


Abstract
Hfq is a posttranscriptional riboregulator and RNA chaperone that binds small RNAs and target mRNAs to effect their annealing and message-specific regulation in response to environmental stressors. Structures of Hfq-RNA complexes indicate that U-rich sequences prefer the proximal face and A-rich sequences the distal face; however, the Hfq-binding sites of most RNAs are unknown. Here, we present an Hfq-RNA mapping approach that uses single tryptophan-substituted Hfq proteins, all of which retain the wild-type Hfq structure, and tryptophan fluorescence quenching (TFQ) by proximal RNA binding. TFQ properly identified the respective distal and proximal binding of A15 and U6 RNA to Gram-negative Escherichia coli (Ec) Hfq and the distal face binding of (AA)3A, (AU)3A and (AC)3A to Gram-positive Staphylococcus aureus (Sa) Hfq. The inability of (GU)3G to bind the distal face of Sa Hfq reveals the (R-L)n binding motif is a more restrictive (A-L)n binding motif. Remarkably Hfq from Gram-positive Listeria monocytogenes (Lm) binds (GU)3G on its proximal face. TFQ experiments also revealed the Ec Hfq (A-R-N)n distal face-binding motif should be redefined as an (A-A-N)n binding motif. TFQ data also demonstrated that the 5′-untranslated region of hfq mRNA binds both the proximal and distal faces of Ec Hfq and the unstructured C-terminus.
INTRODUCTION
Hfq is a pleiotropic posttranscriptional regulator found in many Gram-negative and Gram-positive bacteria (1–5). Hfq was first identified as the Escherichia coli (Ec) host factor necessary for replication of the RNA bacteriophage Qβ (6,7) but its more significant role in bacterial physiology has become clear over the past decade (8,9). Hfq acts as an RNA chaperone by binding to non-coding small RNAs (sRNAs) and their target mRNAs, facilitating the annealing of their complementary trans-encoded sequences and effecting message-specific regulation (10–16). Hfq appears to down-regulate most targeted messages; however, some mRNAs, such as rpoS, which encodes the stationary phase sigma factor σs, require Hfq for efficient translation (17). Hfq regulates multiple pathways involved in stress responses, membrane integrity, quorum sensing and virulence (3,18–21). This protein is an essential virulence factor in Ec, Salmonella typhimurium (St), Pseudomonas aeruginosa, Vibrio cholerae and other pathogenic bacteria (20,22–25). Recently, Hfq has been found to be involved in the multidrug resistance mechanisms of Ec and Salmonella enterica serovar typhimurium (26,27). This resistance is conferred by Hfq regulation of the expression of the AcrAB multidrug efflux pump in Ec (26) and the SmvA efflux pump in S. enterica (27). Hfq has also been shown to autorepress its expression by binding to at least two sequences, site A and site B, in the 5′-untranslated region (5′-UTR) of hfq mRNA (28–31).
Currently two non-exclusive models of Hfq function in riboregulation are supported by experimental data (17). One emphasizes the chaperone activity of Hfq, which actively unfolds the secondary structures of mRNAs or sRNAs to facilitate sRNA-mRNA annealing or to alter access to the ribosome binding site. The second model assumes that Hfq binds to mRNA and sRNA at the same time, thereby increasing the local concentration and facilitating annealing.
The structures of full-length Staphylococcus aureus (Sa) Hfq and the conserved core of Ec Hfq (residues 2–69), as well as those of the Hfq proteins from other bacteria, have revealed a hexameric toroid and an Sm fold (32,33). Each subunit consists of a highly bent five-stranded antiparallel β sheet whereby β strands 1 through 3 comprise the Sm1 motif and β strands 4 and 5, the Sm2 motif. In most eubacteria, an N-terminal α-helix sits on one side of each subunit. This helix-containing face is designated the proximal face; the opposite side is called the distal face. Interestingly, Hfq proteins contain a highly variable C-terminus, e.g. Ec Hfq has a 44-residue-long C-terminal tail beyond its Sm core, whereas Sa Hfq has <10 residues beyond the Sm core (32). To date the C-terminus has not been visible in high-resolution crystal structures, most likely due to its inherent flexibility (32,34).
Several structures of Hfq bound to shorter RNA sequences have been determined. The structure of Sa Hfq bound to an AU5G RNA showed the RNA oligomer bound in a circular manner in the pore on the proximal face (35). Other structures have been solved with RNA bound to the proximal face, two in which the Ec Hfq is bound to AU6A (36,37) and the St Hfq-U6 complex (38). These structures reveal a similar, but non-identical, binding mechanism with a single nucleotide bound per subunit. The base of the nucleotide stacks between adjacent aromatic residues (F42 in Ec Hfq) and sequence specificity is conferred mainly by hydrogen bonds between the base and either side chain or backbone atoms of nearby residues. In the St Hfq-U6 structure, the RNA backbone adopts a constrained conformation, which differs from that observed in Sa Hfq-AU5G complex (38). The former conformation allows specific recognition of the free terminal 3′-hydroxyl group, thus explaining the preferential binding of Hfq to U-rich 3′ ends. The Ec Hfq-AU6A complexes show either an adenosine and three uridines or four uridines bound in the pore in a manner more similar to AU5G binding to Sa Hfq (36). These structures support the idea that U-rich RNA sequences, often found in sRNAs, will bind preferentially to the proximal face.
Four structures of RNA bound to the distal face have also been determined. These are Ec Hfq-A15 (39), Bacillus subtilis (Bs) Hfq-(AG)3A aptamer (40), Sa Hfq-A7 (41) and the Ec Hfq-A7-AU6A ternary complex (37). The Ec Hfq-A15 and Ec Hfq-A7 complex structures show three nucleotides bound per subunit of Hfq and support the hypothesis that (A-R-N)n sequences, where A is an adenine nucleotide, R is any purine nucleotide and N is any nucleotide, bind to the distal face. The (A-R-N)n motif has been shown to be critical in the proper sRNA regulation of several mRNAs including rpoS, fhlA and glmS (42–46). The A-site specificity of Ec Hfq is conferred by peptide backbone hydrogen bonding (Q33 in Ec Hfq). The R-site of Ec Hfq appears to be able to accommodate both adenosine and guanosine whereby the purine ring sticks into a pocket and packs against a series of aromatic and non-polar residues (Y25, L26, I30, L32 in Ec Hfq). The adenosine N3, N6, N1 and ribosyl 2′-hydroxyl atoms hydrogen bond to Nδ of residue N28, Oε atom of residue Q52, Oγ of residue T61 and the carbonyl oxygen of residue G29, respectively. The N site has no protein–nucleic acid interactions and likely represents the entrance or exit point for RNA. By contrast, the Bs Hfq-(AG)3A and Sa Hfq-(AA)3A complex structures show a significantly different distal-face binding mode, designated as the (R-L)n binding motif, where R is a purine nucleotide and L is the R site linker and can be any nucleotide (41,47). The (R-L)n binding motif presents 12 possible binding sites per hexamer versus the possibility of 18 binding sites of the (A-R-N)n motif found in the Gram-negative Ec Hfq. The presence of an (A-R-N)n or an (R-L)n binding mode can be attributed to subtle sequence differences between Hfq proteins from Gram-negative and Gram-positive bacteria (41). Consequently, the R sites of the Ec, Bs and Sa Hfqs are equivalent but not identical and the mode of protein-nucleobase stacking differs so that residues F25 and Q31 are stacking with adenines in the Sa Hfq-A4 structure. However, the altered sequences also preclude the formation of the A-site in the Bs or Sa Hfq proteins (41).
In total, the published structures of these Hfq-RNA complexes establish unequivocally two RNA binding faces on Hfq and suggest where and how certain longer physiologically relevant RNA sequences may interact with Hfq. Yet, they do not provide a complete view of the binding modes of larger target mRNAs or sRNAs, which display a variety of secondary structures, e.g. DsrA, RhyB, MicA, OxyS and Spot42 (48). To help fill this gap a series of small angle x-ray scattering studies (SAXS) on full-length Hfq bound to intact sRNAs (34,49,50) have been carried out and suggested that a single RNA sequence will interact with Hfq on only one face of the hexamer at a time (49). In these SAXS structures, the RNA sequences lie across the face of the molecule such that not all subunits contact the RNA; additionally the RNA does not appear to be bound circularly, as observed with the shorter RNA sequences used in crystal structures. Further, two SAXS studies suggest conflicting roles for the Hfq C-terminus, with one study concluding that this region may interact with RNA (50), whereas the other study sees no interaction (49). This indicates that further studies on the role of the C-terminus in RNA binding need to be conducted.
One current impediment to understanding the full function of Hfq is the dearth of structural information on longer and physiologically relevant RNAs. To begin to address this problem we have developed a rapid, structure-guided nucleobase-intrinsic tryptophan fluorescence quenching (TFQ) approach to map the region or regions to which an RNA sequence binds on Hfq and to identify likely interacting amino acid residues. This methodology takes advantage of the lack of Trp residues in the Ec, Sa and Listeria monocytogenes (Lm) Hfq, our ‘test’ proteins, but clearly is not limited to only Hfq proteins that do not contain Trp residues. Thus, we have created multiple Ec, Sa and Lm Hfq proteins that contain a single engineered Trp residue, which has been placed throughout the entire length of the full-length Ec Hfq sequence, at selected proximal locations in full-length Sa Hfq and a distal side location in Lm Hfq. This technique allows us to identify correctly the binding locations of a series of ‘control’ sequences to Ec, Sa and Lm Hfq, but more important, to redefine the Ec Hfq (A-R-N)n as well as the Sa Hfq (R-L)n distal-face binding motifs more precisely and to confirm that (A-A-N)n and (A-L)n stretches bind the distal faces of Ec and Sa Hfq, respectively. We also explore the interactions between Ec Hfq with the 5′-UTR of its own mRNA to provide a fuller understanding of how the two previously identified binding sites (site A and site B) contact Hfq to effect its translational autoregulation. Finally, with this set of Trp-substituted Hfq proteins we begin to evaluate whether or not the C-terminus of Ec Hfq interacts with any RNA. Our results demonstrate that intrinsic TFQ is a valuable and readily utilizable tool for elucidating Hfq-RNA interaction in the absence of high-resolution structural information and can be combined with other lower resolution structural or biochemical data to define the Hfq-binding modes of short oligoribonucleotides and longer physiologically relevant RNA sequences.
MATERIALS AND METHODS
Protein over expression, purification and site-directed mutagenesis
Ec, Sa and Lm Hfq were overexpressed in Ec strain ER2566 Δhfq using the pTYB11 vector. The cells were grown in Luria Broth containing 50 µg/ml ampicillin at 37°C to an OD600 between 0.4 and 0.6. Expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 20 h at 18°C. Cells were harvested at 4°C and stored at −80°C or lysed immediately using a microfluidizer. To remove contaminating RNA and DNA, 10 µg/ml DNAse and 10 µg/ml RNAse were added to crude lysate and stirred at 4°C for 2 h before clarification by centrifugation at 17 500 rpm for 30 min at 4°C. All full-length Hfq proteins and the Ec Hfq truncate, which is composed of residues 2–69 (the core), were purified using the IMPACT-CN system as described (10). Wild type (WT) Ec and Sa Hfq and their tryptophan mutants were buffer exchanged into 20 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES)-Na, pH 8.0, 150 mM NaCl, 0.5 mM EDTA and concentrated from 100 to 200 µM (hexamer). WT Lm Hfq and its Trp mutants were concentrated to 100 µM in 20 mM HEPES-Na, pH 8.0, 500 mM NaCl, 0.5 mM EDTA and stored at 25°C due to cold denaturation. WT Ec and Sa Hfq were stored at 4°C, whereas their mutants were stored at 25°C due to cold denaturation.
Crystallization, data collection, structure determination and refinement
All Hfq Trp mutants used for crystallization were C-terminally truncated at residue 69. Hfq Trp mutant crystals were obtained by the hanging-drop vapour diffusion method. Drops contained 1 µl of protein to 1 µl of well solution with a range of protein concentrations (5–9 mg/ml). Proteins were crystallized from solutions containing 22–28% polyethlyene glycol (PEG) 3350, 26–32% isopropanol and 0.1 M Tris base, pH 8.0–9.0. X-ray intensity data were collected under cryo-conditions at the Advanced Photon Source (Beamline 22-ID or 22-BM) in Argonne, IL. Crystals were flash frozen directly from the crystallization drops in a nitrogen stream at 100 K. Data were processed using HKL-2000 or HKL-3000 (51). Structure determination via molecular replacement was carried out in Phaser (52) using apo Hfq (PDB ID: 1HK9) as the search model. Structure building and refinement were carried out in Coot (53) and Phenix (54), respectively. Selected data collection, reduction and refinement statistics are shown in Supplementary Table S1. Root Mean Square Deviation (RMSD) calculations were done using secondary structure matching and Cα atom alignment via SSM superpose (55).
Determination of Hfq-RNA binding affinity by fluorescence polarization
The RNA binding affinities of Ec and Sa Hfq and the Trp mutants were determined using a fluorescence polarization-based binding assay and a PanVera Beacon 2000 instrument (Invitrogen, Madison, WI, USA). Hfq was serially diluted into 100 µl of binding buffer containing 20 mM HEPES-Na, pH 8.0, 0.5 mM EDTA, 200 mM NaCl and 1 nM 5′-fluorescein-labelled RNA. Samples were excited at 490 nm and emission was detected at 530 nm and data were collected at 295 K. Data were analysed assuming a 1:1 binding stoichiometry between one Hfq hexamer and one molecule of RNA. The data were plotted using KaleidoGraph (Synergy Software) and the generated curves were fit using non-linear least squared analysis, assuming a bimolecular model such that the Kd values represent the protein concentration at half maximal RNA binding (56). The binding isotherms were fit to the equation, P = {(Pbound-Pfree) [protein]/(KD+[protein])}+Pfree, where Pbound is the maximum polarization at saturation, P is the polarization at a given protein concentration, Pfree is the polarization of free fluorescein-labelled RNA and Kd is the equilibrium dissociation constant. At a minimum all values were independently determined in triplicate.
Tryptophan fluorescence quenching
TFQ measurements were performed using an RF-5301PC spectrofluorophotometer (Shimadzu, Nakagyo-ku, Kyoto, Japan) at 298 K. TFQ was done by exciting the single Hfq Trp residue at 298 nm and scanning the emission fluorescence spectrum from 320–400 nm. A 1 ml of sample containing 1 µM of each Hfq protein in binding buffer (20 mM HEPES-Na, pH 8.0, 200 mM NaCl, 0.5 mM EDTA) was scanned and followed by addition of a specific RNA sequence. Initially two concentration ranges were examined, 10 and 40 µM RNA, to determine the overall sensitivity of the system. However, subsequent quenching studies were done using 1 and 4 µM RNA to increase the physiological relevance of the result and to decrease any ambiguity that might result from secondary, lower affinity binding. Each titration was done at least three times (Standard deviations are listed in Supplementary Table S2). Data were analysed using Microsoft Excel. Quenching was determined using the arbitrary fluorescence maximal height for each Trp mutant. Quenching percentage was calculated using the following equation:
(1 − ((FR−FB)/(F0−FB))) × 100, where FR is the fluorescence value after addition of RNA to the Hfq solution, F0 is the initial fluorescence value of the Hfq solution without RNA and FB is the fluorescence of buffer without RNA or Hfq.
RNA samples and generation of the 5′-UTR of hfq mRNA
Oligoribonucleotides were purchased from IDT (Coralville, IA) or Oligos Etc (Wilsonville, OR) and used without further purification. The 64-nucleotide 5′-UTR of the hfq mRNA of Ec was synthesized biochemically. Briefly, the pMCSG7 vector was modified to include the DNA sequence 5′-TAA TAC GAC TCA CTA TAG GAT TTT TTC AGA ATC GAA AGG TTC AAA GTA CAA ATA AGC ATA TAA GGA AAA GAG AGA ATG GGA TCC-3′ using standard ligation-independent cloning techniques. To prepare RNA, the modified vector was midiprepped, phenol:chloroform extracted and 8–20 µg of clean DNA vector was digested using BamH1-HF (NEB, Ipswich, MA) at 37°C for 16 h. The T7 RNA polymerase reaction [100 µl of 10× Buffer (0.5 M Tris base, pH 7.5, 0.25 M MgCl2, 0.05 M EDTA), 40 µl of 50 mM spermidine, 200 µl of rNTP mix (20 mM/rNTP), 30 µl of 100 units/ml inorganic pyrophosphatase (NEB), 80 µl of 1 mg/ml T7 RNA polymerase, 60 µl of 8–20 µg/ml digested DNA vector, 440 µl of dH2O) was carried out at 37°C for 16 h and quenched with 2 ml of 0.45 M EDTA. The reaction was then purified by acidic phenol:chloroform extraction and ethanol precipitated at −80°C for 16 h. The RNA pellet was further purified using 70% ethanol and air dried before resuspending in 100 µl of 10 mM sodium cacodylate, pH 6.5.
RESULTS AND DISCUSSION
TFQ identifies known binding faces
Germane crystal structures demonstrate A15 RNA binds to the distal face of the Ec Hfq (39) and U6 RNA binds to the proximal face of Ec and St Hfq (37,38). These RNA sequences were therefore used as controls to test the capability of TFQ to discern Ec Hfq distal face binding from proximal face binding, thus validating TFQ as an Hfq–RNA interaction-mapping technique. Using the Ec Hfq-A15 crystal structure as a guide, tryptophan mutations were introduced at positions Y25, K31, which is proximal to but does not interact with A15, and Q33, which interacts with A15 through its backbone carbonyl and amide groups (Figure 1). Similar contacts are seen in the Ec Hfq-A7 complex structure (37). These mutants were designed to allow us to confirm the general position in which A15 bound as well as refine the RNA binding site on the distal face. Similarly, we chose to make an F42W mutation on the opposite Hfq face to assess proximal face binding (Figure 1). An F39W mutation was made to investigate a proximal face ‘edge’ binding site based on its recently reported importance in binding the sRNAs ArcZ and McaS [(57), http://dx.doi.org/10.1016/j.jmb.2013.01.006] and its observed uridine-triphosphate binding (58). To explore potential binding to the positively charged cleft on the surface of Ec Hfq we chose to make an F11W mutation (Figure 1). On the basis of the identification of a lateral rim binding site for RhyB RNA to St Hfq (59) and that the lateral rim residues play a role in Hfq-RpoS binding (60) we used an R17W mutation, to study whether any of our selected RNAs bind to this site on Ec Hfq (Figure 1). Finally, as the role of the Hfq C-terminus in RNA binding remains unclear (43,61,62) but in specific cases necessary for mRNA regulation (43), we created three individual Trp mutations, G77W, Y83W and Q95W, in this unstructured region (Figure 1). All mutations were generated in the full-length Ec hfq sequence.
Figure 1.
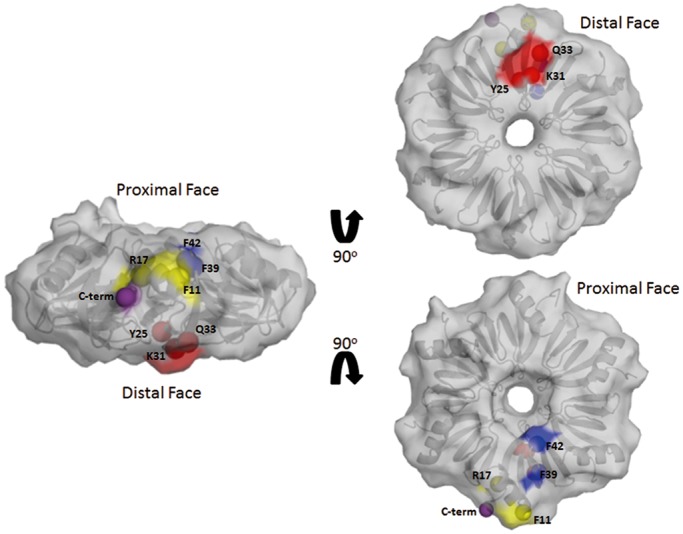
Surface representation of the WT Ec Hfq structure and the positions of the tryptophan-substituted residues. Highlighted in red are residues on the distal face that were mutated to tryptophan (Trp); in yellow are the lateral residues that were mutated to Trp, in blue the proximal face residues that were mutated to Trp and in purple the position to indicate the beginning to the C-terminal 44 residues of Ec Hf. A cartoon of the underlying hexamer is shown in grey. PDB accession number for the WT Ec Hfq structure is 1HK9 (33).
TFQ experiments were carried out by titrating RNA into 1 μM protein (hexamer), which is ∼10-fold below the reported cellular concentration of the Hfq hexamer (63), to reduce the major effects of non-specific and lower affinity binding. The samples were excited at 298 nm and the emission spectra scanned from 320–400 nm (Figure 2). After each titration was completed, the amount of TFQ was quantified. We define a significant Hfq-RNA interaction having occurred when the total quenching at 4 µM RNA concentration exceeds 10% and this quenching is 2-fold greater than the quenching at 1 µM RNA concentration. If quenching of Trp residues inserted individually on both faces meets these criteria then we would conclude the RNA ligand is able to bind both faces. If one face is quenched at least 1.5-fold greater than the other, then that face is defined as being the preferred binding face.
Figure 2.

Representative intrinsic TFQ titration experiment. To provide the initial value of tryptophan fluorescence, 1 μM Hfq mutant F42W in the absence of RNA was excited at 298 nm and the emission scanned from 320–400 nm. The maximum fluorescence intensity is found at 343 nm (denoted by a solid vertical red line). RNA quenching of the tryptophan fluorescence is calculated by measuring the intensity differences at wavelength 343 nm after addition of an RNA aliquot, employing the equation Quenching (%) = (1 − ((FR−FB)/(F0−FB))) × 100, and carrying out the appropriate corrections as described in the ‘Materials and Methods’ section.
The initial TFQ experiments with A15 RNA showed that Ec Hfq mutants Y25W and Q33W were quenched significantly at the RNA concentrations examined (Figure 3A, Supplementary Figure S1A). Some quenching was also observed with mutant F11W, suggesting a linkage between these sites, although structural artefacts of the F11W change might be responsible for this quenching (see below). Intriguingly, residue G77W showed small but significant quenching with A15, indicating that this C-terminal residue may interact with longer A-tracts in solution, possibly with a flipped out N-site nucleotide. As expected, neither proximal-face mutant F39W nor F42W is quenched by A15 in the 1–4 µM range; however, they begin to quench in the less physiologically relevant 10–40 µM range (Supplementary Figure S1A), indicating low affinity or non-specific binding at these high RNA concentrations. Ec Hfq mutants R17W, K31W, Y83W and Q95W are not quenched in the lower micromolar ranges, indicating no specific interaction occurs between these residues and A15. The finding that the Q33W but not the K31W residue is quenched by A15 is particularly important as those data indicate that RNA must be near the residue under study: residue K31W can be modelled to be as close as 6 Å to the RNA but is not quenched, whereas residue Q33W can be modelled to stack directly against the adenine ring and is quenched (Supplementary Figure S2).
Figure 3.

Control TFQ experiments for Ec, Sa and Lm Hfq Trp mutants using A15, a distal face-binding RNA, or U6, a proximal face-binding RNA. Panels (A) (left) and (B) (left) show A15 quenching of Ec and Sa/Lm Hfq Trp mutants, respectively. Panels (A) (right) and (B) (right) show U6 quenching of Ec and Sa/Lm Hfq Trp mutants, respectively. The x-axis labels under each bar graph refer to the tryptophan-substituted residue within that Hfq protein. The percent quenching is shown on the y-axis. The bar graphs are coloured by location on Hfq as shown in Figure 1. The solid bar represents the percent quenching by 1 µM RNA while the diagonal striped bar above the solid bar represents the percent quenching by 4 µM RNA. (C) A sequence alignment of the Sa and Lm Hfq proteins. The key proximal-face U6 binding residues Sa Y42 and Lm F43 are boxed. Identical residues between the two proteins are shown below the alignment.
Analogous TFQ experiments with U6 RNA show that Ec Hfq mutant F42W, located on the proximal face, is quenched significantly at all RNA concentrations used, whereas mutant F11W has a fluorescence signal that is enhanced equally at all concentrations (Figure 3A; Supplementary Figure S1B). No other mutant is quenched at the lower concentration ranges (1–4 μM). In the 10–40 µM RNA range, the F39W mutant shows some quenching, although this is not as significant as that observed for Ec mutant F42W, and suggests a second, weaker affinity binding site, which is consistent with the quenching of the nearby Hfq ‘rim’ mutant S38W by DsrA and other RNA molecules (60), and that this residue binds RNA when a U-rich sequence is present at a high local concentration such as might occur in SgrS (64) (Supplementary Figure S1B).
In addition to mapping the RNA binding sites on a Gram-negative Hfq homologue and to show the general utility of this approach, we extended our studies to Hfq homologues from the Gram-positive bacteria Sa and Lm. Using the structure of the Sa Hfq-A4 complex (41) we created mutants F25W and Q31W. Due to the inability to produce Sa Hfq Y42W, a proximal face mutant, we chose to study the Lm Hfq after a sequence alignment of Sa and Lm Hfq revealed 40% sequence identity and 81% sequence homology between the two proteins and that Lm residue F43 resides in the same location as Y42 from Sa (Figure 3C). Additionally, Hfq has been shown to be functionally important in sRNA regulation in this pathogenic bacterium (65). Therefore mutation F43W was made in the 77 residue full-length Lm Hfq. As anticipated, Sa distal face mutants F25W and Q31W are quenched in the presence of A15 but are not quenched by U6 (Figure 3B). By contrast, proximal face mutant F43W is quenched by U6 but not quenched by A15 (Figure 3B).
These results confirm that Trp residues show robust intrinsic fluorescence quenching when the appropriate RNA sequences are bound nearby at a distance <6 Å. To ensure further that the observed quenching is not simply an effect of non-specific quenching of Trp by RNA nucleotides, we titrated A15 or U6 into 6 μM N-acetyl-l-tryptophanamide, a Trp residue mimic. Neither A15 nor U6 induced quenching (Supplementary Figure S3), indicating that a specific interaction must occur to result in TFQ. Since significant quenching is observed only for residues that are known from crystal structures to interact directly with a particular RNA sequence, we conclude that TFQ can be used to map accurately RNA binding sites on Hfq.
Tryptophan mutants do not create false binding sites
To ensure that the designed mutations did not significantly impact the biochemical function and biologically relevant RNA binding sites of Hfq, fluorescence polarization-based RNA binding assays were used to determine the equilibrium dissociation constants (Kd) of each mutant for the control RNA sequences (Supplementary Figure S4). It is important to emphasize here that due to the high protein and RNA concentrations necessary for each TFQ experiment, TFQ could not be used to determine the equilibrium dissociation constants of a particular mutant for a particular RNA sequence. Calculated binding affinities are shown in Table 1.
Table 1.
Equilibrium dissociation constants (Kd) for Trp mutants to A15 and U6
| A15 |
U6 |
|||
|---|---|---|---|---|
| E. coli Hfq | ||||
| WT Hfq S | 1.4 nM ± 0.9 a,b | Fold changec | 766.0 nM ± 73.7 | Fold change |
| F11W | 7.1 nM ± 0.6 | 5.0 | 502.7 nM ± 55.5 | 0.7 |
| R17W | 3.7 nM ± 1.5 | 2.6 | 1.6 µM ± 0.08 | 2.1 |
| Y25W | 258.2 nM ± 32.7 | 184.4 | 836.8 nM ± 62.5 | 1.1 |
| K31W | 1.4 nM ± 0.5 | 1.0 | 1123.3 ± 73.0 | 1.5 |
| Q33W | 15.9 nM ± 3.6 | 11.4 | 404.7 nM ± 182.4 | 0.5 |
| F39W | 0.59 nM ± 0.35 | 0.4 | 2.0 µM ± 0.4 | 2.6 |
| F42W | 0.28 nM ± 0.09 | 0.2 | 4.8 µM ± 782.5 | 6.2 |
| G77W | 2.9 nM ± 1.7 | 2.1 | 379.9 nM ± 93.2 | 0.5 |
| Y83W | 7.4 nM ± 2.8 | 5.3 | 777.9 nM ± 254.1 | 1.0 |
| Q95W | 2.6 nM ± 1.1 | 1.9 | 848.3 nM ± 289.4 | 1.1 |
| S. aureus Hfq | ||||
| WT Hfq | 4.2 nM ± 0.5 d | 69.8 nM ± 7.0 d | ||
| F25W | 11.3 µM ± 5.0 | 2700 | 41.9 nM ± 25.1 | 0.6 |
| Q31W | 1.6 µM ± 0.08 | 386.7 | 114 nM ± 23.2 | 1.6 |
As anticipated, the bulkier Trp mutants can lower the binding affinity in some cases. Specifically, Ec Hfq mutant Y25W reduces the binding affinity of A15 by 184-fold (from 1.4 to 258 nM) and Q33W reduces the binding affinity of A15 by 12-fold (from 1.4 to 15.9 nM). Sa Hfq mutant F25W reduces A15 binding by 2700-fold (from 4.2 nM to 11.3 µM) and Sa Q31W reduces A15 binding by 387-fold (from 4.2 nM to 1.6 µM). However, all other mutations do not affect A15 binding adversely (Table 1). We also observed that Ec Hfq mutant F42W reduces U6 binding affinity, but by only ∼6-fold (from 766 nM to 4.8 μM), whereas the other Trp-substituted residues show no significant effects on binding this ribooligonucleotide (Table 1). Interestingly, the affinity of U6 for WT Ec Hfq, which to the best of our knowledge has not been reported previously, is ∼30-fold lower than that observed for U6 binding to St Hfq (38). By contrast, the binding affinity of Ec Hfq for longer U-tracts is much stronger with a Kd = 6.6 nM for Ec Hfq binding to U16 (Supplementary Figure S4C), which is similar to the previously calculated Ec Hfq binding affinity for U18 (48). This indicates that the binding affinity for poly-U sequences by Ec Hfq is strengthened significantly as the sequence is lengthened and suggests the recruitment of other residues such as F39 in binding. Our experimentally obtained value for the Ec Hfq-U6 binding is further supported by the finding that when the same stock of fluoresceinated U6 is used, we obtain the same nanomolar binding affinity for Sa Hfq-U6 as has been reported previously (35) and thus conclude that our experimentally determined Kd for Ec Hfq-U6 is correct.
Hence, we are able to conclude that mutations involved in RNA binding show a decreased binding affinity for those RNA sequences they bind; however, because the protein–RNA interaction is still observed via fluorescence polarization, this loss of affinity is not detrimental to our interpretation of the preferred location of RNA binding as ascertained by our TFQ experiments. We are cognizant that false negatives may occur, particularly when binding to WT Hfq is already weak and a particular tryptophan mutant lowers the affinity beyond the experimental detection limits of TFQ. Hence the need to probe a potential RNA binding site by the creation and TFQ measurements of multiple tryptophan mutants in the area of interest, e.g. the Y25W, K31W and Q33W substitutions to measure distal face binding. Additionally, we find that Trp mutations proximal to a particular binding site do not affect the affinity of distant sites for their preferred RNA sequence. For example, the Ec Hfq F39W mutant binds A15 with a Kd = 0.6 nM, and the Ec Hfq Y25W mutant binds U6 with a Kd = 836 nM: these are the same values observed for WT Hfq binding to these oligoribonucleotides (see Table 1). Thus, at worst, the tryptophan mutations lower the affinity of a few RNAs for their native binding sites, potentially resulting in a false negative binding result for a given single substitution; however, in no case do the Trp mutations create new RNA binding sites (false positives) for the well-studied ‘control’ sequences on either the Ec or Sa Hfq.
Crystal structures of selected Trp mutants show minor structural perturbation
To verify that the Trp mutations do not have a significant impact on the global structure of the resulting Ec Hfq protein, we determined the crystal structures of the F11W, Y25W, F39W and F42W substituted proteins in the context of the C-terminally truncated construct (residues 2–69) and compared their structures to the WT Ec Hfq structure determined by Sauter et al. (33) (PDB ID: 1HK9). Selected crystallographic data are provided in Supplementary Table S1.
Comparison of mutants Y25W, F39W and F42W to WT Ec Hfq reveals little difference in the overall structure (Figure 4A), with the superposition of a single protomer yielding an RMSD ≤ 1.05 Å in all cases and a hexamer to hexamer RMSD ≤ 1.20 Å (Table 2). Superposition of the F11W mutant hexamer and the hexamer of the WT Hfq results in an RMSD of 2.48 Å, indicating a change in the quaternary structure (Figure 4A and C). This difference results from the significant shortening of two β strands due to the adjustment of the backbones of residues Q35 to F39 on β2 and residues I59 to V62 on β5 that is caused by the presence of the bulkier indole side chain. These strands form part of the distal-side intersubunit interface, which is now perturbed in this tryptophan-substituted protein. The remodelling of the β2 and β5 strands and their altered interactions with the adjacent subunit is the likely origin of our F11W fluorescence quenching data.
Figure 4.

(A) Overlays of the structures of WT Ec Hfq and distal (Y25W), lateral (F11W), proximal edge (F39W) and proximal pore (F42W) Hfq tryptophan mutants. All proteins are shown as cartoons with WT Hfq coloured cyan, Y25W coloured green, F11W coloured red, F39W coloured yellow and F42W coloured blue. The calculated RMSD (listed within the grey box) reveals that each mutation does not affect the protomeric structures significantly. (B) Two close up views of the overlay of the structures of the WT Ec Hfq bound to A15 (blue carbon sticks) and the (Y25W) distal face mutation (green carbon sticks) near the position of the substitution and R-site. Y25W takes two conformations, one of which occurs in 2 out of 6 protomers (left). This conformation would block adenine insertion into the R-site (red box). The second conformation (right) occurs in 5 out of 6 protomers and allows base stacking with adenine. However the 2′ oxygen of the ribose clashes with the indole ring (red arc), requiring adjustment of either the phosphodiester or polypeptide backbone or both to relieve the clash. These two structural problems are likely the cause for the significant reduction in A15 binding to this distal face mutant. (C) Close up of the area about the β2 and β5 strands after overlaying WT Ec Hfq and the F11W hexamers. The two major conformational differences between the F11W protein and WT Hfq are enclosed within the red boxes with position 11 shown and numbered in the rightmost figure.
Table 2.
RMSD of superpositions of WT and mutant Ec Hfq protomers or hexamers
| WT:Mutant RMSD | ||
|---|---|---|
| Monomer:Monomer | Hexamer:Hexamer | |
| F11W | 0.30–1.05 Å | 2.48 Å |
| Y25W | 0.27–0.64 Å | 0.72 Å |
| F39W | 0.37–0.54 Å | 1.00 Å |
| F42W | 0.31–0.52 Å | 1.20 Å |
The only other structural difference of significance was found by the alignment of the Y25W mutant with WT Ec Hfq bound to A15. The observed difference is confined to the site of the mutation but provides a structural rationale for the significant change we observe in the Kd of the Y25W protein for A15 (Figure 4B, Table 1). In the Y25W protein, W25 takes two conformations, one of which would block adenine insertion into the R-site and would require the indole ring to flip before adenosine could bind and a second conformation, which although would allow adenine-indole ring stacking, results in steric clash between the 2′ ribose oxygen and the indole ring (Figure 4B). This would require the adjustment of either the RNA phosphodiester or protein polypeptide backbone to relieve the clash. Either alone or combined, these properties of residue W25 are the likely causes for the significant reduction in A15 binding to the distal face mutant but do not obliterate binding (Table 1). The structure of the Ec Hfq (Y25W) protein bound to A15 will be necessary to obtain a complete molecular understanding of the diminished affinity of this protein. The structural changes observed in Y25W, and indeed F11W, would not have been anticipated from modelling studies and underscore the importance of having the high-resolution structures of those mutants that have unusual TFQ properties and significantly lowered RNA binding affinities.
Ec Hfq distal face binding is restricted to (A-A-N)n motifs
The single-stranded A-tract RNA binding mode observed in the Ec Hfq-A15 complex structure suggested that the distal face could bind (A-R-N)n tracts, where A = an adenine nucleotide, R = any purine nucleotide and N = any nucleotide (39). This motif is not found in Hfq proteins from Gram-positive bacteria in that the A site is not present and the observed distal-face RNA binding motif was consistent with an (R-L)n motif, where R = any purine nucleotide and L = a linker nucleotide (41). Although the Ec Hfq R-site was posited to be able to bind guanine nucleotides, no experimental data confirming such binding currently exists. Using our TFQ approach, we tested the hypothesis that both (A-R-N)n sequences, AAGAAGAAGAAGAAG, (A-A-G)5, and GGAGGAGGAGGAGGA, (G-G-A)5, would bind to the distal face with guanine occupying the R-site by necessity in the latter sequence. Our results reveal that rather than binding to the same face, these sequences bind to different faces of Ec Hfq (Figure 5). (G-G-A)5 clearly interacts with residue Ec F42W on the proximal face and quenches none of the distal face tryptophan residues, supporting proximal face binding only (Figure 5A). By contrast, (A-A-G)5 quenches mutants F11W and Q33W but not mutants F39W or F42W, indicating distal face binding only (Figure 5B). Somewhat surprising at first was the weak quenching of R-site mutant Y25W by (A-A-G)5. However, given that this mutation can occlude the R-site resulting in a 180-fold reduction in binding affinity of A15, RNA sequences with intrinsically weaker binding affinity, such as (A-A-G)5, are likely to be unable to bind well even at 4 μM and hence quench weakly this particular R-site substitution. Regardless, distal face binding by (A-A-G)5 is demonstrated by strong Q33W quenching and the ability of this substituted protein to bind an (A-A-N)4 tract with near WT Hfq affinity (39,44). Interestingly, C-terminal mutants, G77W, Y83W and Q95W are not quenched by (A-A-G)5, which differs from G77W quenching by (A-A-A)5 and suggests an alternative entrance/exit pathway for this A-rich polypurine. Further, in the presence of (G-G-A)5, residue Y83W quenches, albeit weakly, whereas the other C-terminal region residues do not, indicating a role for the C-terminal tail in binding to specific RNA sequences.
Figure 5.

TFQ of Ec Hfq by two (A-R-N)n motif RNAs. (A) TFQ by (GGA)5. (B) TFQ by (AAG)5. The x-axis labels under each bar graph refer to the tryptophan-substituted residue within that Hfq protein. The percent quenching is shown on the y-axis. The bar graphs are coloured by location on Hfq and are defined at the bottom of the figure. The solid bar represents the percent quenching by 1 µM RNA, while the diagonal bar above the solid bar represents the observed quenching by 4 µM RNA.
These data reveal that the R-site is actually a physiologically relevant adenine nucleotide-only binding site, and thus the Ec Hfq distal face binding motif is the more restrictive (A-A-N)n motif rather than an (A-R-N)n motif. The (A-R-N)n distal face binding motif has been studied extensively, but until now it has not been possible to confirm that these sequences are binding to the expected face. By using TFQ we have been able to demonstrate that while sequences, which have an (A-R-N)n motif, do indeed bind to Hfq, they do not necessarily bind where expected. This conclusion is supported by the observation that the ARN-2 binding site of the glmS mRNA, which fulfills an (A-A-N)n motif, is the more functionally important distal-face binding site as compared with the ARN-1 site, which only fulfills an (A-R-N)n motif, binding site (43). These results also demonstrate that overall, C-terminal residues do not make strong interactions with (A-A-N)n tracts but appear to interact better with longer (G-G-A)n tracts, implying the C-terminal region of Hfq can interact with longer proximal face-binding RNA.
The Sa Hfq distal face is restricted to (A-L)n motifs
Using our TFQ approach we also tested the hypothesis that the Sa Hfq distal face binding motif is not an (R-L)n motif but a more restrictive (A-L)n binding motif as suggested by our analyses of the Sa Hfq-A4 (41) and Bs Hfq-(AG)3 A (47) complex structures. In these structures each has one adenine ring inserted into the R-site stacked against residue 25, whereas the following 3′-nucleotide, an A or G, is stacked directly over the L-site forming residue Q31. If the R-site is only an adenosine-binding site then the sequences 5′-AUAUAUA-3′, (A-U)3 A, and 5′-ACACACA-3′, (A-C)3 A, but not 5′-GGGGGGG-3′ (G7), and 5′-GUGUGUG-3′, (G-U)3 G, should quench the distal side Trp replacements at positions 25 and 31. Both the (A-U)3 A and (A-C)3 A sequences quenched Sa mutants F25W and Q31W (Figure 6A and B). Further, the (A-C)3 A sequence did not quench Lm F43W, whereas the (A-U)3 A sequence quenches this mutant to a level equal to that seen for Sa F25W and Q31W. The ability of (A-U)3A to bind to both faces is expected, as adenosine has been observed in crystal structures to bind on both faces of Sa Hfq and the (R-L)n distal face binding motif will accommodate uracil at the L-site. Also, based on previously reported binding data, it is known that Sa K33A significantly weakens but does not abolish (A-U)3A binding, whereas it completely abolishes (A-C)3A binding (41). This suggests that the previously reported binding affinity of (A-U)3A for Sa Q33A reflects the binding affinity for the proximal rather than the distal face (41). By contrast, neither the G7 nor (G-U)3G sequence quenches the fluorescence of the F25W and Q31W proteins, but surprisingly the (G-U)3G sequence quenches Lm F43W, indicating that this sequence can bind to the proximal face (Figure 6C and D). G7 does not quench any residues and therefore does not interact with Hfq at even high concentration, a finding consistent with our earlier fluorescence polarization-based binding studies (28). Thus, we conclude that the distal face (R-L)n motif of Sa Hfq, Bs Hfq and most likely Hfq proteins from most if not all Gram-positive bacteria is a more restrictive (A-L)n motif.
Figure 6.

TFQ of Sa or Lm Hfq by (R-L)n motif containing RNAs. (A) TFQ by (AU)3 A. (B) TFQ by (AC)3 A. (C) TFQ by (GU)3 G. (D) TFQ by (G7). The x-axis labels under each bar graph refer to the tryptophan-substituted residue within that Hfq protein. The percent quenching is shown on the y-axis. The bar graphs are coloured by location on Hfq and are defined at the bottom of the figure. The solid bar represents the percent quenching by 1 µM RNA, while the diagonal bar above the solid bar represents the observed quenching by 4 µM RNA.
These quenching data also indicate that Lm Hfq and likely Hfq proteins from other Gram-positive bacteria can bind specific (R-L)n motifs to their proximal faces. The observation that only (A-L)n sequences bind to the distal face demonstrates the impact that TFQ has on understanding how a particular RNA sequence binds Hfq. Until now it had been expected that sequences that fulfilled an (R-L)n motif would bind the distal face based on crystal structures and binding studies. These crystal structures all have adenosine bound in the R-site binding pocket and as such have led to the hypothesis that the R-site is really an adenine nucleotide selective site; however, since it is possible to model a guanine base in the R-site with some rearrangement of the pocket residues, the site could not be labelled unambiguously as adenine specific. Also, while the binding studies indicated that (R-L)n motifs bind Hfq, they provided insight into where the interaction occurs. TFQ thus allows us to redefine experimentally the proposed (R-L)n distal face binding motif to a more restrictive (A-L)n motif, which has been previously hypothesized but never tested (41). Finally, the ability to detect RNA interacting with residue Q31W in Sa Hfq but not with the corresponding Ec Hfq residue illustrates the power of the TFQ approach, as modelling shows that residue Ec Hfq K31W would be close to the RNA but unable to interact with the base, whereas Sa Hfq residue Q31W stacks with the Linker site nucleotide and hence is quenched (Supplementary Figure S2B and D).
Ec Hfq binds the 5′-UTR of Ec hfq mRNA on the proximal and distal sides
Ec Hfq has been shown previously to regulate the amount of Ec hfq mRNA that is present in the cell (28) and to prevent translation initiation of the hfq mRNA, suggesting that the Hfq interaction with hfq mRNA blocks its ribosome binding site (29). Furthermore, previous studies on the Hfq:hfq interaction identified two binding sites for Hfq in the 5′-UTR of the hfq mRNA, which were termed ‘site A’ and ‘site B’ (29) (Figure 7A). To test the ability of our TFQ assay to determine the binding modes of longer pieces of RNA of physiological relevance, we assayed two 24-nucleotide fragments and one 64-nucleotide fragment of the 5′-UTR of Ec hfq mRNA. On the basis of the previous findings we tested the sequences 5′-AUUUUUUCAGAAUCGAAAGGUUCA-3′, which contains site A and an adjacent stem loop; 5′-GCAUAUAAGGAAAAGAGAGAAUGG-3′, which contains site B; and an RNA fragment that contains both sites A and B (Figure 7A). The site A containing oligoribonucleotide significantly quenches the Q33W, F39W, F42W, G77W and Y83W proteins, indicating that site A binds both faces. However, because F42W has 2-fold greater TFQ and is 33% quenched, a preference for the proximal face is strongly suggested and the likely consequence of the AU6A stretch at its 5′ end. The weaker distal face quenching is the likely result of the presence of two AAN triplets within site A. The site A sequence also interacts with two residues of the C-terminal tail (Figure 7B), one of which, Y83W, was also seen to be quenched by (G-G-A)5.
Figure 7.

TFQ of Ec Hfq by the 5′-UTR of hfq mRNA and its Site A and Site B components. (A) The sequence and proposed secondary structure of the 5′-UTR of Ec hfq mRNA. The red lines identify the previously identified Site A, which includes a hairpin structure, and the AG-rich Site B Hfq binding sites. (B) TFQ by hfq mRNA site A. (C) TFQ by hfq mRNA site B. (D) TFQ by the 5′-UTR of hfq mRNA. The x-axis labels under each bar graph refer to the tryptophan-substituted residue within that Hfq protein The percent quenching is shown on the y-axis. The bar graphs are coloured by location on Hfq and are defined at the bottom of the figure. The solid bar represents the percent quenching by 1 µM RNA, while the diagonal bar above the solid bar represents the percent quenching by 4 µM RNA.
The site B containing oligoribonucleotide quenches the F11W, Y25W, Q33W, F39W, F42W and Y83W proteins but prefers the distal face, as strong TFQ is observed using 1 μM site B, whereas proximal face quenching is only seen to occur at four times this concentration (Figure 7C). Further, distal face quenching is 1.7-fold greater than proximal face quenching when 4 μM is used. Regardless, at higher concentrations, site B appears to bind to both faces and to interact with at least one residue of the C-terminal tail (Figure 7C). Using the 64-nucleotide 5′-UTR of the hfq mRNA, we observed that when Site A and Site B are connected physically, there is binding to both faces as residues Q33W and F42W are both quenched (Figure 7D). We also see increased quenching of the lateral and charged cleft residues, which are located in the N-terminal helix, and the C-terminal residues. The charged cleft residue, R17W, has not been seen to be quenched in any of our previous experiments, indicating that longer and more structured RNA sequences may be required for binding to occur at this proposed lateral/rim binding site (59). These data illustrate that Hfq is capable of binding a longer RNA to both sites at the same time, supporting current models that hypothesize Hfq can bind to multiple, different RNA sequences simultaneously with U-rich sRNAs preferring the proximal face and (A-A-N)n containing mRNAs binding to the distal face (Figure 8A). However, we currently cannot rule out the possibility that there are two Hfq populations, one with Site A bound to the proximal face only and the other with Site B bound to the distal face only (Figure 8B). Structural studies are underway to discern the binding mode of the 5′-UTR of hfq mRNA.
Figure 8.

Potential models for Ec Hfq binding to the 5′-UTR of hfq mRNA. (A) hfq mRNA binding such that a single mRNA uses all sets of identified binding sites to interact with both faces of Ec Hfq with the linker between the two binding sites wrapping around the outside of the Hfq hexamer. (B) Two hfq mRNA binding to the proximal and distal faces of a single Hfq hexamer. The 5′-UTR of the hfq mRNA is shown as a ribbon with its 5′ and 3′ ends labelled. The 5′ end of the mRNA contains Site A, which should wrap about the proximal pore and lead to the rim/lateral surface. The 3′ end of the mRNA contains Site B, which has a preference for the distal face and C-terminal region of Hfq. The 5′-UTR of the hfq mRNA is modelled onto a surface rendering of Hfq generated in Pymol using PDB ID: 1HK9 (33).
CONCLUSION AND PERSPECTIVE
We have presented data using a straightforward method that can be used to assess the preferred binding site(s) of particular RNA sequences to Ec, Sa and Lm Hfq and can be readily adapted to understand the RNA binding modes of Hfq from other Gram-negative and Gram-positive bacteria. By placing a reasonable number of structure-guided tryptophan residues in a series of specific locations we have shown that TFQ properly identifies where ‘control’ RNA sequences bind (Figure 3). Using TFQ we have demonstrated that the (A-R-N)n distal face-binding motif of Ec Hfq and likely Hfq proteins from all Gram-negative bacteria is a more restrictive (A-A-N)n binding motif, and the (R-L)n distal face-binding motif of Sa Hfq and presumably all Gram-positive Hfq homologues is a more restrictive (A-L)n binding motif. Surprisingly, the (A-R-N)n motif sequence (G-G-A)5 and the (R-L)n motif sequence (G-U)3 G still bind but to the proximal faces of the Ec Hfq and Sa/Lm Hfq proteins, respectively. Their detailed binding mechanisms will require high-resolution structural studies that are underway. Regardless, our TFQ findings can be used to hone computational searches for potential Hfq-binding RNA sequences and to assign their likely binding sites. We have illustrated also that our TFQ approach can be used to study interactions between Hfq and longer, physiologically relevant RNA sequences. Specifically, Ec Hfq is capable of binding to hfq mRNA site A and site B and that Hfq uses both faces and its C-terminus to bind the 64-nucleotide 5′-UTR of this mRNA simultaneously. One potential limitation of this approach is the inability to synthesize either biochemically or chemically longer RNA sequences in the quantities that are needed for each quenching study. However, the upper limit of such biosynthesis is well beyond the 64 nucleotides of the 5′-UTR of hfq, allowing the evaluation of the binding of full-length sRNAs and the longer, relevant parts of their mRNA targets to Hfq both singly and in combination. The creation of more tryptophan-substituted Hfq proteins should refine further the ability of TFQ to reveal primary and any secondary RNA binding sites. TFQ has also revealed that several residues of the C-terminal region indeed interact with certain RNAs and suggests a functional importance of this region in riboregulation for at least some RNA species. Finally, this method is not limited to the identification of RNA binding loci and should be useful in the analysis of Hfq-protein binding.
ACCESSION NUMBERS
F11W, 4JRK; Y25W, 4JUV; F39W, 4JRI; F42W, 4JLI.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Funding for open access charge: Duke University School of Medicine (to R.G.B.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ms Ivy Paw for her help with protein purification and Kd calculations. The authors would also like to thank Mr Kyle Daniels for providing the T7 RNA Polymerase used to synthesize the 5′-UTR of hfq mRNA. X-ray diffraction data were collected remotely at the Southeast Regional Collaborative Access Team 22-BM and 22-ID beamlines at the Advanced Photon Source, Argonne National Laboratory, which is supported by the US Department of Energy, Office of Science and the Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
REFERENCES
- 1.Sun X, Zhulin I, Wartell RM. Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res. 2002;30:3662–3671. doi: 10.1093/nar/gkf508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valentin-Hansen P, Eriksen M, Udesen C. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 2004;51:1525–1533. doi: 10.1111/j.1365-2958.2003.03935.x. [DOI] [PubMed] [Google Scholar]
- 3.Tsui HC, Leung HC, Winkler ME. Characterization of broadly pleiotropic phenotypes caused by an hfq insertion mutation in Escherichia coli K-12. Mol. Microbiol. 1994;13:35–49. doi: 10.1111/j.1365-2958.1994.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 4.De Lay N, Schu DJ, Gottesman S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013;288:7996–8003. doi: 10.1074/jbc.R112.441386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mura C, Randolph PS, Patterson J, Cozen AE. Archaeal and eukaryotic homologs of Hfq: a structural and evolutionary perspective on Sm function. RNA Biol. 2013;10:636–651. doi: 10.4161/rna.24538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franze de Fernandez MT, Eoyang L, August JT. Factor fraction required for the synthesis of bacteriophage Qbeta-RNA. Nature. 1968;219:588–590. doi: 10.1038/219588a0. [DOI] [PubMed] [Google Scholar]
- 7.Franze de Fernandez MT, Hayward WS, August JT. Bacterial proteins required for replication of phage Q ribonucleic acid. Pruification and properties of host factor I, a ribonucleic acid-binding protein. J. Biol. Chem. 1972;247:824–831. [PubMed] [Google Scholar]
- 8.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 10.Moller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol. Cell. 2002;9:23–30. doi: 10.1016/s1097-2765(01)00436-1. [DOI] [PubMed] [Google Scholar]
- 11.Hori K, Yanazaki Y. Nucleotide sequence specific interaction of host factor I with bacteriophage Q beta RNA. FEBS Lett. 1974;43:20–22. doi: 10.1016/0014-5793(74)81095-1. [DOI] [PubMed] [Google Scholar]
- 12.Senear AW, Steitz JA. Site-specific interaction of Qbeta host factor and ribosomal protein S1 with Qbeta and R17 bacteriophage RNAs. J. Biol. Chem. 1976;251:1902–1912. [PubMed] [Google Scholar]
- 13.de Haseth PL, Uhlenbeck OC. Interaction of Escherichia coli host factor protein with oligoriboadenylates. Biochemistry. 1980;19:6138–6146. doi: 10.1021/bi00567a029. [DOI] [PubMed] [Google Scholar]
- 14.de Haseth PL, Uhlenbeck OC. Interaction of Escherichia coli host factor protein with Q beta ribonucleic acid. Biochemistry. 1980;19:6146–6151. doi: 10.1021/bi00567a030. [DOI] [PubMed] [Google Scholar]
- 15.Zhang A, Altuvia S, Tiwari A, Argaman L, Hengge-Aronis R, Storz G. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J. 1998;17:6061–6068. doi: 10.1093/emboj/17.20.6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beisel CL, Updegrove TB, Janson BJ, Storz G. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J. 2012;31:1961–1974. doi: 10.1038/emboj.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Pannekoek Y, Huis in ‘t Veld R, Hopman CT, Langerak AA, Speijer D, van der Ende A. Molecular characterization and identification of proteins regulated by Hfq in Neisseria meningitidis. FEMS Microbiol. Lett. 2009;294:216–224. doi: 10.1111/j.1574-6968.2009.01568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sittka A, Pfeiffer V, Tedin K, Vogel J. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 2007;63:193–217. doi: 10.1111/j.1365-2958.2006.05489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 2010;13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Brown L, Elliott T. Efficient translation of the RpoS sigma factor in Salmonella typhimurium requires host factor I, an RNA-binding protein encoded by the hfq gene. J. Bacteriol. 1996;178:3763–3770. doi: 10.1128/jb.178.13.3763-3770.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonnleitner E, Hagens S, Rosenau F, Wilhelm S, Habel A, Jager KE, Blasi U. Reduced virulence of a Hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 2003;35:217–228. doi: 10.1016/s0882-4010(03)00149-9. [DOI] [PubMed] [Google Scholar]
- 24.Kulesus RR, Diaz-Perez K, Slechta ES, Eto DS, Mulvey MA. Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 2008;76:3019–3026. doi: 10.1128/IAI.00022-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y, Davis BM, Waldor MK. Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 2004;53:345–354. doi: 10.1111/j.1365-2958.2004.04142.x. [DOI] [PubMed] [Google Scholar]
- 26.Yamada J, Yamasaki S, Hirakawa H, Hayashi-Nishino M, Yamaguchi A, Nishino K. Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. J. Antimicrob. Chemother. 2010;65:853–858. doi: 10.1093/jac/dkq067. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi-Nishino M, Fukushima A, Nishino K. Impact of Hfq on the intrinsic drug resistance of Salmonella enterica serovar typhimurium. Front. Microbiol. 2012;3:205. doi: 10.3389/fmicb.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsui HC, Feng G, Winkler ME. Negative regulation of mutS and mutH repair gene expression by the Hfq and RpoS global regulators of Escherichia coli K-12, J. Bacteriol. 1997;179:7476–7487. doi: 10.1128/jb.179.23.7476-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vecerek B, Moll I, Blasi U. Translational autocontrol of the Escherichia coli hfq RNA chaperone gene. RNA. 2005;11:976–984. doi: 10.1261/rna.2360205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobrero P, Valverde C. Evidences of autoregulation of Hfq expression in Sinorhizobium meliloti strain 2011. Arch. Microbiol. 2011;193:629–639. doi: 10.1007/s00203-011-0701-1. [DOI] [PubMed] [Google Scholar]
- 31.Ziolkowska K, Derreumaux P, Folichon M, Pellegrini O, Regnier P, Boni IV, Hajnsdorf E. Hfq variant with altered RNA binding functions. Nucleic Acids Res. 2006;34:709–720. doi: 10.1093/nar/gkj464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beich-Frandsen M, Vecerek B, Sjoblom B, Blasi U, Djinovic-Carugo K. Structural analysis of full-length Hfq from Escherichia coli. Acta Crystallogr. Sect F. Struct. Biol. Cryst. Commun. 2011;67:536–540. doi: 10.1107/S174430911100786X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauter C, Basquin J, Suck D. Sm-like proteins in Eubacteria: the crystal structure of the Hfq protein from Escherichia coli. Nucleic Acids Res. 2003;31:4091–4098. doi: 10.1093/nar/gkg480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vincent HA, Henderson CA, Ragan TJ, Garza-Garcia A, Cary PD, Gowers DM, Malfois M, Driscoll PC, Sobott F, Callaghan AJ. Characterization of Vibrio cholerae Hfq provides novel insights into the role of the Hfq C-terminal region. J. Mol. Biol. 2012;420:56–69. doi: 10.1016/j.jmb.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher MA, Pearson RF, Moller T, Valentin-Hansen P, Brennan RG. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. EMBO J. 2002;21:3546–3556. doi: 10.1093/emboj/cdf322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Wang L, Zou Y, Zhang J, Gong Q, Wu J, Shi Y. Cooperation of Escherichia coli Hfq hexamers in DsrA binding. Genes Dev. 2011;25:2106–2117. doi: 10.1101/gad.16746011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Wang L, Wu J, Gong Q, Shi Y. Hfq-bridged ternary complex is important for translation activation of rpoS by DsrA. Nucleic Acids Res. 2013;41:5938–5948. doi: 10.1093/nar/gkt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauer E, Weichenrieder O. Structural basis for RNA 3′-end recognition by Hfq. Proc. Natl Acad. Sci. USA. 2011;108:13065–13070. doi: 10.1073/pnas.1103420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Link TM, Valentin-Hansen P, Brennan RG. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl Acad. Sci. USA. 2009;106:19292–19297. doi: 10.1073/pnas.0908744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Someya T, Baba S, Fujimoto M, Kawai G, Kumasaka T, Nakamura K. Crystal structure of Hfq from Bacillus subtilis in complex with SELEX-derived RNA aptamer: insight into RNA-binding properties of bacterial Hfq. Nucleic Acids Res. 2012;40:1856–1867. doi: 10.1093/nar/gkr892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horstmann N, Orans J, Valentin-Hansen P, Shelburne SA, III, Brennan RG. Structural mechanism of Staphylococcus aureus Hfq binding to an RNA A-tract. Nucleic Acids Res. 2012;40:11023–11035. doi: 10.1093/nar/gks809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salim NN, Feig AL. An upstream Hfq binding site in the fhlA mRNA leader region facilitates the OxyS-fhlA interaction. PLoS One. 2010;5:e13028. doi: 10.1371/journal.pone.0013028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salim NN, Faner MA, Philip JA, Feig AL. Requirement of upstream Hfq-binding (ARN)x elements in glmS and the Hfq C-terminal region for GlmS upregulation by sRNAs GlmZ and GlmY. Nucleic Acids Res. 2012;40:8021–8032. doi: 10.1093/nar/gks392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soper TJ, Woodson SA. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA. 2008;14:1907–1917. doi: 10.1261/rna.1110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. Positive regulation by small RNAs and the role of Hfq. Proc. Natl Acad. Sci. USA. 2010;107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Updegrove T, Wilf N, Sun X, Wartell RM. Effect of Hfq on RprA-RpoS mRNA pairing: Hfq-RNA binding and the influence of the 5′ RpoS mRNA leader region. Biochemistry. 2008;47:11184–11195. doi: 10.1021/bi800479p. [DOI] [PubMed] [Google Scholar]
- 47.Baba S, Someya T, Kawai G, Nakamura K, Kumasaka T. Expression, crystallization and preliminary crystallographic analysis of RNA-binding protein Hfq (YmaH) from Bacillus subtilis in complex with an RNA aptamer, Acta Crystallogr. Sect F. Struct. Biol. Cryst. Commun. 2010;66:563–566. doi: 10.1107/S1744309110009942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olejniczak M. Despite similar binding to the Hfq protein regulatory RNAs widely differ in their competition performance. Biochemistry. 2011;50:4427–4440. doi: 10.1021/bi102043f. [DOI] [PubMed] [Google Scholar]
- 49.de Almeida Ribeiro E, Jr, Beich-Frandsen M, Konarev PV, Shang W, Vecerek B, Kontaxis G, Hammerle H, Peterlik H, Svergun DI, Blasi U, et al. Structural flexibility of RNA as molecular basis for Hfq chaperone function. Nucleic Acids Res. 2012;40:8072–8084. doi: 10.1093/nar/gks510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vincent HA, Henderson CA, Stone CM, Cary PD, Gowers DM, Sobott F, Taylor JE, Callaghan AJ. The low-resolution solution structure of Vibrio cholerae Hfq in complex with Qrr1 sRNA. Nucleic Acids Res. 2012;40:8698–8710. doi: 10.1093/nar/gks582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 52.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 56.Lundblad JR, Laurance M, Goodman RH. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol. Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]
- 57.Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J. Mol. Biol. 2013;425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murina V, Lekontseva N, Nikulin A. Hfq binds ribonucleotides in three different RNA-binding sites. Acta Crystallogr. D. Biol. Crystallogr. 2013;69:1504–1513. doi: 10.1107/S090744491301010X. [DOI] [PubMed] [Google Scholar]
- 59.Sauer E, Schmidt S, Weichenrieder O. Small RNA binding to the lateral surface of Hfq hexamers and structural rearrangements upon mRNA target recognition. Proc. Natl Acad. Sci. USA. 2012;109:9396–9401. doi: 10.1073/pnas.1202521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Panja S, Schu DJ, Woodson SA. Conserved arginines on the rim of Hfq catalyze base pair formation and exchange. Nucleic Acids Res. 2013;41:7536–7546. doi: 10.1093/nar/gkt521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vecerek B, Rajkowitsch L, Sonnleitner E, Schroeder R, Blasi U. The C-terminal domain of Escherichia coli Hfq is required for regulation. Nucleic Acids Res. 2008;36:133–143. doi: 10.1093/nar/gkm985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arluison V, Folichon M, Marco S, Derreumaux P, Pellegrini O, Seguin J, Hajnsdorf E, Regnier P. The C-terminal domain of Escherichia coli Hfq increases the stability of the hexamer. Eur. J. Biochem. 2004;271:1258–1265. doi: 10.1111/j.1432-1033.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 63.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 1999;181:6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ishikawa H, Otaka H, Maki K, Morita T, Aiba H. The functional Hfq-binding module of bacterial sRNAs consists of a double or single hairpin preceded by a U-rich sequence and followed by a 3′ poly(U) tail. RNA. 2012;18:1062–1074. doi: 10.1261/rna.031575.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Christiansen JK, Larsen MH, Ingmer H, Sogaard-Andersen L, Kallipolitis BH. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 2004;186:3355–3362. doi: 10.1128/JB.186.11.3355-3362.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.