

Abstract
Background
In a classic model, Giα proteins including Gi1α, Gi2α and Gi3α are important for transducing signals from Giα protein-coupled receptors (GiαPCRs) to their downstream cascades in response to hormones and neurotransmitters. Our previous study has suggested that Gi1α, Gi2α and Gi3α are also important for the activation of the PI3K/Akt/mTORC1 pathway by epidermal growth factor (EGF) and its family members. However, a genetic role of these Giα proteins in the activation of extracellular signal-regulated protein kinase 1 and 2 (ERK1/2) by EGF is largely unknown. Further, it is not clear whether these Giα proteins are also engaged in the activation of both the Akt/mTORC1 and ERK1/2 pathways by other growth factor family members. Additionally, a role of these Giα proteins in breast cancer remains to be elucidated.
Results
We found that Gi1/3 deficient MEFs with the low expression level of Gi2α showed defective ERK1/2 activation by EGFs, IGF-1 and insulin, and Akt and mTORC1 activation by EGFs and FGFs. Gi1/2/3 knockdown breast cancer cells exhibited a similar defect in the activations and a defect in in vitro growth and invasion. The Giα proteins associated with RTKs, Gab1, FRS2 and Shp2 in breast cancer cells and their ablation impaired Gab1’s interactions with Shp2 in response to EGF and IGF-1, or with FRS2 and Grb2 in response to bFGF.
Conclusions
Giα proteins differentially regulate the activation of Akt, mTORC1 and ERK1/2 by different families of growth factors. Giα proteins are important for breast cancer cell growth and invasion.
Keywords: Giα proteins, receptor tyrosine kinase (RTK), EGFR, Gab1, Shp2, EGF, Akt, mTORC1, ERK1/2, and breast cancer
Introduction
Giα proteins including Gi1α, Gi2α and Gi3α transduce signals from Giα protein-coupled receptors (GiαPCRs) to downstream pathways in hormone and chemo-attractant signaling. Recent genetic evidence strongly suggests that Giα proteins also play an important role in cell growth and tumorigenesis. Active mutants of Gi2α promote cell transformation and are associated with ovarian cancer [1,2], whereas Gi2α deficiency results in the development of spontaneous colitis and colon cancer in mice [1]. Also, Gi3α ablation impairs the antiautophagic response to insulin in mouse liver, and Gi2α and Gi3α double deficiency leads to severe embryonic growth retardation and the subsequent embryonic lethality [3], which, at least in part, are similar to phenotypes of many growth factor receptor deficiencies. However, how these Giα proteins are implicated in cell growth and tumorigenesis remains poorly understood.
It is known that the extracellular signal-regulated kinases (ERK1/2) and PI3K/Akt/mTORC1 pathways play an essential role in cell growth and malignant transformation. Both GiαPCRs [4] and growth factor receptors, which are also called receptor tyrosine kinases (RTKs), can activate them in response to their respective ligands. The GiαPCRs-triggered activation is mainly mediated by Gβγ, which can either directly bind and activate PI3K [5], or transmit signals to ERK1/2 through Src-, Shc- and Grb2- dependent and independent mechanisms [6-8]. This activation is sensitive to the treatment of Bordetella pertussis toxin (PTX), which blocks the interaction of Giα proteins with GiαPCRs [6,9,10].
The RTK-triggered activation of these two pathways by growth factors, including basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), heparin-binding EGF-like growth factor (HB-EGF), insulin and insulin-like growth factor 1 (IGF-1) [11,12], is mainly mediated by adaptor proteins and the phosphatase Shp2. The established features of ERK1/2 activation by EGF, IGF-1 and insulin are that activated RTKs recruit Grb2, insulin receptor substrates (IRSs) and Grb2-associated binding protein 1 (Gab1) to interact with Shp2 leading to ERK1/2 activation [13-17]. Intriguingly, Gab1 is also recruited to the FGF receptor (FGFR) and forms a complex with Grb2 and the FGF receptor substrate 2 (FRS2), but it is not involved in ERK1/2 activation by FGFs [18]. Instead, FRS2 and Grb2 as well as their interaction are essential for ERK1/2 activation by FGFs. Further, Gab1 is not implicated in the activation of the PI3K/Akt/mTORC1 pathway by insulin or IGF-1. In its place, IRSs are recruited to insulin receptors (IRs) or IGF-1Rs, interact with PI3Kp85 and activate PI3K [19] generating PIP3 and triggering phosphorylation of Akt on threonine (T) 308 by PDK1 and serine (S) 473 by the mammalian target of rapamycin complex 2 (mTORC2). Activated Akt triggers the activation of the mammalian target of rapamycin complex 1 (mTORC1), which in turn phosphorylates 4E-BP1 and S6 kinase (S6K), which phosphorylates S6. Conversely, Gab1 is critical for EGFR- and FGFR-mediated activation of the PI3K/Akt/mTORC1 pathway by EGF or FGFs. Gab1 is tyrosine (Y)-phosphorylated and then interacts with Grb2 and PI3Kp85. These interactions are essential for PI3K activation by EGF and FGFs.
It is known that GiαPCRs can trans-activate RTKs, and that both of them share some downstream adaptor proteins such as Grb2 and Shc to activate ERK1/2. However, PTX and Gβ blocking peptides can inhibit the activation of Akt and ERK1/2 by GiαPCRs ligands, but not EGF [20,21]. Thus, it is unknown whether Giα proteins can regulate RTKs-mediated activation of the ERK1/2 and PI3K/Akt/mTORC1 pathways by grow factors.
We previously reported that loss of Gi1α and Gi3α in mouse embryonic fibroblasts (MEFs) resulted in a defect in Akt and mTORC1 activation by EGF [20]. Yet, it is largely unknown whether these Giα proteins regulate ERK1/2 activation by EGF or ERK1/2, Akt and mTORC1 activation by other growth factors. Additionally, a role of the Giα proteins in breast cancer pathogenesis is unrevealed. In this study, we investigated a role of the Giα proteins in ERK1/2, Akt and mTORC1 activation by different families of growth factors in mouse embryonic fibroblasts and in human breast cancer cells.
Results
Giα proteins differentially regulate the activation of the ERK1/2 and Akt/mTORC1 pathways by growth factors in mouse embryonic fibroblasts
To elucidate a role for Giα proteins in the activation of ERK1/2, Akt and mTORC1 in response to growth factors, we used wild type (WT) and Gi1α and Gi3α double knockout (Gi1/3 DKO) MEFs [20]. First, we examined the expression levels of Gi1α, Gi2α and Gi3α in MEFs. As shown, both Gi2α and Gi3α were relatively abundant, but Gi1α was expressed at a low level and it required a longer-exposure for a clear signal (Additional file 1: Figure S1A). Interestingly, Gi2α expression level was obviously lower in Gi1/3DKO MEFs than that in WT MEFs (Additional file 1: Figure S1A) indicating that loss of Gi1α and Gi3α might subsequently influence the expression of Gi2α in MEFs.
WT and Gi1/3 DKO MEFs were then treated with EGF, HB-EGF, aFGF, bFGF, IGF-1, insulin, and PDGF and the activation statuses of several intracellular signaling molecules were examined. The expression levels of major relay molecules in the ERK1/2 and Akt pathways in Gi1/3 DKO MEFs were similar to WT controls (Additional file 1: Figures S1B to S1D), whereas the phosphorylation levels of ERK1/2, Akt, and mTORC1’s downstream events including 4E-BP1 and S6 were altered (Figure 1). Kinetic experiments showed that Gi1/3 DKO MEFs exhibited a severe defect in phosphorylation of ERK1/2(T202/Y204), Akt (S473) and S6(S235/236) in response to EGF and HB-EGF (Figures 1A and 1B). Consistent with our previous study [20], loss of Gi2α in MEFs did not have an apparent effect on ERK1/2 activation by EGF (Additional file 1: Figure S1E).
Figure 1.

Giα proteins regulate differential activation of Akt, mTORC1 and ERK1/2 by growth factors. (A-F) WT and Gi1/3 DKO MEFs were treated with (A) EGF (100 ng/ml), (B) HB-EGF (100 ng/ml), (C) aFGF (25 ng/ml), (D) bFGF (25 ng/ml), (E) IGF-1 (20 ng/ml), and (F) insulin (1 μg/ml at the indicated time points. p-ERK (T202/Y204), p-AKT (S473), p-S6 (S235/236), or β-actin were detected by immunoblotting (IB) analysis. The experiments were performed more than six times and similar results were obtained. (G) WT and Gi1/3 DKO MEFs were treated with PDGF for 10 minutes (min) at the indicated doses. p-ERK(T202/Y204), p-AKT(S473), p-S6(S235/236), or β-actin were detected by IB. The experiment was repeated three times and similar results were obtained.
Surprisingly, loss of Gi1α and Gi3α largely impaired phosphorylation of Akt(S473) and S6(S235/236) in response to aFGF and bFGF, but had no apparent effect on ERK/12(T202/Y204) phosphorylation (Figures 1C and 1D). In contrast to the FGF response, the Gi1/3 deficiency largely decreased ERK1/2(T202/Y204) phosphorylation in response to IGF-1 and insulin (Figures 1E and 1F), whereas this deficiency only led to a minor defect in Akt(S473) phosphorylation [20]. Interestingly, S6(S235/236) phosphorylation was largely impaired in the deficient cells (Figures 1E and 1F).
To determine if GiαPCRs are involved in the activation of ERK1/2 by EGF, we treated cells with Giα protein inhibitor PTX. Consistent with our previous report, EGF-induced ERK1/2 activation was not reduced by PTX treatment (Additional file 1: Figure S1F) [20]. However, IGF-1-induced ERK1/2 activation was inhibited by PTX treatment (Additional file 1: Figure S1F).
Also, loss of Gi1α and Gi3α clearly reduced phosphorylation of Akt(S473), S6(S235/236), 4E-BP1(S65) and ERK1/2(T202/Y204) in response to PDGF at low doses of 0.12, 0.36 or 1.1 ng/ml (Figure 1G). However, the defect in this phosphorylation was not observed in the deficient cells in response to PDGF at high doses (Figure 1G) [20] indicating that Gi1α and Gi3α proteins regulate the activation of Akt, mTORC1 and ERK1/2 by PDGF in a dose dependent manner.
To confirm the above observations, we used RNA interference (RNAi) and reconstitution strategies. As shown, Gi3α was clearly knocked down, whereas Gi1α was weakly detected in MEFs (Figures 2A to 2C). Consistent with above results in Figure 1, the combined knockdown of Gi1/3 impaired ERK1/2(T202/Y204) phosphorylation in response to EGF and IGF-1, and reduced Akt(S473) and S6(S235/236) phosphorylation in response to EGF and bFGF (Figures 2A to 2C). Reintroduction of both Gi1α and Gi3α, or Gi3α alone partially restored phosphorylation of ERK1/2(T202/Y204), or Akt(S473) and S6(S235/236) in Gi1/3 DKO MEFs in response to EGF, bFGF, or IGF-1 (Figures 2D to 2F).
Figure 2.
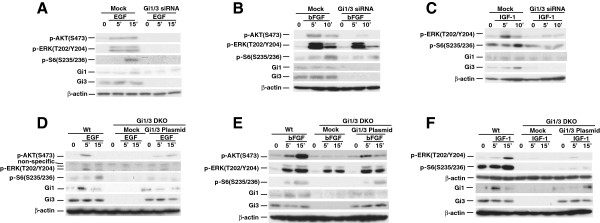
Knockdown of Giα proteins down regulates Akt and ERK activation, and reintroduction of Giα proteins restores their activation by EGF, bFGF and IGF-1. (A-C) WT MEFs transfected with siRNA duplexes specific for Gi1α and Gi3α were treated with (A) EGF (100 ng/ml), (B) bFGF (25 ng/ml), or (C) IGF-1 (20 ng/ml) for the indicated time points. p-AKT(S473), p-ERK(T202/Y204), p-S6(S235/236), β-actin, Gi1α (Gi1) and Gi3α (Gi3) were detected by IB. The experiments were repeated two to three times and similar results were obtained. (D-F) WT MEFs were transfected with empty vector and Gi1/3 DKO MEFs were transfected with WT Gi1α, WT Gi3α or empty expression vectors (5 μg). Cells were treated with (D) EGF (100 ng/ml), (E) bFGF (25 ng/ml) or (F) IGF-1 (20 ng/ml) for 5, 15 min, or left untreated. p-ERK(T202/Y204), p-AKT(S473), p-S6(S235/236), or β-actin, Gi1α and Gi3α were detected by IB. The experiments were repeated two to three times and similar results were obtained.
Taken together, our experimental results demonstrate that Gi1α, Gi2α and Gi3α are functionally distinct in RTKs-mediated activation of ERK1/2, Akt and mTORC1 in growth factor signaling (Table 1).
Table 1.
Pathway activation by growth factors requires Ga proteins
 |
EGF, HB- EGF, PDGF (low doses) | Insulin, IGF | aFGF, bFGF | PDGF (high doses) |
|---|---|---|---|---|
| Akt-mTORC1 pathway |
+ |
- |
+ |
- |
| ERK pathway | + | + | - | - |
+Giα proteins are required; - Giα proteins are not required.
Giα proteins differentially regulate ERK1/2, Akt and mTORC1 activation in breast cancer cells and are important for their in vitro growth and invasion
Growth factor signaling plays a vital role in breast cancer pathogenesis [22-24]. We investigated whether Gi1α, Gi2α and G3α also regulate ERK1/2, Akt and mTORC1 activation by growth factors in breast cancer cells. Knockdown of Gi1/3 in MDA-MB-231-D3H2-LN (MB231) (Caliper Life Science), which expressed abundant Gi2α and Gi3α as well as detectable Gi1α (Figures 3A to 3C), had a less striking effect on phosphorylation of ERK1/2(T202/Y204), Akt(S473) and S6(S235/236) than that in MEFs in response to EGF (Figure 3A). Knockdown of Gi2α reduced Akt(S473) phosphorylation but did not impair phosphorylation of ERK1/2 and S6(S235/236) by EGF (Additional file 2: Figure S2). However, knockdown of Gi1α, Gi2α and G3α (Gi1/2/3) largely impaired this phosphorylation by EGF (Figure 3A) indicating that all three Giα proteins are important for the activation of Akt, mTORC1 and ERK1/2 by EGF. Similarly, knockdown of Gi1/2/3 led to a clear defect in phosphorylation of Akt(S473) and S6(S235/236) but not ERK1/2(T202/Y204) in response to bFGF (Figure 3B). Knockdown of either Gi1/3 or Gi1/2/3 clearly impaired ERK1/2 phosphorylation, and slightly reduced Akt(S473) phosphorylation in response to IGF-1 (Figure 3C).
Figure 3.

Giα proteins regulate differential activation of ERK1/2, Akt and mTORC1 in breast cancer cells in response to EGF, bFGF and IGF-1. MB231 cells were transfected with mock or siRNA duplexes (combination of Gi1α and Gi3α, or Gi1α, Gi2α and Gi3α). Cells were then treated with (A) EGF (100 ng/ml), (B) bFGF (25 ng/ml), or (C) IGF-1 (20 ng/ml) for the indicated time durations. p-AKT (S473), p-ERK (T202/Y204), p-S6 (S235/236), Gi1α (Gi1), Gi2α (Gi2), Gi3α (Gi3) or β-actin were detected by IB. The experiments were repeated two to three times and similar results were obtained.
Because Akt and ERK1/2 signaling is required for cell growth and migration in response to growth factors [25], based on above observations, we reasoned that Giα proteins are involved in growth and invasion of breast cancers in response to EGF, bFGF, IGF-1 and FBS. To test this hypothesis we performed in vitro growth and invasion assays. We observed that knockdown of Gi1/2/3 not only clearly reduced the growth rate of breast cancer cells (Figures 4A and 4B, Additional file 3: Figure S3), but also significantly diminished their invasion ability in response to EGF, bFGF, IGF-1 and FBS (Figures 4C and 4D). Interestingly, knockdown of Gi1/3 reduced the invasion ability of breast cancer cells in response to these growth factors, although the reduction was not significant (Figures 4C and 4D). Additionally, knockdown of Gi2α slightly reduced EGF-, bFGF-, or IGF-1-induced breast cancer invasion (Additional file 4: Figure S4). Taken together, our results suggest that Giα proteins promote growth and invasion of breast cancer cells.
Figure 4.

Giα proteins are critical for breast cancer cell invasion in response to EGF, bFGF and IGF-1. MB231 cells were transfected with scrambled or siRNA duplexes (combination of Gi1α and Gi3α, or Gi1α, Gi2α and Gi3α). After 24 hours, cells were trypsinized. (A-B) Cells were seeded in 6-well plates, grown in DMEM containing 5% or 10% FBS and followed for up to 6 days. (C-D) Cells were seeded in new plates for another 48 hours. Cells were re-trypsinized, seeded into 8 μm transwells pre-coated with matrigel and exposed to EGF (100 ng/ml), bFGF (25 ng/ml), IGF-1 (20 ng/ml), 10% FBS or left untreated (with serum free medium) for 36 hours. Penetrated cells were stained and recorded by an Olympus CKX41 microscopy with an Infinity 2 camera. *P value <0.01 (invaded breast cancer cells transfected with Gi1α, Gi2α and Gi3α siRNA duplexes were compared with cells transfected with scrambled siRNA duplexes). The experiments were repeated three times and similar results were obtained.
To understand how Giα proteins regulate ERK1/2, Akt and mTORC1 activation and breast cancer invasion induced by growth factors in normal and breast cancer cells, we first examined whether Giα proteins are involved in the activation of FAK1, which is important for cell migration, and Shp2, which is critical for the activation of ERK1/2 by EGF and IGF-1. Consistent with our previous report [20] and above results, Gi1/3 DKO MEFs exhibited a defect in phosphorylation of Akt(S473), ERK1/2 and Gab1(Y627) in response to EGF (Figure 5A; Additional file 5: Figure S5A). Also, Gi1/3 DKO MEFs showed impaired S6(S235/236) and Shp2 phosphorylation in response to IGF-1 (Figures 5B; Additional file 5: Figure S5B), and diminished Shp2 phosphorylation in response to EGF (Figure 5A) and bFGF (Figure 5C), whereas they showed a slightly reduced FAK1(Y397) phosphorylation in response to EGF, bFGF and IGF-1 (Figures 5A to 5C).
Figure 5.

Giα proteins are required for phosphorylation of Shp2 but not FAK1 in response to EGF, bFGF and IGF-1. WT and Gi1/3 DKO MEFs were treated with (A) EGF (100 ng/ml), (B) bFGF (25 ng/ml), or (C) IGF-1 (20 ng/ml) for 0, 2, 5 or 15 min. p-FAK(Y397), p-Gab1(Y627), p-Shp2(Y508) and β-actin were detected by IB via anti-p-FAK(Y397), anti-p-Gab1(Y627), anti-p-Shp2(Y508) and anti-β-actin antibodies, respectively. Gab1 and Shp2 were also detected by IB (Additional file 5: Figure S5). Experiments were repeated two times and similar results were obtained.
Next, we examined whether Giα proteins associate with growth factor receptors, their downstream adaptor proteins and Shp2. As shown, bFGF induced Gi3α’s associations with p-FGFR and Gab1 (Figure 6A), but did not further augment the pre-association of Gi3α with FRS2 (Figure 6A). EGF induced the associations of Gi3α with p-EGFR, Gab1 and Shp2 (Figures 6B to 6D) in breast cancer cells.
Figure 6.
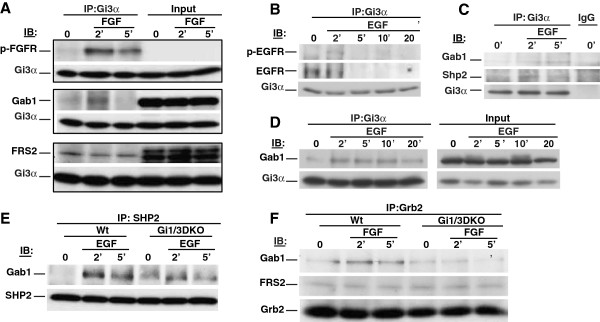
Giα proteins associate with RTKs and their adaptor proteins in breast cancer cells, and are important for interactions of Gab1 with Shp2 or Grb2 in MEFs in response to EGF or bFGF. (A) MB231 cells were treated with bFGF (25 ng/ml) for 0, 2 or 5 min. Whole cell lysates were prepared. The associations of Gi3α with p-FGFR, Gab1 and FRS2 were determined by IB using anti-p-FGFR(Y766), anti-Gab1, anti-FRS2 and anti-Gi3α antibodies, respectively. (B-D) MB231 cells were treated with EGF (100 ng/ml) for 0, 2, 5, 10 or 20 min. The associations of Gi3α with p-EGFR, EGFR (B), Gab1 and Shp2 (C & D) were determined by IB using anti-p-EGFR (Y1173), anti-EGFR, anti-Gab1, anti-Shp2 and anti-Gi3α antibodies, respectively. (E & F) WT and Gi1/3 DKO MEFs were treated with EGF or bFGF for 0, 2 or 5 min. The association of Shp2 with Gab1 in response to EGF (E), and the associations of Grb2 with Gab1 and FRS2 in response to bFGF (F) were determined by IB. Experiments were repeated two to four times. Similar results were obtained.
Because Gab1’s association with Shp2 is critical for ERK1/2 activation by EGF and IGF-1 [15,16,26], and because Grb2’s association with FRS2 is critical for this activation by bFGF [18,27], we determined if Giα proteins are important for these associations. As shown, EGF induced Gab1’s interaction with Shp2 in WT cells, but this interaction was impaired in Gi1/3 DKO MEFs (Figure 6E). The interaction of Gab1 with Grb2, which is critical for activation of the PI3K/Akt pathway by FGF [18], was also impaired in Gi1/3/ DKO MEFs (Figure 6F). Conversely, Grb2’s interaction with FRS2 was intact in Gi1/3 DKO MEFs (Figure 6F). Taken together, our results suggest that Giα proteins are important for Gab1’s interaction with Shp2 in response to EGF and IGF-1, and Grb2’s interaction with Gab1 in response to bFGF.
Discussion
Giα proteins are known to transduce signals from GiαPCRs to their downstream cascades and function at the inner surface of the cell membrane. Here, we demonstrate that Giα proteins also act as transducers for differential RTK-mediated growth factor signaling. Giα proteins form a complex with RTKs and are important for phosphorylation of Gab1 and Shp2, the association of Gab1 with Shp2, as well as the interaction of Gab1 with Grb2 leading to activation of the ERK1/2 and Akt/mTORC1 signaling pathways.
Functional differences among Giα proteins in the regulation of growth factor signaling
Genetic evidence reveals that Gi2/3α DKO mice die between embryonic days 9 and 10 due to severe mass growth retardation [3]. Although the exact death reason is currently unclear, the phenotype of the embryonic lethality strongly suggests that Giα proteins play an essential role in the cell proliferation and growth and survival of embryos.
There are several avenues for Giα proteins to contribute to proliferation and growth. One of them is the activation of GiαPCRs, which transduce signals from their agonists to their downstream PTX-sensitive signal cascades such as the PI3K/Akt/mTORC1 and ERK1/2 signaling pathways [4]. Activated GiαPCRs induce guanine nucleotide exchange at Giα proteins leading to their dissociation from Gβγ subunits. Dissociated Gβγ can trigger the activation of the both pathways [6-8].
Another route is the crosstalk between GiαPCRs and RTKs. RTKs can be transactivated by their agonists, which are either expressively induced by activation of GiαPCRs or secreted via GiαPCR-induced shedding of precursors of growth factors such as pro-HB-EGF [28,29]. Secreted HB-EGF binds to EGFR triggering the activation of multiple important signaling pathways including the PI3K/Akt/mTORC1 and ERK1/2 pathways. The GiαPCR-mediated shedding of HB-EGF is PTX sensitive, whereas EGFR-mediated activation of the PI3K/Akt/mTORC1 and ERK1/2 pathways by EGF is PTX insensitive [20,21]. In this study, we also found that PTX did not block the activation of ERK1/2 by EGF (Additional file 1: Figure S1F).
We demonstrate a possible third avenue in which Giα proteins differentially regulate RTK-mediated activation of the PI3K/Akt/mTORC1 and ERK1/2 signaling pathways. We previously showed that Giα proteins acted downstream of EGFR, but upstream of Gab1, leading to Gab1 phosphorylation and its subsequent interaction with PI3Kp85 in PI3K/Akt/mTORC1 signaling [20]. In this study, we show that Giα proteins are also important for ERK1/2 activation by EGF. Giα proteins formed a complex with the EGFR, its adaptor Gab1 and phosphatase Shp2 in response to EGF, and loss of Giα proteins impaired EGF-induced interaction of Gab1 with Shp2, which is crucial for the activation of ERK1/2 by EGF [30]. As a consequence, loss of Giα proteins led to a defect in the activation of Akt and ERK1/2 by EGF and HB-EGF. However, the relationship between Giα proteins and EGFR is currently unclear. A recent study suggested that EGF induced the formation of a Giα-GIV/Girdin-EGFR complex activating the PI3K-Akt pathway, and the interaction of Gi3α with EGFR was dependent on the guanine exchange factor (GEF) motif in GIV [31]. GIV catalyzed the conversion of GDP-Gi3α to GTP-Gi3α, and its GEF motif was important for autophosphorylation of EGFR and its localization and degradation in response to EGF [31]. However, it is unknown whether the conversion of GDP-Gi3α to GTP-Gi3α by GIV regulates its downstream Gβγ subunits in EGF signaling.
Unlike EGFRs, insulin receptor (IR), IGF-1 receptor (IGF-1R) and FGF receptors (FGFRs) have their own adaptor proteins: FRS2 and IRSs, respectively. FRS2 is crucial for intracellular signaling in response to FGF [18,32] and its interaction with Grb2 is critical for ERK1/2 activation by bFGF [33]. We found that this association was intact in Giα deficient cells. This explains why ERK1/2 activation by aFGF and bFGF was observed in Giα deficient cells. Further, FRS2 interacts with Gab1 and is required for Gab1’s associations with Grb2 and PI3Kp85 as well as PI3K activation [27,34]. We identified that FRS2’s interaction with Gab1 or the interaction of Gab1 with Grb2 was impaired in Giα deficient cells. As a result, the activation of Akt and mTORC1 by aFGF and bFGF was diminished in Giα deficient cells revealing a new Giα/FRS2/Grb2/Gab1-PI3K cascade in FGF signaling.
IR and IGFR bind IRSs, which directly interact with PI3Kp85 leading to the activation of PI3K, Akt and mTORC1 [35]. Therefore, the activation of Akt and mTORC1 by IGF-1 and insulin was not largely reduced in Giα-deficient MEFs or was even enhanced in Gab1-deficient liver in response to insulin [36]. Conversely, the activation of ERK1/2 was not altered in IRS-1-deficient cells in response to IGF-1 [19], indicating that other proteins are required for ERK1/2 activation by IGF-1. Indeed, our data showed that loss of Giα proteins severely impaired ERK1/2 activation by IGF-1 and insulin. Intriguingly, although loss of Giα proteins exhibited a minor defect in Akt activation, the S6 phosphorylation was clearly impaired in Gi1/3-deficient MEFs (Figures 1E and 1F). The possible interpretation is that S6 can also be phosphorylated by other protein kinases such as p90RSK, which is a substrate of ERK1/2 and is able to phosphorylate S6 when mTORC1 activity is inhibited [37]. In addition, our data showed that PTX was able to reduce insulin-induced ERK1/2 activation (Additional 1: Figure S1F). This result suggests that GiαPCRs may be involved in the activation of ERK1/2 by insulin.
Giα proteins in breast cancer
Giα proteins are highly expressed in some breast cancer cells, especially in metastatic breast cancer cells [38]. Giα proteins have been implicated in breast cancer invasion in response to SDF1, which is the only ligand for GiαPCR CXCR4. Upregulation of CXCR4 is vital for Her2-mediated metastasis of breast cancer [39,40]. Interestingly, the association of Giα proteins with Gβγ is considered a prerequisite for functional CXCR4 [38]. In the present study, we found that Giα proteins not only regulated SDF1-mediated invasion of breast cancer (data not shown), but also controlled EGF-, bFGF-, IGF-1- and serum-induced MB231 invasion. Additionally, Giα proteins regulated breast cancer growth.
Overexpression of RTKs such as EGFR family members and FGFRs is a hallmark of breast cancer. This phenotypic overexpression promotes activity of RTKs leading to overactivation of their downstream events and cascades, including Gabs [41,42], Shp2 [43] and the PI3K/Akt/mTORC1 and Ras/ERK1/2 signaling pathways leading to progression, invasion and metastasis of breast cancer [44,45]. We showed that Giα proteins associated with active EGFR and FGFR, as well as their downstream adaptor proteins and Shp2 in response to respective agonists. Further, Giα proteins can differentially regulate the activation of Akt, mTORC1 and ERK1/2 by EGF, bFGF and IGF-1 in breast cancer cells.
In summary, our results demonstrate that Giα proteins are important regulators of RTK signaling in normal cells and in breast cancer cells. Giα proteins differentially regulate the activation of Akt, mTORC1 and ERK1/2 by different growth factors. Given the fact that growth factors and their RTKs are critical for breast cancer progression, invasion and metastasis, pharmacological interference of Giα protein function may provide an alternative therapeutic strategy for breast cancer treatment.
Materials and method
Antibodies
Antibodies raised against EGFR (sc-03), Gi1α (sc-391), Gi2α (sc-7276), Gi3α (sc-262), AKT1/2 (sc-8312), ERK1/2 (sc-94), FRS2 (sc-8318) and SH-PTP2 (Shp2, sc-280) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, Mo). Anti-EGFR, anti-FGFR, anti-Gab1, anti-IGF-1R, and other anti-phospho antibodies were purchased from Cell Signaling Technology (Danvers, MA).
Cell line and culture
WT and Gi1/3 DKO MEFs were characterized and cultured as previous described [20]. MDA-MB-231-D3H2-LN (MB231) cells were purchased from Caliper Life Science (Hopkinton, MA). Cells were cultured in DMEM high-glucose medium supplemented with 10%FBS in a CO2 incubator at 37°C.
Immunoblotting analysis
WT, DKO, transfected or siRNA knockdown cells were starved overnight (o/n), and then treated with bFGF, EGF, HB-EGF (Invitrogen), aFGF (Cell Signaling), IGF-1 (Invitrogen and R & D Systems), or insulin (BD Pharmingen) for the indicated time points. Cells were washed twice with cold 1× phosphate buffered saline (PBS) and lysed with a lysis buffer [1% Triton ×-100, 150 mM NaCl, 50 mM Tris-Cl, pH 7.4, 0.5 mM EDTA, 1 mM sodium orthovanadate, 5 mM beta glycerol phosphate, 1× protease inhibitor cocktail (Roche, Indianapolis, IN)] [46]. Total of 30–60 μg protein lysates were boiled in the sample buffer, separated on 10% SDS-PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA) followed by incubation with antibodies and detection with ECL (Pierce, Rockford, IL).
Immunoprecipitation
Cells were cultured to 70% confluence and then starved o/n. Cells were treated with EGF, or bFGF for different time points and then lysed with the lysis buffer. For each sample, 600–800 μg of proteins were pre-cleared by incubation with 40 μl of protein A/G Sepharose (beads) (Amersham, IL) for 30 minutes at 4°C. After an overnight incubation at 4°C with specific antibodies (2–4 μg/mg protein), 40 μl of protein A/G bead suspensions were added to the complexes and incubated for another 2 hours. Then the beads were washed three times with washing buffer (lysis buffer containing 1 mM PMSF) and once with 1× PBS. The boiled proteins were separated on 10% SDS-PAGE gels and transferred onto PVDF membranes followed by incubation with different antibodies and detection with ECL.
Plasmid transfection
Lipofectamine 2000 (Invitrogen) was used as the transfection reagent. 3.5×105 Gi1/3 DKO cells were seeded in 6-well plates and cultured in DMEM supplemented with 10% FBS without antibiotics. Five μg Gi1α or Gi3α plasmids in 250 μl of serum free medium (SFM) and 10 μl Lipofectamine 2000 in 250 μl of SFM were gently mixed and incubated for 30 min. The plasmid-Lipofectamine complexes were then added to each well. After 5 hours the medium was replaced with complete medium. Cells were cultured for additional 14 hours, starved o/n and then treated with growth factors as described above.
siRNA transfaction
Human and mouse Gi1α, Gi2α, and Gi3α siRNA duplexes were purchased from Santa Cruz Biotechnology. Cells were cultured in 10% FBS DMEM in the absence of antibiotics to 70% confluence at the time of transfection. Lipofectimine RNAiMAX Reagent (7.5 μl, Invitrogen) in 250 μl SFM, and 3.5 μl of Gi1α, Gi2α or Gi3α siRNA duplexes in another 250 μl SFM were gently combined, mixed, and incubated for 25 min at room temperature. The siRNA duplexes-RNAiMAX complexes were added to culture with a final RNA concentration of 10 nM in a total volume of 3 ml. After 16 hours, the medium was replaced by complete medium. Cells were cultured for another 24 hours and then starved o/n followed by treatment for immunoblotting analysis or in vitro invasion assay.
Breast cancer cell in vitro invasion assay
Cell invasion assay was performed using Costar Transwell permeable support (8.0-μm pore size, Corning, NY) in 24-well plates. The starved mock, Gi1/3, Gi1/2/3 siRNA knockdown MB231 cells were harvested with trypsin, resuspended at 5×104 cells/ml in SFM containing 0.1% BSA, and 200 μl of each type of cells were added to the upper chambers coated with Matrigel (1 mg/ml, BD Pharmingen). The lower chambers were filled with 600 μl SFM or SFM containing EGF, IGF-1, bFGF or 10%FBS. After 36 hours culture, cells were stained with HEME 3 Solution (Fisher Diagnostics, Middletown, VA), and non-invaded cells on the top of the transwell were removed with cotton swabs. Invaded cells were counted under an Olympus CKX41 microscopy (Olympus, Japan) and recorded by an Infinity 2 camera (Electron Microscopy Sciences, Hatfield, PA).
Breast cancer cell growth assay
siRNA knockdown MB231 cells (scrambled siRNA duplexes (mock) or Gi1/2/3 siRNA duplexes) (1×104 cells/well) were seeded in 6-well plates in DMEM medium containing 5% or 10% FBS. From day 1, at least three fields in each well were randomly selected for recording images and counting cells under an Olympus CKX41 microscopy with an Infinity 2 camera. Experiments were repeated twice.
Statistical analysis
Statistical difference between two groups was evaluated using Student’s t test. P values less than 0.05 were considered significant. Data were presented as mean ± SD.
Abbreviations
Giα: Inhibitory guanosine nucleotide-binding protein alpha; GiαPCRs: Giα protein-coupled receptors; EGF: Epidermal growth factor; HB-EGF: Heparin-binding EGF-like growth factor; FGF: Fibroblast growth factor; IGF-1: Insulin-like growth factor 1; PDGF: Platelet-derived growth factor; EGFR: EGF receptor; FGFR: FGF receptor; IR: Insulin receptor; IGF-1R: IGF-1 receptor; IRS: Insulin receptor substrate; FRS2: FGF receptor substrate 2; RTK: Receptor tyrosine kinase; Gab1: Grb2-associated-binding protein 1; Grb2: Growth factor receptor-bound protein 2; Shp2: SH2-containing tyrosine-specific protein phosphatase; ERK1/2: Extracellular signal-regulated protein kinase 1 and 2; PI3K: Phosphatidylinositide 3-kinase; mTORC1: Mammalian target of rapamycin complex 1; MEFs: Mouse embryonic fibroblasts; RNAi: RNA interference; siRNA: Small interfering RNA; KO: Knockout; PTX: Bordetella pertussis toxin.
Competing interests
Authors declare that they do not have competing interests.
Authors’ contributions
WMC and MJ designed experiments. WMC, MJ and ZW wrote the paper. LB and GSF edited and revised the paper, and also created key reagents. ZW, RDZ, FJ, SG, JZ, and YW did experiments. All authors read and approved the final manuscript.
Supplementary Material
(A) Left panel: Expression of Gi1α (Gi1), Gi2α (Gi2) and Gi3α (Gi3) in WT and Gi1/3 DKO MEFs. WT and Gi1/3DKO MEFs were lysed and the expression levels of Giα proteins were determined by IB using anti-Gi1α, anti-Gi2α and anti-Gi3α antibodies, respectively. Right panel: Expression of Gi1 and Gi2 in mouse brain tissues. Whole cell lysates of WT, Gi2αF/F/Mx1Cre (Gi2F/F/Mx1CRE) (without induction) and Gi2αF/F/Gi1/3DKO (Gi2F/F/Gi1/3DKO) mouse brain tissues were used to determine the expression levels of Gi1 and Gi2. (B-D) The expression levels of Akt, ERK1/2, Gab1, EGFR, FGFR, IGF-1R and β-actin in WT and Gi1/3 DKO MEFs were determined by IB. (E) Loss of Gi2α (Gi2) did not decrease ERK1/2 activation by EGF. (F) PTX did not block ERK activation by EGF, but reduced ERK1/2 activation by insulin.
Knockdown of Gi2α reduced Akt(S473) phosphorylation, but not did inhibit ERK1/2 and S6 phosphorylation in response to EGF. MB231 cells were transfected with scrambled (mock) or Gi2αsiRNA (Gi2siRNA) duplexes. Cells were treated with EGF for the indicated time points. p-Akt(S473S), p-ERK1/2(T202/Y204), p-S6(S235/236), Gi2α (Gi2) and β-actin were detected by IB.
Knockdown of Gi1α, Gi2α and Gi3α in breast cancer cells impairs their growth ability. MB231 cells were transfected with scrambled or siRNA duplexes [combination of Gi1α, Gi2α and Gi3α (Gi1/2/3)]. After 24 hours, cells were trypsinized, seeded in 6-well plates and cultured in DMEM containing 5% or 10% FBS for up to 6 days. Cells were observed under an Olympus CKX41 microscope and recorded by an Infinity 2 camera.
Knockdown of Gi2α in breast cancer cells did not impair their invasion ability in response to EGF, bFGF and IGF-1. MB231 cells were transfected with scrambled or Gi2αsiRNA (Gi2siRNA) duplexes. Cells were seeded in new plates for another 48 hours. Cells were re-trypsinized, seeded into 8 μm transwells pre-coated with matrigel and exposed to EGF (100 ng/ml), bFGF (25 ng/ml), IGF-1 (20 ng/ml), or left untreated (with serum free medium) for 36 hours. Penetrated cells were stained and recorded.
Loss of Gi1α and Gi3α impairs Akt(S473) and ERK1/2(T202/Y204) phosphorylation in response to EGF, and S6(S235/236) phosphorylation in response to IGF-1.
Contributor Information
Zhanwei Wang, Email: zwang@cc.hawaii.edu.
Rica Dela Cruz, Email: delacruz.rica@gmail.com.
Fang Ji, Email: jtj3210@hotmail.com.
Sheng Guo, Email: sguo@cc.hawaii.edu.
Jianhua Zhang, Email: zjh12195@126.com.
Ying Wang, Email: wangying@ucla.edu.
Gen-Sheng Feng, Email: gfeng@ucsd.edu.
Lutz Birnbaumer, Email: birnbau1@niehs.nih.gov.
Meisheng Jiang, Email: jm@ucla.edu.
Wen-Ming Chu, Email: wchu@cc.hawaii.edu.
Acknowledgements
We are grateful to Drs. Cong Cao, Xuesong Huang and John Marshall for their valuable advice and technical support. We thank Dr. David Ward for critical discussion. This work was supported in part by grants from DOD (PR054819), NIH (AI054128), Hawaii Community Foundation and Leukemia and Lymphoma Society to WMC, NIH (DK069771) to MJ, and in part from the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH (project 201–101643) to LB.
References
- Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Bradley A, Birnbaumer L. Gi2 alpha protein deficiency: a model of inflammatory bowel disease. J Clin Immunol. 1995;15:101S–105S. doi: 10.1007/BF01540899. [DOI] [PubMed] [Google Scholar]
- Gohla A, Klement K, Piekorz RP, Pexa K, Vom Dahl S, Spicher K, Dreval V, Haussinger D, Birnbaumer L, Nurnberg B. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A. 2007;104:3003–3008. doi: 10.1073/pnas.0611434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Kawano T, Nagao M, Kaziro Y, Itoh H. G (i)-dependent activation of c-Jun N-terminal kinase in human embryonal kidney 293 cells. J Biol Chem. 2000;275:7633–7640. doi: 10.1074/jbc.275.11.7633. [DOI] [PubMed] [Google Scholar]
- Coso OA, Teramoto H, Simonds WF, Gutkind JS. Signaling from G protein-coupled receptors to c-Jun kinase involves beta gamma subunits of heterotrimeric G proteins acting on a Ras and Rac1-dependent pathway. J Biol Chem. 1996;271:3963–3966. doi: 10.1074/jbc.271.8.3963. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci U S A. 1994;91:12706–12710. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle JF, Murthy KS. Coupling of the insulin-like growth factor-I receptor tyrosine kinase to Gi2 in human intestinal smooth muscle: Gbetagamma -dependent mitogen-activated protein kinase activation and growth. J Biol Chem. 2001;276:7187–7194. doi: 10.1074/jbc.M011145200. [DOI] [PubMed] [Google Scholar]
- Stryjek-Kaminska D, Piiper A, Zeuzem S. Epidermal growth factor regulates adenylate cyclase activity via Gs and Gi1-2 proteins in pancreatic acinar membranes. Biochem J. 1996;316(Pt 1):87–91. doi: 10.1042/bj3160087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM, McCubrey JA. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY, Wu J. Regulation of the mitogen-activated protein kinase signaling pathway by SHP2. J Biol Chem. 2002;277:9498–9504. doi: 10.1074/jbc.M110547200. [DOI] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Yuan J, Droin N, Long S, Zhang EE, Nguyen TV, Feng GS. Concerted functions of Gab1 and Shp2 in liver regeneration and hepatoprotection. Mol Cell Biol. 2006;26:4664–4674. doi: 10.1128/MCB.02253-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta. 2007;1773:1161–1176. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeper U, Vogel R, Chmielowiec J, Huelsken J, Rosario M, Birchmeier W. Distinct requirements for Gab1 in Met and EGF receptor signaling in vivo. Proc Natl Acad Sci U S A. 2007;104:15376–15381. doi: 10.1073/pnas.0702555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Yuan J, Liu H, Shi Z, Baker K, Vuori K, Wu J, Feng GS. Role of Gab1 in UV-induced c-Jun NH2-terminal kinase activation and cell apoptosis. Mol Cell Biol. 2004;24:1531–1539. doi: 10.1128/MCB.24.4.1531-1539.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamothe B, Yamada M, Schaeper U, Birchmeier W, Lax I, Schlessinger J. The docking protein Gab1 is an essential component of an indirect mechanism for fibroblast growth factor stimulation of the phosphatidylinositol 3-kinase/Akt antiapoptotic pathway. Mol Cell Biol. 2004;24:5657–5666. doi: 10.1128/MCB.24.13.5657-5666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JC, Winnay J, Cheatham B, Kahn CR. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 1997;17:1513–1521. doi: 10.1128/mcb.17.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, Marshall J, Jiang M, Chu WM. Galpha (i1) and Galpha (i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17. doi: 10.1126/scisignal.2000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Biesen T, Hawes BE, Luttrell DK, Krueger KM, Touhara K, Porfiri E, Sakaue M, Luttrell LM, Lefkowitz RJ. Receptor-tyrosine-kinase- and G beta gamma-mediated MAP kinase activation by a common signalling pathway. Nature. 1995;376:781–784. doi: 10.1038/376781a0. [DOI] [PubMed] [Google Scholar]
- Eccles SA. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol. 2011;55:685–696. doi: 10.1387/ijdb.113396se. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17:251–261. doi: 10.1007/s10911-012-9268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M, Elia A, Ong SH, Stambolic V, Pawson T, Tsao MS, Mak TW. grb2 heterozygosity rescues embryonic lethality but not tumorigenesis in pten+/- mice. Proc Natl Acad Sci U S A. 2004;101:15358–15363. doi: 10.1073/pnas.0406613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A. 2001;98:8578–8583. doi: 10.1073/pnas.161259898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. Embo J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Disco. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Cunnick JM, Mei L, Doupnik CA, Wu J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem. 2001;276:24380–24387. doi: 10.1074/jbc.M010275200. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, Ear J, Jung BH, Cabrera B, Carethers JM, Farquhar MG. A G {alpha} i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell. 2010;21:2338–2354. doi: 10.1091/mbc.E10-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh N. Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. 2008;99:1319–1325. doi: 10.1111/j.1349-7006.2008.00840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001;98:6074–6079. doi: 10.1073/pnas.111114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont J, Tesseraud S, Simon J. Insulin signaling in chicken liver and muscle. Gen Comp Endocrinol. 2009;163:52–57. doi: 10.1016/j.ygcen.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Hevener AL, Long S, Zhang EE, Olefsky JM, Feng GS. Deletion of Gab1 in the liver leads to enhanced glucose tolerance and improved hepatic insulin action. Nat Med. 2005;11:567–571. doi: 10.1038/nm1227. [DOI] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JD, Kochetkova M, Akekawatchai C, Dottore M, Lopez A, McColl SR. Differential functional activation of chemokine receptor CXCR4 is mediated by G proteins in breast cancer cells. Cancer Res. 2006;66:4117–4124. doi: 10.1158/0008-5472.CAN-05-1631. [DOI] [PubMed] [Google Scholar]
- Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Ke Y, Wu D, Princen F, Nguyen T, Pang Y, Lesperance J, Muller WJ, Oshima RG, Feng GS. Role of Gab2 in mammary tumorigenesis and metastasis. Oncogene. 2007;26:4951–4960. doi: 10.1038/sj.onc.1210315. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, Harris LN, Richardson A, Neel BG, Gu H. A role for the scaffolding adapter GAB2 in breast cancer. Nat Med. 2006;12:114–121. doi: 10.1038/nm1341. [DOI] [PubMed] [Google Scholar]
- Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1beta in vivo and in vitro. Breast Cancer Res Treat. 2005;89:5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77:400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- Foley J, Nickerson NK, Nam S, Allen KT, Gilmore JL, Nephew KP, Riese DJ 2nd. EGFR signaling in breast cancer: bad to the bone. Semin Cell Dev Biol. 2010;21:95–960. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoi AM, Fu X, Ivanov S, Zhang P, Sheng L, Wu D, Li GC, Chu WM. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J. 2005;24:779–789. doi: 10.1038/sj.emboj.7600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Left panel: Expression of Gi1α (Gi1), Gi2α (Gi2) and Gi3α (Gi3) in WT and Gi1/3 DKO MEFs. WT and Gi1/3DKO MEFs were lysed and the expression levels of Giα proteins were determined by IB using anti-Gi1α, anti-Gi2α and anti-Gi3α antibodies, respectively. Right panel: Expression of Gi1 and Gi2 in mouse brain tissues. Whole cell lysates of WT, Gi2αF/F/Mx1Cre (Gi2F/F/Mx1CRE) (without induction) and Gi2αF/F/Gi1/3DKO (Gi2F/F/Gi1/3DKO) mouse brain tissues were used to determine the expression levels of Gi1 and Gi2. (B-D) The expression levels of Akt, ERK1/2, Gab1, EGFR, FGFR, IGF-1R and β-actin in WT and Gi1/3 DKO MEFs were determined by IB. (E) Loss of Gi2α (Gi2) did not decrease ERK1/2 activation by EGF. (F) PTX did not block ERK activation by EGF, but reduced ERK1/2 activation by insulin.
Knockdown of Gi2α reduced Akt(S473) phosphorylation, but not did inhibit ERK1/2 and S6 phosphorylation in response to EGF. MB231 cells were transfected with scrambled (mock) or Gi2αsiRNA (Gi2siRNA) duplexes. Cells were treated with EGF for the indicated time points. p-Akt(S473S), p-ERK1/2(T202/Y204), p-S6(S235/236), Gi2α (Gi2) and β-actin were detected by IB.
Knockdown of Gi1α, Gi2α and Gi3α in breast cancer cells impairs their growth ability. MB231 cells were transfected with scrambled or siRNA duplexes [combination of Gi1α, Gi2α and Gi3α (Gi1/2/3)]. After 24 hours, cells were trypsinized, seeded in 6-well plates and cultured in DMEM containing 5% or 10% FBS for up to 6 days. Cells were observed under an Olympus CKX41 microscope and recorded by an Infinity 2 camera.
Knockdown of Gi2α in breast cancer cells did not impair their invasion ability in response to EGF, bFGF and IGF-1. MB231 cells were transfected with scrambled or Gi2αsiRNA (Gi2siRNA) duplexes. Cells were seeded in new plates for another 48 hours. Cells were re-trypsinized, seeded into 8 μm transwells pre-coated with matrigel and exposed to EGF (100 ng/ml), bFGF (25 ng/ml), IGF-1 (20 ng/ml), or left untreated (with serum free medium) for 36 hours. Penetrated cells were stained and recorded.
Loss of Gi1α and Gi3α impairs Akt(S473) and ERK1/2(T202/Y204) phosphorylation in response to EGF, and S6(S235/236) phosphorylation in response to IGF-1.