

Abstract
The efficacious delivery of antigens to antigen-presenting cells (APCs), in particular, to dendritic cells (DCs), and their subsequent activation remains a significant challenge in the development of effective vaccines. This study highlights the potential of dissolving microneedle (MN) arrays laden with nanoencapsulated antigen to increase vaccine immunogenicity by targeting antigen specifically to contiguous DC networks within the skin. Following in situ uptake, skin-resident DCs were able to deliver antigen-encapsulated poly-d,l-lactide-co-glycolide (PGLA) nanoparticles to cutaneous draining lymph nodes where they subsequently induced significant expansion of antigen-specific T cells. Moreover, we show that antigen-encapsulated nanoparticle vaccination via microneedles generated robust antigen-specific cellular immune responses in mice. This approach provided complete protection in vivo against both the development of antigen-expressing B16 melanoma tumors and a murine model of para-influenza, through the activation of antigen-specific cytotoxic CD8+ T cells that resulted in efficient clearance of tumors and virus, respectively. In addition, we show promising findings that nanoencapsulation facilitates antigen retention into skin layers and provides antigen stability in microneedles. Therefore, the use of biodegradable polymeric nanoparticles for selective targeting of antigen to skin DC subsets through dissolvable MNs provides a promising technology for improved vaccination efficacy, compliance, and coverage.
Keywords: vaccines, PLGA nanoparticles, dissolvable microneedles, immunotherapy, skin dendritic cells
Successful vaccination relies on the ability of vaccines to activate the adaptive immune system and induce long lasting memory responses. Dendritic cells (DCs) of the immune system are considered the most effective antigen-presenting cell (APC) because of their unique abilities to initiate robust antigen-specific adaptive immune responses. Following activation upon encounter of pathogens, under an inflammatory setting, DCs undergo rapid maturation characterized by the up-regulation of major histocompatibility complex (MHC) and co-stimulatory molecules and migrate to the proximal draining lymph node. The antigen taken up by DCs is then processed and presented to T cells as peptides bound to the MHC class I or II molecules. In this way, DCs can induce and stimulate CD4+ T cells and cytotoxic CD8+ T cells, which have the ability to both prevent and control viral infection and kill tumor cells.1,2
Appropriate vaccine administration is key to ensure successful vaccination. Typically, most vaccines are administered via subcutaneous or intramuscular routes, and although most vaccines administered through hypodermic injection are effective, there remains issues of pain, needle-related diseases or injuries, the requirement of trained personnel, appropriate needle disposal, and suitable storage or transport of vaccines. Many current vaccines must be maintained within specific temperature ranges to retain their potency, and therefore, the associated expense of maintaining the “cold chain” is estimated to cost vaccine programs $200–300 million annually globally.3−5
Recently, intradermal vaccination strategies have highlighted the major potential of skin immunization through this route.6 Clinical trials have demonstrated that epidermal influenza vaccination induced more efficient influenza-specific CD8+ cytotoxic T cell responses compared to the classical intramuscular route.7 Furthermore, more recent studies have also demonstrated that intradermal administration of influenza vaccine initiated stronger immune responses at much lower doses of antigen compared to doses required for intramuscular vaccination.8,9 Therefore, delivery of antigen to the skin, which is highly populated by a large network of epidermal DCs, known as Langerhans cells, and other dermal DC subsets has the potential for greater immunogenicity.
Drug delivery across the stratum corneum (SC) barrier remains an obstacle for efficient transdermal drug delivery to skin DCs. Microneedle (MN) arrays are minimally invasive devices that can be used to bypass the SC barrier and thus achieve enhanced transdermal biomolecule or drug delivery.10,11 Polymeric, water-soluble MN arrays dissolve within minutes in viable skin layers, thereby releasing their payload into skin tissue and leave no residual sharps waste.12 MN arrays are typically fabricated with enough length to traverse the SC and penetrate into the dermis while remaining sufficiently short and narrow enough to avoid stimulation of dermal nerves. Aqueous blends containing the biocompatible, biodegradable, water-soluble polymer, Gantrez AN-139, have proved to be highly suitable for MN fabrication. Therefore, polymeric dissolvable MNs are robust, penetrate skin effectively at relatively low insertion forces, and greatly enhance transdermal delivery.13
In recent years, particle-based vaccines have been proposed for the development of novel immunization-based therapeutic strategies.14 They have been utilized to improve antigen stability in vivo and to ensure controlled and sustained delivery to the vaccination site. Several groups have demonstrated that nanoparticles (NPs) have inherent immunogenic properties comparable with those of traditional vaccine adjuvants, such as aluminum hydroxide (ALUM) or Freund’s complete adjuvant (CFA), and can activate DCs to induce T cell immune responses against encapsulated antigens.15,16 Polymer-based NPs are sub-micrometer-sized polymeric colloidal particles in which a therapeutic agent of interest can be encapsulated within their polymeric matrix or adsorbed or conjugated onto the particle surface.17 Biocompatible PLGA is one of the most successfully used biodegradable polymers for preparation of NPs.18 To date, numerous antigens (proteins, peptides, lipopeptides, viruses, or plasmid DNA) have been successfully encapsulated into PLGA particles.17,19−25 Formulating antigens in PLGA-NPs offers distinct advantages over soluble formulations.26 PLGA-NPs can protect the antigen from proteolytic degradation and enhance uptake by APCs in a targeted and prolonged manner while restricting the entry of encapsulated antigen to the systemic circulation.27 Furthermore, particulate antigens are more efficiently cross-presented via MHCI molecules to CD8+ T cells than soluble antigens.28 This allows the simultaneous induction of both CD4+ as well as robust CD8+ T cell responses.
In this study, we hypothesized that NP-encapsulated antigen delivery specifically to skin DCs through intradermal polymeric dissolvable MNs would lead to robust, antigen-specific T cell immune responses. We encapsulated a model antigen into PLGA nanoparticles in order to prolong the time that vaccine is retained in the skin, therefore, specifically targeting skin-resident DCs. We demonstrate, for the first time, that antigen-carrying NPs can be successfully delivered to skin layers by MN arrays, and following their uptake in situ, skin-derived DCs were able to deliver NPs to cutaneous draining lymph nodes via the afferent lymphatics where they consequently induced potent activation of antigen-specific IFN-γ secreting effector CD4+ and CD8+ T cells. Moreover, we validated the functional importance of this new nanotherapeutic in murine models of para-influenza infection and melanoma cancer.
Results and Discussion
OVA-NPs Activate BMDCs in Vitro and Consequently Stimulate Antigen-Specific T Cells
Chicken ovalbumin (OVA), used as model antigen, was encapsulated into PLGA-NPs by the water-in-oil-in-water (w/o/w) emulsion method, as previously described.29 We characterized our PLGA-NPs by undertaking the analysis of NP size and zeta-potential and evaluating the amount of OVA entrapped in the PLGA (Table 1). It has been reported that PLGA-NPs are efficiently phagocytosed by DCs in culture, resulting in their intracellular localization.30,31 In order to confirm previous findings, we incubated bone-marrow-derived DCs (BMDCs) with fluorescent TRITC-NPs for 30 min. Flow cytometric analysis of BMDC suspensions demonstrated efficient uptake of TRITC-NPs compared to untreated cells (Figure 1A). To validate that TRITC-NPs were indeed internalized by BMDCs, fluorescent micrographs of fixed BMDCs following 30 min incubation with either TRITC-NPs or b-NPs demonstrated efficient internalization of fluorescent NPs by BMDCs, an essential process required for effective antigen processing and presentation following uptake of antigen-encapsulated NPs (Figure 1B).
Table 1. Particle Size, Zeta-Potential, and Antigen Entrapment Efficiency of PLGA-NPs.
| NPs formulation | NP size (nm) | zeta-potential (mV) | antigen entrapment efficiency |
|---|---|---|---|
| b-NPs | 273.1 ± 33.8 | –18.8 ± 4.1 | N/A |
| OVA-NPs | 357.5 ± 45.7 | –20.3 ± 3.5 | 35.8 ± 2.8% |
Figure 1.

OVA nanoparticles activate BMDCs in vitro and consequently stimulate antigen-specific T cells. (A) Flow cytometry dot plot of CD11c versus MHCII expression on FLT3-derived BMDCs (left panel). The percentage of TRITC+ cells within the CD11c+MHCII+ BMDC population was determined for untreated cells (middle panel) and cells incubated with TRITC-NPs (right panel). (B) Fluorescent microscopy image of DAPI-stained BMDCs that were pulsed with fluorescent TRITC-NPs. Image is representative of three independent experiments. (C) Flow cytometry histograms representing expression levels of DC maturation markers by either untreated and treated b-NPs, OVA-NPs or LPS CD11c+ BMDCs for 12 h. (D) Bar graph representation of data from (C) representing mean fluorescence intensity for each DC activation marker. (E) Bar graph of percentages of divided CFSE-labeled OT-II CD4+ T cells following coculture with unpulsed BMDCs or BMDCs pulsed with b-NPs, various concentrations (μg/mL) of OVA-NPs, OVA peptide (OVA323–339), or with anti-CD3 and anti-CD28 antibodies alone (control). (F) Same as in (E), however, demonstrating percentages of proliferation of CFSE-labeled OT-I CD8+ CFSE labeled T cells following coculture with BMDCs unpulsed and pulsed with b-NPs, various concentrations (μg/mL) of OVA-NPs, OVA peptide (OVA257–264), or with anti-CD3 and anti-CD28 antibodies alone (control). Results are representative of two separate experiments.
To become fully activated APCs capable of priming naïve T cells, DCs up-regulate their surface expression of MHC and co-stimulatory molecules. Since conventionally produced NPs may induce DC activation as a result of lipopolysaccharide (LPS) contamination,32 we formulated PLGA-NPs using endotoxin-free OVA. To determine whether the uptake of NPs mediates the phenotypic maturation of DCs in vitro, BMDCs were incubated with b-NPs or OVA-NPs and the levels of surface expression of DC activation molecules (MHCII, CD40, CD80, CD86) were determined by flow cytometry (Figure 1C). Analysis of mean fluorescence intensity reveals significant up-regulation of MHCII, CD40, and CD86 molecules following OVA-NP incubation in comparison to untreated immature BMDCs (Figure 1D). Similar results were obtained by Elamanchili and co-workers, who demonstrated that, following PLGA-NP pulsing, DCs exhibited a modest increase in the expression of MHC class II and CD86 compared to untreated controls.31
To confirm findings that antigen delivery by polymeric NPs to DCs enhanced antigen presentation in vitro,33 unpulsed BMDCs (control) or pulsed with either b-NPs or various concentrations of OVA-NPs were cocultured with purified transgenic OVA-specific T cells. Following washing, DCs were cocultured with CFSE-labeled OVA-specific OT-I (CD8+) or OT-II (CD4+) cells, and after 60 h, T cells were analyzed by flow cytometry to assess their proliferation in vitro. OVA-NPs were capable of efficiently inducing antigen-specific proliferation of both CD4+ (Figure 1E and Supporting Information Figure 1A) and CD8+ (Figure 1F and Supporting Information Figure 1B) OVA-specific T cells. In line with previous studies, these data collectively demonstrate that our PLGA-NP-encapsulated antigen is actively taken up and processed by DCs, and that antigen-derived peptides are efficiently presented onto both MHCI and MHCII molecules to activate antigen-specific T cells in this simple in vitro model. Moreover, antigen-encapsulated NPs were capable of stimulating T cells in a dose response manner, as a significant increase in OT-II and OT-I T cell proliferation was detectable in response to increased concentration of OVA-NPs.
Microneedle Arrays Specifically Target DC-Rich Layers of the Skin and Facilitate Nanoparticle Uptake by Skin DCs
MNs were fabricated and assembled into a multineedle array patch containing 19 × 19 MNs (600 μm needle length) and were formulated to contain 10 μg of NP-encapsulated OVA. In order to confirm that our polymeric MNs could successfully be used for intradermal NP antigen delivery, we availed of optical coherence tomography (OCT) to allow noninvasive visualization of MN arrays while inserted into murine skin in vivo.34 The images confirm that the MNs were capable of successfully traversing the SC barrier, and the needles penetrate into the epidermal and dermal skin layers. Furthermore, we evaluated in situ dissolution kinetics of our biodegradable MNs using OCT and found that the greatest rate of reduction in the height of the MNs penetrating into the skin occurred in the first 5 min, and by 15 min, MNs had both lost their needle profile and efficiently dissolved within the skin layers (Figure 2A).
Figure 2.

Microneedle arrays specifically target DC-rich layers of the skin and induce nanoparticle uptake by skin DCs. (A) Image of the in vivo dissolution profile of MNs laden with OVA-NPs in mice skin over a 15 min period, as assessed by optical coherence tomography. Base plate of MN array patch is indicated, and dashed line represents the level of the epidermis. Image is representative of three separate experiments. (B) Fluorescent microscopy images of mice ear skin cryosections, fixed 4 h following insertion of dissolving MNs laden with b-NPs or TRITC-NPs in vivo. Puncture holes in the skin formed by MNs are indicated (arrows). Image is representative of two separate experiments. (C) Flow cytometry dot plot of DC-enriched cell suspensions from auricular draining lymph nodes isolated 72 h post-immunization of mice with MNs laden with b-NPs or TRITC-NPs. Values represent percentage of rhodamine-positive cells among the total cell population. (D) Flow cytometry contour plot of CD11c versus MHCII expression on DC-enriched cell suspensions from auricular draining lymph nodes of mice immunized with MN-loaded TRITC-NPs (gray contours). Rhodamine+ DCs are represented as an overlay (red contours) to signify that the majority of DCs that have taken up TRITC-NPs were indeed CD11c+MHCIIhigh skin-derived DCs. Data in (D) and (E) are representative of five independent experiments.
We further confirmed in situ distribution of fluorescent TRITC-encapsulated NPs, delivered in vivo following MN application. Four hours following insertion of MNs into murine skin, MN punctures of approximately 70 μm in depth and 20 μm in width remained evident. Fluorescent NPs delivered with MNs were largely deposited within the dermis at a depth between 80 and 130 μm, as punctate fluorescence was observed in close proximity of MN punctures. Importantly, MN puncture-associated fluorescence was not visible in the skin treated with MNs loaded with b-NP, suggesting that the fluorescence observed was not due to autofluorescence (Figure 2B). Overall, these results demonstrate that the geometry and fabrication process of our resulting MNs permitted intradermal targeting without mechanical failure, and their rapid dissolution in the skin layers resulted in the efficient release of encapsulated NPs in close proximity to DC-rich networks that reside within the different murine skin layers.
Local responses to coated metal microneedle-based influenza immunization in the skin have recently been explored, demonstrating correlation of immunization with a local increase of inflammatory cytokines important for recruitment of innate immune cells, such as neutrophils, monocytes, and dendritic cells, at the site of immunization.35 We investigated the events that occur in the skin following polymeric MN application in order to gain insight into the early immunological cellular responses. In this study, empty MNs or MNs laden with b-NPs were applied to the mouse ears, and 24 h later, cell suspensions from ear skin were examined for infiltration of innate inflammatory immune cells by flow cytometry. We observed significant recruitment of circulating monocytes and neutrophils to the ear skin (Supporting Information Figure 2) in response to dissolving MN-mediated immunization. Therefore, intradermal MN application with or without NPs is capable of inducing a localized inflammatory response, leading to the recruitment of innate inflammatory cells to the site of insult, which may be necessary to initiate or enhance efficient immune responses to vaccines delivered via MNs.
Skin DCs play an essential role in the adaptive immune response by integrating both the information of the antigenic threat and the inflammatory state of the tissue where they originally resided and communicating this to T cells in the draining lymph node through chemotactic migration via afferent lymphatics. Therefore, it is now widely accepted that DCs are capable of tailoring the adaptive immune response to exogenous threats by acting as immune sentinels within peripheral tissues of the body.27
To determine whether DCs were capable of uptaking TRITC-NPs following MN vaccination and delivering their cargo to skin-draining lymph nodes, we analyzed DCs from the ear proximal draining lymph nodes 72 h post-MN application. Indeed, we identified over 20% TRITC-positive cells within DC-enriched cell suspensions from auricular draining lymph nodes of mice vaccinated with TRITC-NPs compared to animals receiving MNs loaded with b-NPs (Figure 2C). In Figure 2D, we further characterized these TRITC+ DCs, based on the expression of CD11c and MHCII, and found that they corresponded to emigrating skin DCs, as they expressed the characteristic CD11c+MHCIIhigh profile.36 We could not detect any uptake of TRITC-NPs by lymph-node-resident blood-derived DC subsets, even at 24 h post-MN delivery (Supporting Information Figure 3), implying very low uptake by blood-derived DCs due to systemic delivery of NPs, therefore suggesting that following MN vaccination the majority of the NP payload remains localized to the vaccination site.
Microneedle-Mediated OVA-NP Intradermal Delivery Induces Robust Antigen-Specific Immune Responses by Skin-Resident DC Subsets ex Vivo
Although dissolving polymer microneedle patches have proven successful for influenza vaccination, the contribution of skin DCs in mediating these immune responses following MN vaccination has not been examined in detail.37 Our data suggest that skin-resident DCs, when targeted through MNs, can capture encapsulated antigen and rapidly migrate to the regional draining lymph nodes. To determine whether DCs that had taken up antigen from the MN vaccination site were able to efficiently present their antigenic cargo to antigen-specific T lymphocytes, we isolated total CD11c+ DCs from proximal auricular lymph nodes 72 h post-MN application. Purified ex vivo DCs were cocultured with CFSE-labeled OVA-specific OT-I CD8+ T cells to monitor T cell activation and proliferation. Following 60 h of culture, efficient OT-I proliferation (Figure 3A) and T-cell-derived IFN-γ production (Figure 3B) were observed in response to MN-mediated NP antigen delivery, suggesting that emigrating DCs from the ear following MN application can efficiently prime CD8+ T cells and induce IFN-γ producing effector T cells, a hallmark signature for efficient cytotoxic T cell activation.
Figure 3.

Microneedle OVA nanoparticle intradermal delivery induces robust antigen-specific immune responses by skin-resident DC subsets ex vivo. (A) Bars indicate percentages of divided CFSE-labeled OT-I T cells cocultured with b-NP- or OVA-NP-stimulated DCs or with DCs isolated from untreated mice (control). (B) Corresponding levels of secreted IFN-γ are shown, as analyzed by ELISA. Data are representative of three independent experiments. (C) Bars represent proliferation of CD8+ OT-I T cells cocultured with FACS purified defined subsets of DCs from cLNs, spleen, or inguinal LN tissues 24 or 72 h post-MN immunization. (D) Bars show corresponding levels of IFN-γ secreted by CD8+ OT-I T cells. (E) Same as in (D), but CD4+ OT-II T cell proliferation is shown. (F) Corresponding levels of IFN-γ, released from CD4+ OT-II as analyzed by ELISA. (C–F) Data from two independent experiments.
To determine whether efficient T cell priming was indeed due to skin-derived DCs, we purified by FACS (>95% purity) skin-derived CD11c+MHCIIhigh, and non-skin-derived CD11c+MHCIIlowCD8+ and CD11c+MHCIIlowCD8− DCs (lymph-node resident blood-derived DC subsets) from skin draining LNs of mice immunized with OVA-NPs. In addition, we purified CD11c+ DC cells from the spleen and inguinal LNs of immunized mice to serve as controls for non-skin-draining peripheral tissues or draining lymph nodes that would not drain the vaccination site, respectively, to determine the contribution of non-skin-derived DCs to T cell priming and activation. Although we could not detect fluorescent NP uptake by blood-derived DCs, antigen-encapsulated NPs could potentially enter the blood circulation and/or the lymphatics at very low levels leading to their uptake by blood-derived or lymph-node-resident DCs following NP draining to the lymphatics. Twenty four or 72 h post-MN immunization, FACS-purified DC subsets were cocultured with either OT-I CD8+ or OT-II CD4+ OVA-specific T cells, in the same manner as described above. We found that CD11c+MHCIIhigh skin-derived DCs constituted the only subset capable of presenting exogenous antigen delivered by NPs, as determined by induction of OT-I cell proliferation (Figure 3C). Moreover, we observed that only CD8+ T cells that were cocultured with skin-derived DCs produced elevated levels of IFN-γ (Figure 3D) in the culture medium. No other DC subsets (lymph-node-resident or DCs from non-skin-draining tissues) induced CD8+ proliferation, suggesting that skin-derived DCs are exclusively responsible for efficient CD8+ T cell priming and activation following MN-mediated vaccination. This also suggests that transdermal delivery of NPs via MNs remains generally localized to the skin unless they are actively taken up by DCs in the skin and transported to the proximal draining lymph nodes.
In addition to activating antigen-specific CD8+ T cells, CD11c+MHCIIhigh DCs were capable of priming and activating OVA-specific CD4+ T helper cells (Figure 3E). The rate of proliferation corresponded to the levels of secreted IFN-γ (Figure 3F) detected in culture supernatants. We observed that splenic or inguinal purified DCs did not evoke antigen-specific T cell responses, confirming that spontaneous systemic draining of particulate vaccines is less likely to occur as it has been shown that only nanoparticles <100 nm can leave the interstitial matrix via the interstitial flow and be transported through the lymphatic vessels into the lymph nodes or systemic circulation.38,39
Nanoencapsulation of Antigen Provides Increased Antigen Stability and Induces Prolonged CD8+ T Cell Responses
Numerous studies have demonstrated that controlled delivery systems consisting of nanoparticles can potentially deliver antigens to the desired location at predetermined rates and durations to generate an optimal immune response. The carrier could also potentially protect the vaccine from degradation until it is released.18,40−43 In order to assess the ability of PLGA-NPs to mediate prolonged antigen retention in the skin, we loaded MNs with b-NPs, OVA-NPs, or soluble OVA. Following 3, 4, 7, 14, or 21 days after MN application, we purified total CD11c+ DCs from auricular draining lymph nodes, cocultured them with OVA-specific OT-I T cells, and monitored CD8+ T cell proliferation, as described previously. Our findings revealed that skin DCs isolated 7 days post-antigen delivery were able to evoke significantly increased proliferation of OVA-specific CD8+ T cells following vaccination with OVA-NPs compared to soluble OVA. However, this advantage was lost by 14 days post-MN application (Figure 4A). This result suggests the possibility that antigen nanoencapsulation provides prolonged retention in the skin layers compared to delivery of soluble antigen, or antigen presentation by skin-derived DCs is extended when delivered via PLGA-NPs. Similarly, a previous study also found that presentation of PLGA-encapsulated proteins by monocyte-derived human DCs was markedly prolonged and antigens were presented 50-fold more efficiently on MHC class I molecules compared to soluble proteins.44
Figure 4.

Nanoencapsulation of antigen induces prolonged CD8 T cell responses in vivo and improves antigen stability. (A) Graph representing the percentage of proliferating CFSE-labeled CD8+ OT-I T cells following coculture with DCs purified from auricular LNs 3, 4, 7, 14, or 21 days following MN immunization with b-NPs, OVA-NPs, or soluble OVA. (B) Graph representing the percentage of proliferating CD8+ OT-I T cells following coculture with DCs purified from auricular LNs following immunization with MNs laden with b-NPs, OVA-NPs, or soluble OVA that were stored at ambient conditions for 1 day, or 4, 6, and 10 weeks prior to immunization (left panel). Representative histograms of CFSE dilutions due to cell proliferation after application of MNs prepared 10 weeks prior immunization are also shown (right panel). All data shown are representative from three independent experiments.
In order to examine antigen stability in MNs, we fabricated MNs laden with OVA-NP or soluble OVA. MNs stored at ambient conditions for 4, 6, or 10 weeks were then utilized to immunize mice. Levels of OVA-specific T cell proliferation mediated by skin DCs 3 days post-vaccination, between different MN patches (Figure 4B), were then determined. When used soon after preparation, MNs containing soluble OVA or OVA-NPs exhibited comparable antigen-specific CD8+ response. Thereafter, CD8+ T cell proliferative responses declined when mice were immunized with soluble OVA that was incorporated into MNs >4 weeks prior to vaccination. In contrast, the level of OVA-specific CD8+ T cell proliferation was maintained when nanoencapsulated antigen was formulated into MNs, even at 10 weeks prior to vaccination. This suggested that nanoencapsulation provided increased antigen stability in MNs compared to soluble antigen. Therefore, nanoencapsulated vaccines may provide a solution to the “cold chain” obstacle that many successful vaccine programs face in compliance and penetrance, especially in developing countries. Collectively, these findings highlight the advantage of vaccine encapsulation in NPs as a novel approach for immunization strategies, in particular, with vaccines that have previously demonstrated weak immune responses.
OVA-NP Immunization Induces Robust Antigen-Specific T Cell Responses in Vivo
As antigen-specific T cells proliferated in vitro following immunization with OVA-NPs, we further investigated the efficacy of dissolvable MN immunization with OVA-NPs on the priming and activation of T cells in an in vivo setting. CD45.1 congenic recipient mice were injected with 1 × 106 donor CD45.2+ CFSE-labeled OVA-specific CD8+ or CD4+ T cells and immunized with b-NPs or OVA-NP MNs. This adoptive transfer model facilitates detection of donor T cells in vivo by flow cytometry (CD45.2) among the recipients’ normal compliment of T cells (CD45.1). Analysis of proximal lymph nodes following b-NP immunization demonstrated no significant CD8+ or CD4+ T cell proliferation in vivo. However, following immunization with OVA-NPs, efficient proliferation and activation of both CD8+ (Figure 5A, top panels) and CD4+ (Figure 5A, bottom panels) T cells in vivo were observed. Therefore, nanoencapsulated OVA delivered through MNs is readily presented on both MHC class I and II molecules and induces efficient CD4+ and CD8+ T cell responses in vivo. Although LPS concentrations in OVA-NPs delivered through MNs were measured to be <1 EU/mL by the LAL assay, we examined the prospect that T cell proliferation in vivo was enhanced by minor LPS contamination in OVA-NPs. Therefore, we used LPS-free OVA to formulate OVA-NPs and compared the levels of T cell expansion in vivo between groups immunized with possible LPS-contaminated OVA-NPs and LPS-free OVA-NPs (Supporting Information Figure 4). Although slightly lower proliferation of both CD4+ and CD8+ antigen-specific T cells was observed in a response to LPS-free OVA-NP immunization, we excluded the possibility that antigen-specific T cell activation was largely due to low level LPS contamination during the MN-NP formulation process.
Figure 5.

OVA nanoparticles induce robust antigen-specific T cell responses in vivo. (A) Proliferation of adoptively transferred CD8+ OT-I (top panels) and CD4+ OT-II (bottom panels) cells isolated from the auricular LNs was examined 3 days post-MN immunization with b-NPs or OVA-NPs by flow cytometry. Representative dot plots of CFSE fluorescence versus donor CD45.2+CD8+ or CD45.2+CD4+ represent CFSE dilution following proliferation of T cells. (B) Auricular LN cell suspensions were prepared 72 h following immunization with b-NPs or OVA-NPs and restimulated in vitro with PMA/ionomycin for intracellular IFN-γ staining. FACS dot plots represent percentages of CD8+ T cells producing IFN-γ of gated CD45.2+ donor CD8+ OT-I cells from auricular LNs. Data are from one of three experiments giving similar results. (C) On day 3 following immunization with b-NPs and OVA-NPs, auricular LN cell suspensions were prepared and restimulated in vitro with PMA/ionomycin for intracellular cytokine staining. FACS dot plots represents percentages of IFN-γ or IL-4 producing cells of gated CD45.2+ donor CD4+ OT-II cells from auricular LNs. Representative data from one of five experiments giving similar results are shown.
MN immunization through the skin induced extensive proliferation of donor CD4+ and CD8+ T cells; thus, we next determined whether CD8+ and CD4+ T cells also acquired effector functions. Auricular lymph node cells were harvested 3 days post-MN immunization and restimulated in vitro with PMA/ionomycin to determine their cytokine secretion profile. Indeed, antigen-specific T cells obtained effector functions since roughly half of the injected CD45.2+CD8+ and a third of CD45.2+CD4+ T cells from mice immunized with OVA-NPs produced IFN-γ (Figure 5B,C). In contrast, T cells from mice immunized with b-NPs failed to produce IFN-γ, comparable to unstimulated cultures (data not shown). We did not observe any significant production of IL-4 (Th2 cell-specific cytokine) by OVA-specific CD4+ T cells, indicating that CD4+ T cells displayed mainly Th1 polarization following MN transdermal vaccination, which is necessary for effective cytotoxic T cell immune responses and a desirable attribute for effective vaccine development against viral infections or for inducing efficient antitumor immunity.
Microneedle Immunization Induces Efficient Antitumor Responses
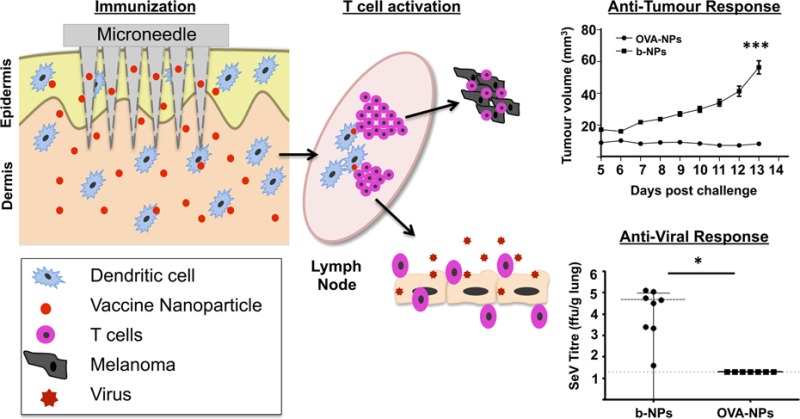
A number of experimental and clinical studies have demonstrated that immunization through the skin induces in vivo activation of CD4+ and CD8+ T cells, antibody production, and systemic and mucosal immunity.37,45,46 For instance, efficient CD8+ T cell responses were induced in melanoma patients through application of peptides on barrier-disrupted skin.47 Thus, we next determined whether OVA-NP immunization through the skin could prevent the growth of B16 melanoma tumor cells expressing OVA (B16.OVA melanoma cells) as a surrogate tumor antigen. We immunized mice with b-NPs or OVA-NPs MNs, and after 3 weeks, animals were injected with 105 B16.OVA tumor cells s.c. into their flanks. None of the mice treated with OVA-NPs developed significant tumor growth over the duration of the experiment (Figure 6A,B), while animals immunized with b-NP displayed significant tumor growth. At the experimental end point, we detected increased levels of IFN-γ in sera of mice immunized with OVA-NPs, suggesting that potent antitumor responses in animals immunized with OVA-NPs was likely producing a dominant Th1 IFN-γ-mediated immune response (Figure 6C).
Figure 6.

Microneedle immunization with OVA nanoparticles induces efficient antitumor responses in vivo. (A) Mice were immunized with MNs loaded with b-NPs or OVA-NPs and inoculated with B16.OVA melanoma cells 3 weeks later. Graph represents tumor volume measurements over 13 days. (B) Images represent differences in tumors sizes among mice treated with b-NPs or OVA-NPs at day 13 post-tumor challenge. (C) Serum levels of IFN-γ detected in tumor-bearing b-NPs or OVA-NPs immunized mice at day 13 measured by ELISA. (D) Representative dot plot representing percentage of OVA-specific CD8+ T cells (upper right quadrant) identified in spleens of b-NP and OVA-NP immunized mice. (E) Representative contour plot of T cell populations of naïve (CD62LhiCD44lo), central memory (CD62LhiCD44hi), and effector memory (CD62LloCD44hi) OVA-specific CD8+ T cells (gate from D) identified in spleens of OVA-NP vaccinated mice. (F) Flow cytometry dot plot analysis representing the percentage of infiltrating CD8+ T cells (upper left quadrant) and B220+ B cells (lower right quadrant) within tumors from mice immunized with b-NPs and OVA-NPs. (G) Dot plot analyses showing the percentage of OVA-specific T cells among the tumor infiltrating CD8+ T cells (gate from F). (H) Contour plot of tumor infiltrating OVA-specific CD8+ T cells (gate from G) for IFN-γ and/or Granzyme B on expression following PMA/ionomycin restimulation. Isotype control staining for IFN-γ and Granzyme B is also shown. All data shown are representative of three independent experiments.
To investigate whether this protection was OVA-specific, we used OVA257–264/H-2Kb pentamers to detect Ag-specific CD8+ T cells in different tissues of tumor-challenged mice. In mice immunized with OVA-NPs, we found a systemic increase of OVA-specific CD8+ T cells, in comparison to the b-NPs immunized group. Clear populations of OVA-specific CD8 T cells were identified in spleens, blood, and draining lymph nodes of OVA-NP-immunized mice (Figure 6D, and data not shown). Importantly, we found that OVA-NP MNs induced efficient antigen-specific effector and memory cells among the OVA-specific CD8+ T cells in the spleens, as the majority of these cells acquired central memory (CD62LhiCD44hi) or effector memory (CD62LloCD44hi) profiles (Figure 6E). Furthermore, among tumor infiltrating cells, we detected an increased infiltration of CD8+ T cells within tumors from animals immunized with OVA-NPs compared to b-NPs-immunized mice (Figure 6F), of which 10% −15% were found to be OVA-specific cytotoxic T cells (Figure 6G), as characterized by their ability to produce IFN-γ and/or Granzyme B (Figure 6H). De Koker et al., who observed protective antitumor immunity following parenteral delivery of OVA-loaded polymeric multilayer capsules, recently reported similar results. However, a two-step immunization regime (“prime and boost”) with a higher dose of antigen (50 μg) was required for effective antitumor protection.27 In contrast, due to direct targeting of the antigen to the dense network of skin dendritic cells through the MN delivery system, our results show that only one inoculation using a significantly reduced dose of nanoencapsulated antigen induces efficient prophylactic antitumor immunity through the activation of tumor-specific killing effector CD8+ cells.
To determine whether MN-delivered NP immunization could induce therapeutic antitumor immunity, mice were injected s.c. with 105 B16.OVA tumor cells, and on days 4 and 9 following tumor inoculation, they were immunized through MNs with b-NPs or OVA-NPs. While tumors grew to the maximal permissible size by day 18 in mice immunized with b-NPs MNs, we observed a marked delay in tumor growth in mice immunized with OVA-NPs. Statistical analysis of tumor volumes between treatment groups performed at day 18 post-tumor injections showed significant inhibition of tumor growth in mice treated with OVA-NPs (Supporting Information Figure 5), suggesting that MN-mediated immunization may be effective for tumor immunotherapy strategies.
Microneedle-Mediated Immunization with OVA-NPs Protects Against Viral Challenge
In order to test the capacity of MN-mediated vaccination to induce protective antiviral immunity in a murine para-influenza virus model, we generated a recombinant Sendai virus (SeV) that expresses a red fluorescent protein/ovalbumin fusion protein (SeV-OVATdT). Mice were immunized with b-NP or OVA-NP MNs and challenged with 2 × 106 pfu SeV-OVATdT intranasally 3 weeks later. All OVA-NP-immunized animals survived the viral challenge, while b-NP-immunized animals did not survive beyond 8 days after challenge (Figure 7A). This demonstrated that antigen NP delivery with dissolving MNs is also capable of inducing robust protective immunity against viral challenge. Taking advantage of the red fluorescent TdTomato reporter gene incorporated within the viral genome, we next investigated the ability of immunized mice to clear virus by flow cytometry following challenge. At day 6 post-challenge, we observed more SeV-OVATdT-positive cells present in total lung cell suspensions of b-NP-immunized mice (Figure 7B) compared to OVA-NP-immunized animals. To confirm that enhanced viral clearance had indeed occurred in OVA-NP-immunized mice, viral titers in the lungs and nasal washes were measured on day 7 following infection. Complete viral clearance from the lungs (Figure 7C) as well as a significant reduction of viral replication in the upper respiratory tracts was detected in mice immunized with OVA-NPs (Figure 7D). Furthermore, lung histopathology at day 6 post-viral challenge demonstrated a mild lymphocytic infiltration in lungs of OVA-NP-immunized mice, while indications of interstitial pneumonia with lymphocytic infiltration in perivascular and peribronchiolar distribution was observed in b-NP-immunized mice (Figure 7E). To address whether these efficient antiviral immune responses were directly due to OVA-NP immunization, we examined infiltrating CD8+ T cells in the lungs for their specificity toward OVA 6 days post-infection. Following immunization with OVA-NPs, over 10% OVA-specific CD8+ T cells could be detected in total lung cell suspension of which a marked proportion were found to be Granzyme B and/or IFN-γ expressing effector CD8+ T cells (Figure 7F) compared to b-NP-immunized animals. Therefore, these data collectively demonstrate that a single immunization with MNs laden with NPs-encapsulated antigen is sufficient to induce protective immunity against respiratory virus challenge and induce effective antiviral adaptive immune responses in vivo.
Figure 7.

Microneedle immunization with OVA nanoparticles protects against viral challenge. (A) Kaplan-Meir graph showing survival of mice challenged with SeV-OVATdT 3 weeks post-immunization. (B) Flow cytometry dot plot of percentage of SeV-OVATdT-positive cells among total lung cell suspensions from b-NP- or OVA-NP-immunized mice 6 days post-infection. Viral titers within lung tissue (C) and nasal washes (D) between b-NP and OVA-NP immunization groups at 7 days post-infection. (E) Lung histology at 6 days post-infection analyzed under light microscope following H&E staining from b-NP- and OVA-NP-immunized animals. (F) Flow cytometry dot plot representing percentage of lung infiltrating OVA-specific CD8+ T cells (top panels) in animals immunized with either b-NPs or OVA-NPs. Percentages of OVA-specific CD8+ T cells producing IFN-γ and/or Granzyme B (bottom panels) following PMA/ionomycin restimulation. All data shown are representative of three independent experiments.
Conclusions
Our findings suggest that polymer MNs dissolve in the skin within minutes and provide a simple and painless means of nanoparticle delivery to skin layers that are enriched with a contiguous network of antigen-presenting DCs. This delivery system facilitates the specific targeting of skin DCs, leading to their efficient activation. It also results in a controlled localized antigen release and distribution. This improved antigen delivery to professional APC augments antigen presentation and supports the generation of potent CD8+ cytotoxic T cell responses and CD4+ Th1 immune responses against an encapsulated antigen. Furthermore, prophylactic vaccination of particulate antigens through MNs protects against both tumor development and viral challenge and also demonstrated attenuated tumor growth in a therapeutic setting. Moreover, our finding that nanoencapsulation improves antigen stability in MNs for a prolonged period at ambient temperatures provides a strong rationale to study the potential of this technology to increase stability of established vaccine antigens at ambient temperatures, thereby obviating the requirement for cold storage. A strategy to circumvent the “cold chain” has enormous potential to conserve resources, improve vaccination coverage, while the self-disabling nature of the dissolving MN will prevent transmission of infection and obviate sharps disposal issues. Overall, our results demonstrate that self-disabling dissolving MN arrays laden with antigen-carrying NPs offer an attractive approach to improve efficacy of vaccine delivery through their capacity to release antigen to skin layers rich in antigen-presenting cells. The activation of DCs in this way consequently cascades an immune response that could be beneficial in the contexts of cancer therapy and infectious diseases.
Materials and Methods
Materials
Microneedle arrays were forumulated with Gantrez AN-139, a copolymer of methylvinylether and maleic anhydride (PMVE/MA) provided by ISP Co. 120 Ltd. (Guildford, UK), PLGA (Resomer RG 502 H) with an acid value of 9 mg of KOH per g of PLGA, molecular weight 9 kDa (Boehringer Ingelheim, Germany).
Preparation of OVA-Loaded NPs (OVA-NPs) or Blank NPs (b-NPs)
Ovalbumin (Sigma Aldrich, grade V purity) or LPS-free EndoGrade ovalbumin (Hyglos GmbH, Germany) was diluted to a concentration of 40 mg/mL in PBS. Ice-cold ovalbumin solution (100 μL) for OVA-NPs or the same amount of ice-cold PBS for preparation of b-NP was carefully mixed with 700 μL of ice-cold organic phase comprising 20 mg of PLGA in 700 μL of dichloromethane (Riedel de Haeen, Germany). The emulsion formed was then injected under moderate stirring into 7 mL of ice-cold solution containing 2.5% (w/v) poly(vinyl alcohol) (Sigma Aldrich). Both phases were then briefly sonicated in an ice bath at 20 mV. Samples were left stirring overnight to allow for organic solvent evaporation. NPs were centrifuged at 17 000g for 30 min at 4 °C and washed three times with PBS. NPs pellets (20 mg) were resuspended in PBS to produce a NP suspension of 160 mg/mL prior to further use. Size and zeta-potential measurements after NP formulation were performed in distilled water on a Malvern Nanosizer ZS. To measure antigen encapsulation, the supernatants obtained after each centrifugation step were collected and measured for their protein content using BCA protein assay (Thermo Scientific). When subtracted from the initial amount of antigen, an encapsulation efficiency of 35.8% for OVA was calculated.
Fabrication of NP-Loaded MNs Prepared from Aqueous Blends of 20% w/w PMVE/MA
MNs prepared from aqueous blends of 20% w/w PMVE/MA were prepared using laser-engineered silicone micromold templates, as described previously.13 Briefly, silicone elastomer was poured into a custom-made aluminum mold and cured overnight at 40 °C. A laser-machine tool (BluLase Micromachining System, Blueacre Technology, Dundalk, Ireland) with a laser (Coherent Avia, Coherent Inc., Pittsburgh, PA, USA) emitting a beam having a wavelength of 355 nm and a pulse length of 30 ns (variable from 1 to 100 kHz) was then employed to produce MN molds (19 × 19 array, 600 μm height). A 30% w/w aqueous solution of PMVE/MA was prepared by adding the required mass of PMVE/MA to ice-cold deionized water, followed by vigorous stirring and heating at 95.0 °C until a clear gel was obtained, due to hydrolysis of the anhydride form of the copolymer to the corresponding acid. Upon cooling, the blend was then readjusted to the final concentration of 30% w/w by addition of an appropriate amount of deionized water. To prepare NP-loaded MNs, the stock solution was diluted with the appropriate amount of NP suspension. Then, 250 mg of this solution, containing 20 mg of b-NPs or OVA-NPs, was poured into each silicone micromold, centrifuged for 15 min at 2200g, and allowed to dry under ambient conditions for 24 h. After drying, another 250 mg of blank 20% w/w PMVE/MA was poured over the first layer to form the MN base plate.
Bone-Marrow-Derived DCs
Bone marrow cells were isolated from femurs and tibias of 6–8 weeks old C57BL/6 mice. They were cultured in RPMI 1640 containing 10% endotoxin-free heat-inactivated FCS, 2 mM l-glutamine, 10 mM HEPES, 50 μM β-mercaptoethanol, 100 U/mL penicillin, and 100 U/mL streptomycin, supplemented supernatant from Flt3 ligand-producing COS cells (kindly provided by N. Nicola, WEHI, Australia). On the 10th day, cells were left unstimulated or stimulated for 12 h with 100 μg/mL of b-NPs, OVA-NPs, or 100 ng/mL LPS, washed, and stained with maturation marker-specific antibodies and subsequently analyzed by the flow cytometer.
For in vitro T cell proliferation assays, BMDCs were left either unstimulated or stimulated with various concentrations (μg/mL) of OVA-NPs or OVA peptides (LSQAVHAA-HAEINEAGR or SIINFEKL, 10 μg/mL) for 3 h. Following stimulation, BMDCs were plated (1:2 dilution) with purified CFSE-labeled CD8+ or CD4+ T cells isolated from spleens of OT-I or OT-II mice, respectively, using magnetic beads (STEMCELL Technologies), according to manufacturer’s instructions. Sixty hours later, T cell proliferation, assayed by CFSE dilutions, was assessed by flow cytometry. As a positive control for T cell proliferation, T cells were stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies (eBioscience).
Mice
Langerin-eGFP (LangGFP) reporter mice (provided by Dr. Bernard Malissen, CIML, France),48 OT-I, and OT-II mice (The Jackson Lab, USA) that express transgenic Vα2 Vβ5.1/5.2 TCR specific for Kb + OVA257–264 and I-Ab + OVA323–339, respectively,49,50 and congenic CD45.1+ (Harlan, UK) mice were housed under pathogen-free conditions. All mice employed for experiments were between 6 and 9 weeks old, aged and sex matched, and were sanctioned in accordance with the UK home office and approved by Queen’s University Belfast Ethical Review Committee.
MN Immunization
LangGFP reporter or congenic CD45.1+ was first anesthetized, and MN arrays laden with bNPs or OVA-NPs were manually inserted onto the untreated dorsal side of both ears. The MNs were held in place for at least 5 min and then removed.
Flow Cytometry
Surface marker expression was assessed by flow cytometry using fluorochrome-conjugated antibodies to CD4, CD8, CD11b, CD11c, CD45.1, CD45.2, MHC class II, Ly6G, Ly6C, and CD207 (BD Biosciences and eBioscience). SIINFEKL/H-2Kb pentamers conjugated to PE staining were used in accordance with manufacturer’s instructions (Pro-Immune). For cytokine staining, cells were restimulated in complete medium at 37 °C for 5 h with 50 ng/mL PMA (Sigma-Aldrich), 500 ng/mL ionomycin (Sigma-Aldrich), with GolgiStop (BD Biosciences) added for the final 4 h. Cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences), in accordance with manufacturer’s instructions, followed by intracellular anti-IL-4, anti-IFN-γ, or anti-Granzyme B (BD Biosciences or eBioscience) staining. Data were collected on FACS Canto II (BD Biosciences) and analyzed using FlowJo software (Tree Star). Identification of different cell subsets isolated from the ear skin was performed on dermal cell suspension, as previously described.36,51
In Vivo Proliferation Assays
Spleens were harvested from OT-I or OT-II mice. CD8+ and CD4+ T cells were enriched from single cell suspension by negative selection using the EasySep mouse CD8+ or CD4+ T cell enrichment kits, respectively (STEMCELL Technologies), and isolated T cells were labeled with 0.2 μM CFSE (Molecular Probes). Groups of CD45.1+ congenic mice were injected intravenously with 106 CFSE-labeled OT-I or OT-II T cells, 24 h prior to MN immunization. Mice were sacrificed 72 h following MN application, and auricular lymph nodes were harvested for FACS analysis. The proliferation of antigen-specific CFSE+ T cells was detected by the decrease of CFSE fluorescence intensity.
Ex Vivo Proliferation Assay
LangGFP reporter mice were immunized as described above. Three days later, auricular lymph nodes were digested with 0.5 mg/mL collagenase D (Sigma Aldrich) for 30 min at 37 °C, pressed through cell strainers, and enriched by positive selection using EasySep mouse CD11c-positive selection kit (STEMCELL Technologies), according to manufacturer’s instructions. For DC subset experiments, cells were first enriched using a Nycodenz gradient, as previously described.52 The cells at the interface were then stained with anti-CD8-PerCP-Cy5.5, anti-MHCII-PE, and anti-CD11c-APC (BD) and sorted on a FACS Aria II instrument (BD Biosciences) for skin-derived CD11c+MHCIIhighCD8– DCs, blood-derived CD8+ DC, and remaining blood-derived CD8– DCs (purity >95%). Then, 105 DCs were cocultured in triplicate with 2 × 105 CFSE-labeled OVA-specific OT-I or OT-II T cells (isolated as described above) for 60 h, and proliferation of T cells was measured by the decrease of CFSE fluorescence intensity.
Tumor Challenge
Three weeks following MN immunization, mice were injected s.c. into the flank with 105 B16.OVA tumor cells53 (kindly provided by Dr. P. Stoitzner, Innsbruck, Austria), mixed with reduced growth factor matrigel (4 mg/mL) (BD Biosciences). Tumor size was assessed every day by measuring the short and long tumor diameters using digital callipers and is expressed by tumor volume (length × width × height/2 × π/6). Animals were euthanized when any tumor diameter reached the maximum allowed dimension of 20 mm according to UK Home Office guidelines.
Challenge of Mice with Recombinant Sendai Virus OVA-TdT (rSeV/OVA-TdT)
To determine virus-specific immune responses, mice were challenged with a recombinant Sendai virus which expressed a fusion protein of full-length ovalbumin (kindly provided by M. Kursar, MPIIB, Germany) and the tandem dimer tomato fluorescent reporter gene (kindly provided by R. Tsien, UCSD, USA). The virus was rescued as previously described.54 Groups of 4–5 mice were challenged 3 weeks after MN immunization by intranasal instillation of 2 × 106 pfu rSeV/OVA-TdT in 30 μL of PBS and monitored for up to 9 days. A weight loss exceeding 20% was used as the experimental end point, at which point mice were humanely euthanized.
Preparation of Lung Homogenates
Mice were sacrificed by cervical dislocation. Lungs were remove and place in 1 mL virus transport medium (Hanks 199 (1×), 25 mM HEPES, 0.05 mg/mL gentamicin, 0.22 M sucrose, 30 mM MgCl2, 0.5 mg/mL amphotericin B), weighed, and 10% (w/v) homogenates prepared using FastPrep tubes containing 250 μL of 1 mm glass beads. Tissues were homogenized in the FastPrep 120 Instrument (Qbiogen, Inc.) with three cycles of 20 s at 4 m/s interspersed with short incubations on ice to avoid overheating. FastPrep tubes were then centrifuged for 1 min at 200g at 4 °C, and supernatants were aliquoted into cryovials, snap frozen, and stored at −80 °C until virus titrations were performed.
Lung single cell suspensions for flow cytometry analyses were performed by enzymatic digestion for 30 min at 37 °C using 1 mg/mL collagenase type 2 (Sigma Aldrich) and 0.02 mg/mL DNase I from bovine pancreas (Sigma Aldrich). After digestion, contaminating erythrocytes present in lung cell suspension were lysed by incubation at 37 °C for 3 min with red cell lysing buffer.
Virus Titer Determination
The rSeV-OVA-TdT titration in experimental samples was performed in LLC-MK2 cells (ECACC 85062804) as previously described,54 except that maintenance medium containing 1% FCS and without acetylated trypsin or methyl cellulose was used to prevent repeated cycles of virus infection. After 48 h incubation at 37 °C in 5% CO2, the numbers of fluorescent cells were counted in appropriate dilutions under a fluorescent microscope (Nikon TE2000-U), and titers were calculated as numbers of fluorescent forming units (ffu)/g of tissue, where a single fluorescent cell constituted a fluorescent forming unit.
Lung Histology
Lobes of lungs were fixed in 10% formalin, embedded in paraffin, and sections of 5 μm were stained with Hematoxilin–Eosin. Images were captured with a microscope (DM5500; Leica) with digital system and LAS AF software (version 1.5.1).
Statistical Analysis
Where appropriate, data were analyzed using a one-way analysis of variance (ANOVA), with post-hoc comparison performed using Tukey’s HSD test. Mann–Whitney test was used to compare percentages of T cell proliferation in response to b-NP and OVA-NP immunization. Unpaired Student’s t test was used to determine the statistical significance of differences in SeV titers in lung homogenates and nasal washes, as well as tumor volumes post-MN treatments between two immunization groups. Probability values are expressed as the following: ***, p < 0.001; **, p < 0.01; and *, p < 0.05.
Acknowledgments
We would like to thank L. Hanna and D. Beattie (Queen’s University Belfast) for animal care, M. McCaigue for technical assistance, and C. McFarlane, A. Fitzsimons, A. Rakha Arora, R. Penalva, and S. Spence for helping with experiments. This work was supported by grants from the Department for Employment and Learning of Northern Ireland (Strengthening the All-Island Research Base) and The Wellcome Trust UK (WT008532).
Supporting Information Available
Supplementary Figures 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Steinman R. M. Lasker Basic Medical Research Award. Dendritic Cells: Versatile Controllers of the Immune System. Nat. Med. 2007, 13, 1155–1159. [DOI] [PubMed] [Google Scholar]
- Steinman R. M. Dendritic Cells in Vivo: A Key Target for a New Vaccine Science. Immunity 2008, 29, 319–324. [DOI] [PubMed] [Google Scholar]
- Hegde N. R.; Kaveri S. V.; Bayry J. Recent Advances in the Administration of Vaccines for Infectious Diseases: Microneedles as a Painless Delivery Devices for Mass Vaccination. Drug Discovery Today 2011, 16, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Koutsonanos D. G.; del Pilar Martin M.; Zarnitsyn V. G.; Sullivan S. P.; Compans R. W.; Prausnitz M. R.; Skountzou I. Transdermal Influenza Immunization with Vaccine-Coated Microneedle Arrays. PLoS One 2009, 4, e4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Pisani E. The Cost of Unsafe Injections. Bull. World Health Org. 1999, 77, 808–811. [PMC free article] [PubMed] [Google Scholar]
- Prausnitz M. R.; Langer R. Transdermal Drug Delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadiere B.; Vogt A.; Mahe B.; Costagliola D.; Hadam S.; Bonduelle O.; Sterry W.; Staszewski S.; Schaefer H.; van der Werf S.; et al. Preferential Amplification of CD8 Effector-T Cells after Transcutaneous Application of an Inactivated Influenza Vaccine: A Randomized Phase I Trial. PLoS One 2010, 5, e10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe R. B.; Newman F. K.; Cannon J.; Duane C.; Treanor J.; Van Hoecke C.; Howe B. J.; Dubin G. Serum Antibody Responses after Intradermal Vaccination Against Influenza. N. Engl. J. Med. 2004, 351, 2286–2294. [DOI] [PubMed] [Google Scholar]
- Kenney R. T.; Frech S. A.; Muenz L. R.; Villar C. P.; Glenn G. M. Dose Sparing with Intradermal Injection of Influenza Vaccine. N. Engl. J. Med. 2004, 351, 2295–2301. [DOI] [PubMed] [Google Scholar]
- Prausnitz M. R. Microneedles for Transdermal Drug Delivery. Adv. Drug Delivery Rev. 2004, 56, 581–587. [DOI] [PubMed] [Google Scholar]
- Al-Zahrani S.; Zaric M.; McCrudden C.; Scott C.; Kissenpfennig A.; Donnelly R. F. Microneedle-Mediated Vaccine Delivery: Harnessing Cutaneous Immunobiology To Improve Efficacy. Expert Opin. Drug Delivery 2012, 9, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. C.; Park J. H.; Prausnitz M. R. Microneedles for Drug and Vaccine Delivery. Adv. Drug Delivery Rev. 2012, 64, 1547–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly R. F.; Majithiya R.; Singh T. R.; Morrow D. I.; Garland M. J.; Demir Y. K.; Migalska K.; Ryan E.; Gillen D.; Scott C. J.; et al. Design, Optimization and Characterisation of Polymeric Microneedle Arrays Prepared by a Novel Laser-Based Micromoulding Technique. Pharm. Res. 2011, 28, 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storni T.; Kundig T. M.; Senti G.; Johansen P. Immunity in Response to Particulate Antigen-Delivery Systems. Adv. Drug Delivery Rev. 2005, 57, 333–355. [DOI] [PubMed] [Google Scholar]
- Gutierro I.; Hernandez R. M.; Igartua M.; Gascon A. R.; Pedraz J. L. Size Dependent Immune Response after Subcutaneous, Oral and Intranasal Administration of BSA Loaded Nanospheres. Vaccine 2002, 21, 67–77. [DOI] [PubMed] [Google Scholar]
- Jaganathan K. S.; Vyas S. P. Strong Systemic and Mucosal Immune Responses to Surface-Modified PLGA Microspheres Containing Recombinant Hepatitis B Antigen Administered Intranasally. Vaccine 2006, 24, 4201–4211. [DOI] [PubMed] [Google Scholar]
- Mahapatro A.; Singh D. K. Biodegradable Nanoparticles Are Excellent Vehicle for Site Directed In Vivo Delivery of Drugs and Vaccines. J. Nanobiotechnol. 2011, 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyam J.; Labhasetwar V. Biodegradable Nanoparticles for Drug and Gene Delivery to Cells and Tissue. Adv. Drug Delivery Rev. 2003, 55, 329–347. [DOI] [PubMed] [Google Scholar]
- Kumari A.; Yadav S. K.; Yadav S. C. Biodegradable Polymeric Nanoparticles Based Drug Delivery Systems. Colloids Surf., B 2010, 75, 1–18. [DOI] [PubMed] [Google Scholar]
- Hunter S. K.; Andracki M. E.; Krieg A. M. Biodegradable Microspheres Containing Group B Streptococcus Vaccine: Immune Response in Mice. Am. J. Obstet. Gynecol. 2001, 185, 1174–1179. [DOI] [PubMed] [Google Scholar]
- Alonso M. J.; Gupta R. K.; Min C.; Siber G. R.; Langer R. Biodegradable Microspheres as Controlled-Release Tetanus Toxoid Delivery Systems. Vaccine 1994, 12, 299–306. [DOI] [PubMed] [Google Scholar]
- Newman K. D.; Elamanchili P.; Kwon G. S.; Samuel J. Uptake of Poly(d,l-lactic-co-Glycolic Acid) Microspheres by Antigen-Presenting Cells in Vivo. J. Biomed. Mater. Res. 2002, 60, 480–486. [DOI] [PubMed] [Google Scholar]
- Putney S. D.; Burke P. A. Improving Protein Therapeutics with Sustained-Release Formulations. Nat. Biotechnol. 1998, 16, 153–157. [DOI] [PubMed] [Google Scholar]
- Uchida T.; Goto S. Oral Delivery of Poly(lactide-co-Glycolide) Microspheres Containing Ovalbumin as Vaccine Formulation: Particle Size Study. Biol. Pharm. Bull. 1994, 17, 1272–1276. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Wonganan P.; Sandoval M. A.; Li X.; Zhu S.; Cui Z. Microneedle-Mediated Transcutaneous Immunization with Plasmid DNA Coated on Cationic PLGA Nanoparticles. J. Controlled Release 2012, 163, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audran R.; Peter K.; Dannull J.; Men Y.; Scandella E.; Groettrup M.; Gander B.; Corradin G. Encapsulation of Peptides in Biodegradable Microspheres Prolongs Their MHC Class-I Presentation by Dendritic Cells and Macrophages in Vitro. Vaccine 2003, 21, 1250–1255. [DOI] [PubMed] [Google Scholar]
- De Geest B. G.; Willart M. A.; Hammad H.; Lambrecht B. N.; Pollard C.; Bogaert P.; De Filette M.; Saelens X.; Vervaet C.; Remon J. P.; et al. Polymeric Multilayer Capsule-Mediated Vaccination Induces Protective Immunity Against Cancer and Viral infection. ACS Nano 2012, 6, 2136–2149. [DOI] [PubMed] [Google Scholar]
- Burgdorf S.; Kurts C. Endocytosis Mechanisms and the Cell Biology of Antigen Presentation. Curr. Opin. Immunol. 2008, 20, 89–95. [DOI] [PubMed] [Google Scholar]
- Abdelghany S. M.; Quinn D. J.; Ingram R. J.; Gilmore B. F.; Donnelly R. F.; Taggart C. C.; Scott C. J. Gentamicin-Loaded Nanoparticles Show Improved Antimicrobial Effects towards Pseudomonas aeruginosa Infection. Int. J. Nanomed. 2012, 7, 4053–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsiak M. E.; Robinson D. R.; Coester C.; Kwon G. S.; Samuel J. Analysis of Poly(d,l-lactic-co-Glycolic Acid) Nanosphere Uptake by Human Dendritic Cells and Macrophages in Vitro. Pharm. Res. 2002, 19, 1480–1487. [DOI] [PubMed] [Google Scholar]
- Elamanchili P.; Diwan M.; Cao M.; Samuel J. Characterization of Poly(d,l-lactic-co-Glycolic Acid) Based Nanoparticulate System for Enhanced Delivery of Antigens to Dendritic Cells. Vaccine 2004, 22, 2406–2412. [DOI] [PubMed] [Google Scholar]
- Vallhov H.; Qin J.; Johansson S. M.; Ahlborg N.; Muhammed M. A.; Scheynius A.; Gabrielsson S. The Importance of an Endotoxin-Free Environment during the Production of Nanoparticles Used in Medical Applications. Nano Lett. 2006, 6, 1682–1686. [DOI] [PubMed] [Google Scholar]
- Uto T.; Wang X.; Sato K.; Haraguchi M.; Akagi T.; Akashi M.; Baba M. Targeting of Antigen to Dendritic Cells with Poly(γ-glutamic acid) Nanoparticles Induces Antigen-Specific Humoral and Cellular Immunity. J. Immunol. 2007, 178, 2979–2986. [DOI] [PubMed] [Google Scholar]
- Donnelly R. F.; Garland M. J.; Morrow D. I.; Migalska K.; Singh T. R.; Majithiya R.; Woolfson A. D. Optical Coherence Tomography Is a Valuable Tool in the Study of the Effects of Microneedle Geometry on Skin Penetration Characteristics and In-Skin Dissolution. J. Controlled Release 2010, 147, 333–341. [DOI] [PubMed] [Google Scholar]
- del Pilar Martin M.; Weldon W. C.; Zarnitsyn V. G.; Koutsonanos D. G.; Akbari H.; Skountzou I.; Jacob J.; Prausnitz M. R.; Compans R. W. Local Response to Microneedle-Based Influenza Immunization in the Skin. MBio 2012, 3, e00012–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henri S.; Poulin L. F.; Tamoutounour S.; Ardouin L.; Guilliams M.; de Bovis B.; Devilard E.; Viret C.; Azukizawa H.; Kissenpfennig A.; et al. CD207+ CD103+ Dermal Dendritic Cells Cross-Present Keratinocyte-Derived Antigens Irrespective of the Presence of Langerhans Cells. J. Exp. Med. 2010, 207, 189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan S. P.; Koutsonanos D. G.; Del Pilar Martin M.; Lee J. W.; Zarnitsyn V.; Choi S. O.; Murthy N.; Compans R. W.; Skountzou I.; Prausnitz M. R. Dissolving Polymer Microneedle Patches for Influenza Vaccination. Nat. Med. 2010, 16, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka Y.; Yoshino H. Lymphatic Targeting with Nanoparticulate System. Adv. Drug Delivery Rev. 2001, 47, 55–64. [DOI] [PubMed] [Google Scholar]
- Reddy S. T.; Rehor A.; Schmoekel H. G.; Hubbell J. A.; Swartz M. A. In Vivo Targeting of Dendritic Cells in Lymph Nodes with Poly(propylene sulfide) Nanoparticles. J. Controlled Release 2006, 112, 26–34. [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Multifunctional Nanocarriers. Adv. Drug Delivery Rev. 2006, 58, 1532–1555. [DOI] [PubMed] [Google Scholar]
- Vasir J. K.; Labhasetwar V. Biodegradable Nanoparticles for Cytosolic Delivery of Therapeutics. Adv. Drug Delivery Rev. 2007, 59, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J.; Steinman R. M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [DOI] [PubMed] [Google Scholar]
- Gamvrellis A.; Leong D.; Hanley J. C.; Xiang S. D.; Mottram P.; Plebanski M. Vaccines That Facilitate Antigen Entry into Dendritic Cells. Immunol. Cell Biol. 2004, 82, 506–516. [DOI] [PubMed] [Google Scholar]
- Waeckerle-Men Y.; Allmen E. U.; Gander B.; Scandella E.; Schlosser E.; Schmidtke G.; Merkle H. P.; Groettrup M. Encapsulation of Proteins and Peptides into Biodegradable Poly(d,l-lactide-co-Glycolide) Microspheres Prolongs and Enhances Antigen Presentation by Human Dendritic Cells. Vaccine 2006, 24, 1847–1857. [DOI] [PubMed] [Google Scholar]
- Partidos C. D.; Beignon A. S.; Mawas F.; Belliard G.; Briand J. P.; Muller S. Immunity under the Skin: Potential Application for Topical Delivery of Vaccines. Vaccine 2003, 21, 776–780. [DOI] [PubMed] [Google Scholar]
- Stoitzner P.; Green L. K.; Jung J. Y.; Price K. M.; Tripp C. H.; Malissen B.; Kissenpfennig A.; Hermans I. F.; Ronchese F. Tumor Immunotherapy by Epicutaneous Immunization Requires Langerhans Cells. J. Immunol. 2008, 180, 1991–1998. [DOI] [PubMed] [Google Scholar]
- Seo N.; Tokura Y.; Nishijima T.; Hashizume H.; Furukawa F.; Takigawa M. Percutaneous Peptide Immunization via Corneum Barrier-Disrupted Murine Skin for Experimental Tumor Immunoprophylaxis. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissenpfennig A.; Henri S.; Dubois B.; Laplace-Builhe C.; Perrin P.; Romani N.; Tripp C. H.; Douillard P.; Leserman L.; Kaiserlian D.; et al. Dynamics and Function of Langerhans Cells in Vivo: Dermal Dendritic Cells Colonize Lymph Node Areas Distinct from Slower Migrating Langerhans Cells. Immunity 2005, 22, 643–654. [DOI] [PubMed] [Google Scholar]
- Hogquist K. A.; Jameson S. C.; Heath W. R.; Howard J. L.; Bevan M. J.; Carbone F. R. T Cell Receptor Antagonist Peptides Induce Positive Selection. Cell 1994, 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Barnden M. J.; Allison J.; Heath W. R.; Carbone F. R. Defective TCR Expression in Transgenic Mice Constructed Using cDNA-Based α- and β-Chain Genes under the Control of Heterologous Regulatory Elements. Immunol. Cell Biol. 1998, 76, 34–40. [DOI] [PubMed] [Google Scholar]
- Tamoutounour S.; Henri S.; Lelouard H.; de Bovis B.; de Haar C.; van der Woude C. J.; Woltman A. M.; Reyal Y.; Bonnet D.; Sichien D.; et al. CD64 Distinguishes Macrophages from Dendritic Cells in the Gut and Reveals the Th1-Inducing Role of Mesenteric Lymph Node Macrophages During Colitis. Eur. J. Immunol. 2012, 42, 3150–3156. [DOI] [PubMed] [Google Scholar]
- Vremec D.; Pooley J.; Hochrein H.; Wu L.; Shortman K. CD4 and CD8 Expression by Dendritic Cell Subtypes in Mouse Thymus and Spleen. J. Immunol. 2000, 164, 2978–2986. [DOI] [PubMed] [Google Scholar]
- Brown D. M.; Fisher T. L.; Wei C.; Frelinger J. G.; Lord E. M. Tumours Can Act as Adjuvants for Humoral Immunity. Immunology 2001, 102, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzelet O.; Loukili N.; Pelet T.; Fairley D.; Curran J.; Power U. F. De Novo Generation of a Non-segmented Negative Strand RNA Virus with a Bicistronic Gene. Virus Res. 2009, 140, 40–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.