

Abstract
In Escherichia coli, the MnmEG complex modifies transfer RNAs (tRNAs) decoding NNA/NNG codons. MnmEG catalyzes two different modification reactions, which add an aminomethyl (nm) or carboxymethylaminomethyl (cmnm) group to position 5 of the anticodon wobble uridine using ammonium or glycine, respectively. In  and
and  , however, cmnm5 appears as the final modification, whereas in the remaining tRNAs, the MnmEG products are converted into 5-methylaminomethyl (mnm5) through the two-domain, bi-functional enzyme MnmC. MnmC(o) transforms cmnm5 into nm5, whereas MnmC(m) converts nm5 into mnm5, thus producing an atypical network of modification pathways. We investigate the activities and tRNA specificity of MnmEG and the MnmC domains, the ability of tRNAs to follow the ammonium or glycine pathway and the effect of mnmC mutations on growth. We demonstrate that the two MnmC domains function independently of each other and that
, however, cmnm5 appears as the final modification, whereas in the remaining tRNAs, the MnmEG products are converted into 5-methylaminomethyl (mnm5) through the two-domain, bi-functional enzyme MnmC. MnmC(o) transforms cmnm5 into nm5, whereas MnmC(m) converts nm5 into mnm5, thus producing an atypical network of modification pathways. We investigate the activities and tRNA specificity of MnmEG and the MnmC domains, the ability of tRNAs to follow the ammonium or glycine pathway and the effect of mnmC mutations on growth. We demonstrate that the two MnmC domains function independently of each other and that  and
and  are substrates for MnmC(m), but not MnmC(o). Synthesis of mnm5s2U by MnmEG-MnmC in vivo avoids build-up of intermediates in
are substrates for MnmC(m), but not MnmC(o). Synthesis of mnm5s2U by MnmEG-MnmC in vivo avoids build-up of intermediates in  . We also show that MnmEG can modify all the tRNAs via the ammonium pathway. Strikingly, the net output of the MnmEG pathways in vivo depends on growth conditions and tRNA species. Loss of any MnmC activity has a biological cost under specific conditions.
. We also show that MnmEG can modify all the tRNAs via the ammonium pathway. Strikingly, the net output of the MnmEG pathways in vivo depends on growth conditions and tRNA species. Loss of any MnmC activity has a biological cost under specific conditions.
INTRODUCTION
Transfer RNAs (tRNAs) are highly and diversely modified, and each has a unique pattern of modification (1–3). These modifications are post-transcriptionally introduced at precise positions by specific enzymes and play important roles in folding, stability, identity and in the functions of tRNAs.
Modifications at the wobble position of the anticodon contribute to the accuracy and efficiency of protein synthesis (1,3,4). Changes in the modification levels of the wobble position affect the synthesis of specific proteins and also lead to complex phenotypes through unknown mechanisms (3,5–10).
Wobble modifications are often complex and require for their synthesis the participation of several enzymes (3). Even though all of the enzymes involved in a specific modification pathway are known, it is unclear how their actions are modulated and coordinated to produce the final modification. Progress in this area may lead to a better understanding of tRNA biology.
Proteins MnmE and MnmG (formerly TrmE and GidA, respectively) are evolutionary conserved from bacteria to eukaryotic organelles. In Escherichia coli, they are involved in the modification of the wobble uridine of  ,
, 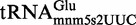 ,
,  ,
,  ,
,  and
and  , all of which read NNA/G codons of the split codon boxes, with the exception of
, all of which read NNA/G codons of the split codon boxes, with the exception of  , which reads codons of the glycine family box (11). MnmE and MnmG are dimeric and form a functional α2β2 heterotetrameric complex (MnmEG) in which both proteins are interdependent (12,13). MnmE is a GTP- and tetrahydrofolate (THF)-binding protein, whereas MnmG is a FAD- and NADH-binding protein (14–18). The MnmEG complex catalyzes the addition of the aminomethyl (nm) and carboxymethylaminomethyl (cmnm) groups to position 5 of the wobble uridine using ammonium and glycine, respectively [Figure 1; (13)]. Both reactions require GTP and FAD as well as NADH if FAD is limiting in the in vitro reaction. Moreover, a THF derivative, likely methylene-THF, serves as the donor of the methylene carbon that is directly bonded to the C5 atom of U34. However, the detailed mechanism of the basic reaction has not yet been demonstrated (11,13). According to current models, GTP hydrolysis by the MnmE G-domain, which is located distant from the MnmEG active center, leads to structural rearrangements in the MnmEG complex, which are essential for tRNA modification (11,19).
, which reads codons of the glycine family box (11). MnmE and MnmG are dimeric and form a functional α2β2 heterotetrameric complex (MnmEG) in which both proteins are interdependent (12,13). MnmE is a GTP- and tetrahydrofolate (THF)-binding protein, whereas MnmG is a FAD- and NADH-binding protein (14–18). The MnmEG complex catalyzes the addition of the aminomethyl (nm) and carboxymethylaminomethyl (cmnm) groups to position 5 of the wobble uridine using ammonium and glycine, respectively [Figure 1; (13)]. Both reactions require GTP and FAD as well as NADH if FAD is limiting in the in vitro reaction. Moreover, a THF derivative, likely methylene-THF, serves as the donor of the methylene carbon that is directly bonded to the C5 atom of U34. However, the detailed mechanism of the basic reaction has not yet been demonstrated (11,13). According to current models, GTP hydrolysis by the MnmE G-domain, which is located distant from the MnmEG active center, leads to structural rearrangements in the MnmEG complex, which are essential for tRNA modification (11,19).
Figure 1.

Synthesis of mnm5(s2)U in E. coli. The MnmEG complex formed by proteins MnmE and MnmG is active on position 5 of the wobble uridine (U34) in  ,
,  ,
,  ,
,  ,
,  and
and  . MnmE is a three-domain protein that binds GTP and methylenetetrahydrofolate (CH2-THF), whereas MnmG is a FAD- and NADH-binding protein. The MnmC(o) activity of the bi-functional enzyme MnmC transforms cmnm5(s2)U to nm5(s2)U and the MnmC(m) activity of MnmC transforms nm5(s2)U to the final product mnm5(s2)U. Thiolation at position 2 of the wobble uridine (U34) in
. MnmE is a three-domain protein that binds GTP and methylenetetrahydrofolate (CH2-THF), whereas MnmG is a FAD- and NADH-binding protein. The MnmC(o) activity of the bi-functional enzyme MnmC transforms cmnm5(s2)U to nm5(s2)U and the MnmC(m) activity of MnmC transforms nm5(s2)U to the final product mnm5(s2)U. Thiolation at position 2 of the wobble uridine (U34) in  ,
,  and
and  is mediated by the protein MnmA. Modifications occur at the 5- and 2-positions independent of each other. TrmL methylates the 2′-OH group of the U-ribose in
is mediated by the protein MnmA. Modifications occur at the 5- and 2-positions independent of each other. TrmL methylates the 2′-OH group of the U-ribose in  (inset panel).
(inset panel).
The wobble uridine (U34) of tRNAs modified by MnmEG may be further modified by other enzymes at position 5 or other positions. In  ,
,  and
and  , thiolation at position 2 of the U34 is performed by MnmA (20), whereas in
, thiolation at position 2 of the U34 is performed by MnmA (20), whereas in  , TrmL methylates the 2′-OH group of the U-ribose (21). Therefore, some tRNA substrates for the MnmEG complex are also substrates for MnmA or TrmL.
, TrmL methylates the 2′-OH group of the U-ribose (21). Therefore, some tRNA substrates for the MnmEG complex are also substrates for MnmA or TrmL.
Moreover, in  ,
,  ,
,  , and
, and  , the resulting products of MnmEG activity are not the final modifications because these tRNAs carry the methylaminomethyl (mnm) group at position 5 of U34. Formation of the mnm5-final group is mediated by the action of the two-domain, bi-functional enzyme MnmC (22–26). The oxidoreductase activity of the C-terminal domain, here designated as MnmC(o), is responsible for the FAD-dependent deacetylation that transforms cmnm5U into nm5U, whereas the methyltransferase activity of the N-terminal domain, designated as MnmC(m), catalyzes the SAM-dependent methylation that transforms nm5U to mnm5U (Figure 1).
, the resulting products of MnmEG activity are not the final modifications because these tRNAs carry the methylaminomethyl (mnm) group at position 5 of U34. Formation of the mnm5-final group is mediated by the action of the two-domain, bi-functional enzyme MnmC (22–26). The oxidoreductase activity of the C-terminal domain, here designated as MnmC(o), is responsible for the FAD-dependent deacetylation that transforms cmnm5U into nm5U, whereas the methyltransferase activity of the N-terminal domain, designated as MnmC(m), catalyzes the SAM-dependent methylation that transforms nm5U to mnm5U (Figure 1).
The crystal structure of MnmC consists of two globular domains interacting with each other (27). The structure also reveals that the two catalytic centers of MnmC(o) and MnmC(m) face opposite sides of the protein, thus favoring a model in which the two domains could function in a relatively independent manner. In fact, when two MnmC mutant proteins possessing a catalytically dead MnmC(o) or MnmC(m) domain were mixed, partial recovery of mnm5-group synthesis was observed (25). However, considering the relatively hydrophilic nature of the domain interface, the possibility that conformational changes within the entire MnmC protein may occur in vivo and affect its functioning, cannot be excluded. A previous attempt to separately express both MnmC domains revealed that MnmC(m) was soluble, whereas MnmC(o) produced inclusion bodies, suggesting that MnmC(m) is required for the correct folding or structural stability of MnmC(o) (25).
How the activities of the MnmEG and MnmC enzymes are organized and modulated remains unclear. Whether tRNAs preferentially utilize one of the two MnmEG pathways, the glycine or the ammonium pathway (Figure 1), in vivo depending on metabolic circumstances has not been investigated. The fact that only cmnm5, but not nm5 or mnm5 has been detected to date at position 5 of U34 in  and
and  suggests that both tRNAs do not use the ammonium pathway and are not substrates for MnmC(o). Accumulation of the nm5s2U nucleoside which may have a dual origin [Figure 1; (13)], has not been observed in total tRNA purified from wild-type E. coli cells (23). Therefore, the mnm5s2U synthesis in
suggests that both tRNAs do not use the ammonium pathway and are not substrates for MnmC(o). Accumulation of the nm5s2U nucleoside which may have a dual origin [Figure 1; (13)], has not been observed in total tRNA purified from wild-type E. coli cells (23). Therefore, the mnm5s2U synthesis in  and
and  appears to be organized to prevent nm5s2U accumulation. However, the presence or absence of cmnm5s2U as an intermediate in mnm5s2U biosynthesis is difficult to determine from nucleoside analysis of bulk tRNA due to the natural occurrence of cmnm5s2U in
appears to be organized to prevent nm5s2U accumulation. However, the presence or absence of cmnm5s2U as an intermediate in mnm5s2U biosynthesis is difficult to determine from nucleoside analysis of bulk tRNA due to the natural occurrence of cmnm5s2U in  . Biosynthetic tuning of mnm5s2U to avoid the accumulation of intermediates could be achieved by either kinetic tuning of the activities of MnmEG and MnmC, which requires that the sequential modifications are performed at similar or increasing rates or selective degradation of partially modified tRNAs. A previous steady-state kinetic analysis of the activities of the full MnmC protein indicated that the MnmC(m)-dependent reaction (nm5→mnm5) occurs faster than the MnmC(o)-dependent reaction (cmnm5→nm5) or at a similar rate at very high substrate (tRNA) concentrations (26). However, this study did not take into account the efficiency of the reactions performed by MnmEG (Figure 1), which is crucial to understand the organization of the mnm5s2U biosynthetic process.
. Biosynthetic tuning of mnm5s2U to avoid the accumulation of intermediates could be achieved by either kinetic tuning of the activities of MnmEG and MnmC, which requires that the sequential modifications are performed at similar or increasing rates or selective degradation of partially modified tRNAs. A previous steady-state kinetic analysis of the activities of the full MnmC protein indicated that the MnmC(m)-dependent reaction (nm5→mnm5) occurs faster than the MnmC(o)-dependent reaction (cmnm5→nm5) or at a similar rate at very high substrate (tRNA) concentrations (26). However, this study did not take into account the efficiency of the reactions performed by MnmEG (Figure 1), which is crucial to understand the organization of the mnm5s2U biosynthetic process.
Modifications located within or adjacent to the anticodon are important for stabilization of codon–anticodon pairing as well as for restricting the dynamics of the anticodon domain and shaping its architecture (1,4). Considering the network of pathways leading to mnm5s2U production (Figure 1), it is important to determine whether mutations affecting the different activities of MnmC [MnmC(o) or MnmC(m)] have distinct biological consequences and to what extent an accumulation of modification intermediates affects bacterial biology.
In this study, we investigated the activities and the tRNA specificity of the MnmEG and MnmC enzymes in vivo and in vitro, analyzed how the U34 modification status is influenced by genetic and physiological conditions in bulk and specific tRNAs and assessed the effects of mnmC mutations on bacterial growth. Our study was facilitated by the cloning and separate expression of the two MnmC domains, MnmC(o) and MnmC(m). In contrast to a previous report (25), we demonstrate that MnmC(o) can fold independently of MnmC(m) and that the separate domains exhibit similar kinetic properties to those of the full protein. We also show that  and
and  are substrates for MnmC(m), but not for MnmC(o). Our data suggest that MnmEG and MnmC are kinetically tuned to produce only the fully modified nucleoside mnm5U in
are substrates for MnmC(m), but not for MnmC(o). Our data suggest that MnmEG and MnmC are kinetically tuned to produce only the fully modified nucleoside mnm5U in  . We demonstrate that all the tRNA substrates of MnmEG are modified in vitro through the ammonium pathway. However, the net output of the ammonium and glycine pathways of MnmEG in vivo depends on growth conditions and tRNA species. Finally, we demonstrate that the loss of any MnmC activity has a biological cost under specific conditions.
. We demonstrate that all the tRNA substrates of MnmEG are modified in vitro through the ammonium pathway. However, the net output of the ammonium and glycine pathways of MnmEG in vivo depends on growth conditions and tRNA species. Finally, we demonstrate that the loss of any MnmC activity has a biological cost under specific conditions.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, media and general techniques
Escherichia coli strains and plasmids are shown in Table 1. A list of the oligonucleotides and primers used in this work is provided in Supplementary Table S1. Transduction with phage P1 was performed as previously described (28). Deletion of the mnmC(o) coding region was performed by targeted homologous recombination (29) using the primers MnmC(o)Δ-F and MnmC(o)Δ-R. The MnmC(o)Δ-F primer introduced a TAA stop codon at the end of the MnmC(m) coding region, which is located in the 5′-terminal region of the mnmC gene. Deletion of the mnmC(o) coding sequence was confirmed by PCR and DNA sequencing. The resulting strain was named IC6629. DNA sequences coding for the MnmC(o) (250–668 a.a.) and MnmC(m) domains (1–250 a.a.) fused to the C-terminal end of a Flag-epitope were amplified by polymerase chain reaction (PCR) from genomic DNA of E. coli MG1655 using the specific primer pairs Flag-MnmC(o)F/Flag-MnmC(o)R and Flag-MnmC(m)F/Flag-MnmC(m)R, respectively. The amplicons were cloned into pBAD TOPO TA for expression under control of the AraC-PBAD system. The resulting plasmids were transformed into a mnmC::kan strain, unless otherwise specified. A DNA fragment encoding the MnmC(m) domain fused at its N-terminal end to a 6x-His-epitope was cloned into the same vector after PCR amplification with the His-MnmC(m)R/His-MnmC(m)F primer pair. LBT (LB broth containing 40 mg/l thymine) and LAT (LBT containing 20 g of Difco agar per liter) were used for routine cultures and plating of E. coli, respectively, unless otherwise specified. When required, antibiotics were added at the following final concentrations: 100 µg/ml ampicillin; 12.5 µg/ml tetracycline; 25 µg/ml chloramphenicol and 80 µg/ml kanamycin. Cell growth was monitored by measuring the optical density of the cultures at 600 nm (OD600).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | References |
|---|---|---|
| Eschirichia coli strains | ||
| DEV16 (IC4385)a | thi-1 rel-1 spoT1 lacZ105UAG valRmnmE-Q192X | (14,48) |
| BW25113 (IC5136)a | lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | (29) |
| TH178 (IC5255)a | mnmA1 fadR::Tn10 | (45) |
| MG1655(IC5356)a | F- | D. Touati |
| TH48 (IC6017)a | ara, Δ(lac-proB), nalA, argEam, rif, thi, fadL::Tn10 | (23) |
| TH49 (IC6018)a | TH48 mnmC(m)-G68D | (23) |
| TH69 (IC6019)a | TH48 mnmC-W131stop | (23) |
| IC4639 | DEV16 mnmE+ bgl (Sal+) | (12) |
| IC5241 | MG1655 mnmG::Tn10 [TetR] | (12) |
| IC5358 | MG1655 mnmE::kan | (43) |
| IC5397 | P1 (IC5255) x IC4639; [IC4639 mnmA-Q233stop] | M. Villarroya |
| IC5827 | BW25113 mnmE::kan | NBRP- Japan |
| IC5854 | BW25113 trmL::kan | (21) |
| IC5937 | IC4639 mnmA::kan [or IC4639 ΔmnmA] | (39) |
| IC5975 | BL21-DE3 mnmG::kan | (17) |
| IC6010 | BW25113 mnmC::Kan [or BW25113 ΔmnmC] | (13) |
| IC6166 | IC5975 carrying pIC1446 | (17) |
| IC6222 | P1 (IC6010) x IC4639; [IC4639 ΔmnmC)] | This study |
| IC6374 | IC4639 trmL::kan [IC4639 ΔtrmL] | (21) |
| IC6411 | P1 (IC5241) x IC6374 (trmL::kan) [IC4639 ΔtrmL ΔmnmG] | (21) |
| IC6424 | IC5358 carrying pIC684 | (14) |
| IC6587 | P1 (TH49) x IC4639; [IC4639 mnmC(m)-G68D] | This study |
| IC6588 | P1 (TH49) x IC5937; [IC4639 ΔmnmA mnmC(m)-G68D] | This study |
| IC6589 | P1 (IC6010) x IC5397; [IC4639 mnmA-Q233stop ΔmnmC] | This study |
| IC6629 | BW25113 mnmC(o)::cat [or BW25113 ΔmnmC(o)] | This study |
| IC6725 | P1 (IC6629) x IC5937 [IC4639 ΔmnmA ΔmnmC(o)] | This study |
| Plasmids | ||
| pIC684 | GST fusion of mnmE cloned in pGEX-2T | (14) |
| pIC1083 |
Eschirichia coli
 cloned in pUC19 cloned in pUC19 |
(34) |
| pIC1253 | flag-mnmC cloned in pBAD-TOPO | (13) |
| pIC1339 | flag-mnmC(o) cloned in pBAD-TOPO | This study |
| pIC1340 | flag-mnmC(m) cloned in pBAD-TOPO | This study |
| pIC1394 |
Eschirichia coli
 cloned into pUC19, SmaI site cloned into pUC19, SmaI site |
This study |
| pIC1395 |
E schirichia coli
 cloned into pUC19, SmaI site cloned into pUC19, SmaI site |
This study |
| pIC1446 | his-mnmG cloned in pET15b | (17) |
| pIC1535 |
Eschirichia coli
 cloned into pBAD-TOPO cloned into pBAD-TOPO |
This study |
| pIC1536 |
Eschirichia coli
 cloned into pUC19, SmaI site cloned into pUC19, SmaI site |
This study |
| pIC1537 |
Eschirichia coli
 cloned into pUC19, SmaI site cloned into pUC19, SmaI site |
This study |
| pIC1550 |
Eschirichia coli
 cloned into pUC19, SmaI site cloned into pUC19, SmaI site |
This study |
| pIC1577 | pBSKrna | (32) |
| pIC1617 |
Eschirichia coli
 cloned in pBSKrna, EcoRV site cloned in pBSKrna, EcoRV site |
This study |
| pIC1664 |
Eschirichia coli
 cloned in pBSKrna, EcoRI/PstI sites cloned in pBSKrna, EcoRI/PstI sites |
This study |
| pIC1665 |
Eschirichia coli
 cloned into pBSKrna, EcoRI–PstI site cloned into pBSKrna, EcoRI–PstI site |
This study |
| pIC1677 | his-mnmC(m) cloned in pBAD-TOPO | This study |
| pIC1714 |
Eschirichia coli
 cloned into pBSKrna, EcoRI–PstI site cloned into pBSKrna, EcoRI–PstI site |
This study |
aName in our collection between brackets.
Growth rate determinations and competition experiments
To determine bacterial growth rates, overnight cultures were diluted 1/100 in fresh LBT medium and incubated at 37°C with shaking. The doubling time was measured by monitoring the optical density of the culture at 600 nm. Samples were taken from exponentially growing cultures after at least 10 generations of steady-state growth. The growth rate was calculated as the doubling time of each culture in the steady-state log phase by linear regression. Competition experiments were performed as previously reported (21). Briefly, the reference strain (BW25113) and the strain to be tested (TH48, TH49 or TH69) were grown separately to stationary phase by incubation at 37°C. Equal volumes of both strains were mixed and a sample was immediately taken to count viable cells on LAT plates with and without the antibiotic required to estimate the content of each strain (tetracycline to identify strains of the TH48 background). Six cycles of 24-h growth at 37°C (with shaking) were performed by diluting mixed cultures of 1/1000 in LBT medium. After the sixth cycle, the mixed culture was analyzed for its strain content as before. The final ratio was calculated as the ratio of the number of colony forming units (CFU) per milliliter recovered on LAT supplemented with tetracycline versus CFU per milliliter on LAT.
Purification of recombinant proteins
Routinely, a single colony was inoculated into 5 ml LBT supplemented with adequate antibiotics and grown overnight at 37°C. The pre-culture was then inoculated (1:100 dilution) into 1000 ml LBT plus antibiotic and grown until the OD600 reached ∼0.5. Protein expression was induced by the addition of 0.2% arabinose (Flag-tagged proteins). The cultures were grown for an additional 3–4 h at 30°C with gentle shaking, harvested by centrifugation (4500g for 15 min at 4°C), washed with TBS buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl and 5 mM MgCl2) and stored at −20°C. For enzyme purification, frozen cells were thawed on ice, resuspended in 10 ml of lysis buffer (TBS containing 1 mM PMSF and 1 mM EDTA) and disrupted by sonication. The lysate was centrifuged at 10 000g for 45 min at 4°C and the supernatant was mixed with 0.3–0.6 ml of pre-equilibrated anti-Flag agarose resin (ANTI-FLAG M2 Affinity Gel, A2220-Sigma) and incubated at 4°C for 1 h with mild shaking. Instructions from the manufacturer were followed for washing and eluting Flag-tagged proteins. The fractions containing the proteins were pooled, concentrated with Amicon Ultra-15 30k devices in TBS buffer and stored in 50-µl fractions with 15% glycerol at −20°C. The His-MnmC(m) domain was purified using Clontech’s TALON Metal Affinity Resin according to the manufacturer’s instructions. The MnmE and MnmG proteins were purified as previously described (14,17). Protein concentrations were determined with a NanoDrop spectrophotometer at 280 nm. The purity of all enzymes was >95% as estimated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and coomassie blue staining.
FAD cofactor analysis
FAD was released from recombinant proteins by heating at 75°C for 5 min in the dark and analyzed by high-performance liquid chromatography (HPLC). Briefly, HPLC separation was achieved with a Synergi 4u Fusion column (25 cm × 4.6 mm, 5 µm) using a gradient of 5 mM ammonium acetate, pH 6.5, to acetonitrile 50% and water 50% over 35 min at a flow rate of 0.6 ml/min. Flavins were detected by fluorescence emission (525 nm) using a Waters 474 scanning fluorescence detector set at 450 nm for excitation.
Stability of MnmC recombinant proteins
Strain IC6010 carrying pIC1253, pIC1339 or pIC1340 was grown overnight in LBT with ampicillin at 37°C. Overnight cultures were diluted 1/100 in the same medium supplemented with 0.1% L-arabinose (inducer of the AraC-PBAD system) and incubated at 37°C for 2 h. To halt the expression of the MnmC recombinant proteins, the cells were recovered by centrifugation at 3000g for 10 min, washed once with fresh LBT medium, resuspended in the same volume of pre-warmed LBT containing ampicillin and 1% glucose (repressor of the AraC-PBAD system) and incubated at 37°C. Samples were taken several times after the addition of glucose. The cells were lysed by brief ultrasonication and the lysate was centrifuged at 10 000g for 10 min at 4°C. The soluble fractions (50–150 μg of total proteins) were analyzed by western blotting using anti-Flag and anti-GroEL antibodies.
Gel filtration analysis of protein interactions
To investigate whether Flag-MnmC(o) interacts with Flag-MnmC(m), each protein was mixed at a final concentration of 5 µM in a final volume of 100 µl in TBS buffer containing 5 mM DTT and 3% glycerol and incubated at room temperature for 2 h. The same procedure was used to study the interaction of the full MnmC protein with MnmE or MnmG. The samples were analyzed by gel filtration using a Superdex 75 HR (MnmC domains) or a Superdex 200 HR (MnmC/MnmE/MnmG) column at a flow rate of 0.7 or 0.3 ml/min, respectively, in TBS Buffer containing 5 mM DTT. Gel filtration markers were used to calibrate the columns. The proteins were detected by ultraviolet (UV) absorbance at 280 nm.
Surface plasmon resonance evaluation of the MnmC(o)–MnmC(m) interaction
Surface plasmon resonance (SPR)-based kinetic analysis was used to determine the affinity between the recombinant MnmC(o) and MnmC(m) proteins. An anti-His monoclonal antibody (Roche, 100 ng/μl in 10 mM sodium acetate, pH 4.5) was immobilized onto a CM-5 sensor chip (Biacore AB, Uppsala, Sweden) at 7000 resonance units (RUs) using an amine coupling kit (Biacore AB) according to the manufacturer’s instructions. His-MnmC(m) at 2 μM in TBS buffer containing 0.005% Tween-20 surfactant was immobilized by capturing (∼600 RUs). Subsequently, various concentrations of Flag-MnmC(o) protein in TBS buffer were passed over the sensor chip at a flow rate of 30 μl/min at 25°C and the interactions were monitored for 1 min. The sensor surface was washed with TBS buffer to detect dissociation and then regenerated with a pulse of 5 mM NaOH (10 s at 60 μl/min). The data were evaluated with BiaEvaluation 3.1 Software (Biacore AB, Uppsala, Sweden).
Isolation of bulk tRNA from E. coli and reverse-phase HPLC analysis of nucleosides
Total tRNA purification and analysis of nucleosides by reverse-phase HPLC were performed as described previously (13,35,36). HPLC analysis was monitored at appropriate wavelengths to achieve optimal adsorption of the target nucleosides, 314 nm for thiolated nucleosides and 254 nm for non-thiolated nucleosides. The nucleosides were identified according to their UV spectra (35) and by comparison with appropriate controls.
Isolation of specific chimeric and native tRNAs
The E. coli
 ,
,  and
and  genes were cloned into pBSKrna (37) digested with either EcoRV (to produce chimeric tRNAs in which the E. coli tRNA is inserted into a human cytosolic
genes were cloned into pBSKrna (37) digested with either EcoRV (to produce chimeric tRNAs in which the E. coli tRNA is inserted into a human cytosolic  sequence that is used as a scaffold) or EcoRI and PstI to produce a tRNA without the scaffold (overexpressed ‘native’ tRNAs). The overproduction of tRNAs in strains transformed with pBSKrna derivative plasmids was performed as described previously (37). Specific chimeric tRNAs were purified from bulk tRNA by the Chaplet Column Chromatography method (38) using a biotinylated DNA probe that is complementary to the scaffold human cytosolic
sequence that is used as a scaffold) or EcoRI and PstI to produce a tRNA without the scaffold (overexpressed ‘native’ tRNAs). The overproduction of tRNAs in strains transformed with pBSKrna derivative plasmids was performed as described previously (37). Specific chimeric tRNAs were purified from bulk tRNA by the Chaplet Column Chromatography method (38) using a biotinylated DNA probe that is complementary to the scaffold human cytosolic  moiety (common to all chimeric tRNAs). The probe was immobilized on a HiTrap Streptavidin HP column. The same approach was used to purify overexpressed ‘native’ and true native tRNAs using biotinylated DNA probes that are complementary to the specific sequence of each E. coli tRNA (Supplementary Table S1).
moiety (common to all chimeric tRNAs). The probe was immobilized on a HiTrap Streptavidin HP column. The same approach was used to purify overexpressed ‘native’ and true native tRNAs using biotinylated DNA probes that are complementary to the specific sequence of each E. coli tRNA (Supplementary Table S1).
In vitro transcription of E. coli tRNAs
The E. coli genes encoding  ,
,  ,
,  ,
,  and
and  were PCR-amplified from genomic DNA using ‘vent’ polymerase and the primers Cys-F/Cys-R, Arg-F/Arg-R, Glu-F/Glu-R, Gly-F/Gly-R and Leu-F/Leu-R, respectively. The amplicons were cloned into a SmaI-linearized pUC19 plasmid to produce pIC1394, pIC1395, pIC1536, pIC1550 and pIC1537. The E. coli gene encoding
were PCR-amplified from genomic DNA using ‘vent’ polymerase and the primers Cys-F/Cys-R, Arg-F/Arg-R, Glu-F/Glu-R, Gly-F/Gly-R and Leu-F/Leu-R, respectively. The amplicons were cloned into a SmaI-linearized pUC19 plasmid to produce pIC1394, pIC1395, pIC1536, pIC1550 and pIC1537. The E. coli gene encoding  was PCR-amplified from genomic DNA using expand-long polymerase and the primers Gln-F and Gln-R and cloned into pBAD-TOPO. The plasmid pIC1083 (containing the
was PCR-amplified from genomic DNA using expand-long polymerase and the primers Gln-F and Gln-R and cloned into pBAD-TOPO. The plasmid pIC1083 (containing the  gene), a derivative of pUC19, was a gift from Dr Tamura (34). Unmodified E. coli tRNAs were prepared by in vitro transcription from BstNI-digested plasmids (pIC1083, pIC1394 and pIC1395) and HindIII-digested plasmids (pIC1535, pIC1536, pIC1550 and pIC1537) using the Riboprobe T7 transcription kit (PROMEGA) and 2–5 μg of each digested plasmid as a DNA template in a 50-μl reaction mix.
gene), a derivative of pUC19, was a gift from Dr Tamura (34). Unmodified E. coli tRNAs were prepared by in vitro transcription from BstNI-digested plasmids (pIC1083, pIC1394 and pIC1395) and HindIII-digested plasmids (pIC1535, pIC1536, pIC1550 and pIC1537) using the Riboprobe T7 transcription kit (PROMEGA) and 2–5 μg of each digested plasmid as a DNA template in a 50-μl reaction mix.
In vitro and in vivo activity of the recombinant MnmC(o) and MnmC(m) proteins
To analyze the in vitro activity of the recombinant proteins, total tRNA (40 µg) from a mnmC-W131stop or mnmC(m)-G68D mutant was incubated in a 200-μl reaction mixture containing 50 mM Tris–Cl (pH 8.0), 50 mM ammonium acetate, 0.5 mM FAD or 0.5 mM S-Adenosyl-L-Methionine (SAM), 5% glycerol and 2 μM of the purified protein [Flag-MnmC, Flag-MnmC(o), or Flag-MnmC(m)] for 40 min at 37°C. tRNA was recovered by phenol extraction and ethanol precipitation and subsequently treated with nuclease P1 and E. coli alkaline phosphatase. The resulting nucleosides were analyzed by reverse-phase HPLC as described (13,30). For in vivo complementation studies, overnight cultures of strains mnmC-W131stop or mnmC(m)-G68D containing pBAD-TOPO or derivative plasmids (pIC1340 and pIC1339) were diluted 1:100 in 100 ml LBT containing 0.2% arabinose and grown to an OD600 of 0.5. The cells were recovered by centrifugation at 4500g for 15 min at 4°C. Bulk tRNA was obtained and analyzed by HPLC as described above.
Kinetic analysis of MnmC- and MnmEG-catalyzed modifications
To assay MnmC(o) or MnmC(m) activity, the reaction mixture (100 μl) contained 50 mM Tris–HCl (pH 8), 50 mM ammonium acetate, 3% glycerol, 2 mM NaCl, 73 µM MgCl2, tRNA (0.5–5 μM), Flag-tagged protein (25 nM) and 100 μM FAD or SAM (depending on the MnmC activity to be determined). The mixtures were pre-incubated at 37°C for 3 min before addition of proteins. All reactions were performed at 37°C. The reactions were halted after 60–90 s of incubation by adding 100 μl of 0.3 M sodium acetate, pH 5.2. To assay the ammonium-dependent MnmEG activity with respect to tRNA concentration, the reaction mixture (100 μl) contained 100 mM Tris–HCl, pH 8, 100 mM ammonium acetate, 5 mM MgCl2, 5% glycerol, 5 mM DTT, 0.5 mM FAD, 2 mM GTP, 1 mM methylene-THF, 10 µg bovine serum albumin (BSA) and tRNA (0.1–2 μM). The reaction was initiated by the addition of 0.1 µM MnmE•MnmG complex obtained as previously described (13) and stopped after 2 min of incubation at 37°C by adding 100 μl 0.3 M sodium acetate, pH 5.2. In all cases, tRNA was finally recovered by phenol extraction and ethanol precipitation and treated with nuclease P1 and E. coli alkaline phosphatase. The resulting nucleosides were analyzed by reverse-phase HPLC. The area of the synthesized nucleoside was calculated and its amount was extrapolated from standard curves that were prepared using chemically synthesized nucleosides (A. Malkiewicz, University of Lodz, Poland) over a range of 0–250 ng. Experiments were performed in triplicate. To determine the Vmax and Km values, the data were fitted to the Michaelis–Menten equation using nonlinear regression (GraphPad Prism v4.0).
Analysis of the tRNA substrate specificity of MnmC(o) and MnmC(m) in vitro
To analyze the specificity of MnmC(o) and MnmC(m) in vitro, the tRNA (10–15 μg) was first modified using the MnmEG complex through the ammonium and glycine pathways as described previously (13). The modified tRNA was phenolized, ethanol precipitated and incubated with 2 μM MnmC(o) or MnmC(m) domains in 200 μl (total volume) of MnmC buffer containing 50 mM Tris–HCl (pH 8.0), 50 mM ammonium acetate, 0.5 mM FAD [for the MnmC(o) assay] or 0.5 mM SAM (for the MnmC(m) assay) and 5% glycerol. After incubating for 40 min at 37°C, the tRNA was recovered by phenolization and ethanol precipitation and the tRNA was then treated with nuclease P1 and E. coli alkaline phosphatase. The resulting nucleosides were analyzed by reverse-phase HPLC as described above.
Acid resistance assay
The acid resistance experiments were performed essentially as previously reported (39). Briefly, strains were grown in LBT containing 0.4% glucose to stationary phase. The cultures were then diluted 1:1000 into EG medium [minimal E medium containing 0.4% glucose; (40)], pH 2.0, supplemented or not with 0.7 mM glutamate. Samples were obtained at 0, 1, 2, 3 and 4 h post acid challenge and spotted on LAT plates.
RESULTS
General roles of the MnmEG and MnmC enzymes in the modification status of tRNAs
In a previous study, we demonstrated that MnmEG catalyzes two different reactions in vitro and produces nm5 and cmnm5 using ammonium and glycine, respectively, as substrates (13). We also reported the presence of cmnm5s2U and nm5s2U in total tRNA purified from an mnmC null mutant. However, it has been suggested that the formation of these intermediates is sensitive to growth conditions and specific strains, which might explain why they were not detected in other studies (23,26,41).
To obtain a clear picture of the functional activity of enzymes MnmEG and MnmC during exponential growth (mid-log phase; OD600 of ∼0.6) in LBT, we analyzed the HPLC profile of bulk tRNA isolated from several strains and different genetic backgrounds (Table 2 and Supplementary Figure S1). In the HPLC analysis, absorbance was monitored at 314 nm to maximize the detection of thiolated nucleosides. We consistently detected a small amount (∼10–20%) of cmnm5s2U in the wild-type strains of three different backgrounds, TH48, BW25113 and MG1655 (Table 2), which may originate from 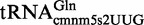 or might be an intermediate of the final product mnm5s2U that is present in
or might be an intermediate of the final product mnm5s2U that is present in  and
and  . Notwithstanding, mnm5s2U was the major final product (∼80–90%). We were unable to identify nucleosides mnm5U and cmnm5Um (which are present in
. Notwithstanding, mnm5s2U was the major final product (∼80–90%). We were unable to identify nucleosides mnm5U and cmnm5Um (which are present in  ,
,  and
and  ) when tRNA hydrolysates were monitored at 254 nm, as a comparison of chromatograms of total tRNA purified from wild-type strains and their mnmE or mnmG derivatives did not allow the detection of any peaks attributable to those nucleosides in the wild-type chromatogram.
) when tRNA hydrolysates were monitored at 254 nm, as a comparison of chromatograms of total tRNA purified from wild-type strains and their mnmE or mnmG derivatives did not allow the detection of any peaks attributable to those nucleosides in the wild-type chromatogram.
Table 2.
Relative distribution of nucleosides in bulk tRNA purified from exponentially growing strains
| Strain | Relative distribution (%) of nucleosidesa |
|||
|---|---|---|---|---|
| nm5s2U | mnm5s2U | cmnm5s2U | s2U | |
| TH48 background | ||||
| wt | 83 ± 5 | 17 ± 5 | ||
| mnmC-W131stop | 31 ± 1 | 69 ± 1 | ||
| mnmC(m)-G68D | 85 ± 5 | 15 ± 5 | ||
| BW25113 background | ||||
| wt | 83 ± 3 | 17 ± 3 | ||
| ΔmnmG or ΔmnmE | 100 | |||
| ΔmnmC | 27 ± 4 | 73 ± 4 | ||
| ΔmnmC(o) | 14 ± 3 | 86 ± 3 | ||
| MG1655 background | ||||
| wt | 85 ± 3 | 15 ± 3 | ||
| ΔmnmG | 100 | |||
atRNA was purified and degraded to nucleosides for HPLC analysis. The percentage of nucleosides represents the distribution of the peak area of each nucleoside compared to the sum of the peak areas of the two nucleosides considered. Each value represents the mean of at least two independent experiments. wt: wild-type.
As expected, the intermediates cmnm5s2U and nm5s2U were observed in mnmC null mutants (carrying mnmC-W131stop or ΔmnmC mutations); these nucleosides were typically distributed in a 70/30 ratio, suggesting that the MnmEG enzyme preferably uses the glycine pathway under the growth conditions used in these experiments (Table 2). This conclusion was consistent with the cmnm5s2U/mnm5s2U ratio observed in the ΔmnmC(o) mutant (∼86/14), which clearly exhibited a MnmC(o)− MnmC(m)+ phenotype. Moreover, the relative distribution of cmnm5s2U and nm5s2U in the mnmC(m)-G68D mutant was ∼15/85, suggesting that most of the cmnm5s2U formed by the glycine pathway is converted to nm5s2U by the MnmC(o) activity present in this strain and that the remaining cmnm5s2U could proceed, at least partially, from  . Taken together, these results clearly support the idea that MnmEG uses ammonium or glycine to modify tRNAs in vivo and that MnmC(m) functions independently of MnmC(o) [see strain ΔmnmC(o) in Table 2] to transform tRNAs modified by MnmEG via the ammonium pathway (Figure 1), which raises the possibility that certain tRNAs may be substrates for MnmC(m), but not for MnmC(o).
. Taken together, these results clearly support the idea that MnmEG uses ammonium or glycine to modify tRNAs in vivo and that MnmC(m) functions independently of MnmC(o) [see strain ΔmnmC(o) in Table 2] to transform tRNAs modified by MnmEG via the ammonium pathway (Figure 1), which raises the possibility that certain tRNAs may be substrates for MnmC(m), but not for MnmC(o).
Expression and purification of the MnmC(o) and MnmC(m) domains
In order to study the functional independence of the two MnmC domains, we decided to express them separately. The full mnmC gene and its two domains, mnmC(o) (encoding for amino acids 250–668 of MnmC) and mnmC(m) (encoding for amino acids 1–250 of MnmC), were N-terminal Flag-tagged by PCR and cloned into the pBAD-TOPO expression vector. Recombinant protein expression was induced in the E. coli mutant mnmC::kan (ΔmnmC) by adding 0.2% arabinose. The Flag-tagged proteins MnmC, MnmC(o) and MnmC(m) were purified close to homogeneity and exhibited apparent molecular masses of 78, 48 and 34 kDa, respectively (Figure 2A).
Figure 2.

Characterization of the MnmC recombinant proteins. (A) Coomassie blue staining of SDS–PAGE containing the purified Flag-MnmC, Flag-MnmC(o) and Flag-MnmC(m) proteins used in this work. (B) HPLC analysis of the Flag-MnmC(o) extract (blue line) and markers (red line). FMN: flavin mononucleotide. (C) Half-life of the Flag-MnmC proteins over time as determined by tracking their decline after the addition of glucose to cultures of IC6010 (ΔmnmC) transformed with the plasmids pIC1253, pIC1339 or pIC1340, which express Flag-MnmC, Flag-MnmC(o) and Flag-MnmC(m), respectively. GroEL was used as a loading control. Protein levels were detected by western blotting. (D) Gel filtration analysis of the purified Flag-MnmC proteins [full MnmC protein: green; MnmC(o): blue and MnmC(m): black] and a Flag-MnmC(o)/Flag-MnmC(m) mix (red). The elution positions of the size markers are indicated on the top. Elution fractions a–d from the chromatography of the Flag-MnmC(o)/Flag-MnmC(m) mix were pooled for further analysis. Inset: SDS–PAGE of elution fractions a–d from the Flag-MnmC(o)/Flag-MnmC(m) mix. Fractions (500 μl) were precipitated with trichloroacetic acid before loading. The gel was stained with coomassie blue. The marker molecular masses are indicated on the left. (E) SPR analysis of the MnmC(o)–MnmC(m) interaction. Flag-MnmC(o) was injected into a solution passing over a sensor chip containing the immobilized His-MnmC(m) (600 RU). Representative sensorgrams for various concentrations of Flag-MnmC(o) are shown. (F) Gel filtration analysis of the purified MnmC protein (blue line) and a mix of MnmC with MnmG (black line) or MnmE (red line). The elution positions of the size markers are indicated on the top. Proteins were detected by UV absorbance at 280 nm.
The Flag-MnmC protein and its C-terminal domain [Flag-MnmC(o)] were yellow, and their spectra showed maximum absorption peaks at ∼375 and 450 nm, indicating the presence of a flavin derivative. The putative cofactor was identified as FAD by its retention time (Figure 2B). Therefore, the isolated MnmC(o) domain contains non-covalently bound FAD, which suggests that it is correctly folded. To our knowledge, this is the first report describing the expression and purification of the soluble form of MnmC(o). A previous attempt to purify this domain was unsuccessful because overexpression of a somewhat different, recombinant MnmC(o) produced inclusion bodies, which led to the hypothesis that MnmC(o) is incapable of folding on its own and requires the presence of MnmC(m) (25). Our data, however, demonstrate that MnmC(o) can fold independently of MnmC(m). Interestingly, the in vivo stability of the separated domains was significantly lower than that of the full protein (Figure 2C), suggesting that the physical interaction of both domains in the full protein confers greater stability.
The full MnmC protein and the separate domains MnmC(o) and MnmC(m) behaved as monomeric proteins when subjected to gel filtration chromatography (Figure 2D). However, a mixture of MnmC(o) and MnmC(m) displayed the same elution profile as the full protein, indicating that the separate domains interact in vitro (Figure 2D). This interaction was further explored by kinetic analysis using surface plasmon resonance (SPR). Flag-MnmC(o) was injected at different concentrations onto the immobilized His-MnmC(m) ligand (Figure 2E). The apparent equilibrium constant KD was 87 ± 15 nM, which is indicative of a strong interaction between MnmC(o) and MnmC(m).
Taking into consideration the functional relationships of MnmC with the MnmEG complex, we explored whether MnmC interacts with members of the complex, i.e. MnmE or MnmG. A final concentration of 5 µM Flag-MnmC was mixed with 5 µM His-MnmG or MnmE and the samples were analyzed by gel filtration. As shown in Figure 2F, no interaction between the full MnmC protein and MnmE or MnmG was observed. The same negative result was obtained by SPR (data not shown).
In vitro and in vivo determination of the enzymatic activities of MnmC(o) and MnmC(m)
To analyze whether the recombinant MnmC(o) and MnmC(m) proteins are functionally active, we first assessed their tRNA modifying capability in vitro using total tRNA purified from exponentially growing cultures as a substrate. The in vitro activity of the recombinant MnmC(o) protein was investigated by incubating tRNA purified from the null mutant mnmC-W131stop (nm5s2U/cmnm5s2U ratio ≈ 31/69, Table 2) with 2 µM Flag-MnmC(o) domain and 0.5 mM FAD. As shown in Figure 3A, a major part of cmnm5s2U was converted to nm5s2U, which demonstrates that Flag-MnmC(o) is catalytically active in the in vitro assay, although a small part of cmnm5s2U, probably proceeding from  , remained unaltered. When tRNA purified from the mnmC(m)-G68D mutant (nm5s2U/cmnm5s2U ratio ≈ 85/15; Table 2) was incubated with 2 µM Flag-MnmC(m) and 0.5 mM SAM, nm5s2U was modified to mnm5s2U, demonstrating the capability of the recombinant protein to perform the methylation reaction in vitro (Figure 3B). As expected, the small peak corresponding to cmnm5s2U (according to its retention time and spectrum) remained unchanged after the in vitro reaction mediated by MnmC(m) (Figure 3B).
, remained unaltered. When tRNA purified from the mnmC(m)-G68D mutant (nm5s2U/cmnm5s2U ratio ≈ 85/15; Table 2) was incubated with 2 µM Flag-MnmC(m) and 0.5 mM SAM, nm5s2U was modified to mnm5s2U, demonstrating the capability of the recombinant protein to perform the methylation reaction in vitro (Figure 3B). As expected, the small peak corresponding to cmnm5s2U (according to its retention time and spectrum) remained unchanged after the in vitro reaction mediated by MnmC(m) (Figure 3B).
Figure 3.

In vitro and in vivo activity of the recombinant proteins MnmC(o) and MnmC(m). (A) and (B) HPLC analysis of total tRNA from a null mnmC mutant (IC6019; panel A) and an mnmC(m)-G68D mutant (IC6018; panel B) before (solid black line) and after in vitro incubation (dotted red line) with purified MnmC(o) and MnmC(m) recombinant proteins, respectively. (C) and (D) HPLC analysis of total tRNA extracted from IC6019 (panel C) and IC6018 (panel D) transformed with pBAD-TOPO (solid black lines) or a pBAD-TOPO derivative expressing Flag-MnmC(o) and Flag-MnmC(m), respectively (dotted red lines). Absorbance was monitored at 314 nm to maximize the detection of thiolated nucleosides. Small variations in elution times between upper and lower panels were probably due to negligible variations in buffers prepared in different days. Note that nucleosides were identified by both elution times and spectra (data not shown).
Then, we assessed the capability of the recombinant MnmC(o) and MnmC(m) proteins to modify tRNA in vivo. The strain with the null mutation mnmC-W131stop (IC6019) was transformed with pBAD-TOPO and its derivative pIC1339 expressing MnmC(o), whereas strain mnmC(m)-G68D (IC6018) was transformed with pBAD-TOPO and its derivative pIC1340 expressing MnmC(m). In the resulting strains, which were grown to exponential phase in the presence of the arabinose inducer, the recombinant MnmC(o) protein catalyzed the conversion of cmnm5s2U into nm5s2U, although a peak of remnant cmnm5s2U was observed (Figure 3C) and nm5s2U was fully transformed to mnm5s2U by MnmC(m) (Figure 3D). Similar results were obtained in the absence of arabinose (data not shown). This finding indicates that recombinant MnmC(o) and MnmC(m) are expressed and accumulate to some extent in the absence of the inducer, although it was not possible to detect them by western blotting with an anti-Flag antibody (data not shown). The fact that both proteins were capable of modifying tRNA despite their very low concentrations suggests that these proteins have high activity in vivo.
Kinetic analysis of the MnmC- and MnmEG-dependent reactions
To explore whether the recombinant MnmC(o) and MnmC(m) proteins display similar kinetic properties to those exhibited by the entire MnmC protein, we conducted steady-state kinetic experiments. The conditions used for each reaction were similar to enable a comparison of the kinetic constants and were based on conditions known to optimize activity (23,26). The substrate for the assays was a chimeric version of E. coli
 expressed from pBSKrna (37,42), which facilitates overproduction and purification of recombinant RNA and has been successfully used by our group in previous studies (21,33). Here, the
expressed from pBSKrna (37,42), which facilitates overproduction and purification of recombinant RNA and has been successfully used by our group in previous studies (21,33). Here, the  gene was inserted into the EcoRV site of the region encoding the scaffold tRNA (a human cytosolic
gene was inserted into the EcoRV site of the region encoding the scaffold tRNA (a human cytosolic  lacking the anticodon region). The resulting chimeric tRNA essentially contained the complete E. coli
lacking the anticodon region). The resulting chimeric tRNA essentially contained the complete E. coli
 sequence fused at its 5′- and 3′-ends to the truncated anticodon stem of human cytosolic
sequence fused at its 5′- and 3′-ends to the truncated anticodon stem of human cytosolic  , producing an RNA of ∼170 nt (Supplementary Figure S2).
, producing an RNA of ∼170 nt (Supplementary Figure S2).
The FAD-dependent-oxidoreductase activity of the recombinant MnmC(o) and MnmC proteins was compared by determining the conversion of cmnm5s2U to nm5s2U on the cmnm5s2U-containing chimeric  (chi-
(chi- ) purified from an mnmC null mutant (IC6010). The SAM-dependent methyltransferase activity of MnmC(m) and MnmC was compared by following the conversion of nm5s2U to mnm5s2U on the nm5s2U-containing chi-
) purified from an mnmC null mutant (IC6010). The SAM-dependent methyltransferase activity of MnmC(m) and MnmC was compared by following the conversion of nm5s2U to mnm5s2U on the nm5s2U-containing chi- extracted from an mnmC(m)-G68D mutant (IC6018). All proteins displayed Michaelis–Menten kinetics with respect to varying concentrations of chi-
extracted from an mnmC(m)-G68D mutant (IC6018). All proteins displayed Michaelis–Menten kinetics with respect to varying concentrations of chi- (Supplementary Figure S3). As shown in Table 3, the enzymatic activity of each isolated MnmC domain had kinetic parameters (kcat and Km) similar to those exhibited by the entire protein, which supports the notion that the MnmC domains function independently.
(Supplementary Figure S3). As shown in Table 3, the enzymatic activity of each isolated MnmC domain had kinetic parameters (kcat and Km) similar to those exhibited by the entire protein, which supports the notion that the MnmC domains function independently.
Table 3.
Kinetic parameters with respect to chi- for reactions catalyzed by MnmC, MnmC(o), MnmC(m), and MnmEG
for reactions catalyzed by MnmC, MnmC(o), MnmC(m), and MnmEG
| Reaction | Km (μM) | Vmax (nmoles min−1 mg−1) | kcat (s−1) | kcat/Km (s−1 μM-1) |
|---|---|---|---|---|
| MnmC(FAD) (cmnm5s2U → nm5s2U) | 15.7 ± 3.4 | 457 ± 12 | 0.59 ± 0.02 | 0.038 |
| MnmC(o) domain(FAD) (cmnm5s2U → nm5s2U) | 6.1 ± 2.1 | 486 ± 69 | 0.39 ± 0.05 | 0.064 |
| MnmC(SAM) (nm5s2U → mnm5s2U) | 4.4 ± 1.1 | 365 ± 77 | 0.46 ± 0.10 | 0.105 |
| MnmC(m) domain(SAM) (nm5s2U → mnm5s2U) | 4.2 ± 1.1 | 895 ± 179 | 0.52 ± 0.10 | 0.124 |
| MnmEG(NH4) (s2U → nm5s2U) | 0.6 ± 0.2 | 5.9 ± 0.3 | 0.012 ± 0.001 | 0.020 |
The values are the mean ± SD of a minimum of three independent experiments.
The kcat constants of the MnmC activities were similar to those of previous studies (23,26), although the Km values were higher than those reported by Pearson and Carell (26), most likely due to the nature of the tRNA substrate used in our experiments. In any case, our data indicated that MnmC(m) displayed a catalytic efficiency (kcat/Km) 2-fold higher than that of MnmC(o) (∼0.1 versus ∼0.05; Table 3), which is in agreement with the proposal that synthesis of the final modification mnm5(s2)U is favored by kinetic tuning of the MnmC activities, thus avoiding accumulation of the nm5(s2) intermediate (26).
We also analyzed the catalytic parameters of the ammonium-dependent reaction mediated by the MnmEG complex, whose activity precedes that of MnmC(m) in the mnm5(s2)U synthesis (Figure 1). We used conditions known to be appropriate for assaying the MnmEG activities in vitro (13) and included the presence of Mg2+ (5 mM), which is required for GTP hydrolysis by MnmE (43). In line with this, the assay buffer differed from that of the MnmC reactions where a relatively low concentration of Mg2+ was used (73 μM), as MnmC activities are known to be severely inhibited at 5 mM of Mg2+ (23). Under the proper in vitro conditions for each enzyme, the modification reaction mediated by MnmEG (incorporation of nm5 into U34) displayed an ∼5-fold lower catalytic efficiency than that of the MnmC(m)-mediated reaction (Table 3). However, this comparison should be handled with caution because the conditions used in the MnmEG and MnmC(m) assays were different, and the Km values may change according to the buffer conditions. In addition, the structure of the chimeric tRNA used as a substrate could affect the catalytic cycle of each enzyme in a different manner.
Therefore, in order to gain information on the efficiency of the mnm5s2U assembly-lines in vivo, we decided to explore the modification status of the native 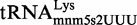 in several strains. The presence of modification intermediates in the tRNA purified from a wild-type strain would be indicative of the MnmEG and MnmC activities not being kinetically tuned. HPLC analysis of native
in several strains. The presence of modification intermediates in the tRNA purified from a wild-type strain would be indicative of the MnmEG and MnmC activities not being kinetically tuned. HPLC analysis of native  purified during the exponential phase revealed no accumulation of the cmnm5s2U intermediate in the wild-type strain (Figure 4A), whereas this nucleoside was predominant in the tRNA extracted from the ΔmnmC strain (Figure 4B). Moreover, we observed no nm5s2U in the native
purified during the exponential phase revealed no accumulation of the cmnm5s2U intermediate in the wild-type strain (Figure 4A), whereas this nucleoside was predominant in the tRNA extracted from the ΔmnmC strain (Figure 4B). Moreover, we observed no nm5s2U in the native 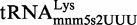 purified from the wild-type strain (Figure 4A), despite this intermediate accumulated in
purified from the wild-type strain (Figure 4A), despite this intermediate accumulated in  of the mnmC(m)-G68D mutant (Figure 4C). These results suggest that
of the mnmC(m)-G68D mutant (Figure 4C). These results suggest that  containing cmnm5s2U or nm5s2U at the wobble position is stable, and consequently that the disappearance of these intermediates in the wild-type strain is probably due to their conversion into mnm5s2U by MnmC activities. Thus, it appears that MnmEG and MnmC are kinetically tuned to produce only the final mnm5 group in
containing cmnm5s2U or nm5s2U at the wobble position is stable, and consequently that the disappearance of these intermediates in the wild-type strain is probably due to their conversion into mnm5s2U by MnmC activities. Thus, it appears that MnmEG and MnmC are kinetically tuned to produce only the final mnm5 group in  .
.
Figure 4.

HPLC analysis of native  purified from different strains at exponential phase. Native
purified from different strains at exponential phase. Native  was purified from the wild-type (A), ΔmnmC (B), mnmC-G68D (C) and ΔmnmG (D) strains and subjected to HPLC analysis, which was monitored at 314 nm. The relevant nucleosides at position 34 (mnm5s2U, s2U, cmnm5s2U and nm5s2U) and position 8 (s4U) of
was purified from the wild-type (A), ΔmnmC (B), mnmC-G68D (C) and ΔmnmG (D) strains and subjected to HPLC analysis, which was monitored at 314 nm. The relevant nucleosides at position 34 (mnm5s2U, s2U, cmnm5s2U and nm5s2U) and position 8 (s4U) of 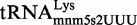 are indicated.
are indicated.
Notably, the intermediate s2U, resulting from the activity of MnmA (Figure 1), was only observed in a ΔmnmG strain, which is defective in the MnmEG activity (Figure 4D). Moreover, we did not detect the non-thiolated intermediates cmnm5U and nm5U when  hydrolysates were monitored at 254 nm (data not shown). These data suggest that the coordination of the MnmA- and MnmEGC-dependent pathways is tightly coupled to synthesize mnm5s2U on native
hydrolysates were monitored at 254 nm (data not shown). These data suggest that the coordination of the MnmA- and MnmEGC-dependent pathways is tightly coupled to synthesize mnm5s2U on native  without a build-up of intermediates.
without a build-up of intermediates.
tRNA substrate specificity of MnmC(o) and MnmC(m) in vivo
Escherichia coli
 and
and  contain cmnm5 at the wobble uridine instead of the mnm5 found in the remaining MnmEG substrates, (http://modomics.genesilico.pl/sequences/list/tRNA). These data suggest that
contain cmnm5 at the wobble uridine instead of the mnm5 found in the remaining MnmEG substrates, (http://modomics.genesilico.pl/sequences/list/tRNA). These data suggest that  and
and  are not substrates for MnmC(o). Moreover, they raise the question of what happens with the ammonium pathway of the MnmEG complex, which theoretically produces nm5U in all its tRNA substrates (Figure 1). Interestingly, the analysis of gln1 tRNA from Salmonella enterica serovar Typhimurium indicated that 80% of the molecules contained cmnm5s2U, whereas the remaining 20% contained mnm5s2U (44). In this case, we speculate that mnm5s2U might be synthesized by MnmC [using the MnmC(m) activity] from the nm5s2U generated via the ammonium-dependent MnmEG pathway. Therefore, we decided to investigate whether a fraction of
are not substrates for MnmC(o). Moreover, they raise the question of what happens with the ammonium pathway of the MnmEG complex, which theoretically produces nm5U in all its tRNA substrates (Figure 1). Interestingly, the analysis of gln1 tRNA from Salmonella enterica serovar Typhimurium indicated that 80% of the molecules contained cmnm5s2U, whereas the remaining 20% contained mnm5s2U (44). In this case, we speculate that mnm5s2U might be synthesized by MnmC [using the MnmC(m) activity] from the nm5s2U generated via the ammonium-dependent MnmEG pathway. Therefore, we decided to investigate whether a fraction of  and
and  in E. coli contains mnm at position 5 in the wobble uridine, and whether both tRNAs function as substrates for MnmC(m) but not MnmC(o) (Figure 1).
in E. coli contains mnm at position 5 in the wobble uridine, and whether both tRNAs function as substrates for MnmC(m) but not MnmC(o) (Figure 1).
To address this question, we constructed a series of tRNA expression plasmids by inserting the coding sequences of 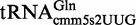 ,
,  and
and  into EcoRI/PstI-digested pBSKrna (37); thus, we eliminated the tRNA scaffold-encoding region (i.e. the DNA region encoding the human cytoplasmic
into EcoRI/PstI-digested pBSKrna (37); thus, we eliminated the tRNA scaffold-encoding region (i.e. the DNA region encoding the human cytoplasmic  ) present in the vector (Supplementary Figure S2). Selected strains were transformed with the resulting plasmids. We then examined the HPLC profile of the overexpressed, ‘native’ tRNAs purified from cultures grown overnight to the stationary phase. It should be noted that the expression of the tRNA genes in these constructs was under the control of the strong constitutive lpp promoter (37). As mutations leading to reduced expression might be selected over time because overexpression ultimately slows growth, the fresh transformation of cells with the pBSKrna derivatives every time and overnight incubation of the transformed cells prior to the purification of the cloned target (in this case, tRNAs) are highly recommended (37).
) present in the vector (Supplementary Figure S2). Selected strains were transformed with the resulting plasmids. We then examined the HPLC profile of the overexpressed, ‘native’ tRNAs purified from cultures grown overnight to the stationary phase. It should be noted that the expression of the tRNA genes in these constructs was under the control of the strong constitutive lpp promoter (37). As mutations leading to reduced expression might be selected over time because overexpression ultimately slows growth, the fresh transformation of cells with the pBSKrna derivatives every time and overnight incubation of the transformed cells prior to the purification of the cloned target (in this case, tRNAs) are highly recommended (37).
The HPLC analysis of  (Figure 5A), used herein as a control, indicated that: (i) mnm5s2U was predominant in a wild-type strain; (ii) both nm5s2U and cmnm5s2U accumulated in the mnmC null strain and (iii) nm5s2U prevailed in the mnmC(m)-G68D strain. Taken together, these results indicate that both MnmEG-MnmC(o)-MnmC(m) and MnmEG-MnmC(m) pathways operate on
(Figure 5A), used herein as a control, indicated that: (i) mnm5s2U was predominant in a wild-type strain; (ii) both nm5s2U and cmnm5s2U accumulated in the mnmC null strain and (iii) nm5s2U prevailed in the mnmC(m)-G68D strain. Taken together, these results indicate that both MnmEG-MnmC(o)-MnmC(m) and MnmEG-MnmC(m) pathways operate on  and that they converge to produce the final modification mnm5s2U in the wild-type strain.
and that they converge to produce the final modification mnm5s2U in the wild-type strain.
Figure 5.

In vivo specificity of MnmC(o) and MnmC(m) for substrate tRNAs. Representative HPLC profiles of  (A),
(A),  (B) and
(B) and  (D) expressed from pIC1664, pIC1714 and pIC1665, respectively. The strains used in panels A and B were IC6017 (WT), IC6018 [mnmC(m)-G68D] and IC6019 (mnmC-W131stop). The strains used in panel D were IC5136 and IC5854. HPLC profiles of commercial markers are shown in panel C. Representative HPLC profiles of native
(D) expressed from pIC1664, pIC1714 and pIC1665, respectively. The strains used in panels A and B were IC6017 (WT), IC6018 [mnmC(m)-G68D] and IC6019 (mnmC-W131stop). The strains used in panel D were IC5136 and IC5854. HPLC profiles of commercial markers are shown in panel C. Representative HPLC profiles of native  purified from strains ΔtrmL (IC6374) and ΔtrmL/ΔmnmG (IC6411) are shown (E).
purified from strains ΔtrmL (IC6374) and ΔtrmL/ΔmnmG (IC6411) are shown (E).
An analysis of overexpressed  (Figure 5B) indicated that cmnm5s2U and nm5s2U accumulated at a ratio of ∼90/10 when tRNA was purified from the mnmC-W131stop mutant. Despite the lower proportion of nm5s2U, the presence of this nucleotide indicates that the MnmEG complex was indeed able to modify
(Figure 5B) indicated that cmnm5s2U and nm5s2U accumulated at a ratio of ∼90/10 when tRNA was purified from the mnmC-W131stop mutant. Despite the lower proportion of nm5s2U, the presence of this nucleotide indicates that the MnmEG complex was indeed able to modify  through the ammonium pathway. Nucleosides cmnm5s2U and mnm5s2U were found at a 90/10 ratio when
through the ammonium pathway. Nucleosides cmnm5s2U and mnm5s2U were found at a 90/10 ratio when  was purified from the wild-type strain. Detection of mnm5s2U in
was purified from the wild-type strain. Detection of mnm5s2U in  clearly indicates that this tRNA is a substrate for MnmC(m).
clearly indicates that this tRNA is a substrate for MnmC(m).
It should be noted that both cmnm5s2U and nm5s2U accumulated in  purified from the mnmC-W131stop mutant, whereas cmnm5s2U practically disappeared when
purified from the mnmC-W131stop mutant, whereas cmnm5s2U practically disappeared when  was purified from the mnmC(m)-G68D strain (Figure 5A). This result was expected, considering that cmnm5s2U was transformed into nm5s2U by the MnmC(o) activity present in the mnmC(m)-G68D strain. Interestingly, the HPLC profiles of
was purified from the mnmC(m)-G68D strain (Figure 5A). This result was expected, considering that cmnm5s2U was transformed into nm5s2U by the MnmC(o) activity present in the mnmC(m)-G68D strain. Interestingly, the HPLC profiles of  purified from the mnmC(m)-G68D and mnmC-W131stop strains were similar (Figure 5B). In both cases, cmnm5s2U was much more abundant than nm5s2U, suggesting that the MnmC(o) activity present in the mnmC(m)-G68D strain did not function in
purified from the mnmC(m)-G68D and mnmC-W131stop strains were similar (Figure 5B). In both cases, cmnm5s2U was much more abundant than nm5s2U, suggesting that the MnmC(o) activity present in the mnmC(m)-G68D strain did not function in  .
.
Overexpression of tRNAs often causes hypomodification. In fact, the hypomodified nucleoside s2U, resulting from the action of MnmA, was relatively abundant in the overexpressed  and
and  (data not shown). Nevertheless, s2U accumulated at a similar proportion in all tested strains so that the ratios of the nucleosides of interest (mnm5s2U, cmnm5s2U and nm5s2U) were not greatly affected. When the
(data not shown). Nevertheless, s2U accumulated at a similar proportion in all tested strains so that the ratios of the nucleosides of interest (mnm5s2U, cmnm5s2U and nm5s2U) were not greatly affected. When the  and
and  hydrolysates were monitored at 254 nm, the non-thiolated nucleosides nm5U, mnm5U, cmnm5U were hardly detected (data not shown). Therefore, we think that the different HPLC pattern of
hydrolysates were monitored at 254 nm, the non-thiolated nucleosides nm5U, mnm5U, cmnm5U were hardly detected (data not shown). Therefore, we think that the different HPLC pattern of  and
and  in the mnmC(m)-G68D strain cannot be attributed to the presence of hypomodified nucleosides. Rather, it reveals that
in the mnmC(m)-G68D strain cannot be attributed to the presence of hypomodified nucleosides. Rather, it reveals that  is not a substrate for MnmC(o).
is not a substrate for MnmC(o).
We also analyzed the HPLC profile of  purified from a trmL strain in which the methylation of the ribose in the wobble uridine is impaired due to the ΔtrmL mutation (21). We adopted this approach to examine the modification of
purified from a trmL strain in which the methylation of the ribose in the wobble uridine is impaired due to the ΔtrmL mutation (21). We adopted this approach to examine the modification of  because we were unable to distinguish nucleosides cmnm5Um, nm5Um and mnm5Um in our HPLC chromatograms. However, we were able to identify the peaks corresponding to cmnm5U, nm5U and mnm5U using proper markers (Figure 5C). As shown in Figure 5D, overexpressed
because we were unable to distinguish nucleosides cmnm5Um, nm5Um and mnm5Um in our HPLC chromatograms. However, we were able to identify the peaks corresponding to cmnm5U, nm5U and mnm5U using proper markers (Figure 5C). As shown in Figure 5D, overexpressed  purified from a ΔtrmL strain contained cmnm5U. However, no traces of mnm5U or nm5U were detected in this tRNA. Similar results were obtained when analyzing native, non-overexpressed
purified from a ΔtrmL strain contained cmnm5U. However, no traces of mnm5U or nm5U were detected in this tRNA. Similar results were obtained when analyzing native, non-overexpressed  purified in the exponential phase (Figure 5E). Altogether, these data suggest that
purified in the exponential phase (Figure 5E). Altogether, these data suggest that  is not modified by the ammonium-dependent MnmEG pathway and that it is not a substrate for MnmC(o).
is not modified by the ammonium-dependent MnmEG pathway and that it is not a substrate for MnmC(o).
tRNA substrate specificity of MnmC(o) and MnmC(m) in vitro
To further explore the specificity of MnmC(o) and MnmC(m) for  ,
, 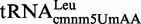 and
and  , we performed in vitro modification reactions using in vitro synthesized tRNAs as substrates. These modification reactions included two steps. First, substrate tRNA was modified by the MnmEG complex through the ammonium (Figure 6A, B, E and F) or the glycine (Figure 6C and D) pathways. The resulting tRNA carrying nm5U or cmnm5U (solid black lines) was subsequently used as a substrate to characterize the activity of the MnmC(m) or the MnmC(o) domains, respectively.
, we performed in vitro modification reactions using in vitro synthesized tRNAs as substrates. These modification reactions included two steps. First, substrate tRNA was modified by the MnmEG complex through the ammonium (Figure 6A, B, E and F) or the glycine (Figure 6C and D) pathways. The resulting tRNA carrying nm5U or cmnm5U (solid black lines) was subsequently used as a substrate to characterize the activity of the MnmC(m) or the MnmC(o) domains, respectively.
Figure 6.

In vitro specificity of MnmC(o) and MnmC(m) for substrate tRNAs. (A–D) The modification reactions were performed using in vitro synthesized tRNAs in two steps. First, the substrate tRNA was modified by the MnmEG complex via the ammonium (A) and (B) or the glycine pathway (C) and (D) and the resulting tRNA (solid black line) carrying nm5U or cmnm5U was used as a substrate to examine the activity of the MnmC(m) or MnmC(o) domain (dashed red line), respectively. (E) HPLC analysis of in vitro synthesized  after in vitro modification by MnmEG (solid black line) and MnmC(m) (dashed red line). (F) Overexpressed
after in vitro modification by MnmEG (solid black line) and MnmC(m) (dashed red line). (F) Overexpressed  purified from the strain IC6411 (ΔtrmL/ΔmnmG) served as the substrate in the ammonium-dependent reaction catalyzed by MnmEG. The resulting tRNA (black line) was used as a substrate for the MnmC(m)-mediated reaction (red line).
purified from the strain IC6411 (ΔtrmL/ΔmnmG) served as the substrate in the ammonium-dependent reaction catalyzed by MnmEG. The resulting tRNA (black line) was used as a substrate for the MnmC(m)-mediated reaction (red line).
The data in Figure 6 indicate that MnmC(m) catalyzed the conversion of nm5U into mnm5U in both  (panel A) and
(panel A) and  (panel B), whereas MnmC(o) catalyzed the conversion of cmnm5 into nm5 in
(panel B), whereas MnmC(o) catalyzed the conversion of cmnm5 into nm5 in  (panel C), but not in
(panel C), but not in  (panel D). These results support the idea that
(panel D). These results support the idea that 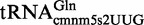 does not function as a substrate for MnmC(o).
does not function as a substrate for MnmC(o).
Surprisingly, we observed that MnmEG catalyzed the formation of nm5U from in vitro synthesized  and, in turn, nm5U was converted into mnm5U through MnmC(m) (Figure 6E). This result contradicts the data provided in Figure 5D and E, where no traces of nm5U or mnm5U were observed in the HPLC analysis of the
and, in turn, nm5U was converted into mnm5U through MnmC(m) (Figure 6E). This result contradicts the data provided in Figure 5D and E, where no traces of nm5U or mnm5U were observed in the HPLC analysis of the  purified from a trmL strain. Considering that the experiments shown in Figure 6E were performed using an in vitro synthesized
purified from a trmL strain. Considering that the experiments shown in Figure 6E were performed using an in vitro synthesized  (therefore lacking modifications), we thought that the presence of some modified nucleoside(s) in the in vivo synthesized
(therefore lacking modifications), we thought that the presence of some modified nucleoside(s) in the in vivo synthesized  (Figure 5D and E) could hinder the recognition of this substrate by MnmEG. However, when we used overexpressed
(Figure 5D and E) could hinder the recognition of this substrate by MnmEG. However, when we used overexpressed  purified from a double ΔtrmL/ΔmnmG mutant as a substrate in the in vitro modification assay, the MnmEG-mediated synthesis of nm5s2U was once again observed (Figure 6F). Moreover, the MnmEG-modified
purified from a double ΔtrmL/ΔmnmG mutant as a substrate in the in vitro modification assay, the MnmEG-mediated synthesis of nm5s2U was once again observed (Figure 6F). Moreover, the MnmEG-modified  was a good substrate for the in vitro synthesis of mnm5 through MnmC(m) (Figure 6F). Altogether, these results suggest that the ammonium pathway of MnmEG is ineffective on
was a good substrate for the in vitro synthesis of mnm5 through MnmC(m) (Figure 6F). Altogether, these results suggest that the ammonium pathway of MnmEG is ineffective on  in vivo, even though it efficiently functions on this tRNA in the in vitro assay.
in vivo, even though it efficiently functions on this tRNA in the in vitro assay.
We also assessed the activity of MnmC(o) on the  purified from a trmL strain and on the in vitro transcribed
purified from a trmL strain and on the in vitro transcribed  previously modified by MnmEG in vitro via the glycine pathway (i.e. on tRNA molecules carrying cmnm5U). The synthesis of nm5 from cmnm5 was not observed under any condition (data not shown). Therefore, we concluded that
previously modified by MnmEG in vitro via the glycine pathway (i.e. on tRNA molecules carrying cmnm5U). The synthesis of nm5 from cmnm5 was not observed under any condition (data not shown). Therefore, we concluded that 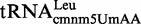 is a substrate in vitro for MnmC(m) (Figure 6E and F) but not MnmC(o).
is a substrate in vitro for MnmC(m) (Figure 6E and F) but not MnmC(o).
Notably, MnmEG also catalyzed the ammonium-dependent synthesis of nm5U in  ,
,  and
and  obtained by in vitro transcription (Supplementary Figure S4). We suspect that modification of these tRNAs follows a pattern similar to that observed in
obtained by in vitro transcription (Supplementary Figure S4). We suspect that modification of these tRNAs follows a pattern similar to that observed in  , because all these tRNA species appear to contain mainly the mnm5 group of U34. However, this hypothesis should be examined in future studies.
, because all these tRNA species appear to contain mainly the mnm5 group of U34. However, this hypothesis should be examined in future studies.
Synthesis of nm5U and cmnm5U is modulated by growth conditions and the tRNA species
The overexpressed  and
and 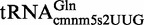 purified from the mnmC-W131stop mutant during the stationary phase exhibited different cmnm5s2U/nm5s2U ratios (∼35/65 and ∼90/10, respectively; Figure 5A and B). Moreover, the ratio in the overexpressed
purified from the mnmC-W131stop mutant during the stationary phase exhibited different cmnm5s2U/nm5s2U ratios (∼35/65 and ∼90/10, respectively; Figure 5A and B). Moreover, the ratio in the overexpressed  (∼35/65) differed from that found in the total tRNA obtained from the mnmC-W131stop strain during exponential growth (∼69/31; Table 2). These results suggest that the glycine pathway (which produces cmnm5s2U) could be less effective on
(∼35/65) differed from that found in the total tRNA obtained from the mnmC-W131stop strain during exponential growth (∼69/31; Table 2). These results suggest that the glycine pathway (which produces cmnm5s2U) could be less effective on  in the stationary phase. These observations prompted us to carefully investigate the efficiency of the glycine and ammonium pathways in total tRNA and specific native tRNAs during the growth curve.
in the stationary phase. These observations prompted us to carefully investigate the efficiency of the glycine and ammonium pathways in total tRNA and specific native tRNAs during the growth curve.
First, we determined the modification status of total tRNA in the wild-type strain and mnmC mutants. In the wild-type or mnmC(m)-G68D strain (Figure 7, panels A and B), we could not identify which pathway contributes to the synthesis of the final modification (mnm5s2U or nm5s2U, respectively) because the activity of MnmC(o) converges both pathways by transforming cmnm5s2U into nm5s2U (Figure 1). However, the strain carrying an mnmC-W131stop or mnmC(o) null mutation (Figure 7, panels C and D) revealed that the level of cmnm5s2U and nm5s2U or mnm5s2U in bulk tRNA depends on the growth phase; nm5s2U (or mnm5s2U) accumulates as optical density increases, whereas cmnm5s2U exhibits the opposite trend and becomes the minor component at an OD600 of ∼2.0. As the data in Figure 7 represent the relative distribution of each nucleoside with respect to the sum of the peak areas of the two nucleosides, it is important to point out that the net decrease in the cmnm5s2U peak area along the growth curve was concomitant with the net increase in the nm5s2U or mnm5s2U area.
Figure 7.

Synthesis of nm5U34 and cmnm5U34 is depending on the growth phase. HPLC analysis of total tRNA purified from TH48 (A), TH49 (B), TH69 (C) and IC6629 (D) during the growth cycle. The strains were grown in LBT. The percentage of nucleosides represents the distribution of the peak area of each nucleoside compared to the sum of the peak areas of the two nucleosides considered. Each time point represents the average from two independent experiments. The standard deviations were within ±20%.
Interestingly, when the ΔmnmC(o) strain was grown in minimal medium instead of LBT, cmnm5s2U was consistently observed as the major intermediate, irrespective of the growth phase (Table 4). Therefore, the growth conditions (growth medium and growth phase) affect the pathway responsible for modification at position 5 of U34.
Table 4.
Reprogramming of the U34 modification depends on the growth conditions
| Nucleoside distributiona | OD600 nm (LBT) |
OD600 nm (MM) |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.6 | 0.9 | 2 | 2.6 | 5 (O/N) | 0.6 | 1.7 | 3 (O/N) | |
| cmnm5s2U | 86 ± 3 | 41 ± 4 | 23 ± 7 | 22 ± 7 | 32 ± 1 | 89 ± 1 | 96 ± 1 | 100 |
| mnm5s2U | 14 ± 3 | 59 ± 4 | 77 ± 7 | 78 ± 7 | 68 ± 1 | 11 ± 1 | 4 ± 1 | 0 |
aRelative distribution (%) of nucleosides. Total tRNAs were purified throughout the growth cycle from strain ΔmnmC(o) (IC6629) growing in LBT or minimal medium (MM; YM9 supplemented with 0.4% glucose) and analyzed by HPLC. The OD600 of the overnight (O/N) cultures was different according to the growth medium.
We subsequently analyzed the behavior of native tRNAs in both the exponential and stationary phases (Table 5 and Supplementary Figure S5). During the exponential growth of the ΔmnmC strain, cmnm5s2U was the major intermediate accumulated in native  (94%), but this nucleoside became the minor intermediate (4%) in the stationary phase. In contrast, HPLC analysis revealed that cmnm5s2U was consistently the major intermediate present in native
(94%), but this nucleoside became the minor intermediate (4%) in the stationary phase. In contrast, HPLC analysis revealed that cmnm5s2U was consistently the major intermediate present in native  (>90%), irrespective of the growth phase. Altogether, these results indicate that
(>90%), irrespective of the growth phase. Altogether, these results indicate that  functions different from
functions different from  and total tRNA when the ΔmnmC strain is grown to stationary phase in LBT (i.e. in growth conditions that favor the ammonium pathway). Therefore, the accumulation of cmnm5 or nm5 depends on the specific characteristics of the tRNA.
and total tRNA when the ΔmnmC strain is grown to stationary phase in LBT (i.e. in growth conditions that favor the ammonium pathway). Therefore, the accumulation of cmnm5 or nm5 depends on the specific characteristics of the tRNA.
Table 5.
Reprogramming of U34 modification also depends on the tRNA species
| Specific tRNAs | Growth Phase | LBTa |
|
|---|---|---|---|
| cmnm5s2U | nm5s2U | ||
 |
Exponential | 94 ± 1 | 6 ± 1 |
| Stationary | 4 ± 2 | 96 ± 2 | |
 |
Exponential | 99 ± 1 | 1 ± 1 |
| Stationary | 93 ± 2 | 7 ± 2 | |
aRelative (%) distribution of the nucleosides in accordance with the growth phase and the tRNA species. Native tRNAs were purified from strain ΔmnmC (IC6010) during the exponential (OD600 ∼0.4) or stationary phase (OD600 ∼3).
Importance of MnmC-mediated modifications for cell growth
The limited data available regarding the effect of mnmC mutations on growth rate are contradictory. No differences between mnmC and wild-type strains were initially reported (22,45). However, more recently, Pearson and Carell (26) measured the growth rate of an mnmC-knockout strain from the Keio collection and observed a larger growth constant for the wild-type strain, in agreement with the hypothesis that accumulation of intermediates in the synthesis of mnm5(s2)U affects the translation process negatively. We investigated the effect of mnmC mutations on growth rate in different genetic backgrounds and did not observe significant differences between the wild-type strain and any derivative carrying an mnmC allele [ΔmnmC, mnmC-W131stop, ΔmnmC(o) and mnmC(m)-G68D; Table 6]. We cannot explain why our results differ from those reported by Pearson and Carell (26), even when the same background (BW25113) was used. Perhaps, the dilution method used to maintain the pre-cultures in exponential growth could explain the different results. We took great care to not dilute the pre-cultures below an OD600 of 0.25 and to not exceed an OD600 of 0.5 in order to avoid major changes under the exponential growth conditions.
Table 6.
Growth rates of mnmC mutants in different genetic backgrounds
| Background | Doubling time (min)a |
|||||||
|---|---|---|---|---|---|---|---|---|
| wt | mnmCb | mnmC(m)c | ΔmnmC(o) | ΔmnmA | mnmA | ΔmnmA | ΔmnmA | |
| ΔmnmCd | mnmC(m)c | ΔmnmC(o) | ||||||
| TH48 | 34 ± 1 | 33 ± 1 | 32 ± 1 | nd | nd | nd | nd | nd |
| BW25113 | 28 ± 1 | 30 ± 1 | nd | 28 ± 1 | 40 ± 1 | nd | nd | 58 ± 3 |
| IC4639 | 28 ± 1 | 29 ± 1 | 30 ± 1 | nd | 56 ± 1 | 73 ± 2 | 78 ± 2 | nd |
aDoubling-time values of strains (wild-type or carrying the indicated mutations) are the mean ± SD of at least two independent experiments.
bThe null mnmC alleles were mnmC-W131stop (TH48 background) and ΔmnmC (IC4639 and BW25113 backgrounds).
cThe mnmC(m) point mutation was mnmC(m)-G68D. nd, not done.
dThe mnmA point mutation was mnmA-Q233stop.
The lack of effect in our hands of mnmC mutations during the exponential growth of cells contrasts with the behavior of mnmE- and mnmG-knockout strains, which, as we have previously demonstrated, grow slower than the wild-type strain (12,32). These data suggest that the lack of any modification at the wobble uridine is more deleterious to the cell than the presence of any intermediate of mnm5(s2) synthesis. Supporting this conclusion, we also observed that the mnmC mutations, unlike the mnmE mutations, did not affect resistance to acid stress of E. coli (Figure 8). This resistance depends on several systems, one of which involves proteins whose translation likely depends on tRNAs modified by MnmEG (39,46).
Figure 8.

The effect of mnmE and mnmC mutations on acid resistance. Strains IC5136 (wild-type), IC5827 (ΔmnmE), IC6010 (ΔmnmC) and IC6629 [ΔmnmC(o)] were grown in LBT containing 0.4% glucose to stationary phase and then diluted 1:1000 into EG pH 2.0 medium supplemented or not with glutamate. The samples were spotted after 0, 1, 2, 3 and 4 h post acid challenge on LAT plates.
The MnmA protein catalyzes the last step of thiolation at position 2 of U34 in  ,
, 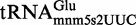 , and
, and 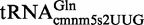 (Figure 1). Inactivation of the mnmA gene has been reported to increase the doubling time (31,47). Interestingly, combination of the mnmA and mnmC mutations had a synergic effect (Table 6), revealing that loss of any MnmC activity is detrimental under specific conditions. Mutation mnmC(m)-G68D appeared to slow down the growth rate of the mnmA-knockout strain slightly more than the ΔmnmC mutation.
(Figure 1). Inactivation of the mnmA gene has been reported to increase the doubling time (31,47). Interestingly, combination of the mnmA and mnmC mutations had a synergic effect (Table 6), revealing that loss of any MnmC activity is detrimental under specific conditions. Mutation mnmC(m)-G68D appeared to slow down the growth rate of the mnmA-knockout strain slightly more than the ΔmnmC mutation.
We also performed competition experiments between BW25113 (which is a wild-type, tetracycline-sensitive strain) and TH48 (wild-type), TH49 (mnmC(m)-G68D) and TH69 (mnmC-W131stop), which are tetracycline-resistant strains (Table 7). This type of experiment compares not only the growth in exponential phase, but also the ability to sustain and recover from stationary phase. Although TH48 and its derivatives were out-competed by BW25113, it is clear that the impairment of the MnmC functions reduces the ability of the strain to compete. Curiously, in these experiments, mutant mnmC-W131stop was less competitive than mnmC(m)-G68D.
Table 7.
Growth competition assay
| Straina | Ratiob at mix time | Ratiob after six 24-h cycles |
|---|---|---|
| Wild-type (TH48) | 0.56 ± 0.007 | 0.021 ± 0.003 |
| mnmC(m)-G68D (TH49) | 0.53 ± 0.09 | 0.0020 ± 0.0006 |
| mnmC-W131stop (TH69) | 0.56 ± 0.04 | 0.0002 ± 0.0001 |
aBW25113 (tetracycline-sensitive strain) and the indicated TH strain (tetracycline-resistant strain) were mixed 1:1 at the start of the experiment.
bThe ratio was calculated as CFU/ml recovered on LAT supplemented with tetracycline versus CFU/mL recovered on LAT.
DISCUSSION
This study aimed to further explore the activities and specificities of the MnmEG complex and the MnmC domains, the ability of specific tRNAs to follow the ammonium- or glycine-MnmEG pathway under different growth conditions and the effect of mnmC mutations on cell growth.
The fulfillment of the two first aims was facilitated by the cloning and separate expression of the MnmC domains. We demonstrate for the first time that the MnmC(o) domain can fold independently of MnmC(m) and that both domains are catalytically active when expressed separately. Importantly, the domains exhibit similar kinetic properties to those observed in the entire MnmC protein, which suggests that the two domains operate independently of each other. Moreover, MnmC(o) and MnmC(m) differ in terms of their specificity for tRNA substrates. Thus, MnmC(o) modifies  , but not
, but not  and
and 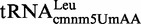 . In contrast, all three tRNAs (
. In contrast, all three tRNAs ( ,
, 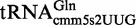 and
and  ) serve as substrates for MnmC(m) not only in vitro, but also in vivo at least in the cases of
) serve as substrates for MnmC(m) not only in vitro, but also in vivo at least in the cases of  and
and  . These data strongly support the notion that the MnmC(o) and MnmC(m) domains function independently. The presence of mnm5s2U in in vivo synthesized
. These data strongly support the notion that the MnmC(o) and MnmC(m) domains function independently. The presence of mnm5s2U in in vivo synthesized  suggests that the nomenclature of this tRNA should be changed to
suggests that the nomenclature of this tRNA should be changed to  .
.
Sequence searches of complete genomes have revealed that the distribution of the putative orthologs of the E. coli bi-functional MnmC protein are conserved only in γ-proteobacteria, with some additional members in other bacterial classes (24). Interestingly, in several genomes, potential orthologs of a single domain have been identified (24), but the biological function has been experimentally demonstrated only in the case of the Aquifex aeolicus DUF752 protein, an MnmC(m) homolog (41). It is unclear from the phylogenetic analysis whether the independent putative orthologs represent the ancestral or derived versions of the bi-functional MnmC enzyme (24). The ability of the E. coli MnmC(o) and MnmC(m) domains to function independent of each other, as shown in this work, suggests that the origin of the full MnmC protein present in γ-Proteobacteria (24) likely occurred by domain fusion. The MnmC(o)–MnmC(m) fusion in E. coli suggests a functional advantage of the spatial proximity of both domains. The fusion could increase the stability of the resulting protein. In fact, our results indicate that the entire MnmC protein is substantially more stable than the separate domains (Figure 2C) or other RNA modification proteins, such as RsmG or MnmG, which have half-lives of ∼17 and 31 min, respectively (48). The possibility that the fusion also facilitates the functional cooperation between the domains during modification of tRNAs following the ‘canonical’ MnmEG-MnmC(o)–MnmC(m) pathway (cmnm5U→nm5U→mnm5U) cannot be ruled out. A better understanding of how the MnmC protein functions will entail determining whether the binding of a tRNA molecule to one domain exerts a negative allosteric effect on the other domain. In this respect, the structural characterization of MnmC–tRNA complexes could prove helpful.
Biosynthesis of complex modifications like mnm5s2U, cmnm5s2U, cmnm5Um and mnm5U requires the participation of at least two specific enzymes (Figure 1). Our in vivo data suggest that MnmEG and MnmC activities are kinetically tuned to produce the final modification mnm5s2U on native  because no intermediates (cmnm5s2U and nm5s2U) were observed in a wild-type strain (Figure 4). Apparently, the intermediates are not subject to active degradation as they were shown to be stable in strains ΔmnmC and mnmC(m)-G68D. Notably, neither s2U nor the non-thiolated nucleosides cmnm5U and nm5U were found in native
because no intermediates (cmnm5s2U and nm5s2U) were observed in a wild-type strain (Figure 4). Apparently, the intermediates are not subject to active degradation as they were shown to be stable in strains ΔmnmC and mnmC(m)-G68D. Notably, neither s2U nor the non-thiolated nucleosides cmnm5U and nm5U were found in native  purified from the wild-type strain (Figure 4; data not shown). Therefore, we believe that the MnmEG–MnmC and MnmA pathways are also coordinated by tuning the activities and relative abundances of the tRNA-modifying enzymes. Modifications of U34 by MnmEG and MnmA occur independently of each other [(30,49); Figure 4D], but whether the presence of the s2 group facilitates the modification of position 5 in
purified from the wild-type strain (Figure 4; data not shown). Therefore, we believe that the MnmEG–MnmC and MnmA pathways are also coordinated by tuning the activities and relative abundances of the tRNA-modifying enzymes. Modifications of U34 by MnmEG and MnmA occur independently of each other [(30,49); Figure 4D], but whether the presence of the s2 group facilitates the modification of position 5 in  ,
,  and
and  by modulating the electron density distribution in the uridine ring remains unclear.
by modulating the electron density distribution in the uridine ring remains unclear.
There are very few data available on the identity determinants required by TrmL. Methylation of the ribose 2′-OH group by this enzyme occurs as a late step in  maturation given that the enzyme fails to methylate
maturation given that the enzyme fails to methylate  without the prior addition of N6-(isopentenyl)-2-methyladenosine (ms2i6A) at position 37 (21). Nucleoside ms2i6A is also present in
without the prior addition of N6-(isopentenyl)-2-methyladenosine (ms2i6A) at position 37 (21). Nucleoside ms2i6A is also present in  . Hence, it is feasible that TrmL also requires ms2i6A as a positive identity determinant to modify
. Hence, it is feasible that TrmL also requires ms2i6A as a positive identity determinant to modify  . Here our data indicate that MnmEG modifies
. Here our data indicate that MnmEG modifies  in the absence of 2′-O-methylation (Figures 5D and E; 6E and F), but we do not know whether TrmL requires the presence of the cmnm5 group to act on
in the absence of 2′-O-methylation (Figures 5D and E; 6E and F), but we do not know whether TrmL requires the presence of the cmnm5 group to act on  . More experiments are needed to unravel the order of action of the modifying enzymes working on U34 and the identity elements required by each enzyme.
. More experiments are needed to unravel the order of action of the modifying enzymes working on U34 and the identity elements required by each enzyme.
Another interesting question concerning tRNA maturation is whether physical interactions among tRNA modification enzymes contribute to the efficiency of complex pathways. In Aquifex aeolicus, an interaction between the E. coli MnmC(m) homolog, protein DUF752 and the A. aeolicus MnmE and MnmG proteins has been observed (41). In E. coli, we did not detect interaction of MnmC with MnmE or MnmG by either fast-performance liquid chromatography (FPLC) analysis (Figure 2F) or SPR (data not shown). However, we cannot rule out the possibility of the MnmEG complex being able to interact with MnmC in vivo. It appears reasonable to hypothesize that the spatial clustering of tRNA-modifying enzymes within the cell can optimize the coordination of their work. Therefore, one challenging aim for future studies is to determine whether tRNA modifying enzymes are spatially organized, as observed for other enzymes involved in RNA biology, such as the components of the RNA degradosome (50).
The use of strains carrying the ΔmnmC(o) or ΔmnmC mutation has allowed us to study the ability of MnmEG to use the ammonium or the glycine pathway under different growth conditions and thus, to detect the reprogramming of U34 base modification in both bulk and specific tRNAs (Tables 4 and 5; Figure 9). The analysis of bulk tRNA indicates that when ΔmnmC(o) or ΔmnmC strains are grown in LBT, the glycine pathway prevails over the ammonium pathway in the exponential phase as cmnm5s2U is the most abundant nucleoside (Figure 9A). However, a gradual change in the output of both pathways is observed along the growth curve so that the nucleosides produced by the ammonium pathway become prevalent during entry into the stationary phase (Figure 9B). In contrast, when cells are grown in minimal medium, the glycine pathway is prevalent in both the exponential and stationary phases (Figure 9A). A preference by MnmEG for the ammonium or the glycine pathway to catalyze tRNA modification may depend on the availability of these substrates, which, in turn depends on the growth medium and cell metabolism.
Figure 9.

Summary scheme of the reprogramming of U34 base modification in both bulk and specific tRNAs. The main and minor modification pathways are indicated by thick and thin arrows, respectively; pathways whose activity was only observed in vitro are indicated by dotted arrows. Activity of TrmL on  has been omitted in panel D. Labels in each panel indicate the type of tRNA (specific or bulk tRNA) following the pathways, and the growth conditions under which the tRNA was obtained. LogP, logarithmic phase; StatP, stationary phase; MM, minimal medium; LBT, LBT medium.
has been omitted in panel D. Labels in each panel indicate the type of tRNA (specific or bulk tRNA) following the pathways, and the growth conditions under which the tRNA was obtained. LogP, logarithmic phase; StatP, stationary phase; MM, minimal medium; LBT, LBT medium.
The nature of tRNA species also appears crucial for the net output of the MnmEG pathways (Table 5 and Figure 9). Thus, whereas  follows the pattern observed in bulk tRNA and is preferably modified via the ammonium pathway during the transition to the stationary phase,
follows the pattern observed in bulk tRNA and is preferably modified via the ammonium pathway during the transition to the stationary phase,  fails to be effectively modified through this pathway, as indicated by the low accumulation of nm5s2U in the ΔmnmC mutant. Moreover, the ammonium pathway proves ineffective in modifying
fails to be effectively modified through this pathway, as indicated by the low accumulation of nm5s2U in the ΔmnmC mutant. Moreover, the ammonium pathway proves ineffective in modifying  at any phase of growth as no nm5U or mnm5U is detected in a trmL strain (Figure 5D and E). Therefore, neither
at any phase of growth as no nm5U or mnm5U is detected in a trmL strain (Figure 5D and E). Therefore, neither  nor
nor  is a good substrate for the ammonium pathway in vivo, even though both tRNAs appear rather well modified by this pathway in vitro (Figure 6B and E; Figure 9C and D).
is a good substrate for the ammonium pathway in vivo, even though both tRNAs appear rather well modified by this pathway in vitro (Figure 6B and E; Figure 9C and D).
Why the behavior of  and
and  is different from that of
is different from that of  remains unresolved. The use of ammonium or glycine by MnmEG could be regulated by interactions of this complex with its tRNA substrates and unknown factors in vivo.
remains unresolved. The use of ammonium or glycine by MnmEG could be regulated by interactions of this complex with its tRNA substrates and unknown factors in vivo.  and
and  may be poor substrates for the ammonium pathway and thus out-competed by other tRNAs in vivo. Alternatively,
may be poor substrates for the ammonium pathway and thus out-competed by other tRNAs in vivo. Alternatively,  and
and  carrying nm5 or mnm5, but not cmnm5 may be unstable and specifically degraded by some nuclease(s) (5,51). Additional experiments are required to test these hypotheses and to elucidate the mechanism underlying the selection by MnmEG of the glycine or the ammonium pathway.
carrying nm5 or mnm5, but not cmnm5 may be unstable and specifically degraded by some nuclease(s) (5,51). Additional experiments are required to test these hypotheses and to elucidate the mechanism underlying the selection by MnmEG of the glycine or the ammonium pathway.
Our results highlight the crucial role of MnmC(o) and Mnm(m) in the biosynthesis of mnm5(s2)U in accordance with growth conditions (Figure 9). During the exponential phase in LBT and at any phase in minimal medium, the oxidoreductase activity of MnmC(o) is required to drive the intermediate generated by the more active glycine pathway [cmnm5(s2)U] toward the MnmC(m)-dependent synthesis of the final modification in  and, presumably, in
and, presumably, in  ,
,  and
and  (Figure 9A). In contrast, MnmC(o) plays a minor role when the ammonium pathway prevails; i.e. during the transition to stationary phase of cultures growing in LBT. In this case, the methylase activity of MnmC(m) functions fairly well alone [i.e. without the participation of MnmC(o)] to drive the production of the final modification mnm5(s2)U by transforming the intermediate nm5(s2) generated by the more active ammonium pathway (Table 4 and Figure 9B). Thus, the incorporation of MnmC(o) and MnmC(m) into the enzyme content of E. coli extends the adaptability of this bacterium to changing growth conditions. How our findings in E. coli apply to other bacteria, especially those lacking certain MnmC activity, is an issue that merits future research.
(Figure 9A). In contrast, MnmC(o) plays a minor role when the ammonium pathway prevails; i.e. during the transition to stationary phase of cultures growing in LBT. In this case, the methylase activity of MnmC(m) functions fairly well alone [i.e. without the participation of MnmC(o)] to drive the production of the final modification mnm5(s2)U by transforming the intermediate nm5(s2) generated by the more active ammonium pathway (Table 4 and Figure 9B). Thus, the incorporation of MnmC(o) and MnmC(m) into the enzyme content of E. coli extends the adaptability of this bacterium to changing growth conditions. How our findings in E. coli apply to other bacteria, especially those lacking certain MnmC activity, is an issue that merits future research.
The lack of modifications at the wobble uridine due to the presence of mnmE or mnmG mutations leads to impaired growth, high sensitivity to acidic pH and defects in translational fidelity(12,35,46,48,52–55). Here our functional studies suggest that MnmC impairment is less detrimental than the inactivation of the MnmEG complex given that mnmC mutations neither reduce the growth rate in rich medium (Table 6) nor affect resistance to acidic pH (Figure 8). Nevertheless, impairment of any MnmC activity has a biological cost. Thus, mnmC mutations diminish the ability of cells to compete (Table 7), suggesting that fully modified tRNAs and, therefore the activities of MnmC, are required for efficient translation of mRNAs involved in the stationary phase stress response. In addition, our data indicate that mnmC mutations slow the growth rate of a mnmA strain, revealing a synergistic effect of mutations mnmC and mnmA. As mnmC mutations alone do not affect the growth rate, we hypothesize that the presence of the MnmA-dependent thiolation at position 2 of U34 in a subset of tRNAs can compensate for the lack of MnmC modifications at position 5 during exponential growth.
In an mnmA background, accumulation of nm5U produced by the mnmC(m)-G68D mutation appears slightly more detrimental than accumulation of cmnm5 caused by a null mnmC mutation (Table 6). In line with this, it has been demonstrated that the reading of an amber codon by an ochre-suppressing derivative of  (supG) is also more affected by the mnmC(m)-G68D mutation than by the null mnmC-W131stop mutation, although this effect is dependent on the codon context (56). Therefore, it appears that cmnm5(s2) confers the cell some advantage over nm5(s2)U to translate specific mRNAs during exponential growth.
(supG) is also more affected by the mnmC(m)-G68D mutation than by the null mnmC-W131stop mutation, although this effect is dependent on the codon context (56). Therefore, it appears that cmnm5(s2) confers the cell some advantage over nm5(s2)U to translate specific mRNAs during exponential growth.
Strikingly, in the competition experiments (Table 7), the mnmC-W131stop mutation appears to be more disadvantageous than the mnmC(m)-G68D allele, which reveals an opposite trend to that observed in the growth rate determination experiments (Table 6). During the transition to the stationary phase, nm5(s2)U is more abundant in mnmC(m)-G68D than in mnmC-W131stop (Figure 7, panels B and C). Therefore, the slightly better ability of the mnmC(m)-G68D mutant to compete might indicate that nm5(s2)U is more effective than cmnm5(s2)U in translating mRNAs required to sustain stationary phase stress conditions. Thus, it appears that the advantages offered by each intermediate, nm5(s2)U and cmnm5(s2)U, depend on the nature of the mRNAs to be translated which, in turn, depend on the growth conditions. Obviously, the synthesis of the final modification mnm5s2U is more advantageous to the cell as the strains expressing a fully active MnmC protein grow better under stressing conditions (i.e. those related to lack of MnmA and competition with other bacteria) than those strains lacking any MnmC activity. In conclusion, adaptation of MnmEG to use ammonium or glycine, together with the acquisition of MnmC(o) and MnmC(m) activities, provide E. coli with significant advantages to synthesize mnm5s2U and to survive under different conditions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Spanish Ministry of Economy and Competitiveness [BFU2007-66509 and BFU2010-19737]; the Generalitat Valenciana [ACOMP/2012/065; PROMETEO/2012/061 to M.-E.A.]; the Swedish Science Research Council [project BU-2930] and Carl Trygger Foundation (to G.R.B.); a pre-doctoral fellowship from the Spanish Ministry of Economy and Competitiveness (to M.-J.G.); I.M. was supported by short-term fellowships from the Ministry of Science and Innovation and Generalitat Valenciana during his stays at the Umea University. Funding for open access charge: Spanish Ministry of Economy and Competitiveness [BFU2010-19737].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful to M. Villarroya, S. Prado, R. Ruiz-Partida and E. Knecht for much valuable advice. We also thank the National BioResource Project (NIG, Japan) for providing some of the E. coli strains used in this study.
REFERENCES
- 1.Björk GR, Hagervall TG. Cellular and Molecular Biology. Washington DC: ASM Press; 2005. Transfer tRNA modification (posting data). Escherichia Coli and Salmonella. [Google Scholar]
- 2.Grosjean H. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Texas, USA: Landes Biosciencies; 2009. Nucleic acids are not boring long polymers of only four types of nucleotides: a guided tour; pp. 1–18. [Google Scholar]
- 3.El Yacoubi B, Bailly M, de Crécy-Lagard V. Biosynthesis and Function of Posttranscriptional Modifications of Transfer RNAs. Annu. Rev. Genet. 2012;46:69–95. doi: 10.1146/annurev-genet-110711-155641. [DOI] [PubMed] [Google Scholar]
- 4.Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Reports. 2008;9:629–635. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, Beinat M, Karicheva O, Mandel H, Ofek N, Segel R, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009;85:401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasarman F, Antonicka H, Horvath R, Shoubridge EA. The 2-thiouridylase function of the human MTU1 (TRMU) enzyme is dispensable for mitochondrial translation. Hum. Mol. Genet. 2011;20:4634–4643. doi: 10.1093/hmg/ddr397. [DOI] [PubMed] [Google Scholar]
- 8.Ghezzi D, Baruffini E, Haack TB, Innvernizzi F, Melchionda L, Dallabona C, Storm TM, Parini R, Burlina AB, Meitinger TM, et al. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am J. Hum. Genet. 2012;90:1079–1087. doi: 10.1016/j.ajhg.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Huang B, Eliasson M, Ryden P, Bystrom AS. Elongator complex influences telomeric gene silencing and DNA damage response by its role in wobble uridine tRNA modification. PLoS Genet. 2011;7:e1002258. doi: 10.1371/journal.pgen.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armengod ME, Moukadiri I, Prado S, Ruiz-Partida R, Benitez-Paez A, Villarroya M, Lomas R, Garzon MJ, Martinez-Zamora A, Meseguer S, et al. Enzymology of tRNA modification in the bacterial MnmEG pathway. Biochimie. 2012;94:1510–1520. doi: 10.1016/j.biochi.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Yim L, Moukadiri I, Björk GR, Armengod ME. Further insights into the tRNA modification process controlled by proteins MnmE and GidA of Escherichia coli. Nucleic Acids Res. 2006;34:5892–5905. doi: 10.1093/nar/gkl752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moukadiri I, Prado S, Piera J, Velázquez-Campoy A, Björk GR, Armengod ME. Evolutionarily conserved proteins MnmE and GidA catalyze the formation of two methyluridine derivatives at tRNA wobble positions. Nucleic Acids Res. 2009;37:7177–7193. doi: 10.1093/nar/gkp762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabedo H, Macián F, Villarroya M, Escudero JC, Martínez-Vicente M, Knecht E, Armengod ME. The Escherichia coli trmE (mnmE) gene, involved in tRNA modification, codes for an evolutionarily conserved GTPase with unusual biochemical properties. EMBO J. 1999;18:7063–7076. doi: 10.1093/emboj/18.24.7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scrima A, Vetter IR, Armengod ME, Wittinghofer A. The structure of the TrmE GTP-binding protein and its implications for tRNA modification. EMBO J. 2005;24:23–33. doi: 10.1038/sj.emboj.7600507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osawa T, Inanaga H, Numata T. Crystallization and preliminary X-ray diffraction analysis of the tRNA-modification enzyme GidA from Aquifex aeolicus. Acta Crystallogr. Sec.t F Struct. Biol. Cryst. Commun. 2009;65:508–511. doi: 10.1107/S1744309109013591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi R, Villarroya M, Ruiz-Partida R, Li Y, Proteau A, Prado S, Moukadiri I, Benítez-Páez A, Lomas R, Wagner J, et al. Structure-function analysis of Escherichia coli MnmG (GidA), a highly conserved tRNA-modifying enzyme. J. Bacteriol. 2009;191:7614–7619. doi: 10.1128/JB.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer S, Scrima A, Versees W, Wittinghofer A. Crystal structures of the conserved tRNA-modifying enzyme GidA: implications for its interaction with MnmE and substrate. J. Mol. Biol. 2008;380:532–547. doi: 10.1016/j.jmb.2008.04.072. [DOI] [PubMed] [Google Scholar]
- 19.Prado S, Villarroya M, Medina M, Armengod ME. The tRNA-modifying function of MnmE is controlled by post-hydrolysis steps of its GTPase cycle. Nucleic Acids Res. 2013;41:6190–6208. doi: 10.1093/nar/gkt320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeuchi Y, Shigi N, Kato J, Nishimura A, Suzuki T. Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol. Cell. 2006;21:97–108. doi: 10.1016/j.molcel.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Benítez-Páez A, Villarroya M, Douthwaite S, Gabaldon T, Armengod ME. YibK is the 2'-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNA(Leu) isoacceptors. RNA. 2010;16:2131–2143. doi: 10.1261/rna.2245910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagervall TG, Bjork GR. Genetic mapping and cloning of the gene (trmC) responsible for the synthesis of tRNA (mnm5s2U)methyltransferase in Escherichia coli K12. Mol. Gen. Genet. 1984;196:201–207. doi: 10.1007/BF00328051. [DOI] [PubMed] [Google Scholar]
- 23.Hagervall TG, Edmonds CG, McCloskey JA, Bjork GR. Transfer RNA(5-methylaminomethyl-2-thiouridine)-methyltransferase from Escherichia coli K-12 has two enzymatic activities. J. Biol. Chem. 1987;262:8488–8495. [PubMed] [Google Scholar]
- 24.Bujnicki JM, Oudjama Y, Roovers M, Owczarek S, Caillet J, Droogmans L. Identification of a bifunctional enzyme MnmC involved in the biosynthesis of a hypermodified uridine in the wobble position of tRNA. RNA. 2004;10:1236–1242. doi: 10.1261/rna.7470904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roovers M, Oudjama Y, Kaminska KH, Purta E, Caillet J, Droogmans L, Bujnicki JM. Sequence-structure-function analysis of the bifunctional enzyme MnmC that catalyses the last two steps in the biosynthesis of hypermodified nucleoside mnm5s2U in tRNA. Proteins. 2008;71:2076–2085. doi: 10.1002/prot.21918. [DOI] [PubMed] [Google Scholar]
- 26.Pearson D, Carell T. Assay of both activities of the bifunctional tRNA-modifying enzyme MnmC reveals a kinetic basis for selective full modification of cmnm5s2U to mnm5s2U. Nucleic Acids Res. 2011;39:4818–4826. doi: 10.1093/nar/gkr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura A, Sengoku T, Nishimoto M, Yokoyama S, Bessho Y. Crystal structure of the bifunctional tRNA modification enzyme MnmC from Escherichia coli. Protein Sci. 2011;20:1105–1113. doi: 10.1002/pro.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JH. A Short Course in Bacterial Genetics: A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 29.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elseviers D, Petrullo LA, Gallagher PJ. Novel E. coli mutants deficient in biosynthesis of 5-methylaminomethyl-2-thiouridine. Nucleic Acids Res. 1984;12:3521–3534. doi: 10.1093/nar/12.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilsson K, Lundgren HK, Hagervall TG, Bjork GR. The cysteine desulfurase IscS is required for synthesis of all five thiolated nucleosides present in tRNA from Salmonella enterica serovar typhimurium. J. Bacteriol. 2002;184:6830–6835. doi: 10.1128/JB.184.24.6830-6835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Vicente M, Yim L, Villarroya M, Mellado M, Perez-Paya E, Bjork GR, Armengod ME. Effects of mutagenesis in the switch I region and conserved arginines of Escherichia coli MnmE protein, a GTPase involved in tRNA modification. J. Biol. Chem. 2005;280:30660–30670. doi: 10.1074/jbc.M503223200. [DOI] [PubMed] [Google Scholar]
- 33.Benítez-Páez A, Villarroya M, Armengod ME. The Escherichia coli RlmN methyltransferase is a dual-specificity enzyme that modifies both rRNA and tRNA and controls translational accuracy. RNA. 2012;18:1783–1795. doi: 10.1261/rna.033266.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Himeno H, Asahara H, Hasegawa T, Shimizu M. In vitro study of E.coli tRNA(Arg) and tRNA(Lys) identity elements. Nucleic Acids Res. 1992;20:2335–2339. doi: 10.1093/nar/20.9.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gehrke CW, Kuo KC. Ribonucleoside analysis by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1989;471:3–36. doi: 10.1016/s0021-9673(00)94152-9. [DOI] [PubMed] [Google Scholar]
- 36.Emilsson V, Kurland CG. Growth rate dependence of transfer RNA abundance in Escherichia coli. EMBO J. 1990;9:4359–4366. doi: 10.1002/j.1460-2075.1990.tb07885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ponchon L, Beauvais G, Nonin-Lecomte S, Dardel F. A generic protocol for the expression and purification of recombinant RNA in Escherichia coli using a tRNA scaffold. Nat. Protoc. 2009;4:947–959. doi: 10.1038/nprot.2009.67. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T, Suzuki T. Chaplet column chromatography: isolation of a large set of individual RNAs in a single step. Methods Enzymol. 2007;425:231–239. doi: 10.1016/S0076-6879(07)25010-4. [DOI] [PubMed] [Google Scholar]
- 39.Gong S, Ma Z, Foster JW. The Era-like GTPase TrmE conditionally activates gadE and glutamate-dependent acid resistance in Escherichia coli. Mol. Microbiol. 2004;54:948–961. doi: 10.1111/j.1365-2958.2004.04312.x. [DOI] [PubMed] [Google Scholar]
- 40.Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- 41.Kitamura A, Nishimoto M, Sengoku T, Shibata R, Jager G, Bjork GR, Grosjean H, Yokoyama S, Bessho Y. Characterization and structure of the Aquifex aeolicus protein DUF752: a bacterial tRNA-methyltransferase (MnmC2) functioning without the usually fused oxidase domain (MnmC1) J. Biol. Chem. 2012;287:43950–43960. doi: 10.1074/jbc.M112.409300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ponchon L, Dardel F. Recombinant RNA technology: the tRNA scaffold. Nat. Methods. 2007;4:571–576. doi: 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- 43.Yamanaka K, Hwang J, Inouye M. Characterization of GTPase activity of TrmE, a member of a novel GTPase superfamily, from Thermotoga maritima. J. Bacteriol. 2000;182:7078–7082. doi: 10.1128/jb.182.24.7078-7082.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P, Crain PF, Nasvall SJ, Pomerantz SC, Bjork GR. A “gain of function” mutation in a protein mediates production of novel modified nucleosides. EMBO J. 2005;24:1842–1851. doi: 10.1038/sj.emboj.7600666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinus MG, Morris NR, Soll D, Kwong TC. Isolation and partial characterization of three Escherichia coli mutants with altered transfer ribonucleic acid methylases. J. Bacteriol. 1975;122:257–265. doi: 10.1128/jb.122.1.257-265.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanjee U, Houry WA. Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 2013;67:65–81. doi: 10.1146/annurev-micro-092412-155708. [DOI] [PubMed] [Google Scholar]
- 47.Kruger MK, Pedersen S, Hagervall TG, Sorensen MA. The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998;284:621–631. doi: 10.1006/jmbi.1998.2196. [DOI] [PubMed] [Google Scholar]
- 48.Benítez-Páez A, Villarroya M, Armengod ME. Regulation of expression and catalytic activity of Escherichia coli RsmG methyltransferase. RNA. 2012;18:795–806. doi: 10.1261/rna.029868.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan MA, Cannon JF, Webb FH, Bock RM. Antisuppressor mutation in Escherichia coli defective in biosynthesis of 5-methylaminomethyl-2-thiouridine. J. Bacteriol. 1985;161:368–376. doi: 10.1128/jb.161.1.368-376.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bandyra KJ, Bouvier M, Carpousis AJ, Luisi BF. The social fabric of the RNA degradosome. Biochim Biophys. Acta. 2013;1829:514–522. doi: 10.1016/j.bbagrm.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phizicky EM, Alfonzo JD. Do all modifications benefit all tRNAs? FEBS Lett. 2010;584:265–271. doi: 10.1016/j.febslet.2009.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bregeon D, Colot V, Radman M, Taddei F. Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev. 2001;15:2295–2306. doi: 10.1101/gad.207701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yim L, Martinez-Vicente M, Villarroya M, Aguado C, Knecht E, Armengod ME. The GTPase activity and C-terminal cysteine of the Escherichia coli MnmE protein are essential for its tRNA modifying function. J. Biol. Chem. 2003;278:28378–28387. doi: 10.1074/jbc.M301381200. [DOI] [PubMed] [Google Scholar]
- 54.Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001;20:4863–4873. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brierley I, Meredith MR, Bloys AJ, Hagervall TG. Expression of a coronavirus ribosomal frameshift signal in Escherichia coli: influence of tRNA anticodon modification on frameshifting. J. Mol. Biol. 1997;270:360–373. doi: 10.1006/jmbi.1997.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagervall TG, Bjork GR. Undermodification in the first position of the anticodon of supG-tRNA reduces translational efficiency. Mol. Gen. Genet. 1984;196:194–200. doi: 10.1007/BF00328050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.