

Cells depleted of Ska arrest at metaphase. Chromosome-associated APC/C shows high ubiquitylation activity. Ska complex helps to localize APC/C on chromosomes. Forced localization of Ska complex to kinetochores enhances APC/C localization to chromosomes and promotes APC/C activity.
Abstract
The spindle and kinetochore–associated (Ska) protein complex is a heterotrimeric complex required for timely anaphase onset. The major phenotypes seen after small interfering RNA–mediated depletion of Ska are transient alignment defects followed by metaphase arrest that ultimately results in cohesion fatigue. We find that cells depleted of Ska3 arrest at metaphase with only partial degradation of cyclin B1 and securin. In cells arrested with microtubule drugs, Ska3-depleted cells exhibit slower mitotic exit when the spindle checkpoint is silenced by inhibition of the checkpoint kinase, Mps1, or when cells are forced to exit mitosis downstream of checkpoint silencing by inactivation of Cdk1. These results suggest that in addition to a role in fostering kinetochore–microtubule attachment and chromosome alignment, the Ska complex has functions in promoting anaphase onset. We find that both Ska3 and microtubules promote chromosome association of the anaphase-promoting complex/cyclosome (APC/C). Chromosome-bound APC/C shows significantly stronger ubiquitylation activity than cytoplasmic APC/C. Forced localization of Ska complex to kinetochores, independent of microtubules, results in enhanced accumulation of APC/C on chromosomes and accelerated cyclin B1 degradation during induced mitotic exit. We propose that a Ska-microtubule-kinetochore association promotes APC/C localization to chromosomes, thereby enhancing anaphase onset and mitotic exit.
INTRODUCTION
The metaphase–anaphase transition is a decision node for launching the irreversible events of chromatid segregation and mitotic exit. If metaphase is unusually prolonged by any one of several defects or interventions, chromosomes may undergo cohesion fatigue, by which the pulling forces of intact spindle microtubules interacting with kinetochores cause chromatids to separate asynchronously (Daum et al., 2011; Stevens et al., 2011). Once separated, single chromatids can exhibit unstable attachments to spindle microtubules, reigniting spindle checkpoint signaling and causing persistent mitotic arrest (Lara-Gonzalez and Taylor, 2012).
Progressive activation of Cdk1 during entry into M phase establishes the mitotic state of chromatin and cytoplasm (Gavet and Pines, 2010). High levels of Cdk1 also prime the machinery that catalyzes subsequent mitotic exit. A key component of this machinery is the anaphase-promoting complex/cyclosome (APC/C), a multisubunit E3 ubiquitin ligase. Bound with its mitotic cofactor, Cdc20, the APC/C catalyzes ubiquitylation of proteins to target them for destruction by the proteasome (King et al., 1995; Sudakin et al., 1995). APC/C-Cdc20 is primed by mitotic phosphorylations catalyzed by Cdk1 and other mitotic kinases (Lahav-Baratz et al., 1995; Kraft et al., 2003). During prometaphase, before chromosomes are fully aligned, APC/C-Cdc20 has selective activity against certain targets such as cyclin A and Nek2A (den Elzen and Pines, 2001; Geley et al., 2001; Hames et al., 2001; Hayes et al., 2006). Its activity against its anaphase targets, securin and cyclin B1, is repressed by the spindle checkpoint, an evolutionarily conserved signaling cascade catalyzed by kinetochores lacking full attachment to spindle microtubules (Musacchio and Salmon, 2007). The spindle checkpoint can be inactivated by treating cells with reversine, a chemical inhibitor of the checkpoint kinase Mps1. Prometaphase or metaphase cells treated with reversine rapidly undergo anaphase and mitotic exit (Santaguida et al., 2010). Cells in mitosis can also be induced to exit mitosis downstream of the spindle checkpoint by direct inhibition of Cdk1, the primary mitotic kinase. Of interest, treatment of cells near or at metaphase with small-molecule inhibitors of Cdk1 drives most of the morphological events of mitotic exit, including cytokinesis, chromosome decondensation, and reorganization of the interphase microtubule network (Potapova et al., 2006, 2009, 2011). Addition of Cdk1 inhibitor to cells in mitosis also drives the biochemical changes characteristic of mitotic exit, including degradation of securin and cyclin B1 by APC/C-Cdc20 (Potapova et al., 2011).
The spindle and kinetochore–associated (Ska) protein complex is a heterotrimeric complex (Ska1, Ska2, Ska3) that accumulates on spindle microtubules and at kinetochores after nuclear envelope breakdown, becoming most enriched on kinetochores at metaphase. In vitro studies show that domains on Ska1 and possibly Ska3 bind to microtubules (Welburn et al., 2009; Jeyaprakash et al., 2012; Schmidt et al., 2012). Previously we and others reported that the major phenotypes seen after small interfering RNA (siRNA)–mediated depletion of the Ska3 protein in cultured cells were transient chromosome alignment defects followed by arrest or delay at metaphase (Daum et al., 2009; Theis et al., 2009). We also found that most cells arrested at metaphase undergo cohesion fatigue (Daum et al., 2011). However, reports from other laboratories suggested that the primary defect in Ska-depleted cells is a persistent failure to align chromosomes at the metaphase plate (Gaitanos et al., 2009; Raaijmakers et al., 2009; Welburn et al., 2009). Thus the roles of the Ska complex in mitosis are controversial. In a proteomic analysis in chicken DT40 cells, deletion of the Ska3 gene resulted in a significant decrease of APC/C components associated with chromosomes (Ohta et al., 2010). These findings suggested that Ska complex might have additional roles in mitotic cell cycle control related to regulation of APC/C activity. Here we show in mammalian cells that kinetochore association of the Ska complex increases binding of APC/C to chromosomes and enhances the efficiency of the degradation machinery to sharpen the metaphase–anaphase transition and promote mitotic exit.
RESULTS
The phenotype of Ska complex depletion
Previously we reported that the predominant phenotype resulting from RNA interference (RNAi)–mediated depletion of Ska3 was metaphase arrest, often followed by the asynchronous separation of sister chromatids (cohesion fatigue; Daum et al., 2009). In addition, we noted defects that slowed but generally did not block chromosome alignment to the metaphase plate. However, some laboratories report that the primary phenotype in Ska-depleted cells is failure of chromosomes to align at metaphase (Gaitanos et al., 2009; Welburn et al., 2009). The cohesion fatigue phenotype produces a scattering of chromatids along the spindle that is easily confused with failed alignment, particularly in endpoint studies of fixed cells, in which the complete history of chromosome behavior is not known. Second, metaphase delays and cohesion fatigue are often accompanied by rotation of the spindle within the cell, further confounding interpretation of fixed samples. To resolve this issue, we used video microscopy with a large set of samples to track phenotypes in cells treated with pooled, multiple siRNAs targeting Ska1, Ska2, and Ska3 alone and in all combinations. We also carried out experiments testing siRNA reagents and cells generously provided by Iain M. Cheeseman (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA). All depletions produced nearly identical results, with the dominant phenotype being metaphase arrest or delay (Figure 1, A and B, Supplemental Figure S1A, and Supplemental Movie S1). Cells arrested at metaphase then underwent cohesion fatigue and chromatid scattering. In early mitosis most chromosomes in each cell aligned rapidly, although a few chromosomes sometimes exhibited delayed alignment (Figure 1A and Supplemental Figure S1B). In addition, some cells exhibited the “escaper” phenotype first described by Hanisch et al. (2006) in their work reporting the discovery of the Ska1 and Ska2 proteins. Escapers are paired whole chromosomes that transiently move off but then return to the metaphase plate (Supplemental Movie S2). However, in all our videos, nearly every cell treated with Ska RNAi ultimately achieved full metaphase alignment of all chromosomes. This alignment sometimes became obscured by rotation of the spindle, but continued tracking through additional video frames nearly always revealed that metaphase alignment was maintained, usually for hours. At some point cells then underwent cohesion fatigue, which was accompanied by scattering along the spindle of both separated and paired chromatids.
FIGURE 1:

Depletion of Ska complex components slows alignment and arrests cells at metaphase. (A) HeLa H2B-GFP cells transfected with control siRNA or with pools of siRNA against Ska1, Ska2, and Ska3 alone or in combination at 25 nM were imaged approximately 27 h after transfection. The time taken to progress through prometaphase and metaphase was determined for every cell and plotted. A strict criterion was used to define metaphase alignment, which required that every chromosome was at the metaphase plate for at least two consecutive frames. The graph depicts the time taken to align chromosomes (blue bar), time spent at metaphase in cells that initiated anaphase (yellow bar), and time spent at metaphase in cells that initiated cohesion fatigue (red bar). The asterisk denotes a cell that exited mitosis after undergoing cohesion fatigue. Ska-depleted cells were delayed in chromosome alignment, although ultimately cells reached metaphase. The majority of Ska-depleted cells delayed or arrested at metaphase. (B) Mitotic phenotypes observed after depletion of Ska proteins. The graph denotes the percentage of cells that initiate anaphase without delay, with delay (>80 min at metaphase), or remain arrested at metaphase, eventually undergoing cohesion fatigue. The majority of Ska-depleted cells either delayed or arrested at metaphase. See also Supplemental Figure S1 and Supplemental Movies S1 and S2.
Because Ska-depleted cells exhibited partial defects in chromosome alignment at metaphase, we sought to determine whether anaphase chromatid movement required normal levels of Ska. Buchholz and colleagues had shown that cells arrested at metaphase by Ska3 depletion could be induced to enter anaphase by addition of a Cdk-inhibitor drug (Theis et al., 2009). We induced anaphase in Ska3-depleted metaphase cells by treating with a Cdk inhibitor and quantified chromatid movements. We found that rates of chromatid separation in anaphase were not detectably affected in cells depleted of Ska3, suggesting that full expression of Ska complex is not required for chromatid separation during anaphase (Supplemental Figure S1C).
Finally, the observation of a mitotic phenotype after RNAi-mediated depletion of a protein does not indicate whether the point of execution of the target protein is before mitosis or during mitosis. We used antibody microinjection to carry out temporally specific inhibition of Ska complex function. Injection of anti-Ska3 antibody into mitotic cells produced phenotypes similar to those obtained by siRNA, allowing us to conclude that the metaphase arrest/cohesion fatigue phenotype reflects a needed role for Ska complex during mitosis rather than being a consequence of an earlier cell cycle function (Supplemental Figure S1D).
The Ska complex potentiates cyclin B1 degradation and the metaphase–anaphase transition
The metaphase–anaphase transition is promoted by APC/C-mediated ubiquitylation and subsequent degradation of cyclin B1 and securin by the proteasome. To understand how cells arrest at metaphase after depletion of Ska proteins, we compared cyclin B1 and securin protein levels in cells arrested in mitosis for 8 h by nocodazole treatment, Ska3 depletion, or both nocodazole plus Ska3 depletion (Figure 2A). In nocodazole-arrested cells with or without Ska3 depletion, cyclin B1 and securin levels remained relatively high. However, in cells arrested by Ska3 depletion alone, cyclin B1 and securin levels were reduced by ∼40% (Figure 2A). The fact that nocodazole treatment counteracted cyclin B1 degradation in Ska3-depleted cells allowed us to characterize the effects of spindle assembly on cyclin B1 breakdown during nocodazole-washout experiments in which control and Ska3-depleted cultures started with approximately equal amounts of cyclin B1 (Figure 2B). After nocodazole release, in control cells Cdk1-associated cyclin B1 and H1 kinase activity declined rapidly. In cells depleted of Ska3 the declines of cyclin B1 and kinase activity were gradual and incomplete (Figure 2B). At 6 h postrelease from nocodazole, Ska3-depleted cells had six times more cyclin B1 associated with Cdk1 than did control cells (Figure 2C).
FIGURE 2:

Ska3-depleted cells are inefficient in cyclin B1 degradation and inactivate Cdk1 incompletely. (A) HeLa cells were transfected with control or Ska3 siRNA at 25 nM final concentration. At 24 h after transfection, control siRNA cultures were treated with 3.3 μM nocodazole for 8 h. Ska3-depleted cultures were washed to remove any preexisting mitotic cells and then treated with dimethyl sulfoxide (DMSO; control) or 3.3 μM nocodazole for 8 h. Mitotic cells were collected and assayed by Western blotting. Ska3-depleted cells degraded 40% cyclin B1 compared with nocodazole-arrested cells. (B) Cultures of control and Ska3-depleted cells were rinsed to remove any preexisting mitotic cells and then blocked in 3.3 μM nocodazole for 4 h. Mitotic cells were collected and then released into fresh medium without drugs. Samples were collected hourly for 6 h. Cdk1 was precipitated using p13-Suc1 beads, and associated cyclin B1 was determined by immunoblotting. Cdk1 kinase activity on histone H1 was also measured. Whole-cell lysates were blotted for Cdk1, cyclin B1, and Ska3. (The ns in the Ska3 blot indicates a nonspecific band that runs between the phosphorylated and dephosphorylated forms of Ska3.) (C) Blot quantification revealed that Cdk1-associated cyclin B1 was sixfold higher in Ska3-depleted cells vs. control cells at 6 h after nocodazole wash out (NWO). (D) Control or Ska3-depleted HeLa H2B-GFP cells were treated with DMSO (control), 3.3 μM nocodazole, or 10 μg/ml cycloheximide (CHX) and then imaged for 24 h. CHX induced mitotic exit in Ska3-depleted cells. The numbers on top of the bars in the graph indicate the number of cells that exited/total number of mitotic cells. (E) Scatterplot depiction of time taken to initiate anaphase in control (25 ± 2.8 min) and Ska3-depleted cells (222 ± 12.9 min) treated with CHX. Each dot represents one cell; long horizontal line depicts mean, and whiskers denote SEM.
Unlike most proteins, cyclin B1 is actively synthesized during early mitosis (Mena et al., 2010). Because cyclin B1 levels declined partially in Ska3-depleted cells, we asked whether protein synthesis was required to maintain mitotic arrest. We found that addition of cycloheximide induced mitotic exit in Ska3-depleted cells with intact spindles after an average of 4 h (Figure 2, D and E). Although the effects of protein synthesis inhibition on other proteins cannot be discounted, this evidence supports the idea that in HeLa cells containing intact spindle microtubules, the reduced degradation of cyclin B1 due to Ska3 depletion is insufficient to drive anaphase onset and mitotic exit in the presence of sustained cyclin B1 synthesis.
The spindle checkpoint normally inhibits anaphase onset in cells in which kinetochores are improperly attached to spindle microtubules. We and others showed that Ska-depleted cells undergo anaphase onset and mitotic exit when spindle checkpoint signaling is abrogated (Hanisch et al., 2006; Daum et al., 2009; Gaitanos et al., 2009; Theis et al., 2009; Welburn et al., 2009). We investigated more carefully the timing of anaphase onset, comparing control siRNA-treated cells and Ska3-depleted cells that were simultaneously depleted of Mad2 and BubR1. As expected, control cells underwent catastrophic anaphase and mitotic exit a short time after nuclear envelope breakdown. In contrast, many Ska3-depleted cells showed partial rescue of normal metaphase alignment and were often able to undergo a more orderly anaphase (Supplemental Figure S2 and Supplemental Movie S3).
In cells with intact spindles, Ska3 depletion could stall anaphase onset and impede cyclin B1 breakdown by at least two mechanisms. Ska3 depletion might partially disable kinetochore–microtubule attachments, leading to stronger spindle checkpoint signaling. Ska3 depletion might also compromise a downstream pathway that normally promotes securin and cyclin B1 breakdown upon checkpoint silencing. These potential functions for the Ska complex are not mutually exclusive. To test whether the Ska complex might function after checkpoint silencing independent of microtubules, we monitored mitotic exit timing in control and Ska3-depleted cells treated with the Mps1 inhibitor reversine, which inactivates spindle checkpoint signaling and leads to mitotic exit (Santaguida et al., 2010). We treated cells with high concentrations of nocodazole or Taxol to activate the spindle checkpoint and then added reversine. In cultures treated with nocodazole or Taxol, Ska3 depletion slowed mitotic exit after reversine treatment (Figure 3, A and B).
FIGURE 3:
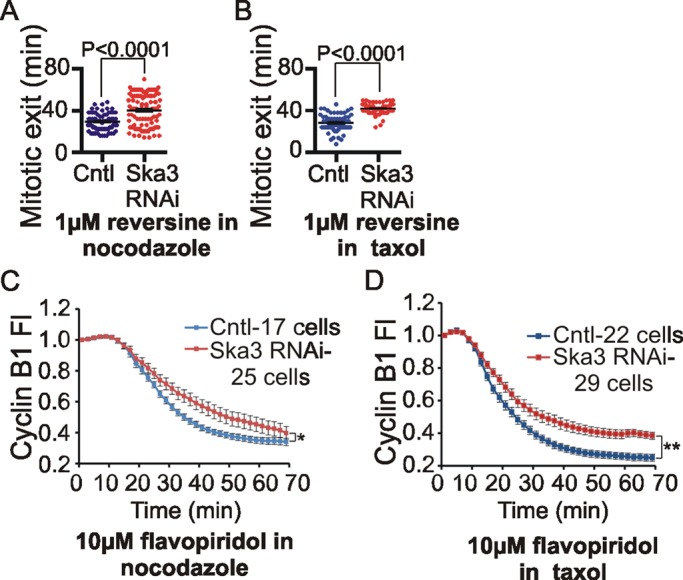
Ska3 depletion delays mitotic exit in cells treated with microtubule drugs. (A) HeLa H2B-GFP cells were transfected with control or Ska3 siRNA at 25 nM for 24 h. After washing off mitotic cells, cultures were treated with 3.3 μM nocodazole for 4 h. Reversine, 1 μM, was added, and time taken to exit mitosis in every cell was determined by imaging. Ska3-depleted cultures were delayed in mitotic exit (40 ± 1.5 min) compared with control cultures (29 ± 0.9 min). (B) Control and Ska3-depleted cells were treated as described but then released from nocodazole arrest into fresh medium for 30 min to allow spindles to form. Cells were then treated with 2 μM Taxol. Reversine, 1 μM, was added, and time taken to exit mitosis in every cell was determined by imaging. Ska3-depleted cells (42 ± 0.5 min) delayed mitotic exit compared with control cells (28 ± 0.9 min). Each dot represents one cell; the long horizontal line depicts mean, and whiskers denote SEM. (C) HeLa H2B-GFP cells were transfected with cyclin B1-mCherry and then transfected with control or Ska3 siRNA at 50 nM. Approximately 27 h posttransfection, cells were treated with 3.3 μM nocodazole. Flavopiridol, 10 μM, was added, and cyclin B1 degradation was recorded by measuring the decay of mCherry fluorescence. Ska3-depleted cells showed slower cyclin B1 degradation (p < 0.05). (D) Control and Ska3-depleted cells were treated as described but were then released from nocodazole arrest into fresh medium for 30 min to allow spindles to form. Cells were then treated with 2 μM Taxol. Then 10 μM flavopiridol was added and cyclin B1-mCherry degradation was measured. Overall, Taxol-arrested cells showed more rapid cyclin B1 degradation compared with nocodazole-arrested cells. Ska3-depleted cells showed slower cyclin B1 degradation (p < 0.005). Error bars indicate SEM, and cells were quantified from at least three independent experiments. The time taken to degrade 50% of cyclin B1 was calculated for every cell and used to determine statistical significance between control and Ska3-depleted cells. See also Supplemental Figures S2 and S3 and Supplemental Movie S3.
Next we tested whether Ska complex functions in promoting mitotic exit further downstream of checkpoint silencing, after Cdk1 inactivation. We previously showed that addition of chemical Cdk1 inhibitors to checkpoint-arrested cells resulted in rapid APC/C-Cdc20–mediated degradation of cyclin B1 and progression to G1 (Potapova et al., 2011). We imaged cells expressing fluorescently tagged cyclin B1 in order to directly analyze the dynamics of cyclin B1 breakdown in individual cells in real time (Clute and Pines, 1999; Bentley et al., 2007). We arrested mock-depleted or Ska3-depleted cells in mitosis with high concentrations of nocodazole or Taxol. Without further treatment, cyclin B1 fluorescence remained constant (unpublished data). When cells were treated with the chemical Cdk1 inhibitor flavopiridol, fluorescent cyclin B1 was degraded after a short delay. In the control situations the lag period for initiation of cyclin B1 degradation after flavopiridol addition was longer, and the rate of cyclin B1 degradation was slower for cells treated with nocodazole than for cells treated with Taxol. However, compared with their respective controls in both nocodazole and Taxol, cyclin B1 degradation lagged in Ska3-depleted cells (Figure 3, C and D). Although the expression level of cyclin B1-mCherry varied somewhat among individual cells, this variation did not significantly affect the kinetics of fluorescent cyclin B1 degradation (Supplemental Figure S3A). However, to avoid transient transfection, we also measured flavopiridol-induced cyclin B1 breakdown kinetics in HeLa cell lines stably expressing the N-terminal 166 amino acids of cyclin B1 fused to green fluorescent protein (GFP; Pfaff and King, 2013). As before, the breakdown of cyclin B1(1-166)-GFP in nocodazole-treated control cells was initiated later than with Taxol-treated control cells. Further, as in cells transiently transfected with full-length cyclin B1, Ska3 depletion slowed cyclin B1(1-166)-GFP breakdown compared with control cells in both nocodazole and Taxol (Supplemental Figure S3, B and C).
Assembled microtubules and the Ska complex foster chromosome association of the APC/C
We investigated whether the Ska complex might have a role in promoting faster degradation of cyclin B1 in cells treated with Taxol versus nocodazole. To confirm that our findings with exogenously expressed, fluorescent cyclin B1 were representative of the endogenous protein, we tested the effects of microtubule drugs on the degradation of endogenous cyclin B1. We arrested cells in mitosis with nocodazole or Taxol and subsequently treated with flavopiridol, taking samples at intervals for Western blotting. The breakdown of endogenous cyclin B1 upon addition of flavopiridol was accelerated in Taxol-treated cells compared with nocodazole-treated cells, again revealing that the presence of assembled microtubules accelerates cyclin B1 breakdown in cells treated with Cdk1 inhibitor (Supplemental Figure S3D). We then compared the level of Ska complex at kinetochores at prometaphase and metaphase with that in cells treated with nocodazole or Taxol. We found that kinetochores of Taxol-arrested cells harbored nearly twice the amount of Ska3 as those with nocodazole-arrested cells (Figure 4A).
FIGURE 4:

Microtubules promote APC/C accumulation on chromosomes. (A) HeLa cells grown on coverslips were treated with DMSO (control) or nocodazole (3.3 μM) for 3 h. To accumulate cells in Taxol, cultures were released after 2 h from nocodazole into fresh media for 30 min to allow spindle assembly and then treated with 2 μM Taxol for 30 min. Immunofluorescence was done, and Ska3 at kinetochores was quantified. Representative images are shown in the image panel. Scale bar, 10 μm. Ska3 was maximal at metaphase kinetochores. Taxol-treated cells accumulated twice as much Ska3 at kinetochores as nocodazole-treated cells. At least five cells in each condition were measured, and mean with SEM was plotted. (B) HeLa cells were released from thymidine block into 3.3 μM nocodazole–containing media. To collect cells in Taxol, cells were released from nocodazole into media without drugs to allow spindle formation before being treated with 2 μM Taxol for 30 min. MG132-treated cells were collected by releasing cells from nocodazole into 25 μM MG132 for 1.5 h. Chromosomes were isolated from nocodazole-, Taxol-, and MG132-treated cells, and associated APC/C components (Cdc27, Cdc16) and MCC components (Mad2, BubR1, Cdc20) were determined by Western blotting. Taxol- and MG132-treated cells show ∼1.5 times more Cdc27 associated with chromosomes than nocodazole-treated cells. Taxol-treated cells also show lower levels of MCC components associated with chromosomes than do nocodazole-treated cells. (C) HeLa cells were treated with 3.3 μM nocodazole as in B, and cytoplasmic and chromosome fractions were isolated. Cell equivalent volumes were loaded on SDS–PAGE gel to compare relative concentration of APC/C in cytoplasmic and chromosome fractions. (D) The cytoplasmic fraction was diluted 15-fold, and fractions were tested for the ability to ubiquitylate myc-cyclin B1(1-102) in vitro. The control lane consisted of ubiquitylation assay reagents lacking an APC/C source. The depletion of the nonubiquitylated myc-cyclin B1(1-102) was quantified from three independent experiments, and mean with SEM is depicted. Although approximately twice the APC/C concentration was present in the cytoplasmic fraction compared with the chromosome fraction, the chromosome fraction exhibited stronger cyclin B1 ubiquitylation activity.
We sought to discover how assembled microtubules and Ska complex might affect cyclin B1 degradation. In a previous study we found that the APC/C was bound to chromosomes and concentrated at kinetochores in cultured mammalian cells (Topper et al., 2002). The concentration of APC/C subunits at kinetochores has also been reported in other studies of mammalian cells and Drosophila (Jorgensen et al., 1998; Huang and Raff, 2002; Acquaviva et al., 2004). We tested whether assembled microtubules might affect the level of chromosome-associated APC/C, which we detected by Western blotting for Cdc27 and Cdc16. Chromosome fractions of cells arrested with Taxol or arrested at metaphase with the proteasome inhibitor MG132 had significantly higher levels of APC/C components than chromosome fractions obtained from cells arrested with nocodazole (Figure 4B). In cells arrested by the spindle checkpoint the primary inhibitor of APC/C activity is the mitotic checkpoint complex (MCC). Recent reports demonstrate that spindle checkpoint signaling is stronger in nocodazole-treated cells than in Taxol-treated cells (Collin et al., 2013; Dick and Gerlich, 2013). Thus we compared the level of MCC components in chromosome fractions isolated from nocodazole-treated or Taxol-treated cells. We found that MCC components Mad2, BubR1, and Cdc20 were increased in chromosome fractions of cells treated with nocodazole and decreased in chromosome fractions of cells treated with Taxol, consistent with the idea that stronger checkpoint signaling correlates with lower APC/C and higher MCC on chromosomes (Figure 4B).
As we showed in a previous study (Topper et al., 2002), the proportion of cellular APC/C bound to chromosomes is small, ∼6% of the total, and primarily in the high-mobility or dephosphorylated form compared with the bulk cytoplasmic APC/C in nocodazole-arrested mitotic cells (Figure 4C). Of interest, in ubiquitylation assays with tagged cyclin B1 protein, we found that chromosome fractions from nocodazole-arrested cells showed significantly higher ubiquitylation activity than did cytoplasmic fractions (Figure 4D).
A proteomics study in chicken DT40 cells reported that the genetic deletion of Ska3 led to decreased levels of APC/C components on chromosome fractions (Ohta et al., 2010). We assessed whether Ska3 depletion might compromise APC/C association with chromosomes in mammalian cells. We discovered that Ska3 depletion diminished the level of Cdc27 and Cdc16 bound to chromosomes in nocodazole-treated (Figure 5A) and MG132-treated cells (Figure 5B). Thus the Ska complex, whose binding to kinetochores is promoted by microtubules, appears to potentiate the association of APC/C on mitotic chromosomes. We also saw diminished accumulation of APC/C on chromosomes of nocodazole-arrested cells when we depleted Hec1, a component of the Ndc80 complex (Figure 5C). Ska complex recruitment was previously shown to be dependent on the Ndc80 complex (Figure 5D; Gaitanos et al., 2009; Raaijmakers et al., 2009; Welburn et al., 2009; Chan et al., 2012). In contrast, chemical inhibition of the kinetochore protein CENP-E using a small-molecule inhibitor, GSK 923295 (Wood et al., 2010; Gudimchuk et al., 2013), had no observable effect on Ska3 localization to the kinetochore (Figure 5D), nor did it affect the level of chromosome-associated APC/C (Figure 5E).
FIGURE 5:

Ska3 promotes APC/C association with chromosomes. (A) HeLa cells transfected with control or Ska3 siRNA at 25 nM for 26 h were treated with 3.3 μM nocodazole for 16 h. Chromosomes were isolated to determine the amount of associated Cdc27 and Cdc16 by Western blotting. Total amounts of APC/C were similar in whole cells, but Ska3 depletion decreased Cdc27 levels on chromosomes by ∼50%. (B) HeLa cells transfected with control or Ska3 siRNA at 25 nM were treated with 2 mM thymidine for 24 h and then released into 3.3 μM nocodazole for 12 h. Mitotic cells were collected and released from nocodazole into media containing 25 μM MG132 for 3 h. Chromosomes were isolated, and the amount of APC/C associated was determined by Western blotting. Total amounts of APC/C in whole cells were similar, but Ska3 depletion decreased Cdc27 levels on chromosomes by ∼60%. (C) HeLa cells were treated with 2 mM thymidine for 12 h to arrest in S phase. Cells were released from the thymidine block and transfected with control or Hec1 siRNA at 75 nM. At 12 h later, cells were transfected again with control or Hec1 siRNA at 75 nM and treated with 2 mM thymidine. At 24 h later, cells were released from the second thymidine block into media containing 3.3 μM nocodazole. At 12 h later, control and Hec1 siRNA–transfected cells were treated with 25 μM MG132 to prevent mitotic exit. After 2 h, mitotic cells were collected, chromosomes were isolated, and the amount of APC/C associated was determined by Western blotting. Total amounts of APC/C in whole cells were similar, but Hec1 depletion decreased Cdc27 levels on chromosomes by 80%. (D) HeLa cells grown on coverslips were treated as in C. On release from second thymidine block, control and Hec1 siRNA–transfected cells were treated with 3.3 μM nocodazole and 25 μM MG132 to prevent mitotic exit. One control coverslip was released into media containing CENP-E inhibitor GSK 923295 at 300 nM and 3.3 μM nocodazole. After 2 h, all coverslips were fixed and immunolabeled with anti-Ska3 antibody and ACA. Ska3 at kinetochores was quantified. Quantification showed that CENP-E inhibition by GSK 923295 did not affect Ska3 levels at kinetochores, whereas Hec1 depletion abolished Ska3 recruitment to kinetochores in the absence of microtubules. Kinetochores in at least five cells in each condition were quantified, and the mean with SEM is plotted. (E) HeLa cells were arrested in S phase by addition of 2 mM thymidine. At 24 h later, the cells were released into media containing 3.3 μM nocodazole and 300 nM GSK 923295. At 12 h later, mitotic cells were collected, and chromosomes were isolated to determine the amount of associated APC/C. CENP-E inhibition did not affect amount of APC/C associated with chromosomes.
Forced localization of Ska complex at kinetochores accelerates cyclin B1 breakdown
Because Ska accumulation at kinetochores in cells with assembled microtubules correlated with increased APC/C association with chromosomes, we carried out experiments designed to tease apart the potential direct roles of the Ska complex on APC/C localization and activity, independent of its possible roles in stabilizing kinetochore–microtubule interactions. We made plasmid expressing a fusion construct of the resident kinetochore protein, Mis12, linked to Ska1-GFP. Control plasmids were expressing Mis12-GFP and Ska1-GFP. All plasmids were mutated to be resistant to a pool of Ska1 siRNAs. To test the functionality of the fusion proteins, we transfected plasmids expressing them into cells that were then treated with control or Ska1 siRNA. Controls (cells treated with nonspecific siRNA) expressing Mis12-GFP, Ska1-GFP, or Mis12Ska1-GFP progressed through mitosis normally (unpublished data). In Ska1-depleted cells expressing Mis12-GFP, the majority of cells delayed or arrested at metaphase. The metaphase arrest phenotype was substantially rescued in cells expressing siRNA-resistant Ska1-GFP or Mis12Ska1-GFP (Supplemental Figure S4A). Both Ska1-GFP and Mis12Ska1-GFP but not Mis12-GFP also rescued APC/C accumulation on chromosomes in nocodazole-arrested cells depleted of endogenous Ska1 (Supplemental Figure S4B).
We next tested whether artificially targeting extra Ska1 protein to kinetochores in cells lacking microtubules would stimulate greater recruitment of the Ska complex. In cultures treated with 3.3 μM nocodazole, immunofluorescence analysis revealed that cells expressing Mis12Ska1-GFP exhibited ∼1.5 times more Ska3 at kinetochores than did cells expressing Mis12-GFP or Ska1-GFP (Figure 6A). Comparison of GFP levels in cells expressing the three plasmids revealed that high levels of untargeted Ska1-GFP produced correspondingly high levels of Ska3 at kinetochores (Supplemental Figure S5A). Of importance, expression of the kinetochore-targeted Mis12Ska1-GFP plasmid increased Ska3 accumulation at kinetochores independent of microtubule or expression level (Supplemental Figure S5A and Figure 6A).
FIGURE 6:

Targeting Ska complex to the kinetochore in the absence of spindle microtubules enhances cyclin B1 degradation. (A) HeLa cells grown on coverslips were transfected with Mis12-GFP, Ska1-GFP, or Mis12Ska1-GFP plasmids. Approximately 27 h posttransfection, 3.3 μM nocodazole was added for 2 h. Cells were fixed and immunolabeled with anti-Ska3 antibody and ACA. Quantification of Ska3 levels showed an ∼1.5-fold increase in kinetochore-associated Ska3 levels upon expression of Mis12Ska1-GFP plasmid. Kinetochores in at least eight cells for each condition were quantified, and the mean with SEM is plotted. Comparison of Ska3 at the kinetochore based on GFP expression levels in cells transfected with the Mis12 plasmids shows increased recruitment of Ska3 to kinetochore in cells expressing high amount of untargeted Ska1-GFP plasmid (Supplemental Figure S5A). (B) HeLa cells transfected with Mis12-GFP, Ska1-GFP, or Mis12Ska1-GFP plasmids were treated with 3.3 μM nocodazole for 15 h. Mitotic cells were collected, and chromosomes were isolated to determine the amount of associated Cdc27 and Cdc16. Blot quantification showed an approximately threefold increase in chromosomal Cdc27 in Mis12Ska1-GFP–expressing cells compared with Mis12-GFP–expressing cells. A lesser increase in chromosomal Cdc27 was also observed by overexpressing untargeted Ska1-GFP plasmid. However, Ska1-GFP plasmid was expressed at higher levels than Mis12Ska1-GFP plasmid (Supplemental Figure S5B). (C) HeLa cells were cotransfected with cyclin B1-mCherry plasmid and either Mis12-GFP, Ska1-GFP, or Mis12Ska1-GFP plasmid. Approximately 36 h after transfections, cells were treated with 3.3 μM nocodazole. Then 10 μM flavopiridol was added to induce cyclin B1 degradation. Kinetochore-targeted Mis12Ska1-GFP–expressing cells showed the strongest enhancement of cyclin B1 degradation (**p < 0.005, ****p < 0.0005 ). Although cells expressing Ska1-GFP were slightly accelerated in cyclin B1 degradation compared with Mis12-GFP–expressing cells, the two populations were not statistically significant (p = 0.26). Comparison of total GFP fluorescence levels at the initial time indicated that Ska1-GFP was expressed on average at higher levels than Mis12Ska1-GFP (Supplemental Figure S5D). Error bars indicate SEM, and cells were quantified from at least three independent experiments. The time taken to degrade 50% of cyclin B1 was calculated for every cell and used to determine statistical significance between different groups.
We tested the ability of the fusion proteins to stimulate APC/C accumulation on chromosomes in the absence of microtubules. Cells expressing Ska1-GFP and Mis12Ska1-GFP both showed increased accumulation of APC/C components Cdc27 and Cdc16 (Figure 6B). Comparison of expression levels in transfected cells by immunoblotting revealed that Ska1-GFP was expressed, on average, at higher levels than Mis12Ska1-GFP (Supplemental Figure S5B). Thus we speculate that overexpression of untargeted Ska1 does stimulate some increased APC/C accumulation on chromosomes, but the kinetochore-targeted Mis12Ska1-GFP expressed at lower levels is more efficient in recruiting APC/C.
Finally, we determined whether exogenous kinetochore-targeted Ska complex would enhance cyclin B1 degradation in the absence of kinetochore–microtubule attachments. We analyzed fluorescent cyclin B1 breakdown driven by flavopiridol treatment in cells arrested in mitosis by treatment with high concentrations of nocodazole. Without flavopiridol addition, cyclin B1 levels remained constant in cells expressing Mis12Ska1-GFP, Ska1-GFP, or Mis12-GFP (unpublished data). On flavopiridol addition, cells expressing Mis12Ska1-GFP showed higher efficiency of cyclin B1 degradation than cells expressing Mis12-GFP or Ska1-GFP (Figure 6C). Cells expressing Ska1-GFP trended toward slightly accelerated cyclin B1 breakdown compared with cells expressing Mis12-GFP, although this difference did not score as significant. Initial levels of cyclin B1-mCherry expression varied somewhat among cells, but the mean fluorescence levels were similar in cells expressing Mis12-GFP, Ska1-GFP, or Mis12Ska1-GFP (Supplemental Figure S5C). Comparison of total cytoplasmic GFP fluorescence expression levels in cells transfected with the three plasmids again showed that Ska1-GFP was expressed at higher levels than Mis12Ska1-GFP (Supplemental Figure S5C, bar graph). Thus, targeting Ska complex to kinetochores yielded a specific increase in both accumulation of chromosome-associated APC/C and cyclin B1 degradation efficiency.
DISCUSSION
Anaphase onset is an important and irreversible transition node in cell cycle progression. Nearly all the events of mitotic exit can be induced in mitotic cells by global inhibition of Cdk1 activity using chemical kinase inhibitors (Potapova et al., 2006, 2011). The exception to this generalization is chromatid separation, which appears to require a “head start” before the other events of mitotic exit (Potapova et al., 2011; Shindo et al., 2012; Yaakov et al., 2012). The key regulator is the APC/C, which fosters a sharp transition by rapidly ubiquitylating late mitotic targets, securin and cyclin B1, at anaphase onset. However, it is simplistic to contend that the APC/C is simply “activated” at the metaphase–anaphase transition, since it shows clear activities before metaphase. The APC/C actively ubiquitylates early mitotic targets such as cyclin A and Nek2A that are largely degraded during prometaphase. Moreover, the APC/C also shows activity against late mitotic targets such as cyclin B1 in cells arrested by the spindle checkpoint after treatment with microtubule drugs. Gradual degradation of cyclin B1 appears to be responsible for the phenomenon called “mitotic slippage” by which cells arrested in M phase with microtubule drugs progress to G1 without completing mitosis (Brito and Rieder, 2006). Thus regulation of APC/C activity in mitosis is complex.
The spindle checkpoint is a signal transduction pathway that restrains anaphase onset in response to suboptimal microtubule attachment and tension at kinetochores and is most readily exhibited in cells treated with microtubule inhibitors. However, in vertebrate cells the spindle checkpoint functions even in an unperturbed mitosis (Musacchio and Salmon, 2007). Abrogation of spindle checkpoint signaling in vertebrate cells induces premature anaphase onset and mitotic exit (Gorbsky et al., 1998). Thus the control of anaphase onset has often been discussed in terms of “silencing the spindle checkpoint” to activate the APC/C. However, perhaps a more accurate description would be to regard the APC/C as the target for several interacting positive or negative regulators whose cumulative tempering of APC/C activities control cell cycle progression in M phase. In cells with intact microtubules, when the Ska complex is depleted, mitotic cells advance with some alignment delay to metaphase but then arrest with fully formed spindles. Unlike control metaphase cells, Ska-depleted metaphase cells cannot rapidly and fully degrade securin and cyclin B1. With time, kinetochore–microtubule attachments in Ska-depleted metaphase cells are robust enough to force cohesion fatigue, chromatid separation, and reactivation of the spindle checkpoint by the separated chromatids (Daum et al., 2009; Lara-Gonzalez and Taylor, 2012).
Here we reexamined the phenotypes of cells depleted of Ska complex proteins. In cells depleted of Ska, most chromosomes align in a timely manner, whereas a minority exhibit transient defects in achieving or maintaining alignment. Ska-depleted cells then commonly arrest at metaphase and undergo cohesion fatigue. We also provide evidence that the Ska complex does not play a major role in anaphase chromatid movement. Although we cannot rule out the possibility that residual Ska proteins present after siRNA-mediated depletion are able to carry out chromosome movement duties of the complex, it is clear that full expression of Ska is required to undergo anaphase onset and mitotic exit. Recently it was shown that Ska complex is regulated by Aurora B kinase activity and binds directly to the Ndc80 kinetochore complex (Chan et al., 2012). In addition to its roles in kinetochore–microtubule attachment, the Ndc80 complex plays important functions as a platform for kinetochore association of spindle checkpoint–signaling proteins (McCleland et al., 2003). Depletion of Ska leads to an accumulation of the checkpoint kinase Bub1 at kinetochores (Daum et al., 2009). Thus depletion of the Ska complex may act to induce mitotic arrest, at least in part, through changes in the spindle checkpoint signaling at kinetochores.
Of importance, in this work we demonstrate a role for Ska complex in the timing of cyclin B1 degradation and mitotic exit in cells treated with high concentrations of nocodazole, in which kinetochore–microtubule interactions are absent. When the potent inhibitory effect of the spindle checkpoint is removed experimentally by treating cells with the Mps1 inhibitor reversine, depletion of the Ska complex delays mitotic exit. Similarly, when mitotic exit is induced downstream of checkpoint inactivation by treatment with the Cdk1 inhibitor flavopiridol, depletion of the Ska complex slows cyclin B1 degradation. In both cases, depletion of the Ska complex slows but does not completely prevent stronger activity of the APC/C toward securin and cyclin B1. Thus the Ska complex is one of several modulators of APC/C activity. However, because the Ska-dependent stimulation of APC/C activity was obtained in cells treated with high concentrations of nocodazole, the results indicate that the Ska complex plays a role in promoting the metaphase–anaphase transition and mitotic exit, independent of any effects on stabilization of kinetochore–microtubule interactions.
Finally, we examined how Ska accumulation at kinetochores might regulate and enhance APC/C function. In addition to temporal control of APC/C activity, substantial evidence indicates that the APC/C is regulated spatially within the cell, potentially as a mechanism for regulating chromatid separation in relation to the other events of mitotic exit (Clute and Pines, 1999; Huang and Raff, 1999; Shindo et al., 2012; Yaakov et al., 2012). APC/C concentrates on chromosomes, particularly at kinetochores (Jorgensen et al., 1998; Huang and Raff, 2002; Topper et al., 2002; Acquaviva et al., 2004). Recent studies in budding yeast and mammalian cells using fluorescent reporters of separase activity show that separase is activated on chromosomes abruptly to ensure synchronous chromatid separation at anaphase (Shindo et al., 2012; Yaakov et al., 2012). In mammalian cells APC/C activity toward securin is initiated first in the cytoplasm and finally on the chromosomes, seemingly to promote the final and abrupt activation of separase on chromosomes just before anaphase onset (Shindo et al., 2012). In addition, cyclin B1 is concentrated on chromosomes until late metaphase and disappears from chromosomes during the metaphase–anaphase transition (Clute and Pines, 1999; Bentley et al., 2007). These studies suggest that chromosomes may be the final site of APC/C activation to ensure the synchronization of chromatid separation with the other events of mitotic exit. Our present data suggest that the minor portion of dephosphorylated APC/C associated with chromosomes has more ubiquitylation activity than the abundant phosphorylated cytosolic APC/C. We also show that depletion of Ska complex results in partial loss of APC/C from chromosomes, consistent with proteomic studies in chicken DT40 cells. (Ohta et al., 2010). Here we show that this loss correlates with sluggish APC/C activity and penetrant mitotic arrest. We do not know the detailed mechanism by which kinetochore accumulation of Ska promotes APC/C concentration on chromosomes. Its role may be direct or indirect and may involve other components of the kinetochore interacting with spindle microtubules. However, taken together, our new results suggest that one role of the Ska complex is to promote chromosomal localization of APC/C, potentiating its swift, complete activation to initiate chromatid separation and hone the anaphase trigger.
MATERIALS AND METHODS
Cell culture
We used HeLa cells stably transfected with GFP fused to histone 2B (HeLa H2B-GFP) and HeLa cells stably transfected with mCherry fused to histone 2B (HeLa H2B-mCherry; Daum et al., 2009). Stable HeLa cells expressing cyclin B1(1-166)-GFP were a kind gift from Randall W. King (Harvard Medical School, Boston, MA; Pfaff and King, 2013). All cell lines were grown in culture flasks or chambered coverslips in DMEM with 10% fetal bovine serum (FBS) supplemented with penicillin and streptomycin in 5% CO2 at 37°C.
To synchronize HeLa cells, cultures were treated with 2 mM thymidine for 24 h and released into media containing 3.3 μM nocodazole. Transient transfection of HeLa cells for expression of cyclin B1-mCherry, Ska1-GFP, Mis12-GFP, and Mis12Ska1-GFP was achieved using the FuGENE 6 (Roche, Indianapolis, IN; Promega, Madison, WI; or Mirus, Madison, WI) transfection reagent according to manufacturer's instructions. Transfection of siRNA was done using Lipofectamine RNAi reagent (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. On-Target Plus SMARTpool siRNA against Ska1, Ska2, and Ska3 was obtained from Dharmacon/Thermo Scientific (Pittsburgh, PA), and these were used at 25–50 nM final concentration. For siRNA-resistant rescue experiments (Supplemental Figure S4), siGenome siRNA against Ska1 was obtained from Dharmacon/Thermo Scientific and used at 50–100 nM final concentration. To deplete Hec1, a single custom Hec1 RNAi that was previously described was purchased from Qiagen (Valencia, CA) and used at 75 nM final concentration (Miller et al., 2008).
Plasmids
Cyclin B1-GFP plasmid was a kind gift from Randall W. King. Cyclin B1-mCherry plasmid was made by inserting cDNA encoding cyclin B1 from cyclin B1-GFP plasmid into mCherry-N1 plasmid. Mis12-GFP, Ska1-GFP, and Mis12Ska1-GFP plasmids were made by inserting the respective cDNAs into pEGFPN1 vectors. GFP-Ska1 siRNA-resistant plasmid (resistant to siGenome Ska1 RNAi) was a kind gift from Iain M. Cheeseman. Ska1-GFP and Mis12Ska1-GFP (siRNA resistant) were made by inserting cDNA encoding siRNA-resistant Ska1 from GFP-Ska1 into pEGFPN1 or Mis12-GFP vector, respectively.
Live-cell imaging
HeLa H2B-GFP/mCherry cells were grown in Nunc chambered coverslips (Thermo Scientific). To maintain appropriate pH levels and avoid evaporation during imaging, culture media was exchanged to Leibovitz's L-15 medium supplemented with 10% FBS, penicillin, and streptomycin. Other drugs were added as indicated, and the medium was overlaid with mineral oil. Time-lapse fluorescence images were collected using 20× or 40× objectives and a Zeiss Axiovert 200M inverted microscope equipped with an objective heater, air curtain, a Hamamatsu Orca-ER camera (Hamamatsu Photonics, Bridgewater, NJ), and MetaMorph software (Molecular Devices, Sunnyvale, CA). Images were captured every 4–15 min for 12–24 h. Time-lapse videos displaying the elapsed time between consecutive frames were assembled using MetaMorph software. The first time frame after nuclear envelope breakdown (NEB), metaphase chromosome alignment, and anaphase onset/mitotic exit was noted in Excel (Microsoft), and mitotic interval from NEB to metaphase (alignment time), metaphase to anaphase (metaphase duration), or NEB to anaphase onset/mitotic exit was calculated. Mitotic duration for every cell was determined, and data were depicted as scatter plots with mean and SEM. In scatter plots, each dot represents one cell; long horizontal line depicts mean, and whiskers denote SEM. The unpaired Student's t test in Prism (GraphPad) was used to assess statistical significance.
Quantitative image analysis
To measure the fluorescent cyclin B1-mCherry or cyclin B1(1-166)-GFP degradation in living cells, time-lapse images were collected at 2- to 4-min intervals. The region was drawn around each cell to be measured, and the identical region was placed in an area without fluorescent objects to be used for background subtraction. The net average fluorescence intensity of a pixel in the region of interest was calculated for each time point. Because cells expressed different levels of fluorescent cyclins, the net average intensity values were normalized to the initial (first time point) value, which was designated as 1. Averages of normalized intensity values of at least five identically treated cells were calculated for each time point and plotted with SEM. For these experiments, all parameters during image acquisition were maintained the same. To calculate statistical significance, the time taken to degrade 50% of initial cyclin B1 was calculated for every cell, and unpaired Student's t test in Prism was used to compute p values between groups (Collin et al., 2013).
Immunofluorescence
HeLa cells were grown on glass coverslips and treated as detailed in the figure legends. Cells were fixed in 2% paraformaldehyde/PHEM solution (60 mM PIPES, pH 6.9, 25 mM HEPES, 10 mM EGTA, 4 mM MgCl2) containing 0.5% Triton X-100 for 15 min. Coverslips were washed in MBST (10 mM MOPS, 150 mM NaCl, 0.05% Tween 20), blocked in 20% boiled normal goat serum (BNGS), and incubated overnight with primary antibodies. Samples were then incubated with secondary antibodies for 1–2 h, stained with DNA dye 4’,6-diamidino-2-phenylindole and mounted using Vectashield (Vector Laboratories, Burlingame, CA). The following primary antibodies were used: ACA/CREST (anti-centromere antibodies from Antibody, Inc.) and rabbit anti-Ska3 (Daum et al., 2009). Secondary antibodies used were goat anti-rabbit antibodies conjugated to Cy3 or fluorescein isothiocyanate (FITC; Jackson ImmunoResearch, West Grove, PA) or goat anti-human antibody conjugated to Cy3 or FITC. The images were acquired using a Zeiss Axioplan II microscope equipped with 100× objectives (numerical aperture, 1.4) and a Hamamatsu Orca 2 camera and processed using MetaMorph software and CorelDraw (Corel). Quantification of the immunofluorescence images was done as described previously (Daum et al., 2009). The graphs depict average fluorescence value with SEM in each condition. An unpaired Student's t test in Prism was used to determine statistical significance between groups.
Western blotting and blot quantification
Whole-cell HeLa cell extracts were prepared by lysis in APCB buffer (20 mM Tris-Cl, pH 7.7, 100 mM KCl, 50 mM sucrose, 1 mM MgCl2, 0.1 mM CaCl2, 0.5% Triton X-100) containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and microcystin (400 nM). For electrophoresis, sample loading buffer (Invitrogen) and dithiothreitol (DTT) to a final concentration of 50 mM were added. Proteins were separated with a NuPAGE gel electrophoresis system (Invitrogen) and transferred to a 0.45-μm polyvinylidene difluoride (PVDF) membrane (Immobilon PVDF; Millipore, Billerica, MA) via a Genie transfer apparatus (Idea Scientific, Minneapolis, MN). Membranes were blocked in 5% nonfat dry milk (NFDM) and 0.05% Tween 20 in Tris-buffered saline (TBS). Primary antibodies included rabbit anti-Ska3 antibody (Daum et al., 2009), mouse anti–β-actin (Abcam, Cambridge, MA), mouse anti–cyclin B1 (BD Biosciences, Chicago, IL), rabbit anti-securin (Invitrogen), rabbit anti–phospho-histone H3 (Ser10; Cell Signaling, Danvers, MA), rabbit anti-Cdk1 (Cell Signaling), rabbit anti-Cdc20 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Cdc27 and rabbit anti-Cdc16 (kind gifts from Philip Hieter, University of British Columbia), rabbit anti–histone H3 (Abcam), mouse anti-GFP (Covance, Richmond, CA), mouse anti-Hec1 (Novus Biologicals, Littleton, CO), rabbit anti-BubR1 (Bethyl Laboratories, Montgomery, TX), and rabbit anti-Mad2 (Bethyl Laboratories). Membranes were washed in TBS/0.05% Tween 20 (TBST) and then incubated with secondary antibodies in 5% NFDM/TBST. Secondary antibodies include horseradish peroxidase goat anti-mouse or anti-rabbit antibodies (Jackson ImmunoResearch). After washes, membranes were developed using West Pico Chemiluminescent reagent (Pierce, Rockford, IL) and imaged using a Kodak 4000M or Alpha Innotech FluorChem HD2 imaging station.
Western blots were quantified by calculating integrated band intensities using MetaMorph software and Excel. To correct for local background, signal intensity of a concentric region was determined and subtracted from original band intensity. For every Western blot quantified, dilution series of appropriate control was loaded on SDS–PAGE gel, and band intensities were quantified and a standard curve was created. This was used to calculate the intensity of the test band reliably. Every experiment was repeated at least three times, and the quantification shows the mean result obtained from all experimental repeats with SEM.
Cdk1/cyclin B1 kinase assays
HeLa cells were grown in 150-mm plates. The cells were transfected with control or Ska3 siRNA for 30 h. Nocodazole was added at 3.3 μM concentration to the cells for ∼4 h. Mitotic cells were collected after incubation, nocodazole was washed off, and cells were released to progress through mitosis. Cells were collected every hour for 6 h, flash-frozen, and stored at −80°C until use. Cell pellets were lysed in APCB buffer supplemented with protease (Sigma-Aldrich) and phosphatase inhibitors (400 nM microcystin). Total Cdk1 was precipitated using p13-Suc1 agarose-conjugated beads (Millipore). For kinase assay, precipitated Cdk1 was incubated in kinase buffer (25 mM Tris-HCl, pH 7.5, 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2). Each reaction contained 1–2 mg/ml histone H1, 200μM ATP, and 1 μCi of [γ32P]ATP. Reactions were incubated at 37°C for 20 min, stopped by addition of sample loading buffer (Invitrogen), and separated by SDS–PAGE in 4–12% Bis-Tris gels (Invitrogen). The gel was dried and exposed to a phosphor screen (Amersham, Piscataway, NJ) that was then scanned with a Storm scanner.
Chromosome preparation and immunoblotting
HeLa cells were grown in 150-mm plates and transfected with control or Ska3 siRNA. Mitotic cells were collected in medium containing 3.3 μM nocodazole. Cells were lysed with ELB buffer on ice (1× PHEM, 0.5% Triton X-100, 1 mM DTT, 10% glycerol, protease inhibitors, and 400 nM microcystin) and centrifuged to separate cytoplasmic fractions from chromosome fractions. Chromosome fractions were further washed with ELB at least five times to remove cytoplasmic contamination and resuspended in 1/4 volume ELB of cytoplasmic fractions. The protein concentration of cytoplasmic fractions was determined using the bicinchoninic acid protein assay kit (Pierce). The chromosomes were DNase treated and resuspended in sample loading buffer (Invitrogen) and 50 mM DTT. Proteins were separated with a NuPAGE gel electrophoresis system (Invitrogen), transferred to a 0.45-μm PVDF membrane (Immobilon PVDF) via a Genie transfer apparatus and immunoblotted with antibody to APC/C components Cdc27, Cdc16, and antibody to histone H3.
In vitro ubiquitylation assay
The cytoplasmic fraction was diluted 15 times to get APC/C amounts more closely equivalent to chromosome fraction. The ubiquitylation activities of chromosome fraction and diluted cytosolic fraction were compared. We performed 40-μl reactions containing 40 mM Tris, pH 7.5, 5 mM MgCl2, 5% (vol/vol) glycerol, 1 mM DTT, 0.5 mg/ml BSA, 2 mM ATP, 2 mg/ml ubiquitin (Boston Biochem, Ashland, MA), purified E1 (100 ng), UbcH10 (200 ng), and Ube2S (200 ng). Recombinant myc-tagged cyclin-B1(1–102) was used at the final concentration of 300 ng/μl as an APC/C substrate, and the chromosome fraction and diluted cytosolic fraction were the source of APC/C. The ubiquitylation reactions were performed at 37°C for 1 h, stopped by the addition of sample loading buffer, resolved by SDS–PAGE, and analyzed by anti-myc immunoblot to probe cyclin B1 ubiquitylation. The experiment was repeated three times, and the mean result obtained from all experimental repeats with SEM was plotted.
Supplementary Material
Acknowledgments
We thank P. Todd Stukenberg for advice and discussion. We thank Iain M. Cheeseman for sharing HeLa H2B-YFP cells, GFP-Ska1 (siRNA-resistant), Ska1 siRNA, and Ska3 siRNA. We thank Randall W. King for the cyclin B1 plasmid and cyclin B1(1-166)-GFP–expressing stable cell lines. We thank Tarun Kapoor for the Mis12Mad1-mCherry plasmid. We thank Philip Hieter for anti-Cdc27 and anti-Cdc16 antibody. We also thank the members of the Gorbsky lab, especially Tamara A. Potapova, and members of the Cell Cycle and Cancer Biology Program at the Oklahoma Medical Research Foundation for discussion. This work was supported by a grant from the National Institute of General Medical Sciences (GM50412) and by the McCasland Foundation.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- Ska
spindle and kinetochore–associated
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-07-0421) on January 8, 2014.
REFERENCES
- Acquaviva C, Herzog F, Kraft C, Pines J. The anaphase promoting complex/cyclosome is recruited to centromeres by the spindle assembly checkpoint. Nat Cell Biol. 2004;6:892–898. doi: 10.1038/ncb1167. [DOI] [PubMed] [Google Scholar]
- Bentley AM, Normand G, Hoyt J, King RW. Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell. 2007;18:4847–4858. doi: 10.1091/mbc.E06-06-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YW, Jeyaprakash AA, Nigg EA, Santamaria A. Aurora B controls kinetochore-microtubule attachments by inhibiting Ska complex-KMN network interaction. J Cell Biol. 2012;196:563–571. doi: 10.1083/jcb.201109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol. 2013;15:1378–1385. doi: 10.1038/ncb2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr Biol. 2011;21:1018–1024. doi: 10.1016/j.cub.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr Biol. 2009;19:1467–1472. doi: 10.1016/j.cub.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen N, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol. 2001;153:121–136. doi: 10.1083/jcb.153.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick AE, Gerlich DW. Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol. 2013;15:1370–1377. doi: 10.1038/ncb2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitanos TN, Santamaria A, Jeyaprakash AA, Wang B, Conti E, Nigg EA. Stable kinetochore-microtubule interactions depend on the Ska complex and its new component Ska3/C13Orf3. EMBO J. 2009;28:1442–1452. doi: 10.1038/emboj.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Pines J. Progressive activation of cyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol. 2001;153:137–148. doi: 10.1083/jcb.153.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky GJ, Chen RH, Murray AW. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol. 1998;141:1193–1205. doi: 10.1083/jcb.141.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudimchuk N, Vitre B, Kim Y, Kiyatkin A, Cleveland DW, Ataullakhanov FI, Grishchuk EL. Kinetochore kinesin CENP-E is a processive bi-directional tracker of dynamic microtubule tips. Nat Cell Biol. 2013;15:1079–1088. doi: 10.1038/ncb2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hames RS, Wattam SL, Yamano H, Bacchieri R, Fry AM. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001;20:7117–7127. doi: 10.1093/emboj/20.24.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch A, Sillje HH, Nigg EA. Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J. 2006;25:5504–5515. doi: 10.1038/sj.emboj.7601426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MJ, Kimata Y, Wattam SL, Lindon C, Mao G, Yamano H, Fry AM. Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat Cell Biol. 2006;8:607–614. doi: 10.1038/ncb1410. [DOI] [PubMed] [Google Scholar]
- Huang J, Raff JW. The disappearance of cyclin B at the end of mitosis is regulated spatially in Drosophila cells. EMBO J. 1999;18:2184–2195. doi: 10.1093/emboj/18.8.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Raff JW. The dynamic localisation of the Drosophila APC/C: evidence for the existence of multiple complexes that perform distinct functions and are differentially localised. J Cell Sci. 2002;115:2847–2856. doi: 10.1242/jcs.115.14.2847. [DOI] [PubMed] [Google Scholar]
- Jeyaprakash AA, Santamaria A, Jayachandran U, Chan YW, Benda C, Nigg EA, Conti E. Structural and functional organization of the Ska complex, a key component of the kinetochore-microtubule interface. Mol Cell. 2012;46:274–286. doi: 10.1016/j.molcel.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Jorgensen PM, Brundell E, Starborg M, Hoog C. A subunit of the anaphase-promoting complex is a centromere-associated protein in mammalian cells. Mol Cell Biol. 1998;18:468–476. doi: 10.1128/mcb.18.1.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598–6609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz S, Sudakin V, Ruderman JV, Hershko A. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc Natl Acad Sci USA. 1995;92:9303–9307. doi: 10.1073/pnas.92.20.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Taylor SS. Cohesion fatigue explains why pharmacological inhibition of the APC/C induces a spindle checkpoint-dependent mitotic arrest. PLoS One. 2012;7:e49041. doi: 10.1371/journal.pone.0049041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT. The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev. 2003;17:101–114. doi: 10.1101/gad.1040903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena AL, Lam EW, Chatterjee S. Sustained spindle-assembly checkpoint response requires de novo transcription and translation of cyclin B1. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Johnson ML, Stukenberg PT. Kinetochore attachments require an interaction between unstructured tails on microtubules and Ndc80(Hec1) Curr Biol. 2008;18:1785–1791. doi: 10.1016/j.cub.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Ohta S, et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell. 2010;142:810–821. doi: 10.1016/j.cell.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff KL, King RW. Determinants of human cyclin B1 association with mitotic chromosomes. PLoS One. 2013;8:e59169. doi: 10.1371/journal.pone.0059169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Daum JR, Byrd KS, Gorbsky GJ. Fine tuning the cell cycle: activation of the Cdk1 inhibitory phosphorylation pathway during mitotic exit. Mol Biol Cell. 2009;20:1737–1748. doi: 10.1091/mbc.E08-07-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Daum JR, Pittman BD, Hudson JR, Jones TN, Satinover DL, Stukenberg PT, Gorbsky GJ. The reversibility of mitotic exit in vertebrate cells. Nature. 2006;440:954–958. doi: 10.1038/nature04652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Sivakumar S, Flynn JN, Li R, Gorbsky GJ. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol Biol Cell. 2011;22:1191–1206. doi: 10.1091/mbc.E10-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers JA, Tanenbaum ME, Maia AF, Medema RH. RAMA1 is a novel kinetochore protein involved in kinetochore-microtubule attachment. J Cell Sci. 2009;122:2436–2445. doi: 10.1242/jcs.051912. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, et al. The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell. 2012;23:968–980. doi: 10.1016/j.devcel.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo N, Kumada K, Hirota T. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev Cell. 2012;23:112–123. doi: 10.1016/j.devcel.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Stevens D, Gassmann R, Oegema K, Desai A. Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS One. 2011;6:e22969. doi: 10.1371/journal.pone.0022969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, et al. Comparative profiling identifies C13orf3 as a component of the Ska complex required for mammalian cell division. EMBO J. 2009;28:1453–1465. doi: 10.1038/emboj.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper LM, Campbell MS, Tugendreich S, Daum JR, Burke DJ, Hieter P, Gorbsky GJ. The dephosphorylated form of the anaphase-promoting complex protein Cdc27/Apc3 concentrates on kinetochores and chromosome arms in mitosis. Cell Cycle. 2002;1:282–292. [PubMed] [Google Scholar]
- Welburn JP, Grishchuk EL, Backer CB, Wilson-Kubalek EM, Yates JR, 3rd, Cheeseman IM. The human kinetochore Ska1 complex facilitates microtubule depolymerization-coupled motility. Dev Cell. 2009;16:374–385. doi: 10.1016/j.devcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KW, et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc Natl Acad Sci USA. 2010;107:5839–5844. doi: 10.1073/pnas.0915068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaakov G, Thorn K, Morgan DO. Separase biosensor reveals that cohesin cleavage timing depends on phosphatase PP2A(Cdc55) regulation. Dev Cell. 2012;23:124–136. doi: 10.1016/j.devcel.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.