, the AP1/SEP/AGL6 superclade of floral MADS-box genes underwent one or two duplication events in the common ancestor of the core eudicots. However, the functional evolution of the AP1/SEP/AGL6 superclade in basal eudicots remains uncharacterized. Epimedium sagittatum is a basal eudicot species valued for its medicinal properties and showing unique floral morphology. In this study, structural and functional variation of FUL-like (AP1 subfamily), SEP-like and AGL6-like genes in this species was investigated to further our understanding of flower evolution in angiosperms. Detailed investigations into the microsynteny and evolutionary history of the floral A and E class MADS-box genes in eudicots were undertaken and used to trace their genomic rearrangements.
Methods
One AP1-like gene, two SEP-like genes and one AGL6-like gene were cloned from E. sagittatum. Their expression patterns were examined using quantitative RT-PCR in different vegetative and reproductive organs at two developmental stages. Yeast two-hybrid assays were carried out among AP1/SEP/AGL6 superclade, AP3/PI and AGAMOUS subfamily members for elucidation of dimerization patterns. In addition, possible formation of a ternary complex involving B class proteins with the A class protein EsFUL-like, the E class SEP-like protein EsAGL2-1 or the AGL6-class protein EsAGL6 were detected using yeast three-hybrid assays. Transgenic Arabidopsis or tobacco plants expressing EsFUL-like, EsAGL2-1 and EsAGL6-like under the cauliflower mosaic virus (CaMV) 35S promoter were generated and analysed. Genomic studies of AP1 syntenic regions in arabidopsis, columbine, strawberry, papaya, peach, grapevine and tomato were conducted for microsyntenic analyses.
Key Results
Sequence and phylogenetic analyses showed that EsFUL-like is a member of the AP1 (A class) subfamily, EsAGL2-1 and EsAGL2-2 belong to the SEP-like (E class) subfamily, and EsAGL6-like belongs to the AGL6 (AGL6 class) subfamily. Quantitative RT-PCR analyses revealed that the transcripts of the four genes are absent, or minimal, in vegetative tissues and are most highly expressed in floral organs. Yeast two-hybrid results revealed that of the eight MADS-box proteins tested, only EsAGL6-like, EsAGL2-1 and EsAGL2 were able to form strong homo- and heterodimers, with EsAGL6-like and EsAGL2-1 showing similar interaction patterns. Yeast three-hybrid analysis revealed that EsFUL1-like, EsAGL6-like and EsAGL2-1 (representing the three major lineages of the Epimedium AGL/SEP/ALG6 superclade) could act as bridging proteins in ternary complexes with both EsAP3-2 (B class) and EsPI (B class), which do not heterodimerize themselves. Syntenic analyses of sequenced basal eudicots, rosids and asterids showed that most AP1-like and SEP-like genes have been tightly associated as neighbours since the origin of basal eudicots. Ectopic expression of EsFUL-like in arabidopsis caused early flowering through endogenous high-level expression of AP1 and formation of secondary flowers between the first and second whorls. Tobacco plants with ectopic expression of EsAGL2-1 showed shortened pistils and styles, as well as axillary and extra petals in the initial flower.
Conclusions
This study provides a description of EsFUL-like, EsAGL2-1, EsAGL2-2 and EsAGL6-like function divergence and conservation in comparison with a selection of model core eudicots. The study also highlights how organization in genomic segments containing A and E class genes in sequenced model species has resulted in similar topologies of AP1 and SEP-like gene trees.
Keywords: Basal eudicots, AP1/SEP/AGL6 superclade, MADS-box, microsynteny analysis, evo-devo, Epimedium sagittatum
INTRODUCTION
MADS-box genes, including type I and type II, encode transcription factors that control diverse developmental processes in flowering plants (Alvarez-Buylla et al., 2000; Masiero et al., 2011). In the four-whorled flower of Arabidopsis, type II MADS box genes (MIKC type) work together to specify the identity of floral organs (reviewed by Krizek and Fletcher, 2005; Theissen and Melzer, 2007). Type II MADS domain proteins consist of the MADS, intervening, keratin-like and C-terminal domains (Egea-Cortines et al., 1999). The highly conserved MADS domain is responsible for DNA binding, and is followed by the weak I domain. The K and C domains are required for protein complexes (Egea-Cortines et al., 1999). The ABCDE model (Pelaz et al., 2000, 2001b; Theissen, 2001) states that these genes fall into different categories based on their spatial–temporal function in floral development. A-function genes [APETALA1 (AP1) and APETALA2 (AP2)] specify sepal identity in the outer domain of the floral meristem; B-function genes [APETALA3 (AP3) and PISTILLATA (PI)] in combination with A-function genes regulate petal identity in the second-whorl; B-function genes together with C-function genes [AGAMOUS (AG)] determine male reproductive organ identity, and C-function genes alone regulate the identity of female organs in the fourth whorl (Coen and Meyerowitz, 1991). In addition, D-class genes determine specification of ovule development (Pinyopich et al., 2003). The SEPALLATA-like genes AtSEP1 (previously AGAMOUS LIKE 2), AtSEP2 (AGL4), AtSEP3 (AGL9) and AtSEP4 (AGL3) are required for organ identity in all whorls of the flower and are collectively called E genes (Pelaz et al., 2000, 2001b). All but one (AP2, AP2/ERF gene family) of the A-, B-, C- and E-function genes characterized in plants are MADS box genes (reviewed by Kaufmann et al., 2005). Together, these proteins form multimetric protein complexes consisting of four proteins (‘Floral Quartet Model’) that determine the identity of floral organ primordia (Honma and Goto, 2001).
Many previous investigations have focused on linking the functional evolution of the floral MADS-box genes to morphological diversification of floral traits (Kim et al., 2005; Becker et al., 2011). Some of the earliest gene duplication, diversification and fixation events occurred within the type II MADS-box genes, resulting in the occurrence of 12 subfamilies (Becker et al., 2003). Moreover, detailed phylogenies of some MADS-box gene subfamilies suggest that a gene duplication event occurred before the radiation of extant angiosperms, producing two intra-subfamily lineages, for example the AP3 and PI lineages within the AP3/PI subfamily, the AG and SEEDSTICK (STK) lineages within the AG subfamily, and the AGL2/AGL3/FBP9 and AGL9 lineages within the SEP subfamily (Litt and Irish, 2003; Kramer et al., 2004; Zahn et al., 2005; Shan et al., 2007). Another one or two successive gene duplications pre-dated the emergence of core eudicots, resulting in the euAP1, euFUL and AGL79 lineages within the AP1 subfamily, the TOMATO MADS6 (TM6) and euAP3 clade within the AP3 lineage, the euAG and PLENA (PLE) clade within the AG lineage, the AGL2/4, AGL3 and FLORAL BINDING PROTEIN9 (FBP9) clades within the AGL2/3/4 lineage, and the euAGL6 and AGL6-like clade within the AGL6 subfamily (Kramer et al., 1998, 2004; Litt and Irish, 2003; Zahn et al., 2005; Viaene et al., 2010). It is suggested that the functional divergence of paralogous MADS-box genes following duplication has been instrumental for the diversity of angiosperm floral development and morphology (Kim et al., 2005). For instance, the duplication and divergence of AP3 and TM6 in the AP3 lineage was probably responsible for clearer separation of sepal and petal identity in core eudicots (Hileman and Irish, 2009).
A close relationship exists between the AP1, SEP and AGL6 lineages based on the phylogenetic analyses of type II MADS-box genes in angiosperms, giving rise to the AP1/SEP/AGL6 superclade (Nam et al., 2003; Zahn et al., 2005). Furthermore, no AP1 or SEP-like genes have been found in extant gymnosperms to date (Zahn et al., 2005). There have been many reports on the isolation and functional characterization of AP1, SEP and AGL6 subfamily genes in core eudicots and monocots, but not in basal eudicots. Most genes in the AP1 subfamily are expressed in developing floral meristems and young flower organ primordia, as has been shown for AP1 (Huijser et al., 1992; Mandel et al., 1992; Mandel and Yanofsky, 1995). The genome of Arabidopsis thaliana harbours four AP1-like genes, AP1, FRUITFULL (FUL), CAULIFLOWER (CAL) and AGL79. Partially redundant activities of AP1, FUL and CAL in controlling floral meristem identity have been characterized by comparing single, double and triple mutants (Ferrandiz et al., 2000). However, a unique role was attributed to AP1 for specification of sepal and petal identity (Mandel et al., 1992), as well as for FUL in fruit development (Gu et al., 1998). The E-class genes SEP1/2/3/4 also function largely redundantly in Arabidopsis and are critical for the identity of all four whorls of floral organs and floral meristem determinacy (Pelaz et al., 2000, 2001a, b; Ditta et al., 2004). However, SEP-like genes were shown to exhibit non-redundant roles in other model plants. For example, LeMADSRIN is specifically required for fruit maturation in tomato (Vrebalov et al., 2002), GhGRCD1 is required for staminode specification in Gerbera (Kotilainen et al., 2000) and PhFBP9 is involved in plant architecture in petunia (Vandenbussche et al., 2003). Despite the lack of an obvious phenotype for agl6 mutants, a recent report on AGL6 function in Arabidopsis revealed that two key regulators of flowering time, FLOWERING LOCUS C (FLC) and FLOWERING LOCUS T (FT), were activated in a 35S::AGL6 overexpressor line and activation tagging mutant (Yoo et al., 2011). In petunia, redundant functions for PhAGL6 and SEP-like genes in petal and anther development were revealed through double and triple mutant analysis (Rijpkema et al., 2009).To date, in-depth investigations of AP1/SEP/AGL6 superclade genes have been largely restricted to the core eudicots, and little is known about their evolution, diversification and function in basal eudicots. In the present study, the basal eudicot Epimedium sagittatum (Berberidaceae) was selected as a model species for better resolving the ancestral relationships and functions of this important MADS-box superclade, prior to their duplication and divergence in the core eudicots. Epimedium plants are an excellent evolutionary model due to their distinctive and diverse floral morphologies, displaying evolutionarily intermediate forms including petaloid sepals and petals with nectariferous (nectar secreting) tissue on their inner face (Stearn, 2002). Petals are variable in form and comprise a blunt nectariferous sac, or a nectariferous spur, at the outermost end (Fig. 1), with some studies referring to these as nectariferous leaves (Hu et al., 2012). In addition to the structural and functional analysis of specific Epimedium MADS-box genes, microsynteny analysis of AP1 and SEP-like genomic regions from basal eudicots, rosids and asterids species was performed to determine the basis for the similar phylogenetic topology of these genes in eudicots as a whole.
Fig. 1.

Photographs of Epimedium flowers. (A) E. acuminatum, (B) E. dolichostemon, (C) E. franchetii, (D) E. leptorrhizum, (E) E. sagittatum and (F) E. pseuwushanense.
MATERIALS AND METHODS
Plant materials
Whole inflorescences, floral buds, roots and leaves were obtained from the following taxa: Epimedium sagittatum, Dysosma pleiantha Woodson, Nandina domestica Thunb. and Mahonia bealei (Fort.) Carrière. All plants were cultivated in WuHan Botanical Garden, the Chinese Academy of Sciences. All the materials for expression assays were immediately frozen in liquid nitrogen and stored at –80 °C.
Cloning and characterization of FUL-like, SEP-like and AGL6-like orthologues in E. sagittatum
Total RNA was isolated from the tissues described above and from the dissected floral organs listed below using trizol reagent (Invitrogen, Carlsbad, CA, USA), for gene isolation and real-time quantitative PCR. First-strand cDNA for cloning was synthesized by Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) with a poly (T) primer. Isolation of the full length of the EsFUL-like homologue from E. sagittatum was performed with the EsFUL-like F and EsFUL-like R primers (Supplementary Data Table S1) designed based on expressed sequence tag data (Zeng et al., 2010).
Partial sequences containing the stop codon and 3′ untranslated regions of EsAGL2-1, EsAGL2-2 and EsAGL6-like genes were obtained by 3′ rapid amplification of cDNA ends (RACE) using degenerate primers and the SMARTII primer (Clontech, Carlsbad, CA, USA). To provide evidence of the duplication event of FUL-like genes in basal eudicots, partial cDNA of FUL-like genes from the additional basal eudicots D. pleiantha, N. domestica and M. bealei were isolated using the above method. Primary PCR products were diluted 1 : 10 and used as a template in a second PCR reaction. All PCR primers are listed in Supplementary Data Table S1. The resulting cDNA fragments were purified from gel and cloned into pMD19-T vectors (Takara, Dalian, China). To discriminate between different gene fragments after cloning FUL-like genes, for each taxon 30 clones were sequenced (Invitrogen, Guangzhou, China), analysed by restriction digestion, or both. For isolation of the EsAGL2-1, EsAGL2-2 and EsAGL6-like gene in E. sagittatum, 30 clones for each gene were sequenced using the M13 universal primers (Invitrogen, Guangzhou, China). Full-length cDNA sequences of EsAGL2-1, EsAGL2-2 and EsAGL6-like were obtained by 5′ RACE.
Phylogenetic analysis
Protein sequence alignments were performed using the MUSCLE server (EMBL), with default settings. Nucleotide alignments were generated using aa2dna based on the amino acid alignment (Shan et al., 2007; Hu et al., 2012). Phylogenetic analyses were conducted in MEGA5 using the maximum-likelihood method. A general time-reversible (GTR) model with a proportion of invariable sites was selected using model test. The resulting tree was subjected to bootstrap analysis based on 1000 replicates (Felsenstein, 1985).
Stable transformation assay of EsFUL-like, EsAGL2-1 and EsAGL6-like
To construct vectors for ectopic expression of EsFUL-like, EsAGL2-1 and EsAGL6-like genes, full-length coding sequences were digested from pMD19-T (Takara), using SalI and KpnI, and ligated behind the 35S constitutive promoter in an XhoI and KpnI digested binary pMV vector (derivative of pBI121 using T4 DNA ligase; Takara). This yielded the constructs p35Spro-EsFUL-like, p35Spro-EsAGL2-1 and p35Spro-EsAGL6-like. All constructs were electroporated into Agrobacterium tumefaciens GV3101. The floral dip method for transforming Arabidopsis was performed as described by Clough and Bent (1998). Transgenic lines surviving on half-strength MS medium with 50 µg mL−1 kanamycin were selected for expression analysis and phenotypic observation. The flowering time of lines with ectopic expression of EsFUL-like, EsAGL2-1 and EsAGL6-like was measured as the number of rosette leaves on the main shoot when the first bolt appeared. To analyse the expression patterns of flowering-time genes in Pro35S-EsFUL-like transgenic lines, we collected wild-type and transgenic young rosette leaves of 4-week-old plants grown under a 16/8-h light/dark photoperiod at 23 °C. At this time point, the wild-type was starting to develop reproductive organs, suggesting both wild-type and transgenic line were in the process of floral transition. The quantitative (q)RT-PCR assays of putative genes including AP1, CONSTANS (CO), FLC, FT, TERMINAL FLOWER (TFL), LEAFY (LFY) and OVEREXPRESSION OF CONSTANS 1 (SOC1) were performed as described below, and the TUBULIN gene was used as a control to normalize expression levels. The primers for qRT-PCR are listed in Supplementary Data Table S1.
Gene expression analysis
Expression of EsFUL, EsAGL2-1, EsAGL2-2 and EsAGL6-like was detected in vegetative tissues (roots and leaves) and floral tissues dissected at both preanthesis (petal expansion initiation) and anthesis (fully expanded petals). Dissected floral tissues included sepals, petals, stamens and carpels. Gene-specific primer pairs (Supplementary Data Table S1) for EsFUL-like, EsAGL2-1, EsAGL2-2 and EsAGL6-like were designed in the C terminus of each gene. Specific Epimedium ACTIN primers (Huang et al., 2012) were used for relative quantification. For qRT-PCR, first-strand synthesis was performed using the PrimeScript RT reagent Kit (Takara). Each amplification reaction was performed with 50 ng of template cDNA. All reactions were performed in 20 µL containing SYBR Premix Ex TaqTM II (Takara) with 100 nm of gene-specific primers or ACTIN control primers. Samples were amplified in triplicate for 40 cycles of 95 °C for 5 s and 60 °C for 34s. Melt curve analysis was used to test whether a single amplification product was formed. Relative mRNA abundance in different organs was determined using the 2−ΔCt method for relative quantification normalized to ACTIN.
Protein–protein interaction studies by yeast two-hybrid and three-hybrid analyses
Yeast two-hybrid analysis was performed using the GAL4 system (Clontech, Mountain View, CA, USA). Full-length cDNAs of EsFUL-like, EsAG, EsAG11, EsAGL2-1, EsAGL2-2 and EsAGL6-like, as well as EsAP3-2 and EsPI without their MADS domain were inserted into the pGADT7 and pGBKT7 vectors (Clontech). All primers with restriction sites used for cloning into pGADT7 and pGBKT7 are listed in Table 1. Due to auto-activation of EsAGL2-2 protein, two types of truncated EsAGL2-2 proteins were tested: EsAGL2-2ΔCI, missing the SEPII motif, and EsAGL2-2ΔCII, missing both the SEPI and the SEPII motifs. Auto-activation was found to be removed in EsAGL2-2ΔCII, and thus yeast two-hybrid analyses of all proteins were performed as described previously to identify homo- and hetero-dimerization (Rijpkema et al., 2009).
Table 1.
Effects of over-expression of EsFUL-like on flowering time as determined by leaf numbers under long day conditions
| Genotype | Rosette leaf number | Cauline leaf number | Total numbers of leaves |
|---|---|---|---|
| 35S::EsFUL | 4·67 (0·52)* | 1·3 (0·52) | 6 (0·7)** |
| WT | 8·0 (0·89) | 2 (0·0) | 10 (0·0) |
* Significance at 1 % level (P < 0·01) compared with wild-type Arabidopsis.
To investigate whether B class proteins interact with AP1/SEP/AGL6 superclade proteins to form a multi-protein complex, the IKC domains of the B class proteins EsAP3-2 and EsPI were expressed along with each of three proteins representing the major lineages of the Epimedium AP1/SEP/AGL6 superclade proteins using the yeast three-hybrid method. To obtain triple transformants, yeast strain AH109 was transformed with EsPIΔM in vector pGBK-T7 as well as one of EsFUL, EsAGL2-1 or EsAGL6-like in vector pGAD-T7. Strains selected as containing each of these three vector combinations were mated to the Y187 strain carrying the pTFT-1 plasmid containing the IKC domain of EsAP3-2. Di-parental mating was performed by mixing strains in liquid yeast extract-peptone-dextrose (YPD) medium. Positive clones containing three vectors were screened on SD-Leu–Trp–Ade (SD medium without leucine, tryptophan and adenine). The yeast three-hybrid interactions were selected on SD-Leu–Trp–Ade–His (SD medium lacking Leu, Trp, Ade and histidine (His)) with 25 mm 3′-aminotriazole (3AT) plates supplemented with X-α-Gal. Three independent clones for every combination and three technical replications for each clone were used to determine the interaction pattern.
Micro-synteny analysis
To study the micro-synteny of A function genes among basal and core eudicot plant genomes, approximately 200 kb of the genomic regions encompassing each of AGL79 (AT3G30260), AGL8 (AT5G60910) and AP1 (AT1G69120) from Arabidopsis were used as queries to find microsyntenic regions of papaya (Carica papaya), strawberry (Fragaria vesca), peach (Prunus persica) and grapevine (Vitis vinifera) using the Plant Genome Duplication Database (PGDD) as described by Causier et al. (2010). Micro-syntenic fragments of tomato (Solanum lycopersicum) and columbine (Aquilegia coerulea) were obtained from the Sol Genomic Network website (http://solgenomics.net/) and phytozome (http://www.phytozome.net/).
RESULTS
Sequence and phylogenetic analysis of FUL-like, SEP-like and AGL6-like genes in E. sagittatum
To gain insight into the evolution of floral structures, we isolated FUL-like, SEP-like and AGL6-like genes from the basal eudicot E. sagittatum. Partial sequences were also obtained for FUL-like genes from the additional basal eudicots D. pleiantha (DpFUL-like), M. bealei (MbFUL-like) and N. domestica (NdFUL-like). Within the full-length sequence of EsFUL-like, we detected an open reading frame encoding 253 amino acids. The FUL-like sequences of D. pleiantha (DpFUL-like), E. sagittatum (EsFUL-like), M. bealei (MbFUL-like) and N. domestica (NdFUL-like) all contain the typical FUL-like gene motifs and paleoAP1 motif (Fig. 2) (Shan et al., 2007; Liu et al., 2010). The full-length cDNAs of EsAGL2-1 and EsAGL2-2 contained open reading frames encoding 241- and 244-amino-acid proteins, respectively. These sequences shared 71·9 % nucleotide identity and 77·5 % amino acid identity in their coding regions, and both deduced proteins contained a conserved SEP I and SEP II motif at their C terminus (Fig. 2; Zahn et al., 2005). Sequence analysis of the EsAGL6-like gene revealed a putative encoded protein of 149 amino acids. Furthermore, alignment to additional dicot AGL6-like sequences indicated that the EsAGL6-like protein contains the highly conserved AGL6 motif I and AGL6 motif II located in the C-terminal region (Fig. 2; Ohmori et al., 2009).
Fig. 2.

Comparison of the deduced amino acid sequences of EsFUL-like, EsAGL2-1, EsAGL2-2 and EsAGL6-like and related MADS-domain proteins. (A) Alignment of domains located in the C terminus of FUL-like proteins. Conserved FUL and paleoAP1 motifs are boxed. (B) The two conserved blocks in the C-terminal region of SEP-like proteins represent the two motifs, SEPI and SEPII. (C) Alignment of the C-terminal regions of AGL6-like proteins. Boxed parts correspond to two highly conserved AGL6-I and AGL6-II motifs. The accession numbers of selected MADS-box proteins are listed in Supplementary Data Table S2.
Phylogenetic analysis was performed for the 30 AP1-like genes and 27 SEP-like genes isolated in this study, supplemented with sequences retrieved from the PGDD and NCBI databases. The AP1-like genes from core eudicots could be assigned to one of three lineages, euAP1, euFUL and AGL79, consistent with previous phylogenetic analyses (Litt and Irish, 2003; Shan et al., 2007). Outside of lineages from the core eudicots, the four AP1-like genes from the family Berberidaceae clustered together with homologues from additional families within the basal eudicot order Ranunculales. However, maximum-likelihood analysis did not support the hypothesized duplication event for AP1-like genes in basal eudicots, possibly as a result of using too few sequences for reconstructing the evolutionary process.
Phylogenetic analysis of SEP-like genes revealed four clades corresponding to AGL2/4, FBP9 and AGL3, in core eudicot species based on previous investigations (Zahn et al., 2005). In addition, the putative EsAGL2-1 and EsAGL2-2 paralogues, which were produced by a gene duplication event within the Ranunculales, were distinct from the core eudicot sequences (Fig. 3). Maximum-likelihood phylogenetic analyses of EsAGL6-like and related genes demonstrated that EsAGL6-like sequences clustered with two additional sequences from A. coerulea and Ranunculus bulbosus.
Fig. 3.



Phylogenetic analysis using nucleotide sequences of selected MADS-box genes. (A) Phylogeny of the AP1 subfamily. The Berberidacea genes together with other sequences from the Ranuancules are located on the basal branch in the tree, representing basal eudicot species. (B) Phylogeny of the SEP subfamily. The SEP-like gene of Amborella trichopoda was used as an outgroup. (C) Phylogenetic tree of selected members of the AGL6-like sequence using Gnetum parvifolum as root. Numbers at nodes are bootstrap values (>50 %) based on 1000 replicates.
Expression pattern of EsFUL-like, EsAGL2-1, EsAGL2-2 and EsAGL6-like genes
To establish the putative functions of EsFUL-like, EsAGL2-1, EsAGL2-2 and EsAGL6-like genes in developmental processes, we examined their tissue-specific expression levels in roots, leaves and dissected floral organs at both late preanthesis and anthesis (Fig. 4). As expected, qRT-PCR analyses on dissected flower and vegetative tissues revealed the highest mRNA abundance for all genes in the floral tissues relative to the vegetative tissues, with all transcripts absent in root tissue. Both EsAGL2-1 and EsAGL6-like were also absent in leaves, while EsAGL2-2 and EsFUL-like were present at relatively low levels in leaves. In addition, most of genes are more strongly expressed in every organ after anthesis, whereas EsFUL-like expression in sepals, stamens and EsAGL2-2 expression in carpels is stronger before anthesis.
Fig. 4.
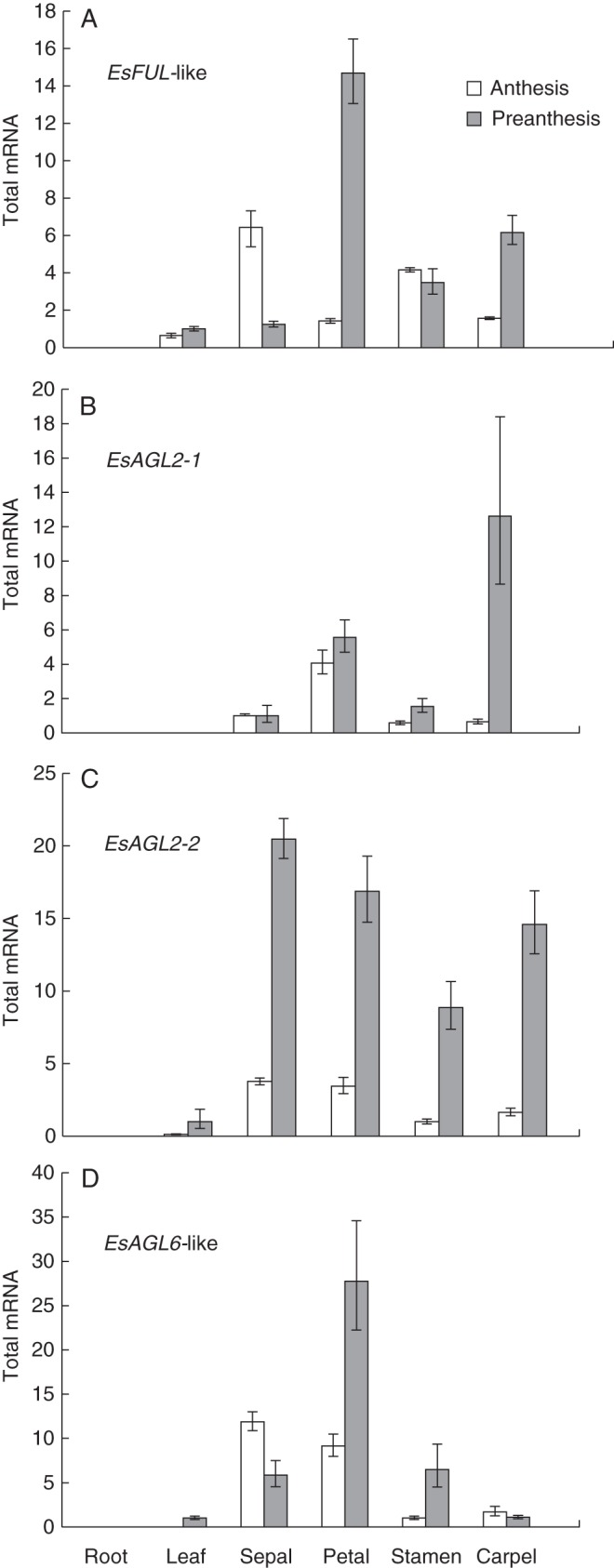
Expression profiles of (A) EsFUL-like, (B) EsAGL2-1, (C) EsAGL2-2 and (D) EsAGL6-like genes in E. sagittatum. Total mRNA was isolated from the roots, leaves, sepals, petals, stamens and carpels. Transcripts at preanthesis and anthesis stages are as indicated in the key. Error bars represent s.e. for three technical replicate reactions. All transcripts were normalized using EsACTIN for E. sagittatum.
Early flowering and flower phenotype caused by constitutive EsFUL-like expression
To further investigate the function of EsFUL-like, EsAGL2-1 and EsAGL6-like genes in Epimedium, we ectopically expressed these three genes in Arabidopsis, driven by the cauliflower mosaic virus (CaMV) 35S promoter. Only EsFUL-like transgenic lines showed obvious phenotypes; EsAGL2-1 and EsAGL6-like transformants were visually indistinguishable from wild-type plants. Fifteen independent kanamycin-resistant transformants of EsFUL-like were obtained and confirmed by PCR screening. Ectopic expression of the gene in each line was also confirmed by qRT-PCR before phenotypic analysis. A significant effect on flowering time was observed in transgenic plants overexpressing EsFUL-like compared with wild-type (Table 1, Fig. 5). In addition, ectopic expression of EsFUL-like showed a strong floral phenotype similar to that observed for the ap1 mutant. Specifically, one or two extra flowers emerged between the first and second whorl, while the morphology of sepals and petals remained unchanged.
Fig. 5.

Functional analysis of the EsFUL-like gene in Arabidopsis. (A) Early-flowering phenotype upon ectopic expression of EsFUL-like (left) and normal vegetative growth of wild-type (right). (B, C) Floral phenotype of EsFUL-like overexpressing lines showing production of a secondary flower as seen from side view (B) and top view (C). Asterisks indicate the secondary flower growing in the axis. (D) Close-up of wild-type flower.
To examine the molecular basis of the early-flowering phenotype of transgenic 35S::EsFUL-like lines, the expression of known flowering-related genes in these lines was analysed by qRT-PCR. This revealed dramatically increased expression of the known floral regulators AP1 and SOC1 in EsFUL-like transgenics compared with the wild-type, while expression of CO, FT and LFY was only slightly increased (Fig. 6). Thus, the perturbed floral phenotype coincided with upregulation of two MADs-box genes. In addition, there was a slight decrease in the transcript abundance of the repressor FLC gene in transgenic plants.
Fig. 6.
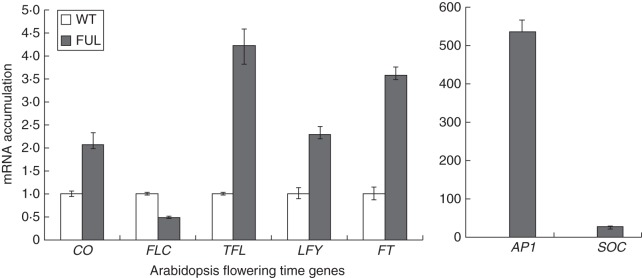
Analysis of flowering time gene expression in 35S::EsFUL-like transgenic Arabidopsis plants. mRNA accumulation for CO, FLC, TFL, LFY, FT, AP1 and SOC1 was determined by qRT-PCR. A fragment of the Tubulin gene was amplified as an internal control. Error bars represent s.e. for three replicate reactions.
Phenotypic changes in over-expression lines of EsAGL2-1 in tobacco
As there were no obvious phenotypic alterations in transgenic Arabidopsis ectopically expressing the EsAGL2-1 and EsAGL6-like genes, their putative function was further investigated by ectopically expressing them in transgenic tobacco, a more closely related solanaceous species. No phenotypic alterations resulted from ectopic EsAGL6-like expression; however, over-expression of EsAGL2-1 caused pistils and stamens to be curled compared with control lines. Furthermore, 35S::EsAGL2-1 plants developed terminal flowers with ectopic organ proliferation in the form of abnormal, petal-like organs protruding from sepals and petals of the initial flower (Fig. 7).
Fig. 7.

Floral phenotype observed upon ectopic expression of EsAGL2-1 in tobacco. (A) Compared with the wild-type tobacco (right), the EsAGL2-1 transgenic tobacco (left) showed extra and abnormal petals produced inside the sepal and petal organs. (B) Side view of the inner organ alterations in EsAGL2-1 transgenic tobacco. Petals were removed to reveal the stamens and pistil. The style and pistil were curled and their length is smaller when compared with the wild-type tobacco.
Protein–protein interaction patterns detected in yeast
Pairwise interactions among MADS box proteins are the molecular basis for the Floral Quartet Model (Theissen, 2001), and thus pairwise interactions between MADS box proteins from different species or lineages of angiosperm may provide insight into the functional diversification and specificity of these protein–protein interactions (Ferrario et al., 2003). In this study, the yeast two-hybrid system was used to detect protein–protein interactions both among members of the AP1 (FUL, A class)/SEP (E class)/AGL6 (AGL6 class) superclade and between these members and B and C class proteins (Table 2). When EsFUL-like, EsAP3-2, EsPI, EsAG, EsAG11, EsAGL2-1, EsAGL2-2 and EsAGL6-like were fused with the GAL4 binding domain and then introduced into the AH109 yeast strain, only EsAGL2-2 showed auto-activation. To prevent this we modified the EsAGL2-2 coding sequence by deleting the SEPI and SEPII motifs in the C terminus. Both the truncated version (in pGBKT7) and the full-length version (pGADT7) were used in subsequent experiments. To analyse the extent of functional conservation and divergence between the paralogous EsAGL2-1 and EsAGL2-2, we first examined their ability to interact with different protein partners. Yeast two-hybrid results showed that both EsAGL2-1 and EsAGL2-2 interacted with EsAP3-2 and EsAGL6, and also interacted with each other and themselves. However, EsAGL2-1, but not EsAGL2-2, interacted with EsFUL-like and EsAG, while EsAGL2-2, but not EsAGL2-1, interacted with EsAG11. In addition to binding EsAGL2-1 and EsAGL2-2, EsAGL6-like was able to dimerize with EsAP3-2, EsAG and EsAG11. No interaction was detected between EsFUL-like and EsAP3-2, EsAG or EsAG11. Furthermore, heterodimers between EsAP3-2 and EsPI were not detected in the yeast two-hybrid system when truncated IKC proteins (missing the MADS domain) were used. Weak homodimerization was detected for EsFUL-like and EsAGL6-like proteins.
Table 2.
Yeast two-hybrid analysis of the E. sagittatum AP1/SQUA, SEP-like and AGL6-like MADS domain proteins
| AD plasmid | pAD-FUL-like | pAD-AP3-2 | pAD-PI | pAD-AG | pAD-AG11 | pAD-AGL2-1 | pAD-AGL2-2 | pAD-AGL6 | |
|---|---|---|---|---|---|---|---|---|---|
| BD plasmid | – | – | – | – | – | – | – | – | – |
| pBD-FUL-like | – | –/ + | – | – | – | – | + | – | – |
| pBD-AP3-2ΔM | – | – | N | – | N | N | – | – | – |
| pBD-PIΔM | – | – | – | N | N | N | – | – | – |
| pBD-AG | – | – | N | N | N | N | + | – | + |
| pBD-AG11 | – | – | N | N | N | N | – | + | + |
| pBD-AGL2-1 | – | – | + | – | – | – | + | + | + |
| pBD-AGL2-2 | – | – | + | – | – | – | – | + | – |
| pBD-AGL6 | – | – | – | – | – | – | + | + | –/ + |
Interaction between selected proteins was tested by growing yeast containing these genes on selection plates SD-Leu–Trp–His–Ade +10 mm 3-AT. Protein interactions detected by yeast two-hybrid assay were classified as absent (–), weak (–/+) or strong (+).
As floral homeotic function requires the formation of higher order protein complexes (Honma and Goto, 2001), yeast three-hybrid analysis was used to test ternary complex formation between the B class protein (AP3-PI heterodimer) and A, C and E class proteins from Arabidopsis, petunia, chrysanthemum and tomato (Honma and Goto, 2001; Ferrario et al., 2003; Shchennikova et al., 2004; Leseberg et al., 2008; Broholm et al., 2010). In this study, we examined the interaction of the B class proteins EsAP3-2 and EsPI, with the A-class protein EsFUL-like, E-class protein EsAGL2-1 and the AGL6 class protein EsAGL6 (Table 3). EsFUL-like, EsAGL2-1 and EsAGL6-like proteins had the ability to form multimeric complexes with a combination of EsAP3-2 and EsPI.
Table 3.
Molecular interactions between EsAP3-2, EsPI and AP1/SEP/AGL6 superclade proteins tested by the yeast three-hybrid method
| Ternary complex | -Leu Trp His + 25 mm 3AT | Ternary complex | -Leu Trp His + 25 mm 3AT |
|---|---|---|---|
| AD-EsFUL + pTFT1-EsAP3-2 + BD-EsPI | + | AD-EsAGL2-1 + pTFT1-AP3-2 BD-EsPI | + |
| AD-EsAGL6-like + pTFT1-EsAP3-2 + BD-EsPI | + | AD + pTFT1-AP3-2 + BD-EsPI | – |
| AD-EsFUL + pTFT1-EsAP3-2 + BD | – | AD-EsAGL2-1 + pTFT1-AP3-2 + BD | – |
The class B genes EsAP3-2 and EsPI were inserted into pTFT-1 and pGBKT7 vectors, respectively, whereas the EsFUL-like, EsAGL2-1 and EsAGL6-like genes were inserted into the pGADT7 vector. +, strong interaction; –/ + , weak interaction; –, no interaction; N, no test.
The euAP1/AGL79/euFUL and AGL3/AGL2, 4/FBP synteny in rosids, asterids and basal eudicots
To further decipher the basis for the similar phylogenetic structure of euAP1/AGL79/euFUL and AGL3/AGL2, 4/FBP9, we used PGDD (Causier et al., 2010) to explore micro-synteny among the Arabidopsis, grapevine, strawberry, peach and papaya genomic regions. Tomato chromosome data available from the Sol Genomic Network website and Columbine (Aquilegia sp.) sequence data from phytozome were also analysed, providing a data set spanning the major eudicot rosid and asterid clades, as well as basal eudicots. The 200-kbp genomic regions of euAP1, AGL79 and euFUL extracted from the core eudicots shared a number of genes, including a universal stress protein (USP), a SEP-like gene and the REVERSIBLY GLYCOSYLATABLE POLYPEPTIDE (RGP) gene. This predicted a USP–AP1–SEP–RGP ancestral genetic composition, which pre-dated the divergence of the AP1, euFUL and AGL79 gene lineages. To test this prediction, two putative segments containing FUL-like genes from the basal eudicot Aquilegia coecula were also explored. One segment also contained an SEP-like gene, FUL-like gene, RGP gene and USP gene, supporting the predicted ancestral state and segmental duplication or polyploidization events giving rise to the three AGL3/AGL2,4/FBP9 lineages following Aquilegia divergence.
Specific collinear genes in different homologous segments of euAP1/AGL79/euFUL
The euAP1 genomic region of all plants analysed showed strong micro-collinearity with the exception of Arabidopsis. At least four genes – NOP14-like (here designated NOP14), a protein of unknown function (here designated UN), USP and KINASE PROTEIN gene (KIN) – were found conserved among all the species we examined (Fig. 8). Moreover, the SQUAMOSA and PROMOTER-BINDING-like 6 (SPL6) genes were present in both the euAP1 and the euFUL blocks in the same orientation for all species. The CRABS CLAW (CRC) and BINDING PROTEIN (DNA BL) genes from Arabidopsis show collinearity in euAP1 genomic fragments in all species except papaya. Comparative analysis of the Arabidopsis genomic regions containing AP1 and CAL suggest that these resulted from a Brassicaceae-specific duplication, followed by subsequent gene loss and rearrangement in the CAL region.
Fig. 8.

Micro-synteny in genomic regions containing the AP1/FUL/AGL79 MADS-box genes in plants. Shown is the genomic region of the core eudicots A. thaliana, P. persica, F. vesca, V. vinifera, C. papaya and S. lycopersicum. The euAP1 region is depicted at the top of the panel; the AGL79 region is shown in the middle of the panel and the euFUL region is shown at the bottom. The two loci in A. coerulea containing the FUL-like gene are shown at the base of the figure, as well as the presumptive genomic region of the common ancestor. The distribution of genes is not representative of the physical distance. Genes are represented by arrows with vertical black lines representing intergenic sequence. Circles represent non-syntenic genes where a black circle indicates one gene, pink indicates two genes, brown indicates three genes, red indicates four genes, white indicates five genes, green indicates seven genes and blue indicates nine genes. Inverted genes of strawberry are shown in a red box; landmark genes discussed in text are indicated. The red box suggests inversion of genes during evolutionary process.
Within the AGL79 region, the DSBA OXIDOREDUCTASE family-like gene (DSBA), REGULATOR OF CHROMOSOME CONDENSATION PROTEIN (RCC1-like), MYB DOMAIN PROTEIN 121 (MYB121) and BRASSINOSTEROID-6-OXIDASE (BR6OX1) genes were strictly conserved in the same order and orientation in rosid and asterid species, while both DSBA-like and RCC1-like were absent in Arabidopsis and BR6OX1 was missing in F. vesca. Also specific to F. vesca, this syntenic block showed inversion of the BR60X1 flanking genes, RGP-like and PHOSPHORIC DIESTER HYDROLASES-like gene (PDH).
Close examination of the euFUL genomic regions revealed that four genes, namely CARRIER protein-like gene (CA), GLYCOSYLTRANSFERASE-like gene (GLY), BETA-N-ACETYLGLUCOSAMINE-like (ACE) and ATP-DEPENDENT RNA-HELICASE (ARH), were syntenic within grapevine, strawberry, peach and papaya. A WD-40 repeat family member (WD-40) was highly syntenic within the euFUL blocks of Arabidopsis, grapevine, peach, strawberry and papaya. The PENTATRICOPEPTIDE (PPR) repeat-containing protein and COBRIA (CO) genes were also syntenic in rosids and asterids except for grapevine and papaya. Interestingly, two separate genomic regions in papaya were found to share collinearity with the euFUL-containing regions of rosids and asterids; however, in both of these blocks, the euFUL-like and FBP9-like genes were absent, suggesting papaya may lack syntenic orthologues for these proteins. To infer whether the putative AGL3 and FBP9 orthologues are absent in the genome of papaya, we searched the PGDD and found two genes (evm.TU.supercontig_660·1 and evm.TU.supercontig_3·196) encoding SEP-like proteins, but lacking syntenic relationship with other species.
DISCUSSION
Previous studies in Arabidopsis and other model species have demonstrated that four pathways in combination regulate the transition to flowering: the photoperiodic, vernalization, autonomous and gibberellic acid pathways (Boss et al., 2004; Searle and Coupland, 2004; Adam et al., 2006). MADS-box genes play critical roles in flowering and floral evolution by integrating these different pathways to form a regulatory network (Michaels and Amasino, 1999; Zhao et al., 2005; Adamczyk et al., 2007). For example, the MADS-box SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) acts as a common target for all four flowering pathways and is transcriptionally controlled by two antagonistic flowering regulators, namely CONSTANS (CO) and FLOWERING LOCUS C (FLC), to accelerate flowering (Onouchi et al., 2000; reviewed by Lee and Lee, 2010). Another key MADS-box gene, AP1, has dual roles in regulating the identity of floral organs in whorl 1 and 2 of the meristem, mainly by activating additional MADS-box genes including AP3, SHORT VEGETATIVE PHASE (SVP), AGAMOUS-LIKE 24 (AGL24) and SOC1 (Liu et al., 2008). The latter three proteins act as suppressors of floral repressors and shoot identity genes (Liu et al., 2008).
Ectopic expression of AP1-like genes from monocots and core eudicots has been shown to induce an early-flowering phenotype (Chang et al., 2009; Lin et al., 2009; Varkonyi-Gasic et al., 2011). Here we have shown that ectopic expression of EsFUL-like from the basal eudicot Epimedium sagittatum, pre-dating the duplication and divergence of euAP1, euFUL and AGL79, mimics the early-flowering phenotypes of the euAP1 and FUL-like ectopic expressors from core eudicots (Elo et al., 2001; Shchennikova et al., 2004) and monocots, respectively (Fornara et al., 2004; Chang et al., 2009; Lin et al., 2009; Kinjo et al., 2012) (Fig. 5). This suggests that euAP1 (not AGL79) from core eudicot species has maintained the ancestral function of the FUL-like gene from basal eudicots in regulating the floral transition. In Arabidopsis rosette leaves, ecoptic expression of the MADS-box genes MADS1 (Lin et al., 2009) and SOC1 leads to AP1 upregulation and a resulting early-flowering phenotype. Consistent with this, a significantly large-fold upregulation of endogenous AP1 occurred in 35S::EsFUL-like lines (Fig. 6). However, these plants also displayed some ap1 mutant phenotypes, including supernumeral growth of axillary organs in primary flowers (Fig. 5). Interestingly, this phenotype was not caused by down-regulation of endogenous AP1 as AP1 expression was detected in transgenic 35S::EsFUL-like Arabidopsis (Fig. 5). One possibility is that the ectopically expressed EsFUL-like has a dominant negative effect on endogenous AP1 levels by sequestering AP1 in a MADS-box multimeric protein complex, which may show altered activity and thus perturbed floral meristem identity (Fornara et al., 2004). However, a dominant-negative effect on AP1 is not consistent with the early-flowering phenotype of 35S::EsFUL-like plants. In wild-type Arabidopsis, suppression of SOC1, SVP and AGL24 by AP1 is required to inhibit shoot and inflorescence formation (Liu et al., 2008). In ectopic EsFUL-like expressor lines, AP1 transcripts were significantly upregulated, but so too were the additional floral regulators SOC1, CO, TFL, LFY and FT (Fig. 6), which may also explain the development of ectopic floral organs and terminal flowers in transgenic plants (Gregis et al., 2009).
Ancestrally conserved functions for FUL-like genes in floral development
The diversification and neofunctionalization of MADS-box transcription factors throughout evolution has contributed to the extensive diversity in plant form and function that exists today. Within the AP1/SEP/AGL6 (APETALA1/SEPATELLA/AGAMOUS-Like6) superclade of floral MADS-box proteins in angiosperms, the AP1/SQUA subfamily has undergone three duplication events giving rise to the three eudicot subfamilies: euAP1, euFUL and AGL79 (Litt and Irish, 2003; Shan et al., 2007). Lineage-specific changes in coding and regulatory sequences following gene duplication events are central to their functional diversification. Analysis of AP1/SQUA-like gene expression patterns in angiosperms indicates that euAP1 lineage genes were transcriptionally restricted to floral tissues following divergence from the euFUL/euAGL79 eudicot lineages, which show broader expression patterns, also shared by their counterparts in basal angiosperms, monocots and basal eudicots (Fornara et al., 2004; Kim et al., 2005; Shan et al., 2007; Chang et al., 2009; Lin et al., 2009; Liu et al., 2010; Pabón-Mora et al., 2012, 2013). Transcripts of EsFUL-like in Epimedium sagittatum were detected in vegetative (leaf) and floral tissues both before and after anthesis (Fig. 4), consistent with broad transcription of FUL-like genes in basal eudicots. This further supports a broad ancestral function for FUL-like genes in flowering and floral meristem determination in angiosperms, which may have been later partitioned between different paralogous gene copies following duplication and neofunctionalization in the core eudicots.
A role for E class and AGL6 genes in Epimedium
Alignment of Epimedium EsAGL2-1 and EsAGL2-2 to other plant E class MADS-box proteins revealed the conservation of the common SEP I and SEP II motifs in the otherwise divergent C terminus (Fig. 2). Phylogenetic analysis, combined with tissue-profiling of transcript abundance (Fig. 3), revealed that these putative paralogues may have arisen by gene duplication, followed by slight modification of expression profile and also preferred protein interactive partners. E class proteins form homodimers or heterodimers with other MADs-box proteins in several plant species (Pelaz et al., 2001a; Liu et al., 2010; Ruokolainen et al., 2010). Consistent with this, yeast two-hybrid assays revealed that EsAGL2-1 and EsAGL2-2 interacted broadly with proteins from the A, B, C, E and AGL6 classes (Table 2). Furthermore, co-expression of interacting partners in all floral organs (Fig. 3) supports these protein interactions, and suggests a functional requirement for protein partners within the same spatiotemporal boundaries. Together, this indicates that the conserved pattern referring to broadly interactive ability of E class proteins was established in basal eudicots before diversification of the core eudicots. Similar evidence from other basal eudicots, including Euptelea pleiospermum and Akebia trifoliate, also supports this conclusion (Liu et al., 2010).
Functional studies in Arabidopsis and tomato revealed essential roles of SEP-like E class genes in promoting the identity of floral organs in all whorls (Goto et al., 2001). Ectopic expression of AGL2-like genes from lily (Tzeng et al., 2003) and orchid (Chang et al., 2009) in Arabidopsis caused early-flowering phenotypes. In this study, ectopic expression of EsAGL2-1 resulted in malformed stamens and pistils, and produced extra petaloid organs, suggesting a role for EsAGL2-1 in regulating the development of petals, stamens and carpels. However, an early-flowering phenotype was not visible in the 35S::EsAGL2-1 transgenic lines in this study, suggesting that EsAGL2-1 may not be involved in regulating the timing of the floral transition. In Epimedium, the similar sequence structure, expression profiles and shared protein interactions of EsAGL2-1 and EsAGL2-2 may make them partially redundant, similar to SEP genes in Arabidopsis (Ditta et al., 2004).
Within the AGL6 subfamily of MADS-box genes, the divergence of the two AGL6-like and euAGL6 lineages in eudicots was associated with expansion of the AGL6-like expression domain to include vegetative tissues, and putative neofunctionalization of the two clades (Viaene et al., 2010). In Epimedium, EsAGL6-like gene expression was confined to reproductive tissues (Fig. 4), supporting reports that the ancestral AGL6 subfamily was restricted to reproductive organs (Viaene et al., 2010). Yeast two-hybrid analysis revealed that, similarly to the SEP proteins, the EsAGL6-like protein can interact with proteins of other lineages such as FUL-like and AG lineage members. It has been suggested that AGL6 and SEP share similar functions on the basis of screening their interactive partners in Arabidopsis and Petunia (de Folter et al., 2005; Rijpkema et al., 2009). Therefore, it is possible that similar functions for both classes are evolutionarily conserved in basal eudicots. Overexpression of EsAGL6-like in transgenic Arabidopsis and tobacco revealed no obvious phenotype in this study. Further functional analysis using gene knock-down techniques, such as virus-induced gene silencing or RNAi, should facilitate improved functional analysis of EsAGL6-like genes.
Comparative protein–protein interactions (PPIs) and multicomplex formation in E. sagittatum
Similar PPI combinations were found in Epimedium as demonstrated for other basal eudicots (Liu et al., 2010; Pabón-Mora et al., 2013): for instance, homodimers formed for EsFUL-like, EsAGL2-1 and EsAGL2-2, and heterodimers formed between EsAGL2-1 and EsAGL2-2. Therefore, conserved PPIs within basal eudicots may play an important role in the establishment of conserved floral architectures (Shan et al., 2009; Liu et al., 2010). In accordance with the results from Arabidopsis, chrysanthemum and tomato (Shchennikova et al., 2004; Piwarzyk et al., 2007; Leseberg et al., 2008), the direct interaction of a B class protein with an E class protein was demonstrated in Epimedium, suggesting such heterodimerization may be universal in eudicots. Proper flower development is essential for angiosperm reproduction and survival. Specific combinations of higher order MADS-box complex formation may be essential for precisely regulating target genes for floral organ identity (Theissen, 2001; Melzer et al., 2009). The Antirrhinum MADS proteins, DEF (AP3), GLO (PI) and SQUA (AP1), were the first reported to have the capacity for higher-order complex formation in binding the CArG box of downstream genes (Egea-Cortines et al., 1999). The majority of MADS box higher-order complexes consist of at least one protein belonging to the A class or E class together with a combination of B class proteins. For instance, heterodimers between AP3 and PI can interact with SEP1 or SEP3 in yeast three-hybrid assays (Yang and Jack, 2004; Piwarzyk et al., 2007). Furthermore, A class proteins including CDM8 (euFUL clade), CDM41 (FBP29 clade) and CDM111 (AP1 clade), and the E class protein CDM44 form ternary complexes with the presumed B class protein heterodimer CDM115–CDM86 in Chrysanthemum (Shchennikova et al., 2004). Our study indicates that A, E and AGL6 class proteins are able to interact with the B class proteins to form ternary complexes in Epimedium, suggesting this capacity may have been established before evolution of the core eudicots. Additionally, EsFUL, EsAGL2-1 and EsAGL6-like were able to bridge the interaction between EsAP3-2 and EsPI. Nonetheless, it is difficult to assign a biological role for ternary complexes involving EsFUL, EsAGL2-1 and EsAGL6-like proteins identified in this study. In Arabidopsis, ecoptic expression of AP3, PI and SEP3 together converted leaves to petals, a phenotype that was further pronounced when AP1 was simultaneously over-expressed (Honma and Goto, 2001). Interaction of a B class protein heterodimer with an A class protein that is referred to as the third protein can enhance the binding capability of the transcription factor complex (Egea-Cortines et al., 1999). We therefore speculate that an important function of EsFUL-like, EsAGL2-1 and EsAGL6 proteins are to add transcriptional activity to multimeric MADS transcription factor complexes.
Duplication of A/E probably resulted from genome triplication
The AP1/SQUA subfamily contains three lineages in core eudicots (euAP1, euFUL and AGL79) as a result of two gene duplication events (Litt and Irish, 2003; Shan et al., 2007). Comparative genomic analysis of publically available genome sequences indicates that a shared ancient polyploidization event, named γ, contributed to triplication of the genomes of asterids and rosids (Jaillon et al., 2007; Velasco et al., 2007; Jiao et al., 2011; Vekemans et al., 2012). Our synteny analysis of the AP1/SQUA locus in core eudicots identified three preserved syntenic groups of genes, indicating that a genome triplication event contributed to the occurrence of the three lineages (euAP1, euFUL and AGL79). In addition, tight association between adjacent A-type and E-type MADS box genes on the same chromosome were observed. Thus, it is likely that the similar phylogenetic topology shared within euAP1/euFUL/AGL79 and AGL2/AGL3/FBP9 is a result of the γ event. Interestingly, this linkage is not found in the euAP3/TM6 and euAG/PLE lineages (Causier et al., 2010), suggesting independent loss of one copy after consecutive whole genome triplication. The majority of plant transcription factors are derived from processes of exponential genome expansion after polyploidization (Tang et al., 2008). Therefore, increased numbers and complexity of transcription factors following ancient genome duplication events are suggested to have had a major impact on species diversification (Vekemans et al., 2012). For MADS-box genes, relaxed selection on some branches at the base of the AGL79 and euFUL lineage, the euAP3 lineage, the euAG lineage, as well as the AGL3 and FBP9 lineages, may have contributed to non-functionalization, neofunctionalization or subfunctionalization of genes, and enhanced complexity of the protein interaction network (Shan et al., 2009). Furthermore, divergence of regulatory regions may also be involved in diversification of MADS-box gene function (Shan et al., 2007).
SUPPLEMENTARY DATA
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Natural Science Foundation of China (31000919), CAS/SAFEA International Partnership Program for Creative Research Teams Project, and Knowledge Innovation Project of The Chinese Academy of Sciences [KSCX2-EW-J-20]. We thank Dr Tieyao Tu for discussions relating to synteny analysis, Dr Richard Immink for discussions of yeast two-hybrid and three-hybrid assys, and Drs Mei Zhang and Keqiang Wu for help with transforming Arabidopsis. We also thank Drs Erik Souer and Yaowu Yuan for critical editing of the manuscript.
LITERATURE CITED
- Adam H, Jouannic S, Morcillo F, Richaud F, Duval Y, Tregear JW. MADS box genes in oil palm (Elaeis guineensis): patterns in the evolution of the SQUAMOSA, DEFICIENS, GLOBOSA, AGAMOUS, and SEPALLATA subfamilies. Journal of Molecular Evolution. 2006;62:15–31. doi: 10.1007/s00239-005-0333-7. [DOI] [PubMed] [Google Scholar]
- Adamczyk BJ, Lehti-Shiu MD, Fernandes DE. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant Journal. 2007;50:1007–1019. doi: 10.1111/j.1365-313X.2007.03105.x. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla ER, Pelaz S, Liljegren SJ. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proceedings of the National Academy of Sciences. 2000;97:5328–5333. doi: 10.1073/pnas.97.10.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Saedler H, Theissen G. Distinct MADS-box gene expression patterns in the reproductive cones of the gymnosperm Gnetum gnemon. Development Genes and Evolution. 2003;213:567–572. doi: 10.1007/s00427-003-0358-0. [DOI] [PubMed] [Google Scholar]
- Becker A, Alix K, Damerval C. The evolution of flower development: current understanding and future challenges. Annals of Botany. 2011;107:1427–1431. doi: 10.1093/aob/mcr122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss PK, Bastow RM, Mylne JS, Dean C. Multiple pathways in the decision to flower: enabling, promoting, and resetting. Plant Cell. 2004;16:18–31. doi: 10.1105/tpc.015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broholm SK, Pöllänen E, Ruokolainen S, et al. Functional characterization of B class MADS-box transcription factors in Gerbera hybrida. Journal of Experimental Botany. 2010;61:75–85. doi: 10.1093/jxb/erp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causier B, Castillo R, Xue Y, Schwarz-Sommer Z, Davies B. Tracing the evolution of the floral homeotic B- and C-function genes through genome synteny. Molecular Biology Evolution. 2010;27:2651–2664. doi: 10.1093/molbev/msq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Chiu Y, Wu J, Yang C. Four orchid (Oncidium Gower Ramsey) AP1/AGL9-like MADS box genes show novel expression patterns and cause different effects on floral transition and formation in Arabidopsis thaliana. Plant and Cell Physiology. 2009;50:1425–1438. doi: 10.1093/pcp/pcp087. [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant Journal. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- Ditta G, Pinyopich A, Robles P, Pelaz S, Yanofsky MF. The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Current Biology. 2004;14:1935–1940. doi: 10.1016/j.cub.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Egea-Cortines M, Saedler H, Sommer H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. The EMBO Journal. 1999;18:5370–5379. doi: 10.1093/emboj/18.19.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elo A, Lemmetyinen J, Turunen ML, Tikka L, Sopanen T. Three MADS-box genes similar to APETALA1 and FRUITFULL from silver birch (Betula pendula) Physiologia Plantarum. 2001;112:95–103. doi: 10.1034/j.1399-3054.2001.1120113.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Ferrandiz C, Gu Q, Martienssen R, Yanofsky MF. Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development. 2000;127:725–734. doi: 10.1242/dev.127.4.725. [DOI] [PubMed] [Google Scholar]
- Ferrario S, Immink RG, Shchennikova A, Busscher-Lange J, Angenent GC. The MADS box gene FBP2 is required for SEPALLATA function in petunia. Plant Cell. 2003;15:914–925. doi: 10.1105/tpc.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Folter S, Immink RG, Kieffer M, et al. Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell. 2005;17:1424–1433. doi: 10.1105/tpc.105.031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornara F, Parenicova L, Pelucchi N, et al. Functional characterization of OsMADS18, a member of the AP1/SQUA subfamily of MADS box genes. Plant Physiology. 2004;135:2207–2219. doi: 10.1104/pp.104.045039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Kyozuka J, Bowman JL. Turning floral organs into leaves, leaves into floral organs. Current Opinion in Genetics and Development. 2001;11:449–456. doi: 10.1016/s0959-437x(00)00216-1. [DOI] [PubMed] [Google Scholar]
- Gregis V, Sessa A, Dorca-Fornell C, Kater MM. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant Journal. 2009;60:626–637. doi: 10.1111/j.1365-313X.2009.03985.x. [DOI] [PubMed] [Google Scholar]
- Gu Q, Ferrandiz C, Yanofsky MF, Martienssen R. The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development. 1998;8:1509–1517. doi: 10.1242/dev.125.8.1509. [DOI] [PubMed] [Google Scholar]
- Hileman LC, Irish VF. More is better: the uses of developmental genetic data to reconstruct perianth evolution. American Journal of Botany. 2009;96:83–95. doi: 10.3732/ajb.0800066. [DOI] [PubMed] [Google Scholar]
- Honma T, Goto K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature. 2001;409:525–529. doi: 10.1038/35054083. [DOI] [PubMed] [Google Scholar]
- Hu J, Zhang J, Shan H, Chen Z. Expression of floral MADS-box genes in Sinofranchetia chinensis (Lardizabalaceae): implications for the nature of the nectar leaves. Annals of Botany. 2012;110:57–69. doi: 10.1093/aob/mcs104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sun W, Wang Y. Isolation and molecular characterisation of flavonoid 3′-hydroxylase and flavonoid 3′, 5′-hydroxylase genes from a traditional Chinese medicinal plant, Epimedium sagittatum. Gene. 2012;497:125–130. doi: 10.1016/j.gene.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Huijser P, Klein J, Lonnig WE, Meijer H, Saedler H, Sommer H. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO Journal. 1992;11:1239–1249. doi: 10.1002/j.1460-2075.1992.tb05168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury JM, Noel B, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Melzer R, Theissen G. MIKC-type MADS domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene. 2005;347:183–198. doi: 10.1016/j.gene.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Kim S, Koh J, Yoo M-J, et al. Expression of floral MADS-box genes in basal angiosperms: implications for the evolution of floral regulators. Plant Journal. 2005;43:724–744. doi: 10.1111/j.1365-313X.2005.02487.x. [DOI] [PubMed] [Google Scholar]
- Kinjo H, Shitsukawa N, Takumi S, Murai K, et al. Diversification of three APETALA1/FRUITFULL-like genes in wheat. Molecular Genetics and Genomics. 2012;287:283–294. doi: 10.1007/s00438-012-0679-7. [DOI] [PubMed] [Google Scholar]
- Kramer EM, Dorit RL, Irish VF. Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics. 1998;149:765–783. doi: 10.1093/genetics/149.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EM, Jaramillo MA, Di Stilio VS. Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in angiosperms. Genetics. 2004;166:1011–1023. doi: 10.1534/genetics.166.2.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizek BA, Fletcher JC. Molecular mechanisms of flower development: an armchair guide. Nature Reviews Genetics. 2005;6:688–698. doi: 10.1038/nrg1675. [DOI] [PubMed] [Google Scholar]
- Kotilainen M, Elomaa P, Uimari A, Albert VA, Teeri TH. GRCD1, an AGL2-like MADS box gene, participates in the C function during stamen development in Gerbera hybrid. Plant Cell. 2000;12:1893–1902. doi: 10.1105/tpc.12.10.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lee I. Regulation and function of SOC1, a flowering pathway integrator. Journal of Experimental Botany. 2010;61:2247–2254. doi: 10.1093/jxb/erq098. [DOI] [PubMed] [Google Scholar]
- Leseberg CH, Eissler CL, Wang X, Johns MA, Duvall MR, Mao L. Interaction study of MADS-domain proteins in tomato. Journal of Experimental Botany. 2008;59:2253–2265. doi: 10.1093/jxb/ern094. [DOI] [PubMed] [Google Scholar]
- Lin EP, Peng HZ, Jin QY, et al. Identification and characterization of two Bamboo (Phyllostachys praecox) AP1/SQUA-like MADS-box genes during floral transition. Planta. 2009;231:109–120. doi: 10.1007/s00425-009-1033-0. [DOI] [PubMed] [Google Scholar]
- Litt A, Irish VF. Duplication and diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: implications for the evolution of floral development. Genetics. 2003;165:821–833. doi: 10.1093/genetics/165.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen H, Er HL, et al. Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development. 2008;135:1481–1491. doi: 10.1242/dev.020255. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhang J, Zhang N, et al. Interactions among proteins of floral MADS-box genes in basal eudicots: implications for evolution of the regulatory network for flower development. Molecular Biology and Evolution. 2010;27:1598–1611. doi: 10.1093/molbev/msq044. [DOI] [PubMed] [Google Scholar]
- Mandel MA, Yanofsky MF. The Arabidopsis AGL8 MADS box gene is expressed in inflorescence meristems and is negatively regulated by APETALA1. Plant Cell. 1995;7:1763–1771. doi: 10.1105/tpc.7.11.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MA, Gustafson-Brown C, Savidge B, Yanofsky MF. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- Masiero S, Colombo L, Grini PE, Schnittger A, Kater MM. The emerging importance of type I MADS Box transcription factors for plant reproduction. Plant Cell. 2011;23:865–872. doi: 10.1105/tpc.110.081737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer R, Verelst W, Theissen G. The class E floral homeotic protein SEPALLATA3 is sufficient to loop DNA in ‘floral quartet’-like complexes in vitro. Nucleic Acids Research. 2009;37:144–157. doi: 10.1093/nar/gkn900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels SD, Amasino RM. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell. 1999;11:949–956. doi: 10.1105/tpc.11.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J, dePamphilis CW, Ma H, Nei M. Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Molecular Biology and Evolution. 2003;20:1435–1447. doi: 10.1093/molbev/msg152. [DOI] [PubMed] [Google Scholar]
- Ohmori S, Kimizu M, Sugita M, et al. MOSAIC FLORAL ORGANS1, an AGL6-like MADS box gene, regulates floral organ identity and meristem fate in rice. Plant Cell. 2009;21:3008–3025. doi: 10.1105/tpc.109.068742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onouchi H, Igeno MI, Perilleux C, Graves K, Coupland G. Mutagenesis of plants overexpressing CONSTANS demonstrates novel interactions among Arabidopsis flowering-time genes. Plant Cell. 2000;12:885–900. doi: 10.1105/tpc.12.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabón-Mora N, Ambrose BA, Litt A. Poppy APETALA1/FRUITFULL orthologs control flowering time, branching, perianth identity, and fruit development. Plant Physiology. 2012;158:1685–1704. doi: 10.1104/pp.111.192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabón-Mora N, Sharma B, Holappa LD, Kramer EM, Litt A. The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. The Plant Journal. 2013;74:197–212. doi: 10.1111/tpj.12113. [DOI] [PubMed] [Google Scholar]
- Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;405:200–203. doi: 10.1038/35012103. [DOI] [PubMed] [Google Scholar]
- Pelaz S, Gustafson-Brown C, Kohalmi SE, Crosby WL, Yanofsky MF. APETALA1 and SEPALLATA3 interact to promote flower development. Plant Journal. 2001a;26:385–394. doi: 10.1046/j.1365-313x.2001.2641042.x. [DOI] [PubMed] [Google Scholar]
- Pelaz S, Tapia-Lopez R, Alvare-Buylla ER, et al. Conversion of leaves into petals in Arabidopsis. Current Biology. 2001b;11:182–184. doi: 10.1016/s0960-9822(01)00024-0. [DOI] [PubMed] [Google Scholar]
- Pinyopich A, Ditta GS, Savidge B, et al. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature. 2003;424:85–88. doi: 10.1038/nature01741. [DOI] [PubMed] [Google Scholar]
- Piwarzyk E, Yang Y, Jack T. Conserved C-terminal motifs of the Arabidopsis proteins APETALA3 and PISTILLATA are dispensable for floral organ identity function. Plant Physiology. 2007;145:1495–1505. doi: 10.1104/pp.107.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijpkema AS, Zethof J, Gerates T, Vandenbussche M. The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant Journal. 2009;60:1–9. doi: 10.1111/j.1365-313X.2009.03917.x. [DOI] [PubMed] [Google Scholar]
- Ruokolainen S, Ng YP, Albert VA, Elomaa P, Teeri TH. Large scale interaction analysis predicts that the Gerbera hybrida floral E function is provided both by general and specialized proteins. BMC Plant Biology. 2010;10:129. doi: 10.1186/1471-2229-10-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle I, Coupland G. Induction of flowering by seasonal changes in photoperiod. EMBO Journal. 2004;23:1217–1222. doi: 10.1038/sj.emboj.7600117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H, Zhang N, Liu C, et al. Patterns of gene duplication and functional diversification during the evolution of the AP1/SQUA subfamily of plant MADS-box genes. Molecular Phylogenetics and Evolution. 2007;44:26–41. doi: 10.1016/j.ympev.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Shan H, Zahn L, Guindon S, et al. Evolution of plant MADS box transcription factors: evidence for shifts in selection associated with early angiosperm diversification and concerted gene duplications. Molecular Biology and Evolution. 2009;26:2229–2244. doi: 10.1093/molbev/msp129. [DOI] [PubMed] [Google Scholar]
- Shchennikova AV, Shulga OA, Immink R, et al. Identification and characterization of four chrysanthemum MADS-box genes, belonging to the APETALA1/FRUITFULL and SEPALLATA3 subfamilies. Plant Physiology. 2004;134:1632–1641. doi: 10.1104/pp.103.036665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearn WT. The genus Epimedium and other herbaceous Berberidaceae. Portland, OR: Timber Press; 2002. [Google Scholar]
- Tang H, Wang X, Bowers JE, et al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Research. 2008;18:1944–1954. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theissen G. Development of floral organ identity: stories from the MADS house. Current Opinion in Plant Biology. 2001;4:75–85. doi: 10.1016/s1369-5266(00)00139-4. [DOI] [PubMed] [Google Scholar]
- Theissen G, Melzer R. Molecular mechanisms underlying origin and diversification of the angiosperm flower. Annals of Botany. 2007;100:603–619. doi: 10.1093/aob/mcm143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng T, Hsiao C, Chi P, Yang C. Two lily SEPALLATA-like genes cause different effects on floral formation and floral transition in Arabidopsis. Plant Physiology. 2003;133:1091–1101. doi: 10.1104/pp.103.026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbussche M, Zethof J, Souer E, et al. Toward the analysis of the petunia MADS box gene family by reverse and forward transposon insertion mutagenesis approaches: B, C, and D floral organ identity functions require SEPALLATA-like MADS box genes in petunia. Plant Cell. 2003;15:2680–2693. doi: 10.1105/tpc.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkonyi-Gasic E, Moss SM, Voogd C, et al. Identification and characterization of flowering genes in kiwifruit: sequence conservation and role in kiwifruit flower development. BMC Plant Biology. 2011;11:72. doi: 10.1186/1471-2229-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekemans D, Proost S, Vanneste K, et al. Gamma paleohexaploidy in the stem lineage of core eudicots: significance for MADS-box gene and species diversification. Molecular Evolution and Biology. 2012;29:3793–3806. doi: 10.1093/molbev/mss183. [DOI] [PubMed] [Google Scholar]
- Velasco R, Zharkikh A, Troggio M, et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One. 2007;2 doi: 10.1371/journal.pone.0001326. e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrebalov J, Ruezinsky D, Padmanabhan V, et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science. 2002;296:343–346. doi: 10.1126/science.1068181. [DOI] [PubMed] [Google Scholar]
- Viaene T, Vekemans D, Becker A, Melzer S, Geuten K. Expression divergence of the AGL6 MADS domain transcription factor lineage after a core eudicot duplication suggests functional diversification. BMC Plant Biology. 2010;10:148. doi: 10.1186/1471-2229-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jack T. Defining subdomains of the K domain important for protein–protein interactions of plant MADS proteins. Plant Molecular Biology. 2004;55:45–59. doi: 10.1007/s11103-004-0416-7. [DOI] [PubMed] [Google Scholar]
- Yoo SK, Hong SM, Lee JS, Ahn JH. A genetic screen for leaf movement mutants identifies a potential role for AGAMOUS-LIKE 6 (AGL6) in circadian-clock control. Molecules and Cells. 2011;31:281–287. doi: 10.1007/s10059-011-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn LM, Kong H, Leebens-Mack J, et al. The evolution of the SEPALLATA subfamily of MADS-box genes: a preangiosperm origin with multiple duplications throughout angiosperm history. Genetics. 2005;169:2209–2223. doi: 10.1534/genetics.104.037770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng S, Xiao G, Guo J, et al. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics. 2010;11:94. doi: 10.1186/1471-2164-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Yu Y, Meyer D, Wu C, Shen W. Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3 K36. Nature Cell Biology. 2005;7:1256–1260. doi: 10.1038/ncb1329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.