

Summary
Tumor-propagating cells in acute leukemia maintain a stem/progenitor-like immature phenotype and proliferative capacity. Acute myeloid leukemia (AML) and acute T-lymphoblastic leukemia (T-ALL) originate from different lineages through distinct oncogenic events such as MLL fusions and Notch signaling, respectively. We found that Zfx, a transcription factor that controls hematopoietic stem cell self-renewal, controls the initiation and maintenance of AML caused by MLL-AF9 fusion and of T-ALL caused by Notch1 activation. In both leukemia types, Zfx prevents differentiation and activates gene sets characteristic of immature cells of the respective lineages. In addition, endogenous Zfx contributes to gene induction and transformation by Myc overexpression in myeloid progenitors. Key Zfx target genes include the mitochondrial enzymes Ptpmt1 and Idh2, whose overexpression partially rescues the propagation of Zfx-deficient AML. These results show that distinct leukemia types maintain their undifferentiated phenotype and self-renewal by exploiting a common stem cell-related genetic regulator.
Introduction
Acute leukemia is characterized by the rapid overproduction of malignant immature hematopoietic cells that inhibit normal hematopoiesis in the bone marrow (BM) and invade peripheral organs. T cell acute lymphoblastic leukemia (T-ALL) accounts for 15–20% of acute leukemia cases in adults and children, whereas acute myeloid leukemia (AML) is the most common acute leukemia found in adults. Both types of leukemia are associated with a high risk of relapse after chemotherapy treatment. The development of T-ALL and AML is driven by distinct oncogenic pathways that "hijack" normal molecular mechanisms operating in the respective T cell and myeloid progenitors.
Aberrant activation of the NOTCH1 receptor plays a major role in the pathogenesis of T-ALL, with activating NOTCH1 mutations occurring in >50% of human T-ALL cases (Weng et al., 2004). Notch1 is essential for early development of T cell progenitors in the thymus, but becomes dispensable for T cell development after the CD4+CD8+ double-positive (DP) stage (Pui et al., 1999; Radtke et al., 1999; Wolfer et al., 2001). The activation of Notch receptor releases its intracellular domain (NotchIC), which translocates to the nucleus, forms a complex with transcription factor CSL and activates transcription of target genes. Hes1 is a canonical direct target of Notch/CSL that is required both for normal T cell development and for Notch-induced T-ALL (Wendorff et al., 2010). Overexpression of NotchIC in murine hematopoietic progenitors is sufficient to initiate transplantable T-ALL, which originates from highly proliferative CD4−CD8− double-negative (DN) stage 4 (DN4) and CD4−CD8+ immature single-positive (ISP) thymocytes (Li et al., 2008).
Chromosomal translocations involving the mixed lineage leukemia gene (MLL) with multiple fusion partners are common in human AML (Liedtke and Cleary, 2009). Experimental overexpression of MLL fusion proteins such as MLL-AF9 (MA9) causes transformation of murine myeloid progenitors (Krivtsov et al., 2006; Somervaille and Cleary, 2006). The resulting AML cells can be propagated in cytokine-supplemented cultures and cause serially transplantable AML in recipient mice. These leukemias are hierarchically organized and include cells with immature c-Kit+ phenotype that can propagate the disease. MLL is a histone methyltransferase that is required for normal HSC function (Jude et al., 2007; McMahon et al., 2007). Oncogenic MLL fusion proteins recruit endogenous nuclear protein complexes to facilitate the transcription of target genes such as Hoxa9 and Meis1 (Muntean et al., 2010), which are necessary (Ayton and Cleary, 2003; Wong et al., 2007) and sufficient for the transformation (Kroon et al., 1998). Additional transcription factors that facilitate MLL-induced transformation, such as Myb, have also been identified (Zuber et al., 2011a).
A common feature of many cancers, including acute leukemia, is their dependence on the cellular proto-oncogene c-Myc (Myc). Myc is a transcription factor that induces multiple target genes such as metabolic enzymes and cell cycle regulators to promote the survival and proliferation of transformed cells. Myc and its regulator Brd4 have been shown to be important for AML propagation (Wong et al., 2010; Zuber et al., 2011b). In T-ALL, Myc represents a direct target of Notch signaling that contributes to leukemia growth (Palomero et al., 2006; Weng et al., 2006) and maintains the leukemia-initiating capacity of undifferentiated leukemic cells (King et al., 2013). However, common factors that cooperate with and/or act downstream of Myc in different leukemia types have not been fully elucidated.
ZFX is a transcription factor that is encoded on the X chromosome and contains an acidic transcriptional activation domain and a DNA-binding zinc finger domain. Murine and human ZFX are expressed ubiquitously, yet the function of Zfx appears cell type-specific. Thus, murine Zfx is generally dispensable for embryonic development and for the growth of multiple cell types including embryonic fibroblasts, myeloid progenitors and neural stem/progenitor cells (Galan-Caridad et al., 2007). However, Zfx is necessary for the self-renewal and survival of adult hematopoietic stem cells (HSCs) in vivo and of embryonic stem cells (ESCs) in vitro. Zfx is highly conserved in vertebrates, and similarly controls the self-renewal of human ESCs (Harel et al., 2012). Given its essential and specific role in normal stem cell self-renewal, we hypothesized that Zfx might regulate the aberrant self-renewal of leukemia cells in T-ALL and AML.
Results
Zfx contributes to the development of Notch1-induced T-ALL
First, we tested the role of Zfx in normal T cell development in the thymus. Pan-hematopoietic Zfx deletion using Tie2-Cre (Fig. S1A) delayed the DN to DP transition in the fetal thymus, and reduced proliferation of DN4 and ISP thymocytes; however, it did not preclude normal thymocyte development (Fig. S1B and S1C). Furthermore, Zfx deletion at the DN to DP transition using CD4-Cre did not impair thymocyte development in any way (Fig. S1D and S1E). We conclude that Zfx facilitates the massive proliferation of DN4 and ISP thymocytes, but is largely dispensable for this process and for T cell development in general.
To study the role of Zfx in the development of spontaneous T-ALL in vivo, we used the CD4-Cre deleter combined with a Cre-inducible Eef1a1-NotchIC allele (Buonamici et al., 2009). These mice were crossed with a conditional null Zfx allele (Zfxfl/y), so that Cre would induce NotchIC and delete Zfx in the same cell. Cre-induced NotchIC induction in both control Zfxwt/y and Zfxfl/y mice initiated in DN thymocytes and was complete by the DP stage (Fig. 1A). The Eef1a1-NotchIC+ CD4-Cre+ Zfxwt/y mice had abnormal DP T cells in the blood (Fig. 1B), developed extreme splenomegaly (~750×106 splenocytes, Fig. 1C), and 100% of them succumbed to T-ALL by 2–4 months of age (Fig. 1D). In contrast, the Eef1a1-NotchIC+ CD4-Cre+ Zfxfl/y mice never showed DP T cells in the periphery (Fig. 1B), had spleens of the normal size (~60×106 splenocytes, Fig. 1C), and ~30% of them survived for >7 months. The remaining animals succumbed to an inflammatory disease characterized by wasting and skin inflammation, which was caused by NotchIC activation (Fig. 1D) but was clearly distinct from T-ALL. This phenotype likely reflects the pro-inflammatory effector T cell differentiation induced by activated Notch1 (Alam et al., 2010). We conclude that the loss of Zfx completely abrogates the development of Notch-induced T-ALL from immature thymocytes.
Figure 1. Zfx contributes to the development of Notch-driven T-ALL.
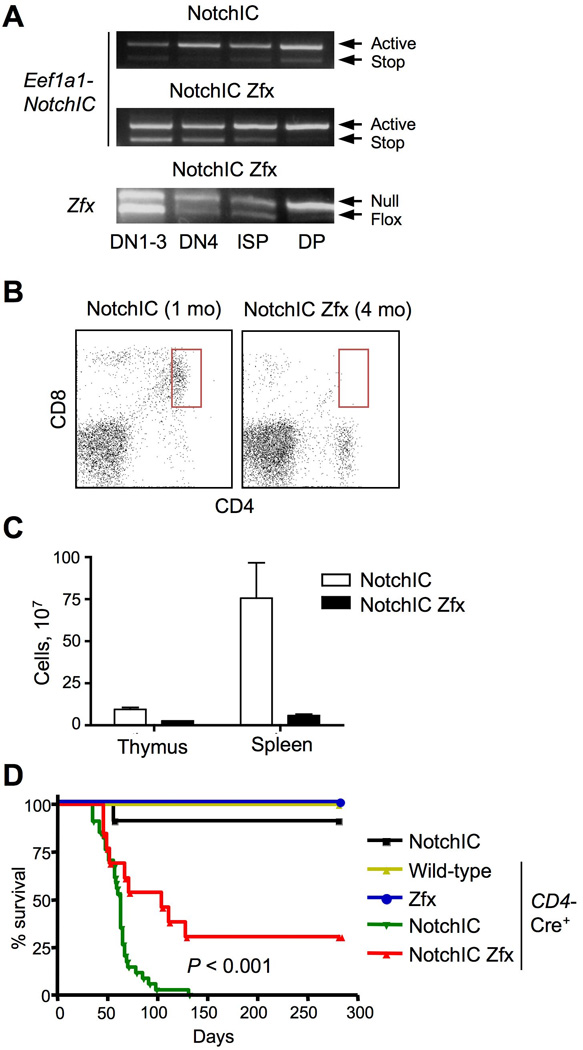
Mice carrying T cell-specific Cre transgene (CD4-Cre) and Cre-inducible activated Notch1 (Eef1a1-NotchIC) with (NotchIC Zfx) or without (NotchIC) the conditional Zfxfl allele were analyzed.
(A) Recombination kinetics of the Eef1a1-NotchIC allele during T cell development. The indicated thymocyte subsets from pre-leukemic 4-wk old animals were sorted and analyzed by genomic PCR.
(B) Representative staining profiles of T cells in the peripheral blood; the abnormal DP population associated with T-ALL is highlighted.
(C) The cellularity of the thymus and spleen from moribund mice (mean ± SEM of 5–6 animals).
(D) The survival of experimental animals and of the indicated control mice (Eef1a1-NotchIC only; CD4-Cre only; CD4-Cre+ Zfxfl without Eef1a1-NotchIC) (n =12–30).
See also Fig. S1.
Zfx facilitates propagation and prevents differentiation of T-ALL
To examine the role of Zfx in the maintenance of pre-established T-ALL, we transduced retroviral NotchIC-IRES-GFP into hematopoietic progenitors carrying the Zfxwt/y or Zfxfl/y allele and the tamoxifen-inducible Cre recombinase (R26-CreER) (Fig. 2A). Each transduced culture was transferred into an individual recipient animal to generate multiple independent leukemia lines of each genotype. Four months after the transfer, all recipients succumbed to GFP+ T-ALL with extensive infiltration into the BM and spleen (Fig. S2A). These primary T-ALL lines were then transplanted into secondary recipient mice, which were treated 2 days later with either tamoxifen (Tmx) or vehicle (oil). Tmx treatment led to efficient recombination of Zfxfl into the null ZfxΔ allele (Fig. S2B), and did not affect leukemia development from the control Zfxwt/y cells (Fig. 2B). All seven transplanted T-ALL lines produced fulminant leukemia in vehicle-treated recipients, but only one produced a delayed leukemia with the recombined ZfxΔ allele in Tmx-treated recipients (Fig. 2B). The remaining leukemias were also delayed and carried the unrecombined Zfxfl allele, revealing strong selection for the rare T-ALL cells that escaped Zfx deletion. The expansion of T-ALL was abrogated by Tmx treatment as late as 6 days post-transfer (Fig. 2C), confirming that the loss of Zfx impairs T-ALL propagation independently of its initial engraftment.
Figure 2. Zfx contributes to the propagation of pre-established T-ALL.
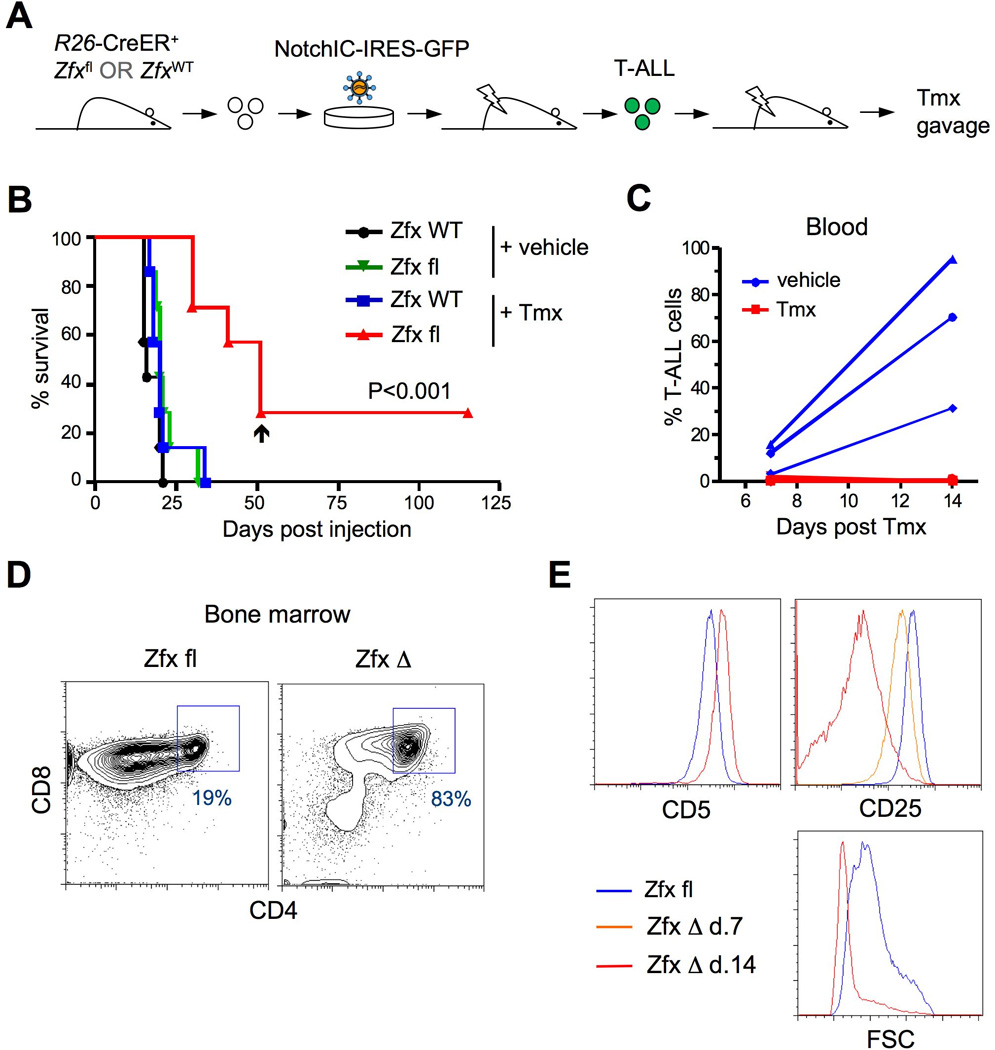
(A) Schematic of experimental approach to test the role of Zfx in pre-established NotchIC dependent T-ALL.
(B) The survival of mice transplanted with T-ALL cells followed by inducible Zfx deletion. Independent primary R26-CreER+ T-ALL lines carrying wild-type (ZfxWT) or conditional (Zfxfl) Zfx allele (7 of each genotype) were transplanted into recipient mice, which were treated 2 days later with either Tmx or vehicle. Arrowhead indicates the only recipient of R26-CreER+ Zfxfl cells that died from Zfx-deficient leukemia.
(C) The propagation of secondary T-ALL after delayed Zfx deletion. Three primary R26-CreER+ Zfxfl/y T-ALL lines were transplanted into recipient mice, which were treated 6 days later with either Tmx or vehicle. Shown is the fraction of GFP+ T-ALL cells in the peripheral blood at the indicated time points after Tmx treatment.
(D–E) The phenotype of T-ALL cells after Zfx deletion. Secondary recipients of T-ALL cells described in panel C were sacrificed at 7 or 14 days after vehicle/Tmx treatment, and their BM were analyzed by flow cytometry. Panel D shows the CD4/CD8 expression profile of gated GFP+ T-ALL 14 days after treatment with vehicle (Zfx fl) or Tmx (Zfx Δ). Panel E shows the staining level of indicated markers or forward scatter (FSC) of gated GFP+ T-ALL after the treatment with vehicle on day 14 or with Tmx on day 7 (for CD25) or 14.
See also Fig. S2.
The analysis of ex vivo T-ALL cells shortly after Zfx deletion revealed normal expression of functional Notch target genes Hes1 and Myc (Fig. S2C), suggesting that Zfx does not directly affect the Notch pathway activity. To analyze the fate of T-ALL after Zfx deletion, the recipients of primary R26-CreER+ Zfxfl/y T-ALL were treated with vehicle or Tmx and analyzed 1–2 weeks later. All vehicle-treated recipients were moribund by 14 days, with a high fraction of T-ALL in the blood (Fig. 2C), BM and spleen. Although Tmx treatment abrogated T-ALL expansion in the peripheral blood (Fig. 2C), a small fraction of GFP+ T-ALL cells could be detected in the BM and spleens at these time points. One week after Tmx, Zfx-deficient T-ALL cells in the BM manifested the same immature phenotype, high proliferation rate and minimal apoptosis as control T-ALL cells (Fig. S2D-E). Two weeks after Zfx deletion, the residual T-ALL cells shifted from the CD4low to DP phenotype (Fig. 2D) and upregulated DP marker CD5 (Fig. 2E). Furthermore, they showed a dramatic reduction of DN marker CD25 and of the forward scatter parameter indicative of cell size (Fig. 2E). The reduction of CD25 could be detected as early as one week after Zfx deletion (Fig. 2E and data not shown). Thus, defective propagation of Zfx-deficient T-ALL cells is associated with their progressive differentiation into the more mature DP thymocyte-like cells.
Zfx facilitates the initial transformation and propagation of AML
To test the role of Zfx in AML, we induced Zfx deletion in vivo and transduced myeloid progenitors with a retrovirus encoding the MA9 oncogene (Fig. 3A). MA9-transduced common myeloid progenitors (CMPs) and granulocyte/macrophage progenitors (GMPs) from control animals displayed efficient serial replating in semi-solid media (Fig. 3B). In contrast, CMPs and GMPs from Zfx-deficient BM formed normal colonies on the first passage but failed at serial replating (Fig. 3B). These data suggest that Zfx is dispensable in normal myeloid progenitors as described (Galan-Caridad et al., 2007), but becomes essential in MA9-transformed progenitors that acquire the capacity for self-renewal.
Figure 3. Zfx contributes to the initiation and propagation of MA9-induced AML.
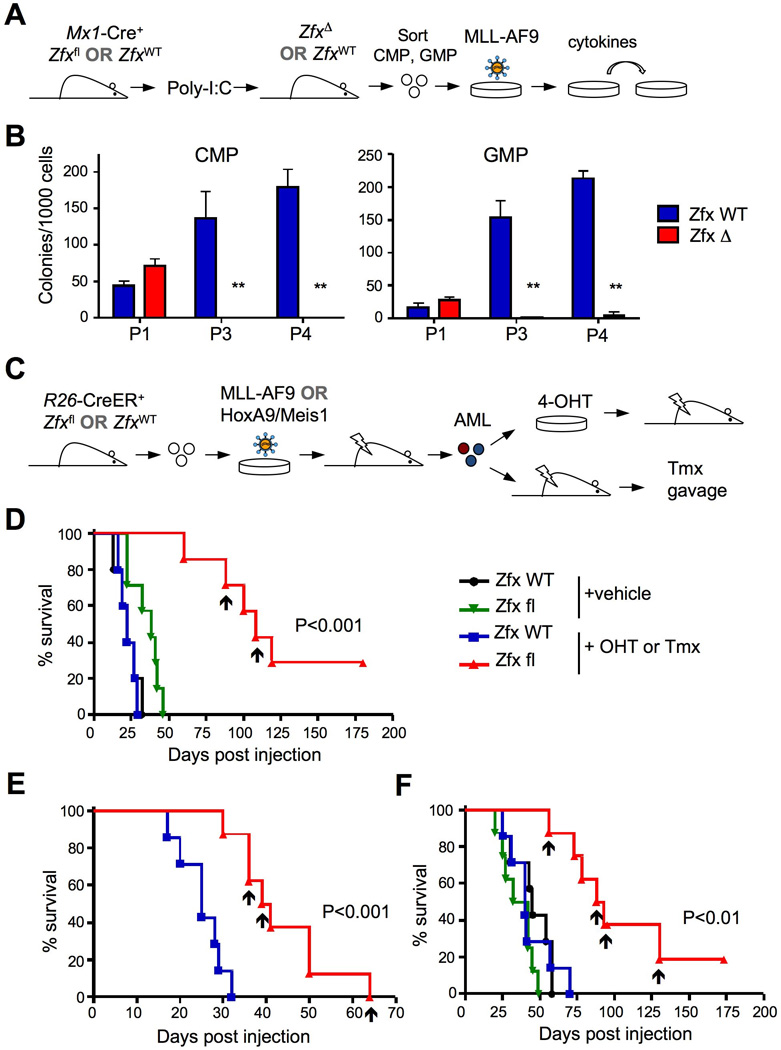
(A) Schematic of experiment to test the transformation of Zfx-deficient myeloid progenitors by the MLL-AF9 (MA9) retrovirus.
(B) Clonal outgrowth of Zfxwt/y and ZfxΔ/y common myeloid progenitors (CMP) and granulocyte-monocyte progenitors (GMP) transduced with MA9. Shown are colony yields at the indicated passages (P) in semi-solid medium (mean S.E.M. of 3–5 independent parallel cultures). **P<0.01.
(C) Schematic of experimental approach to test the role of Zfx in pre-established AML in vivo.
(D) The effect of Zfx deletion on leukemia initiation by primary MA9-induced AML. Independent primary R26-CreER+ AML lines carrying wild-type (ZfxWT) or conditional (Zfxfl) Zfx allele (7–8 of each genotype) were incubated with 4-OHT or vehicle for 3 days and transplanted into secondary recipients. Shown is the Kaplan-Meier survival plot of recipient mice; arrowheads indicate recipients of R26-CreER+ Zfxfl cells that died from Zfx-deficient leukemia.
(E) The effect of Zfx deletion in vivo on the progression of MA9-induced AML. Untreated primary R26-CreER+ AML lines carrying ZfxWT or Zfxfl alleles were transplanted into secondary recipients, which were treated with Tmx 10 days later. The results are shown as in panel (D).
(F) The effect of Zfx deletion on leukemia initiation by Hoxa9/Meis1-induced AML. The experiment was performed and is presented as in panel (D), except that AML was induced by Hoxa9/Meis1 instead of MA9 retrovirus. The results are shown as in panel (D).
See also Fig. S3.
To assess the role of Zfx in the propagation of pre-established AML, we used retroviral MA9 to transduce hematopoietic progenitors from Zfxwt/y or Zfxfl/y R26-CreER+ mice. Upon transfer into primary recipients, these cells caused fatal AML that could be transplanted into secondary recipients or propagated in cytokine-enriched medium (Fig. 3C). AML cells from moribund primary recipients were cultured with 4-hydroxytamoxifen (4-OHT) for 72 hours to induce Zfx deletion, and then transplanted into secondary recipients. Prior to the transplantation, the ZfxΔ/y cells displayed no significant changes in the cell cycle or surface marker expression (Fig. S3A-B). The treatment had no effect on leukemia development from the Zfxwt/y cells, but it significantly delayed AML development from the Zfxfl/y cells (Fig. 3D). Next, we transferred untreated primary AML cells directly into secondary recipients, and administered Tmx in vivo 10 days later. The development of AML was significantly delayed in the recipients of Zfxfl/y cells (Fig. 3E), confirming that Zfx controls the propagation rather than engraftment of AML. We also tested the role of Zfx downstream of Hoxa9 and Meis1, the key transcriptional targets of MA9. The deletion of Zfx did not affect the expression of Hoxa9, Meis1 or Myb in MA9 AML cells (Fig. S3C). Furthermore, Zfx deletion significantly delayed leukemic outgrowth from primary Hoxa9/Meis1-induced AML cells (Fig. 3F), suggesting that Zfx does not act upstream of these factors.
Among the 7–8 independent primary Zfxfl/y leukemia lines analyzed in each experiment, the majority either did not grow after Zfx deletion or gave rise to delayed AML that retained the unrecombined Zfx allele. The 2 MA9 AML lines and 4 Hoxa9/Meis1 AML lines that grew in the absence of Zfx showed delayed growth kinetics and more differentiated phenotype (Fig. 3D-F and data not shown). The analysis of one such line showed unique overexpression of several genes including transcription factor Six1 (Fig. S3D), which has been implicated in MLL-mediated transformation (Wang et al., 2011). Thus, partial resistance to Zfx deletion in rare AML lines is associated with potential compensatory changes in their expression profile. Altogether, our data suggest that Zfx is important for the propagation of AML caused by MA9 or its effectors Meis1/Hoxa9.
Zfx maintains clonogenic growth and prevents differentiation of AML
To test the in vitro clonogenic growth of Zfx-deficient AML, MA9-transformed Zfxwt/y and Zfxfl/y R26-CreER+ AML cells were treated with 4-OHT and propagated in semi-solid medium with cytokines (Fig. 4A). Colonies derived from single ZfxΔ/y cells displayed less compact morphology in the first passage (Fig. 4B), and the frequency of colony-forming cells was decreased ~5-fold in subsequent passages (Fig. 4C). Thus, Zfx supports the optimal frequency of clonogenic cells in cytokine-driven AML cultures.
Figure 4. Zfx controls the clonogenic growth and immature phenotype of murine MA9 AML.
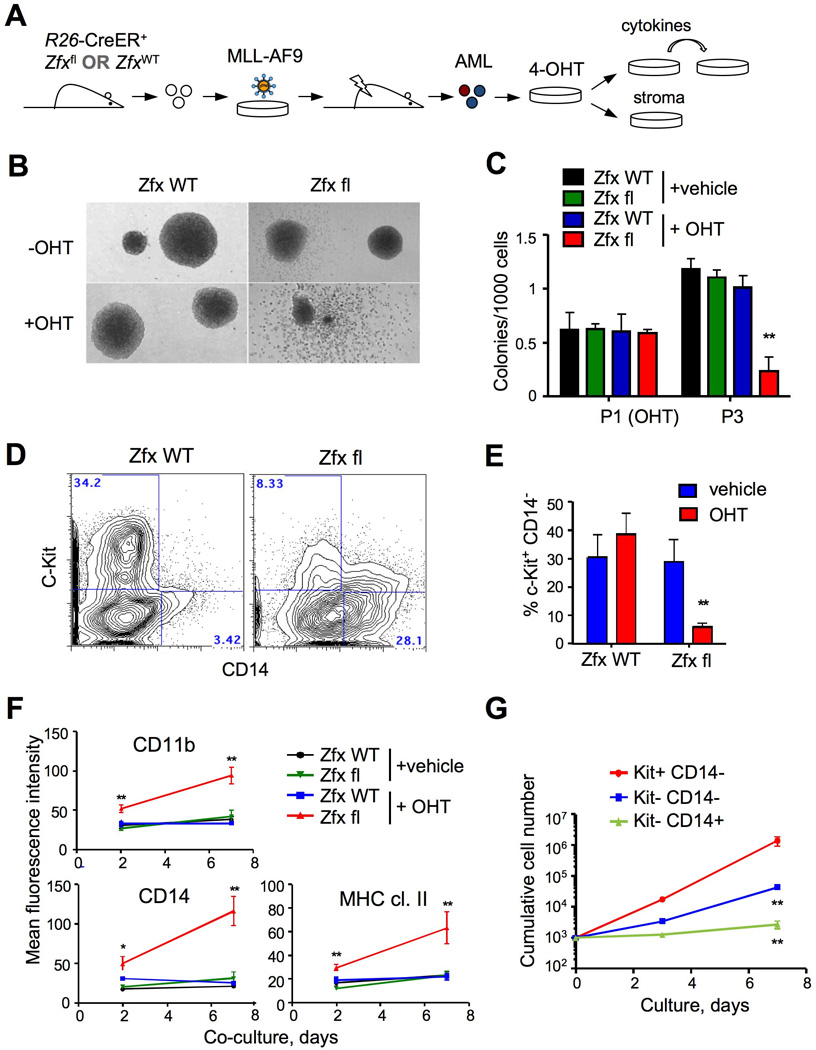
(A) Schematic of experimental approach to test the role of Zfx in the growth and phenotype of pre-established AML.
(B–C) The effect of Zfx loss on MA9 AML cells grown in cytokine-supplemented culture. Primary R26-CreER+ AML lines carrying wild-type (ZfxWT) or conditional (Zfxfl) Zfx allele were incubated with 4-OHT or vehicle for 4 days (passage 1, P1) and passaged in semi-solid medium. Shown are representative microphotographs of colonies at P1 (panel B) and colony yields at P3 (mean ± SEM of 6 independent cultures). **P<0.01.
(D–G) The effect of Zfx loss on MA9 AML cells co-cultured with BM stromal cells. Primary AML lines described above were cultured with 4-OHT or vehicle in cytokine-supplemented liquid culture and plated on stromal cells without cytokines.
(D) Representative staining profiles of OHT-treated cells after 4 days of stromal co-culture
(E) The fraction of cells with progenitor phenotype (c-Kit+ CD14−) after 7 days of stromal co-culture (mean ± SEM of 6 independent cultures). **P<0.01.
(F) Mean fluorescence intensity of myeloid differentiation markers during stromal co-culture (mean ± SEM of 7–8 independent cultures). *P<0.05, **P<0.01.
(G) The growth potential of Zfx-deficient AML cells after stromal co-culture. Cell fractions of the indicated phenotype were sorted from OHT-treated R26-CreER+ Zfxfl AML cells grown in stromal co-culture, and replated into cytokine-supplemented liquid culture (mean ± SEM of 3 independent cultures). **P<0.01.
See also Fig. S4.
To model AML propagation in a native BM environment, we grew 4-OHT-treated Zfxwt/y and Zfxfl/y R26-CreER+ MA9 AML cells on a BM stromal cell layer in the absence of exogenous cytokines (Sykes et al., 2011). After several days of culture, control AML cultures maintained a prominent fraction of c-Kit+ cells that lacked the myeloid marker CD14. Zfx deletion caused the loss of this c-Kit+CD14− subset, whereas differentiated c-Kit−CD14+ cells accumulated (Fig. 4D,E). Furthermore, the expression levels of several myeloid differentiation markers (CD11b, MHC class II, CD80) progressively increased in Zfx-deficient cells (Fig. 4F). The resulting Zfx-deficient c-Kit− CD14+ cells grew poorly in cytokine-supplemented liquid culture, confirming the loss of proliferative capacity (Fig. 4G). These results suggest that Zfx opposes the differentiation of murine AML cells grown in the presence of BM stroma.
Human leukemia samples show a significant increase in the expression of ZFX and its expressed human gametolog ZFY (Fig. S4A). Several Zfx-specific lentiviral shRNA constructs impaired cell growth in the human NotchIC-dependent T-ALL line RPMI-8402, MA9-expressing AML cell line NOMO-1 and in three additional leukemia cell lines (Fig. S4B-E). Like murine MA9 AML cells, a distinct fraction of NOMO-1 cells express c-Kit; ZFX knockdown depleted this population and increased the expression of myeloid marker CD14 (Fig. S4F). Thus, ZFX facilitates the growth of human leukemia cell lines and helps prevent their phenotypic differentiation.
Zfx controls the gene expression programs of undifferentiated cells
To establish the transcriptional program controlled by Zfx in leukemic cells, we performed genome-wide expression profiling on murine T-ALL and AML cells shortly after Zfx deletion. Genes that were positively and negatively regulated by Zfx were defined by comparing ZfxΔ/y samples to the respective control Zfxfl/y samples (and, in the case of AML, to Zfxwt/y samples) (Table S1). In parallel, we performed ChIP-seq analysis of ZFX binding to chromatin in the human ZFX-dependent cell lines RPMI-8402 (T-ALL) and NOMO-1 (AML). ZFX binding regions in both lines showed striking enrichment for the proximal (<1 Kb) promoters and 5' UTRs of genes (Table S2), suggesting that ZFX binds close to the transcription start site (TSS). As outlined in Fig. 5A, the sets of Zfx-regulated genes in T-ALL and AML were compared to ChIP targets in the respective human leukemia, as well as to ChIP targets in murine ESCs (Chen et al., 2008) (Table S3). The majority (>80%) of genes that were decreased in Zfx-deficient T-ALL or AML had Zfx binding regions within 1 Kb of the TSS in either murine ESCs or in the corresponding human leukemia line (Fig. 5B). Of these Zfx binding regions, 44% and 78% were conserved between murine ESCs and RPMI-8402 and NOMO-1 cells, respectively (Fig. 5B). Most of ZFX binding sites within Zfx-regulated genes were tightly clustered within 300 bp of the TSS (Fig. S5A). In contrast, the majority of genes that were increased in Zfx-deficient T-ALL or AML did not contain Zfx binding sites within 1 Kb of the TSS (Fig. 5B). Thus, Zfx in leukemic cells directly activates its target genes by binding within their proximal promoter regions, whereas Zfx-mediated gene repression appears indirect.
Figure 5. Characterization of gene expression program controlled by Zfx in T-ALL and AML.
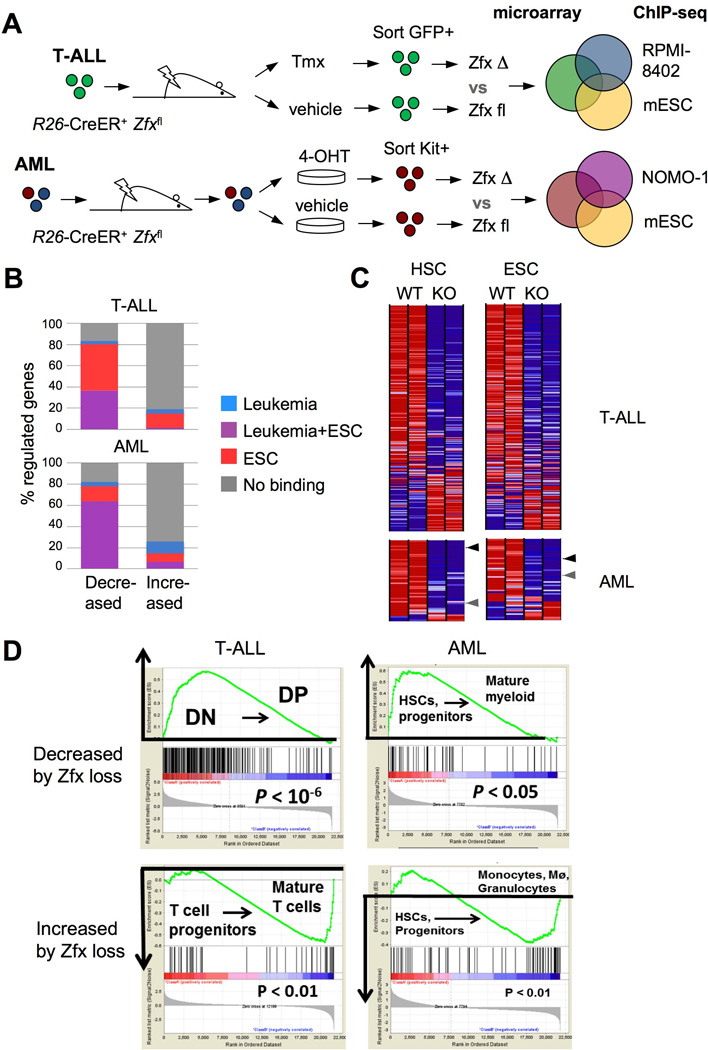
(A) Schematic of the approach to define Zfx-controlled gene expression programs in leukemia, using the overlap between Zfx-regulated gene sets and genomic Zfx binding regions (as determined by microarrays and ChIP-seq, respectively).
(B) The identification of direct Zfx target genes in T-ALL and AML. Shown is the percentage of Zfx-regulated genes in murine T-ALL and AML that had significant Zfx binding regions within 1 Kb of the TSS in murine ESCs and/or the respective human leukemia cell line.
(C) The expression of leukemia-derived Zfx target gene sets in normal Zfx-deficient stem cells. Shown are expression levels (as heat maps) in control vs Zfx-deficient HSCs and ESCs (Galan-Caridad et al., 2007) of direct Zfx target genes defined in T-ALL and AML. Arrowheads highlight Zfx target genes Ptpmt1 (black) and Idh2 (grey).
(D) The expression of leukemia-derived Zfx target genes during normal development of the respective lineages. Genes that were decreased or increased by Zfx loss in T-ALL were analyzed for their expression in normal DN versus DP thymocytes; genes that were decreased or increased by Zfx loss in AML were analyzed for their expression in normal HSCs and myeloid progenitors versus mature myeloid cells. Shown is the output of gene set enrichment analysis (GSEA), with the enrichment score graphs on top.
See also Fig. S5 and Tables S1–S5.
The overlap of expression analysis in murine leukemias and ChIP-Seq in the corresponding human leukemia lines yielded sets of direct target genes that were activated by Zfx in T-ALL and AML (Table S4). Most of these genes were also reduced in Zfx-deficient ESCs and HSCs, suggesting a shared regulation by Zfx in normal stem cells and leukemic cells (Fig. 5C). Importantly, Zfx target gene sets in T-ALL or AML did not overlap with the proliferation-associated gene sets (Fig. S5B and Table S5), indicating that the Zfx gene expression program is not a mere reflection of cell proliferation. Within the normal differentiation hierarchy, the expression of Zfx T-ALL target set was enriched in normal DN thymocytes compared to the more differentiated DP thymocytes (Fig. 5D). This enrichment was not due to the higher proliferation rate of DN cells, because it was observed both in the quiescent (DN3a) and in proliferating (DN3b/DN4) DN subsets (Fig. S5C). Similarly, the expression of the Zfx AML target set was enriched in the HSC/myeloid progenitor BM compartment compared to mature myeloid cells (Fig. 5D). In contrast, the genes that were increased in Zfx-deficient T-ALL and AML were enriched in mature T cells and myeloid cells, respectively (Fig. 5D). Thus, in both T-ALL and AML cells, Zfx directly activates gene sets enriched in immature cells of the respective lineages, and indirectly prevents the induction of differentiation programs.
Zfx facilitates Myc-induced gene expression and transformation
Given the important role of Myc in both T-ALL and AML, we asked whether Zfx cooperated with Myc in leukemia development. Retroviral overexpression of Myc in myeloid progenitors is sufficient to induce their transformation and serial replating capacity (Luo et al., 2005). To test the role of Zfx in Myc-induced transformation, we treated Zfxfl/y R26-CreER+ mice or control Zfxwt/y R26-CreER+ mice with Tmx to induce Zfx deletion in the BM. Myeloid progenitors from these mice were transduced with retroviral vectors encoding GFP alone or GFP and Myc, and GFP+ cells were sorted and plated in semi-solid medium (Fig. 6A). Myc-encoding retrovirus supported serial replating of control progenitors, whereas Zfx-deficient progenitors failed to propagate beyond the first passage (Fig. 6B). Thus, endogenous Zfx facilitates the propagation of Myc-transformed progenitors, suggesting that it acts downstream or in parallel to Myc in leukemogenesis.
Figure 6. Zfx contributes to Myc-induced progenitor transformation.
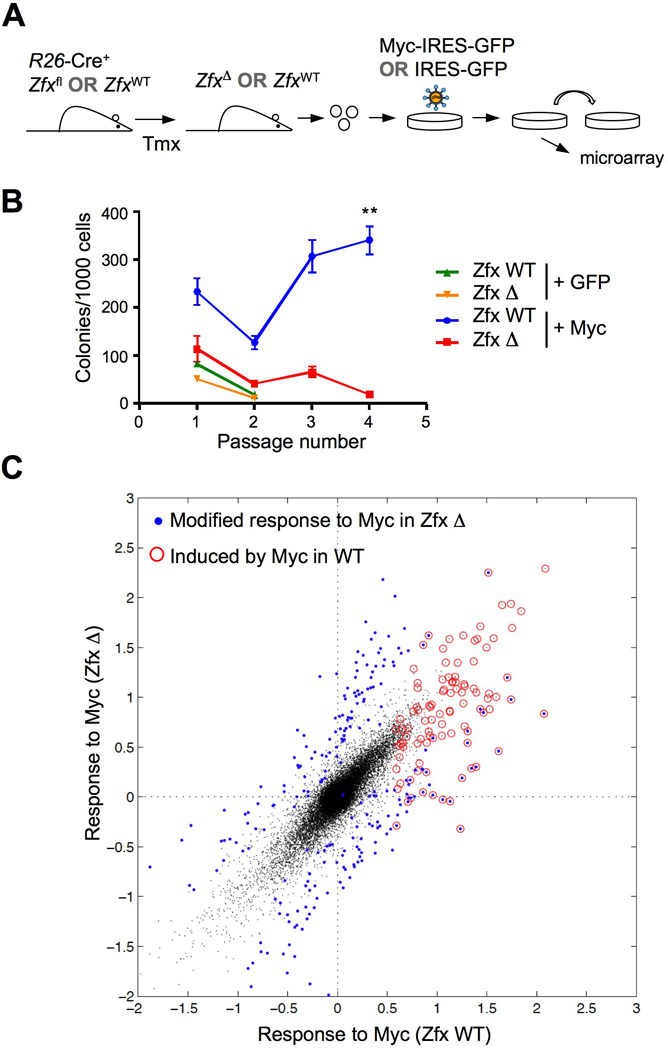
(A) Schematic of experiment to test the role of Zfx in the transformation of myeloid progenitors by Myc.
(B) Effect of Zfx deletion on clonogenic growth of Myc-transformed myeloid progenitors. Myeloid progenitors from mice with Tmx-induced Zfx deletion (ZfxΔ/y) or from control Tmx-treated mice (Zfxwt/y) were transduced with Myc-GFP or GFP only retroviruses, and GFP+ progenitors were sorted and propagated in semi-solid medium. Shown are the colony yields over serial passages (mean ± SEM of 6 independent cultures) ** P<0.01.
(C) Response to Myc overexpression in Zfx-deficient progenitors. Wild-type or Zfxdeficient myeloid progenitors transduced with Myc/GFP or GFP only as above were cultured for 4 days and analyzed by microarray (3 independent cultures for each sample). Average differential expression in Myc-expressing versus GFP-expressing cells of each Zfx genotype (Myc response) was calculated for each probe. Shown is the pairwise comparison of Myc responses in wild-type versus Zfx-deficient cells, with the probes showing differential responses in blue. The probes whose levels were increased by Myc in wild-type cells are circled in red.
See also Fig. S6 and Tables S6–S7.
To test whether Zfx modulates the Myc-dependent gene expression program, we performed microarray analysis of wild-type or Zfx-deficient progenitors at the first passage after transduction with Myc or GFP. This analysis identified a distinct set of genes that were induced by Myc in wild-type progenitors (Fig. 6C, circled dots, and Table S6). Nearly a quarter of these genes (21 out of 94, 22%) were induced to a lower extent in Zfx-deficient progenitors (Fig. 6C, blue circled dots below the diagonal), compared to only 3 genes (3.2%) that were over-induced (blue circled dots above the diagonal). Conversely, the decrease of genes in response to Myc overexpression was affected weakly and non-directionally in Zfx-deficient progenitors (Fig. S6A and Table S6). Furthermore, the decrease of Zfx target genes in Zfx-deficient progenitors was not directionally affected by Myc overexpression (Fig. S6B and Table S7). These results suggest that endogenous Zfx facilitates the optimal induction of Myc-induced expression program that may ultimately lead to progenitor transformation.
Mitochondrial enzymes PTPMT1 and IDH2 are functional Zfx target genes
Direct Zfx target sets in T-ALL and AML included both lineage-specific as well as common target genes, the latter comprising 43% of Zfx targets in AML (Fig. 7A and Table S4). This incomplete overlap suggests the tissue specificity of some Zfx targets, but may also reflect technical limitations of target identification. Common Zfx targets included mitochondrial isocytrate dehydrogenase (IDH2) and mitochondrial protein tyrosine phosphatase 1 (PTPMT1) genes, which were also activated by Zfx in normal HSCs and ESCs (Fig. 5C). The deletion of Zfx decreased the expression of Idh2 and Ptpmt1 in murine T-ALL (5-fold and 12-fold, respectively) and in AML (2-fold and 4-fold, respectively) (Fig. 7B). The expression of Idh2 and Ptpmt1 was increased by Myc overexpression in wild-type progenitors, but did not reach the same levels in the absence of Zfx (Fig. 7C). The promoters of Idh2 and Ptpmt1 are bound by Myc along with Zfx in murine ESC and in human leukemia cells (Fig. S7A,B), raising the possibility of direct regulation by both factors.
Figure 7. Zfx activates the expression of Ptpmt1 and Idh2 in leukemia cells.
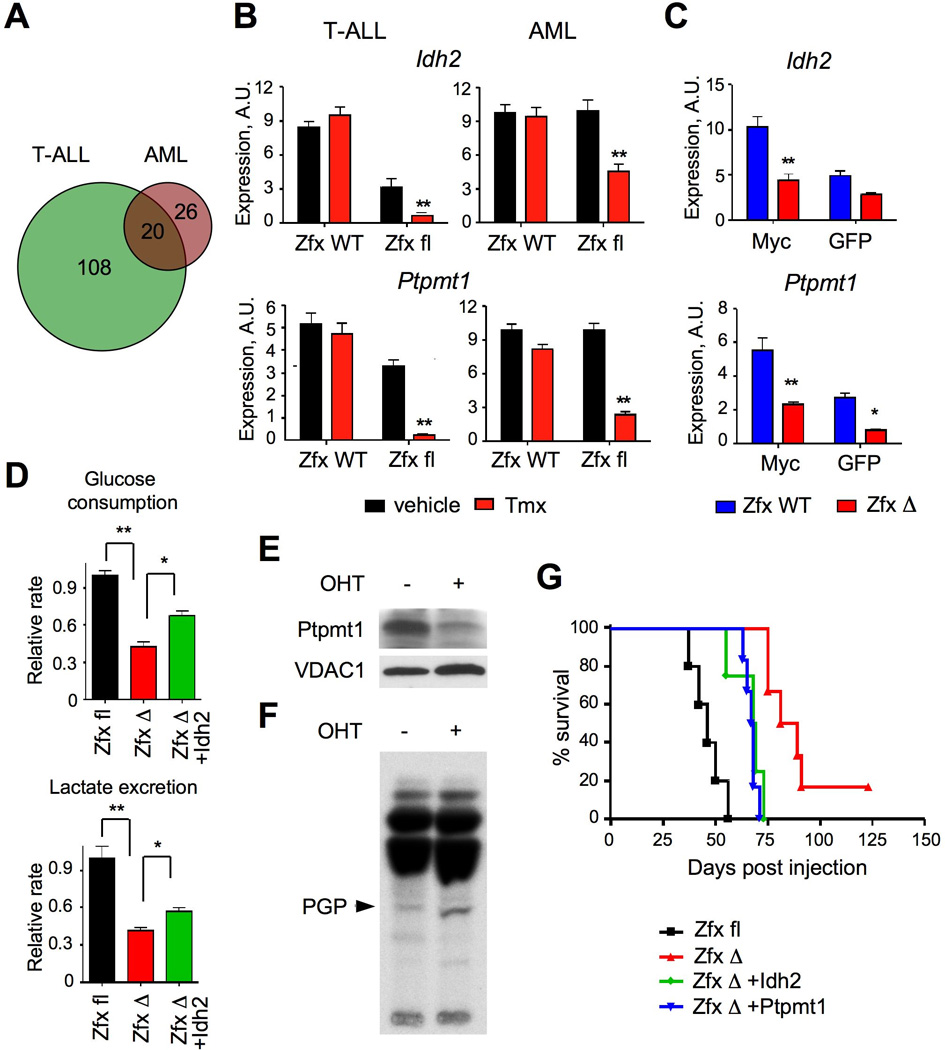
(A) The overlap between conserved direct targets of Zfx activation in T-ALL and AML.
(B) The expression of Idh2 and Ptpmt1 in murine leukemia cells after Zfx deletion. The deletion was induced by Tmx (or vehicle as a control) in the R26-CreER+ conditional (Zfxfl/y) or control (Zfxwt/y) leukemia cells. NotchIC-transformed T-ALL cells were sorted from secondary recipients 5 days after Tmx treatment; MA9-transformed primary AML were incubated with 4-OHT for 3 days. Shown are relative expression levels as determined by qRT-PCR (mean ± SEM of 5 independent lines for T-ALL and 7–8 independent lines for AML). **P<0.01.
(C) The expression of Idh2 and Ptpmt1 in Zfx-deficient progenitors during transformation by Myc. Shown are relative transcript levels determined by qRT-PCR in ZfxΔ/y or Zfxwt/y progenitors 4 days after transduction by Myc-GFP or GFP only (mean ± SEM of 6 independent cultures) * P<0.05, ** P<0.01.
(D) Effect of Zfx deletion on the glycolysis rate of murine AML cells grown in liquid culture with cytokines. MA9-transformed R26-CreER+ Zfxfl/y AML line was transduced with retroviral vectors expressing Idh2 or GFP alone. The cells were incubated with 4-OHT to induce Zfx deletion, propagated for 4 days in liquid culture and analyzed for the steady-state glucose consumption and lactate excretion rates (mean ± SEM of triplicate cultures). *P<0.05, **P<0.01.
(E–F) Effect of Zfx deletion on the expression and function of Ptpmt1. MA9-transformed R26-CreER+ Zfxfl/y AML line was incubated with 4-OHT for 5 days to induce Zfx deletion, and analyzed 4 days later by Western blotting for Ptpmt1 (panel D) and by thin layer chromatography for 32P-labeled mitochondrial lipids (panel E). The position of PGP was confirmed by parallel analysis of pure 14C-labeled PGP.
(G) Effect of Idh2 and Ptpmt1 overexpression on the growth of Zfx-deficient AML. MA9-transformed R26-CreER+ Zfxfl/y AML line was transduced with retroviral vectors expressing Idh2, Ptpmt1 or GFP alone. The cells were incubated with 4-OHT to induce Zfx deletion, and the resulting ZfxΔ/y cells were transferred into secondary recipients. Each recipient received one cell line resulting from an independent transduction with the respective vector. Shown are Kaplan-Meier survival plots of the recipient groups; the difference between Idh2 or Ptpmt1-expressing and control GFP-expressing ZfxΔ/y AML is significant (P<0.01). The results represent a summary of 3 independent experiments involving 1–3 lines of each genotype.
See also Fig. S7.
IDH2 was shown to be important for human leukemia growth (Ward et al., 2010), and we further confirmed that its knockdown impaired the growth of murine MA9 AML cells (Fig. S7C). IDH2 may drive biosynthesis in leukemia cells in part by facilitating conversion of glutamine into citrate (Ward et al., 2010; Wise et al., 2011). Zfx-deficient AML cells showed reduced glucose consumption and lactate production rates, consistent with impaired glycolytic flux in the mitochondria (Fig. 7D). These defects were partially rescued by Idh2 overexpression, suggesting that Zfx-dependent Idh2 expression facilitates glycolysis in AML cells. Ptpmt1 facilitates the production of mitochondrial structural lipid cardiolipin by dephosphorylating its precursor phosphatidylglycerophosphate (PGP) (Zhang et al., 2011). Indeed, Zfx-deficient AML cells expressed lower levels of Ptpmt1 protein (Fig. 7E), and showed the accumulation of PGP (Fig. 7F), consistent with rapid PGP accumulation in Ptpmt1-deficient cells (Zhang et al., 2011).
To test whether the overexpression of Idh2 or Ptpmt1 could rescue the propagation of Zfx-deficient AML, we transduced Zfxfl/y R26-CreER+ AML cells with GFP-marked retroviral vectors expressing these genes. Transduced cells were treated with vehicle or 4-OHT for 72 hours to delete Zfx prior to transplantation into recipients. Zfx-deficient AML cells with retroviral overexpression of either Ptpmt1 or Idh2 caused leukemia more quickly than those transduced with GFP-only retrovirus (Fig. 7G), while remaining Zfx-negative (Fig. S7D). Even though retroviral overexpression results in supraphysiological expression levels, the observed partial rescue of Zfx-deficient AML cells identifies Idh2 and Ptpmt1 as functional targets of Zfx in leukemia.
Discussion
We report that the transcription factor Zfx is important for the emergence and propagation of two acute leukemia types, T-ALL and AML. In normal conditions, Zfx is necessary for the long-term propagation of self-renewing HSCs and ESCs, but is dispensable in a variety of proliferating cells that do not self-renew. Indeed, Zfx plays a limited role in the immature DN4/ISP thymocytes (this study) and in myeloid progenitors (Galan-Caridad et al., 2007), the normal counterparts of leukemia-propagating cells in T-ALL and AML. Thus, Zfx is not universally required for the proliferation of hematopoietic progenitors but becomes important for the self-renewal of their transformed counterparts. The dependence of NotchIC- and MA9-driven leukemias on Zfx is notable given the strength of these oncogenic stimuli; for instance, the propagation of MA9 AML appears independent of Bmi1, an important regulator of normal and leukemic stem cell self-renewal (Smith et al., 2011). Our results reveal a common molecular network operating in distinct types of acute leukemia and highlight the importance of “non-oncogene addiction” of transformed cells to a non-oncogenic, native cellular factor (Luo et al., 2009).
Acute leukemia cells appear “locked” in an undifferentiated state characterized by immature phenotype and clonogenic growth. These characteristics are often restricted to a subset of leukemic cells, which can initiate leukemia upon adoptive transfer and are therefore termed "leukemia-initiating cells" (LICs). The LIC compartment in AML comprises c-Kit+ cells, and the blockade of major oncogenic pathways often leads to their depletion and/or myeloid differentiation. For instance, AML differentiation was observed after the genetic or pharmacological disruption of MLL-containing protein complexes (Bernt et al., 2011; Harris et al., 2012) or of FOXO transcription factors (Sykes et al., 2011). The LICs in Notch-induced T-ALL were recently shown to comprise cells with the immature DN phenotype (King et al., 2013); however, the differentiation of T-ALL into the more mature DP-like cells has not been commonly observed. We found that the loss of Zfx did not directly affect cell survival, proliferation or a proliferation-associated gene expression signature. On the other hand, it induced the differentiation of AML and T-ALL, along with the depletion of cells with the respective immature phenotypes. In each leukemia type, Zfx directly activated sets of genes that were enriched in immature myeloid cells and thymocytes. These gene sets were also regulated by Zfx in the normal ESCs and HSCs, revealing a common Zfx-dependent gene expression program. These observations further support the similarity of gene expression programs underlying the propagation of cancers and normal ESCs (Kim et al., 2010; Somervaille et al., 2009) and HSCs (Eppert et al., 2011).
We found that Zfx contributes to the transformation of myeloid progenitors by Myc, and facilitates the induction of Myc-dependent genes at the early stages of transformation. Zfx binds many actively transcribed promoters in murine ESCs jointly with Myc (Chen et al., 2008), and was proposed as a component of the Myc-driven network that facilitates the growth of ESCs and cancer cells (Kim et al., 2010). However, the specificity of Zfx-Myc cross-talk remains to be elucidated, given that Myc binds broadly to the chromatin and generally amplifies multiple expression programs (Lin et al., 2012; Nie et al., 2012). Furthermore, Zfx has been recently implicated as a potential tumor suppressor in Myc-induced experimental hepatomas (O'Donnell et al., 2012), suggesting that the effect of Zfx on Myc activity may be context-dependent. Importantly, Myc facilitates LIC maintenance in Notch1-induced T-ALL (King et al., 2013), and opposes differentiation of MLL fusion-driven AML (Schreiner et al., 2001; Zuber et al., 2011b). Thus, Myc maintains the propagation and opposes differentiation of both T-ALL and AML, and its function in acute leukemia appears to be dependent on endogenous Zfx.
Among the genes activated by Zfx in the two leukemia types and in normal stem cells, we focused on mitochondrial enzymes Idh2 and Ptpmt1. Because Idh2 is important for the production of citrate from glutamine in cancer cells (Ward et al., 2010; Wise et al., 2011), Zfx-mediated activation of Idh2 in leukemic cells may contribute to their Warburg metabolism and facilitate growth in hypoxic conditions in vivo. Ptpmt1 catalyzes the synthesis of mitochondrial cardiolipin, and is required for HSC/progenitor maintenance (Yu et al., 2013) and for the growth of multiple cancer cell lines (Niemi et al., 2013). It is therefore likely that reduced Ptpmt1 expression, such as observed after Zfx loss, would be particularly detrimental in leukemic cells that undergo continuous self-renewal. Altogether, Zfx-induced expression of Idh2 and Ptpmt1, and likely other targets, facilitates the function and integrity of mitochondria to promote the sustained growth of leukemic cells.
In conclusion, our results establish Zfx as an important endogenous regulator of two disparate, highly aggressive acute leukemia types. While the loss of Zfx also impairs the self-renewal of normal HSCs, the resulting gradual loss of HSCs allows erythro/myelopoiesis to proceed for weeks (Galan-Caridad et al., 2007). This is in contrast to rapid and pronounced effects of induced Zfx deletion on the mortality from acute T-ALL and AML. This kinetic difference, further amplified by combination with other therapy forms, might permit ample window for potential therapeutic targeting of Zfx in acute leukemia.
Experimental Procedures
Animal studies
All mouse studies were performed according to the investigator's protocol approved by the Institutional Animal Care and Use Committee. Mice with the conditional Zfxfl allele and their crosses to pan-hematopoietic Tie2-Cre, interferon-inducible Mx1-Cre and tamoxifen-inducible R26-CreER deleters have been described previously (Galan-Caridad et al., 2007). Spontaneous T-ALL was induced by crossing Eef1a1-NotchIC strain (Buonamici et al., 2009) with CD4-Cre deleter (Jackson Labs), with or without the Zfxfl allele. For in vitro transformation of Zfx-deficient BM progenitors, Zfx deletion was induced by poly-I:C in Zfxfl/y Mx1-Cre+ animals or with Tmx in Zfxfl/y R26-Cre+ animals as described (Galan-Caridad et al., 2007). For in vivo Cre induction in recipients of R26-CreER+ AML or T-ALL, they were administered Tmx at the indicated days after leukemia transfer. For leukemia propagation, sublethally irradiated B6129F1 recipients syngeneic to the R26-CreER+ AML cells were used.
Cell culture and analysis
Murine MA9 and Hoxa9/Meis1 AML cells were maintained in medium with 20% fetal calf serum (FCS) and recombinant murine IL-3, IL-6 (10 ng/ml) and SCF (20 ng/ml) (Peprotech) (R20 AML media). Human NOMO-1 and RPMI-8402 cells were obtained from the Leibniz Institut DSMZ (Braunschweig, Germany) and cultured in medium with 10% FCS.
To induce recombination in R26-CreER+ AML cells, they were treated with 10 nM 4-OHT (Sigma-Aldrich) for 72 hours. BM stromal cells for co-culture experiments were obtained by serially passaging adherent cells isolated from crushed bones of GFP transgenic mice. For assays of clonogenic growth, AML cells were plated in semi-solid Methocult media (M3234, StemCell Technologies) supplemented with cytokines as above, and colony formation was evaluated 5–7 days later. For the analysis of mitochondrial metabolism, murine AML cells grown in cytokine-supplemented liquid culture were incubated with 4-OHT as above or left untreated, and analyzed 4 days later. Glucose consumption and lactate production were analyzed by incubating murine AML cells at high density (2×106/ml) and measuring changes in supernatant glucose and lactate concentration using colorimetric assays (Sigma-Aldrich) over a period of 6 hours. Western blotting of Ptpmt1 and thin layer chromatography of mitochondrial lipids were performed as described (Zhang et al., 2011).
MSCV-based retroviral constructs encoding oncogenes are described in Supplemental Methods. Bicistronic retroviral constructs encoding Ptpmt1 and Idh2 were constructed by cloning open reading frames of mouse Ptpmt1 and Idh2 into MSCV-IRES-GFP. Lentiviral constructs expressing shRNAs to human ZFX have been described (Harel et al., 2012).
Hematopoietic progenitors or primary MA9 AML cells were transduced with these retroviruses and injected i.v. into sublethally irradiated recipient mice or cultured as above. For shRNA expression in human cells, concentrated lentiviral supernatants were applied in triplicate at MOI of 1 followed by selection in puromycin or (for GFP-expressing constructs) by FACS.
Flow cytometry analysis was performed on an LSR II flow cytometer and cell sorting was performed on a FACSAria or Influx flow sorters (BD Immunocytometry Systems). Data were analyzed using FlowJo software (Treestar Inc.). Myeloid progenitor populations were isolated by FACS for the following immunophenotypes: CMP, c-Kit+, Sca1−, Lin−, CD34+, CD16/32lo; GMP, c-Kit+, Sca1−, Lin−, CD34+, CD16/32hi. LICs in AML were sorted based on the following immunophenotypes: Linlo CD16/32hi, CD34+ c-Kit+ and Linlo CD16/32hi CD34− c-kit+.
Expression analysis and ChIP
Genome-wide expression analysis was done using Mouse Gene 1.0 ST microarrays (Affymetrix). Microarray hybridization, scanning and data extraction using the Expression Console software package was according to the manufacturer's instructions. Quantitative RT–PCR analysis was performed using open reading frame-specific primers (sequences available upon request) and the ΔΔCT method as described (Galan-Caridad et al., 2007).
For ChIP, nuclei from 107 formaldehyde-fixed NOMO-1 and RPMI-8402 cells were isolated, lysed and ultrasonically sheared using the TRUChIP High Cell Chromatin Shearing Kit (Covaris). ChIP was performed using anti-ZFX rabbit polyclonal antibody or non-specific rabbit IgG as described (Galan-Caridad et al., 2007). After eluting the sheared immunoprecipitated chromatin, crosslinking were reversed and DNA was recovered by phenol-chloroform extraction. Samples of ‘input’ (sheared but not immunoprecipitated) chromatin were used as controls. Library construction and sequencing were performed by the Yale Center for Genome Analysis (New Haven, CT). The analysis of microarray and ChIP data is described in Supplemental Methods.
Statistical analysis
Statistical significance was estimated using log-rank test for Kaplan-Meier survival plots; two-tailed Student's t-test for the comparison of two groups; or two-way ANOVA for multivariate analysis such as gene expression levels in 4 samples.
Supplementary Material
Highlights.
* Zfx is important for the initiation and propagation of AML and T-ALL
* The loss of Zfx caused differentiation in both leukemia types
* Zfx facilitated leukemic transformation and gene induction by Myc
* Key Zfx target genes included the mitochondrial enzymes Ptpmt1 and Idh2
Acknowledgements
We thank M. Cleary, T. Somervaille, G. Sauvageau, A. Ferrando and T. Ludwig for reagents. Supported by the American Cancer Society (S.P.W., B.R.), Leukemia and Lymphoma Society (B.R.), NIH grants HL084353 (B.R.) and DK18849 (J.E.D.), and NIH training grants HD055165 (S.P.W.) and CA009503 (C.M.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
All microarray and ChIP data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE43022.
References
- Alam MS, Maekawa Y, Kitamura A, Tanigaki K, Yoshimoto T, Kishihara K, Yasutomo K. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2010;107:5943–5948. doi: 10.1073/pnas.0911755107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, Klinakis A, Lukyanov Y, Tseng JC, Sen F, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. 2009;459:1000–1004. doi: 10.1038/nature08020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011 doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- Galan-Caridad JM, Harel S, Arenzana TL, Hou ZE, Doetsch FK, Mirny LA, Reizis B. Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell. 2007;129:345–357. doi: 10.1016/j.cell.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel S, Tu EY, Weisberg S, Esquilin JM, Chambers SM, Liu B, Carson CT, Studer L, Reizis B, Tomishima MJ. Zfx controls the self-renewal of human embryonic stem cells. PLoS One. 2012 doi: 10.1371/journal.pone.0042302. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda- Orgilles B, Perez-Garcia A, Shi J, Vakoc C, et al. The Ubiquitin Ligase FBXW7 Modulates Leukemia-Initiating Cell Activity by Regulating MYC Stability. Cell. 2013;153:1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17:3714–3725. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gounari F, Protopopov A, Khazaie K, von Boehmer H. Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med. 2008;205:2851–2861. doi: 10.1084/jem.20081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–6068. doi: 10.1182/blood-2008-12-197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Li Q, O'Neal J, Kreisel F, Le Beau MM, Tomasson MH. c- Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood. 2005;106:2452–2461. doi: 10.1182/blood-2005-02-0734. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, Basrur V, Elenitoba-Johnson KS, Hess JL. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17:609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi NM, Lanning NJ, Westrate LM, MacKeigan JP. Downregulation of the mitochondrial phosphatase PTPMT1 is sufficient to promote cancer cell death. PLoS One. 2013;8:e53803. doi: 10.1371/journal.pone.0053803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Keng VW, York B, Reineke EL, Seo D, Fan D, Silverstein KA, Schrum CT, Xie WR, Mularoni L, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109:E1377–E1386. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, et al. NOTCH1 directly regulates c- MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC, et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. 1999;11:299–308. doi: 10.1016/s1074-7613(00)80105-3. [DOI] [PubMed] [Google Scholar]
- Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–558. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- Schreiner S, Birke M, Garcia-Cuellar MP, Zilles O, Greil J, Slany RK. MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res. 2001;61:6480–6486. [PubMed] [Google Scholar]
- Smith LL, Yeung J, Zeisig BB, Popov N, Huijbers I, Barnes J, Wilson AJ, Taskesen E, Delwel R, Gil J, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell. 2011;8:649–662. doi: 10.1016/j.stem.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, Chang HY, Shurtleff SA, Downing JR, Cleary ML. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129–140. doi: 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM, et al. AKT/FOXO Signaling Enforces Reversible Differentiation Blockade in Myeloid Leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QF, Wu G, Mi S, He F, Wu J, Dong J, Luo RT, Mattison R, Kaberlein JJ, Prabhakar S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117:6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendorff AA, Koch U, Wunderlich FT, Wirth S, Dubey C, Bruning JC, MacDonald HR, Radtke F. Hes1 is a critical but context-dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity. 2010;33:671–684. doi: 10.1016/j.immuni.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer A, Bakker T, Wilson A, Nicolas M, Ioannidis V, Littman DR, Lee PP, Wilson CB, Held W, MacDonald HR, et al. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat Immunol. 2001;2:235–241. doi: 10.1038/85294. [DOI] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TC, Ficara F, Carico C, Arnold C, Chen CZ, Cleary ML. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010;70:3833–3842. doi: 10.1158/0008-5472.CAN-09-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, Qu CK. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011a;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011b;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.