

Myofibroblast transition from mesenchymal or epithelial precursors triggers loss of the primary cilium. In epithelial cells this requires contact injury and TGFβ and is mediated by Rac- and Smad3-dependent myosin phosphorylation. Thus the myofibroblast is a unique cilium-less cell with markedly reprogrammed ciliary signaling.
Abstract
Myofibroblasts, the culprit of organ fibrosis, can originate from mesenchymal and epithelial precursors through fibroblast–myofibroblast and epithelial–myofibroblast transition (EMyT). Because certain ciliopathies are associated with fibrogenesis, we sought to explore the fate and potential role of the primary cilium during myofibroblast formation. Here we show that myofibroblast transition from either precursor results in the loss of the primary cilium. During EMyT, initial cilium growth is followed by complete deciliation. Both EMyT and cilium loss require two-hit conditions: disassembly/absence of intercellular contacts and transforming growth factor-β1 (TGFβ) exposure. Loss of E-cadherin–dependent junctions induces cilium elongation, whereas both stimuli are needed for deciliation. Accordingly, in a scratch-wounded epithelium, TGFβ provokes cilium loss exclusively along the wound edge. Increased contractility, a key myofibroblast feature, is necessary and sufficient for deciliation, since constitutively active RhoA, Rac1, or myosin triggers, and down-regulation of myosin or myocardin-related transcription factor prevents, this process. Sustained myosin phosphorylation and consequent deciliation are mediated by a Smad3-, Rac1-, and reactive oxygen species–dependent process. Transitioned myofibroblasts exhibit impaired responsiveness to platelet-derived growth factor-AA and sonic hedgehog, two cilium-associated stimuli. Although the cilium is lost during EMyT, its initial presence contributes to the transition. Thus myofibroblasts represent a unique cilium-less entity with profoundly reprogrammed cilium-related signaling.
INTRODUCTION
The myofibroblast, a highly contractile mesenchymal cell whose hallmark is the expression of α-smooth muscle actin (SMA), plays a pivotal role in wound contraction and healing but has also been identified as the culprit in the pathogenesis of fibrotic and fibrocontractile diseases (Hinz et al., 2012). Organ fibrosis is a dysregulated form of tissue repair triggered by chronic epithelial injury (Wynn and Ramalingam, 2012). The epithelium is believed to play a dual role in the generation and tissue accumulation of myofibroblasts. First, it is the source of fibrogenic cytokines that can induce the transformation of myofibroblast precursors such as resident tissue fibroblasts and pericytes (epithelial–mesenchymal cross-talk; Chapman, 2011; Hutchison et al., 2013). Second, the epithelium itself can give rise to myofibroblasts through epithelial–myofibroblast transition (EMyT), an advanced form of epithelial–mesenchymal transition (EMT), characterized by SMA expression (Iwano et al., 2002; Zeisberg and Neilson, 2009; Liu, 2010). The relative contribution of the various sources to myofibroblast generation likely varies in different disease conditions and is the subject of intense debate (Zeisberg and Duffield, 2010; Quaggin and Kapus, 2011; LeBleu et al., 2013). However, there is firm consensus that epithelial cells possess the potentiality to mobilize a myogenic program and thereby transdifferentiate into myofibroblasts (Ng et al., 1998; Masszi et al., 2003; Kim et al., 2009a; Humphreys et al., 2010). Thus, from a cell biological viewpoint, the main question concerns the inputs and signaling pathways that can unleash such a dramatic phenotypic reprogramming.
Transforming growth factor-β1 (TGFβ) has long been known as the main fibrogenic cytokine and prime inducer of EMT and EMyT. However, previous studies by us (Masszi et al., 2004, 2010; Fan et al., 2007) and others (Kim et al., 2009a; Zheng et al., 2009) established that although TGFβ is capable of triggering fibroblast–myofibroblast transition, it is often not sufficient to provoke EMyT. The other prerequisite is an injury (absence or uncoupling) of intercellular contacts. The state of cell contacts regulates EMyT by a variety of mechanisms. Contact disruption activates Rho and Rac (Fan et al., 2007; Samarin et al., 2007; Busche et al., 2008; Sebe et al., 2008), which in turn enhance F-actin polymerization. The ensuing decrease in monomeric actin induces nuclear translocation of myocardin-related transcription factor (MRTF; Fan et al., 2007; Busche et al., 2008), which, in conjunction with serum response factor, drives a multitude of cytoskeletal genes, including SMA (Olson and Nordheim, 2010). Our previous work also revealed that another contact element, β-catenin, is also essential for SMA expression (Masszi et al., 2004), as it maintains MRTF stability and activity (Charbonney et al., 2011). Contact-dependent pathways then synergize with TGFβ signaling to induce EMyT. For example, TGFβ prolongs nuclear accumulation of MRTF (Masszi et al., 2010), whereas contact injury facilitates nuclear retention of Smad3, a major TGFβ signal transducer (Varelas et al., 2010). Smad3 plays a dual role in the transition: it is essential for the induction of many mesenchymal genes (Moustakas and Heldin, 2012), but it is an inhibitor of MRTF (Masszi et al., 2010). However, after initially enhanced Smad3 signaling, Smad3 degrades in the contact-deprived epithelium (Masszi et al., 2010). Thus Smad3 is a critical timer of EMyT, and the process can be dissected into a Smad3-promoted mesenchymal and a Smad3-inhibited myogenic phase. Together these studies revealed that cell contacts are not only targets but also active mediators of EMyT and established a two-hit paradigm in which contact injury and TGFβ are both necessary for the process. This suggests that the injured epithelium is topically susceptible to the transforming effect of TGFβ. Moreover, cytoskeleton reorganization proved to be major input for the transcriptional reprogramming underlying EMyT.
During our studies on the role of the actin skeleton in EMyT, we observed that the microtubule cytoskeleton also exhibits substantial changes. We found that the primary cilium, a microtubule-based organelle, which functions as a mechanosensory and chemosensory antenna of all nucleated cells, undergoes major alterations during EMyT. This observation is of interest for several reasons. The loss or dysfunction of the cilium has been associated with fibrosis. For example, polycystic kidney disease, the prototypic ciliopathy, culminates in tubulointerstitial fibrosis and involves myofibroblast accumulation and, presumably, EMT (Okada et al., 2000; Togawa et al., 2011). Further, genetic loss of the cilium was reported to predispose the endothelium to shear stress–induced endothelial–mesenchymal transition (Egorova et al., 2011). On the other hand, key components of several signaling pathways with known roles in fibrogenesis, such as the hedgehog, platelet-derived growth factor (PDGF), and Wnt pathways (Seeger-Nukpezah and Golemis, 2012), localize to the primary cilium. Thus the primary cilium might be affected during EMyT, and/or, conversely, ciliary changes might affect fibrogenesis. However, neither the fate of the cilium during EMyT nor its potential role in EMyT has been elucidated.
The aim of the present work is to explore the effect of TGFβ-promoted myofibroblast transition on the primary cilium and define the underlying mechanism and ensuing consequences. We also asked how loss of the primary cilium before EMyT induction affects the process. Our results show that myofibroblast transition is associated with initial growth, followed by complete loss of the primary cilium. Ciliary loss is mediated by Smad3-, Rac-, and MRTF-dependent myosin activation, which involves reactive oxygen species (ROS).
RESULTS
Biphasic changes in the primary cilium during EMyT
To characterize the fate of the primary cilium during EMyT, we used our two-hit model, in which a confluent epithelial layer is treated with low-calcium medium (LCM) to uncouple the intercellular contacts and is exposed to TGFβ. This powerful model allowed us to induce synchronized transition and dissect the effects of the two critical inputs (contact disruption and TGFβ) and their synergy during EMyT (Masszi et al., 2004, 2010). We followed the effect of the individual and combined stimuli at early (12 h) and late (48 h) phases of the process using acetyated tubulin (Ac-tub) as a cilium marker (Figure 1). TGFβ added to a confluent, serum-deprived monolayer did not elicit any change in cilium length after 12 h of treatment, whereas LCM induced a significant (on average twofold) increase in this parameter, which was similar after the combined treatment (LCM + TGFβ) as well (Figure 1, A and B). Because uncoupling of the contacts initiates E-cadherin internalization and degradation in tubular cells (Ivanov et al., 2004; Masszi et al., 2004), we asked whether reduced E-cadherin expression might contribute to the observed cilium growth. To test this, we transfected cells with a small interfering RNA (siRNA) against E-cadherin (Charbonney et al., 2011), which effectively diminished the expression of this protein (Figure 1C), and determined cilium length after 24 h of serum deprivation under normal calcium conditions. E-cadherin silencing caused significant (twofold) ciliary elongation, with a large drop in the number of cells with short (<4 μm) and an increase in cells with long (>12 μm) cilia (Figure 1, D and E). Together these findings imply that the early phase of EMyT is associated with cilium lengthening, and this effect may be related to loss of adherens junctions.
FIGURE 1:

The primary cilium undergoes a biphasic change during EMyT. (A) Confluent LLC-PK1 cells were treated as indicated for 12 h (TGFβ, 4 ng/ml) and stained for acetylated tubulin (Ac-tub). (B) Quantification of cilium length for the indicated conditions (mean ± SEM, n = 3, >350 cells/experiment). (C) LLC-PK1 cells were transfected with nonrelated (NR) or E-cadherin (E-cad) siRNA at 40% confluence, serum starved for 24 h after reaching confluence, and then stained for Ac-tub. E-cad knockdown was confirmed by Western blotting. (D) Quantification of cilium length as in B. (E) Distribution of cilium length in control and E-cadherin–depleted cells (n = 3, >350 cells/experiment). (F) LLC-PK1 cells were treated as indicated for 48 h and stained for Ac-tub or polycystin-2 (PC2) or processed for scanning electron microscopy. (G) Quantification of ciliated cells by Ac-tub staining (n = 3, ∼400 cells/experiment).
A dramatically different scenario was seen upon long-term (48 h) treatment, with the actual emergence of the myofibroblast phenotype. Qualitatively, TGFβ or LCM alone caused no change in the pattern of Ac-tub staining, whereas LCM plus TGFβ provoked a dramatic loss of the primary cilium concomitant with a large increase in cytosolic Ac-tub labeling (Figure 1F, top). Increased cytosolic tubulin acetylation often accompanies cilium loss (Overgaard et al., 2009; Pitaval et al., 2010), likely due to the cytosolic relocalization of the ciliary acetyl transferase (Shida et al., 2010). To avoid any confounding effect of higher intracellular Ac-tub staining, we used an alternative cilium marker, polycystin-2, as well as scanning electron microscopy and confirmed that the transformed cells do not possess a cilium (Figure 1F, bottom). Of note, the microvilli are also lost due to loss of epithelial polarity in the transformed cells, which, however, is not directly associated with cilium loss since most nonpolarized (e.g. mesenchymal) cells also possess a primary cilium (e.g., Figure 2, E and F). Next we quantified these changes by comparing the percentage of ciliated cells under each condition after short- and long-term treatment (Figure 1G). Neither TGFβ nor LCM alone altered ciliation within 12 h, whereas the combined treatment caused a slight decrease. After 48 h, the percentage of control (untreated) cells with cilium slightly increased (reaching ∼80%), and this was not altered by TGFβ. Cells exposed to LCM failed to further ciliate between 12 and 48 h but kept their cilium. In contrast, TGFβ acting on contact-uncoupled cells caused a dramatic decrease in the number of ciliated cells: only 5% of the cells preserved their cilium, corresponding to an eightfold loss compared with the level obtained by LCM treatment alone, indicating a net loss of the primary cilium during myofibroblast transition.
FIGURE 2:

Epithelial cells along a wound edge and nonepithelial progenitors lose their primary cilium as they transition into myofibroblasts. (A) Confluent LLC-PK1 layers were wounded, treated as indicated for 48 h, and stained for Ac-tub. (B) Percentage of cells with a primary cilium was determined 24 h after the indicated treatment as a function of the cell row (1–5) from the wound edge (n = 4, 25 cells/experiment for each row). (C) Human skin fibroblasts (HSFs; left) and 10T1/2 cells (right) were serum starved or treated with TGFβ (5 ng/ml) for 72 h, followed by Western blotting for the indicated proteins. (D) The same two cell types were treated as in C and then stained for Ac-tub. Nuclei were visualized with 4′,6-diamidino-2-phenylindole. (E) 10T1/2 cells were treated as in C and then processed for scanning electron microscopy and visualized using magnification 2500× (left) and 15,000× (right). Right, enlarged area indicated by boxes on the left. Bars, 10 and 1 μm for upper and lower panels, respectively.
Because cell cycle reentry is associated with deciliation (Kim and Tsiokas, 2011), we tested whether the EMyT-related loss of the cilium could be due to a change in cell proliferation. Immunostaining for Ki67, a sensitive proliferation marker, revealed that nuclear Ki67 labeling was actually less in TGFβ plus LCM–treated cells than under any other condition, indicating that the loss of the cilium was not due to cell proliferation (Supplemental Figure S1). Taken together, the results indicate that EMyT is associated with a biphasic change in the primary cilium: an initial lengthening is followed by dramatic loss, which is independent of the cell cycle. Because the overall transition was accompanied by the loss of the cilium, we decided to further characterize this phenomenon and the underlying mechanisms.
Locus-specific, TGFβ-induced deciliation of epithelial cells at the wound
An epithelial wound—that is, a discontinuity in the monolayer—is a pathopysiologically more relevant model of cell contact disruption than the application of LCM. Our previous work showed that the wound edge represents a special locus where cells are topically susceptible to the transforming effect of TGFβ (Speight et al., 2013). We therefore checked whether wound-adjacent cells might exhibit ciliary changes in the absence or presence of TGFβ. After scratch wounding of a confluent monolayer, quiescence was achieved by serum deprivation, and then the cultures were left untreated or exposed to TGFβ for 48 h. In the absence of TGFβ, cells were ciliated both in the intact (inner) regions of the monolayer and the wound-adjacent areas (Figure 2A). In contrast, TGFβ caused robust loss of the cilium concomitant with increase in cytosolic Ac-tub staining. However, this effect was fully restricted to cells located in the wound region, whereas cells in the intact part of the layer remained ciliated (Figure 2A). To quantify this effect, we determined the percentage of ciliated cells as a function of location, that is, directly at the wound edge and in several rows behind it (Figure 2B). In the absence of TGFβ, slightly fewer cells possessed a cilium in the first row than in the next, with no further change inward. Thus, wounding per se had only a marginal effect on cilium homeostasis. The addition of TGFβ had no effect in rows four and higher but caused a significant and progressive loss of the cilium in cells located in the first three rows, attaining approximately fourfold decrease in the first row compared with untreated cells at the same locus. Thus TGFβ induced a site-specific (wound-restricted) loss of the primary cilium. Of importance, this effect mirrors the locus-specific EMyT-provoking effect of TGFβ (Fan et al., 2007).
Fibroblast–myofibroblast transition is also associated with loss of the cilium
Myofibroblasts originate from mesenchymal cells such as fibroblasts and pericytes, the latter of which were proposed to be their major precursor in kidney fibrosis models (Humphreys et al., 2010). To assess whether the loss of the primary cilium is specific for EMyT or is a general feature of the myofibroblast phenotype, we treated human skin fibroblasts and 10T1/2 cells, a pericyte-like cell line, with TGFβ, which is sufficient to increase SMA expression in these cells (Figure 2C). Under resting conditions both mesenchymal cell types developed primary cilia (arrows) and contained little intracellular Ac-tub. TGFβ induced loss of the cilium concomitant with substantial increase in cytosolic Ac-tub, recapitulating the effects observed in transitioning epithelial cells (Figure 2D). Because 10T1/2 cells have short cilia and an increase in cytosolic Ac-tub might confound the visualization of the cilium, we verified these findings by scanning electron microscopy (Figure 2E). Taken together, the results show that formation of the myofibroblast phenotype was accompanied by loss of the cilium, irrespective of whether the precursors were epithelial or mesenchymal cells.
Smad3 signaling is necessary but not sufficient to induce cilium loss in epithelial cells
TGFβ mobilizes Smad-dependent and -independent pathways, and Smad3 plays a dual role in EMyT. It is necessary for the mesenchymal phase but delays the myogenic phase (Masszi et al., 2010). We therefore asked whether Smad3 signaling is necessary for loss of the cilium in our epithelial two-hit model. We used two approaches. First we expressed various forms of the TGFβ receptor I (TGFβRI), which show differential activity toward the Smad3 pathway. Cells were transfected with the Flag epitope–tagged wild-type (WT) receptor, the K232R mutant (KR), which is inactive toward Smads but activates noncanonical TGFβ pathways (e.g., p38), or the T204D mutant (TD), which is constitutively active and turns on both Smad and non-Smad pathways (Sorrentino et al., 2008; Kim et al., 2009b). Expression of receptors was followed by Flag immunostaining, and their expected activity toward Smad3 was verified by a Smad3-specific luciferase reporter (Smad binding element 4 [SBE4]) assay. As expected, overexpression of WT or KR did not induce the reporter, whereas TD caused substantial stimulation (Supplemental Figure S2). Next we tested their effect on the primary cilium. Under control conditions neither construct induced any ciliary change, as revealed by double staining for Flag and Ac-tub (Figure 3A) and quantified as percentage of cells with a cilium (Figure 3B). This result implies that TGFβ signaling alone is not sufficient to provoke loss of the cilium. Next we exposed control and receptor-transfected cells to LCM. Remarkably, under these conditions only cells expressing TD exhibited robust (fourfold) decrease in ciliation (Figure 3, C and D). As a second approach, cells were transfected with siRNA against Smad3 and then exposed to TGFβ plus LCM for 48 h. The siRNA efficiently down-regulated Smad3 (Figure 3E, top) and almost completely prevented loss of the cilium compared with LCM-treated cells (Figure 3, E, bottom, and F). Together these results suggest that Smad3 activation is a critical albeit insufficient mediator of cilium loss in the epithelium.
FIGURE 3:

Smad3 signaling is necessary for deciliation. (A–D) LLC-PK1 cells transfected with kinase-dead (KR; A) or constitutively active (TD; C) TGFβR1 grown to confluence, serum starved for 48 h in normal or LCM medium, and stained for the FLAG epitope and Ac-tub. For B and D, ciliation was quantified also for nontransfected and WT TGFβR1-expressing cells (n = 4, 20 cells/condition). (E, F) Cells transfected with nonrelated or Smad3 siRNA, grown to confluence, and then treated with LCM plus TGFβ for 48 h, stained for Ac-tub (A), and quantified for ciliation (F; n = 3, ∼150 cells/condition).
A sustained increase in myosin light chain phosphorylation is necessary and sufficient for loss of the primary cilium
Our previous studies showed that synergy between TGFβ-induced and contact-dependent signaling is necessary for the sustained cytoskeletal remodeling and transcriptional reprogramming (Masszi et al., 2010). Because increased contractility is a major attribute of myofibroblasts (Wipff et al., 2007) and a report proposed that high cell contractility inhibits ciliogenesis (Pitaval et al., 2010), we asked whether changes in myosin light chain (MLC) phosphorylation (a chief marker of contractility) might underlie loss of the cilium and explain the need for both contact disassembly and TGFβ for this process in epithelium. Consistent with previous findings (Fan et al., 2007), acute contact uncoupling (induced by 30 min of LCM treatment) caused a robust increase in MLC phosphorylation, which was restricted to the cell periphery under the disassembled contacts (Figure 4, A and C). TGFβ failed to induce MLC phosphorylation and had no effect on the localization or magnitude of MLC phosphorylation provoked by short-term LCM treatment (Figure 4, A and C). Long-term (48 h) LCM only marginally increased MLC phosphorylation, indicating that it has a transient effect. Long-term TGFβ treatment caused a slight increase in phospho-MLC (pMLC), which was detectable by Western blotting but hardly visible by immunofluorescence. In contrast the combined application of LCM and TGFβ resulted in a robust increase in pMLC, which was apparent both at the cell periphery and in stress fibers (Figure 4, B and D). Of importance, under these conditions, not only the phosphorylated, but also the total MLC level was substantially increased, suggesting that both MLC activation and expression contribute to the overall effect (Figure 4D). Thus EMyT-inducing conditions resulted in prolonged increase in MLC phosphorylation. We argued that if MLC phosphorylation is causally linked to loss of the cilium, then correlation should exist between wound-adjacent distribution of pMLC signals and absence of the cilium. To test this assumption, confluent monolayers were scratch wounded and then left untreated or exposed to TGFβ, followed by dual staining for pMLC and acetylated tubulin (Figure 4E). Forty-eight hours after wounding in the absence of TGFβ, only sporadic and weak pMLC labeling was seen, which was confined to some cells located directly at the wound edge. As mentioned before, cells at the edge remain ciliated. In contrast, in the presence of TGFβ, cells at the wound edge and several rows behind exhibited substantially increased pMLC staining, indicating a spatially restricted increase in contractility. Of importance, these pMLC-positive cells lost their primary cilium and showed increased cytosolic Ac-tub labeling (Figure 4E). Finally we tested whether TGFβ, which is sufficient to elicit myofibroblast transition in pericyte-like 10T1/2 cells (Figure 2D), could also induce sustained MLC phosphorylation. Indeed, 48-h TGFβ exposure resulted in a strong increase in pMLC content (Figure 4F). Taken together, the results show that prolonged MLC phosphorylation coincided with loss of the cilium in both models of EMyT, as well as during pericyte–myofibroblast transition.
FIGURE 4:

Sustained increase in contractility correlates with loss of the primary cilium. (A, B) LLC-PK1 cells were treated as indicated for 30 min (A) or 48 h (B) and stained for pMLC. (C, D) Cells treated as in A and B, respectively, were processed for Western blotting for the indicated proteins. (E) Confluent monolayers were wounded, treated as indicated for 48 h, and stained for pMLC and Ac-tub. (F) 10T1/2 cells were serum starved or treated with TGFβ for 48 h, followed by Western blotting for the indicated proteins.
Next we investigated whether this coincidence reflects a cause–effect relationship and whether MLC phosphorylation is sufficient to provoke loss of the cilium. Cells were transfected with a constitutively active, diphosphomimetic (T18D, S19D, designated as DD) mutant of MLC, tagged with the Myc epitope, and then doubly stained for Myc and Ac-tub. The majority of DD-MLC–expressing cells lost their primary cilium and showed enhanced intracellular Ac-tub staining (Figure 5A). There was a fourfold reduction in percentage of ciliated DD-MLC–expressing cells compared with their untransfected neighbors (Figure 5B). To assess whether myosin is needed for EMyT-associated loss of the cilium, we down-regulated nonmuscle myosin heavy chain (isoform IIA, Myh9) using an effective siRNA (Figure 5C, bottom). Myosin silencing caused near-complete rescue of the cilium in TGFβ plus LCM–treated cells (Figure 5, C and D). Having seen that the two-hit condition elevated total MLC expression as well (Figure 4D), we asked whether MRTF is also critical for loss of the cilium. This possibility was raised by our previous finding that MRTF, which controls expression of various cytoskeletal proteins including myosin, is translocated to the nucleus upon contact disassembly, and its nuclear retention is augmented by TGFβ (Fan et al., 2007; Masszi et al., 2010). Silencing of MRTF caused a reduction in the level and stimulus-induced MLC expression and accordingly substantially reduced the total pMLC content of the stimulated cells (Figure 5E). Concomitantly, MRTF down-regulation completely prevented EMyT-associated cilium loss and increase in cytosolic Ac-tub (Figure 5, F and G). Collectively these data show that high levels of myosin phosphorylation can induce and is necessary for loss of the cilium during EMyT and that MRTF plays an important (presumably permissive) role in this process.
FIGURE 5:

Prolonged increase in myosin activity is sufficient and necessary for cilium loss. (A) Confluent LLC-PK1 cells were transfected with the constitutively active Myc-tagged (DD) MLC II in serum-free medium and stained 24 h later for Myc and Ac-tub. (B) The presence of the cilium was quantified in DD-MLC–expressing cells and their nontransfected neighbors on the same coverslip (mean ± SEM, n = 3, 25 cells/condition). (C) LLC-PK1 cells were transfected with nonrelated (NR) or myosin heavy chain II (MHC II)–specific siRNA, grown to confluence, treated as indicated for 48 h, and then stained for Ac-tub. MHC II knockdown was verified by Western blotting. (D) Ciliation percentage was quantified (mean ± SEM, n = 3, ∼150 cells/experiment). (E) LLC-PK1 cells were transfected with siRNAs against MRTF A and MRTF B or NR siRNA. After reaching confluence, cells were treated as shown for 48 h and then subjected to Western blotting for the indicated proteins. (F) LLC-PK1 cells treated as in E were stained for Ac-Tub. (G) Ciliation percentage for E was quantified (mean ± SEM, n = 3, ≈150 cells/experiment).
Rac1 but not RhoA is critical for EMyT-associated loss of the primary cilium
Contact disassembly activates both RhoA and Rac1 (Busche et al., 2008; Sebe et al., 2008). Of these small GTPases, RhoA is the “classic” inducer of MLC phosphorylation. Moreover, in certain cell types TGFβ prolongs RhoA activation in a Smad3-dependent manner by inducing the expression of Rho guanine nucleotide exchange factors (Lee et al., 2010; Papadimitriou et al., 2012). On the other hand, Rac1 also enhances MLC phosphorylation (through p21-activated kinase) in certain cell types (Chew et al., 1998), although it also indirectly suppresses MLC phosphorylation by inhibiting MLC kinase (Raymond et al., 2011). Cognizant of this scenario, we initially assessed the effects of active RhoA and Rac1 on the status of the cilium and MLC phosphorylation in LLC-PK1 cells. Overexpression of Myc-tagged active RhoA (Q63L; Figure 6, A and B) or Myc-tagged active Rac1 (Q61L; Figure 6, C and D) caused strong MLC phosphorylation and led to the loss of the cilium. To discern whether RhoA or/and Rac1 might play a role in the TGFβ plus LCM–induced deciliation, we separately down-regulated these small GTPases by specific siRNAs, followed by 48-h exposure to the combined stimuli. To our surprise, elimination of RhoA only marginally mitigated cilium loss. In contrast, Rac1 down-regulation provided strong protection against the deciliating effect of TGFβ plus LCM (Figure 6, E and F). We performed Western blot analysis to follow the changes in MLC and the small GTPases after the individual and combined treatments (Figure 6G). Of interest, RhoA was down-regulated by the two-hit condition (48 h) itself, which explains how, although it could play a role in the early phase, it is unlikely to contribute to sustained responses. In contrast, Rac1 expression did not decrease. In fact, in some (but not all) experiments it increased after the combined treatment. Consistent with these finding, siRNA-mediated down-regulation of RhoA only slightly reduced pMLC levels attained after LCM plus TGFβ treatment (Figure 6G), whereas silencing of Rac1 strongly suppressed the rise in pMLC and somewhat mitigated the increase in total MLC. (In addition, we observed that Rho silencing also reduced Rac levels.) Taken together, the results show that two hit-induced suppression of the primary cilium required Rac1, and this small GTPase is critical to sustain MLC phosphorylation in the stimulated cells.
FIGURE 6:

Rac1 is required for increased myosin activity and deciliation during EMyT. (A–D) Confluent LLC-PK1 cells were transfected with constitutively active, Myc-tagged Q63L RhoA (Myc CA-RhoA; A, B) or active, Myc-tagged Q61L Rac1 (Myc CA-Rac1; B, D) and 24 h later doubly stained for the Myc epitope and either pMLC or Ac-tub. Quantification of percentages of ciliated cells for A and C are shown in B and D, respectively. Myc-positive cells were compared with their Myc-negative neighbors on the same coverslips (n = 3, 20 cells/experiment). (E, F) LLC-PK1 cells were transfected at 60% confluence with siRNA against RhoA or Rac1 or nonrelated (NR) siRNA and after reaching confluence treated with LCM plus TGFβ for 48 h, stained for Ac-tub (E), and quantified for ciliation (F; n = 3, ∼150 cells/experiment). (G) LLC-PK1 cells were transfected with Rac1, RhoA, or NR siRNA at 60% confluence and, when confluent, treated as indicated for 48 h and subjected to Western blotting for the indicated proteins.
Increased MLC phosphorylation involves Smad3 and depends on ROS
Rac1 might modify long-term MLC phosphorylation by various mechanisms. Besides its direct effects, Rac1 activation is associated with ROS generation. Namely, Rac1 is a component of the NADPH oxidase (Hordijk, 2006) and was also shown to promote mitochondrial ROS generation (Osborn-Heaford et al., 2012). ROS in turn can increase MLC phosphorylation (Tsai and Jiang, 2010). Of importance, TGFβ induces expression of the NADPH oxidase isoform Nox4 in a Smad3-dependent manner, and this process is required for both fibroblast–myofibroblast transition and EMyT (Cucoranu et al., 2005; Hecker et al., 2009; Boudreau et al., 2012). Together these findings raise the possibility of a unifying mechanism that may involve both Rac1 and Smad3 and lead to increased MLC phosphorylation and consequent loss of the cilium in a ROS-dependent manner. To assess this possibility, initially we tested whether myofibroblast transition induced by TGFβ plus LCM treatment is indeed associated with increased ROS production in LLC-PK1 cells. As shown in Figure 7A and quantified in Figure 7B using the nitro blue tetrazolium assay, transformed cells produced significantly higher amounts of ROS. To test whether ROS contribute to MLC phosphorylation and the ciliary changes, we pretreated cells with apocynin, a potent scavenger and inhibitor of the NADPH oxidase (Petronio et al., 2013), and then subjected them to TGFβ plus LCM stimulation. Apocynin substantially reduced the ensuing MLC phosphorylation and caused a threefold increase in the number of cells that kept their cilium (Figure 7, C–E). Next we tested whether down-regulation of Smad3 before EMyT induction interferes with MLC phosphorylation (this timing should be distinguished from the gradual down-regulation of Smad3 seen during EMyT). siRNA-mediated Smad3 knockdown before TGFβ plus LCM treatment strongly suppressed MLC phosphorylation (Figure 7F). Moreover, EMyT was associated with a robust increase in Nox4 expression, and this response was also significantly inhibited by Smad3 down-regulation (Figure 7, G and H). Taken together, the results show that Smad3 contributes to Nox4 induction and is required for sustained, ROS-dependent MLC phosphorylation, which is a prerequisite for the loss of the cilium.
FIGURE 7:

Reactive oxygen species are necessary for enhanced contractility and deciliation. (A, B) LLC-PK1 cells were grown to confluence and treated with serum-free medium (control) or LCM plus TGFβ for 48 h. Cells were then exposed to NBT for 45 min and processed as described in Materials and Methods. Reduced NBT (formazan) particles were first visualized by light microscopy (A) and then extracted, solubilized, and quantified (B; n = 5). (C) LLC-PK1 cells were treated as in A in the presence of dimethyl sulfoxide (DMSO) or 600 μM apocynin and processed for Western blotting for the indicated proteins. (D) Confluent LLC-PK1 cells were treated with LCM plus TGFβ for 48 h in the presence of DMSO or 600 μM apocynin and stained for Ac-tub. (E) Ciliation for B was quantified (n = 3, ∼150 cells/experiment). (F, G) LLC-PK1 cells were transfected at 60% confluence with nonrelated or Smad3 siRNA and, upon reaching confluence, treated as indicated for 48 h and processed for Western blotting for the indicated proteins. (H) Densitometric quantification of Nox4 expression in control and Smad3-depleted cells upon LCM plus TGFβ treatment, as shown in E (n = 6).
EMyT is associated with profound changes in responsiveness to cilium-specific signals
Next we investigated whether EMyT-associated loss of the cilium translates into altered responsiveness to stimuli that are normally processed through ciliary signaling pathways. Two such ligands are platelet-derived growth factor-AA (PDGF-AA), which acts through PDGF receptor α, a receptor localized to the cilium (Schneider et al., 2005), and sonic hedgehog (Shh), which acts through patched, another cilium-resident receptor (Rohatgi et al., 2007). To assess the effect of EMyT specifically on ciliary signals, we compared the effect of PDGF-AA with that of epidermal growth factor (EGF), whose receptor is nonciliary, using a common downstream response, the phosphorylation of the mitogen-activated protein kinase Erk. Cells were left untreated or transitioned to myofibroblasts by TGFβ plus LCM for 48 h and then exposed to EGF or PDGF-AA for 5 min (Figure 8A). EGF provoked strong Erk phosphorylation in both nontransformed and transformed cells. In contrast, PDGF-AA induced robust Erk phosphorylation in nontransformed cells, but this effect was completely lost in transformed cells. The effect of Shh was tested in 10T1/2 cells, which were either left untreated or transformed to myofibroblasts by TGFβ alone. Cells were transfected with a luciferase reporter construct responsive to Gli, a patched-induced transcription factor, and then stimulated with Shh (Figure 8B). Whereas Shh caused a sevenfold activation of the reporter in nontransformed cells, it had negligible effect in transformed cells. Thus myofibroblast transition of both an epithelial and a mesenchymal precursor, induced by different means, was associated with the loss of responsiveness to different ciliary signaling pathways.
FIGURE 8:
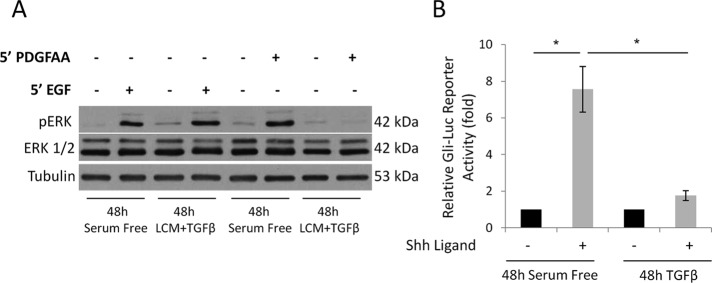
Cilium-related signaling is altered in myofibroblasts. (A) Confluent LLC-PK1 cells were serum starved or treated with LCM plus TGFβ for 48 h. Subsequently, cells were incubated for 5 min with EGF or PDGF-AA and subjected to Western blotting for the indicated proteins. (B) 10T1/2 cells were transfected with the Gli-luciferase reporter construct at 60% confluence. After reaching full confluence, cells were serum starved for 24 h and then left in serum-free medium or treated with TGFβ for 72 h. During the last 24 h of the treatment, Shh ligand was added as indicated, and luciferase activity was measured. Bars, fold increase in firefly luciferase activity normalized to Renilla activity (n = 3).
The primary cilium contributes to the initial phase of EMyT
So far our data show that full-blown EMyT is associated with the loss of the cilium and some cilium-related signaling events. However, a variety of cilium-associated pathways (e.g., PDGF, hedgehog [Hh], Wnt) have EMT-promoting or fibrogenic effects (Seeger-Nukpezah and Golemis, 2012) and may also synergize with TGFβ signaling. Therefore we asked whether the cilium, at least initially, might be involved in the mediation of EMyT or fibroblast–myofibroblast transition. To address this point, we interfered with the cilium before the induction of the transition and then assessed whether this alters the stimulus-induced expression of SMA, the chief myofibroblast marker. First, we treated the cells with HPI-4, a compound that inhibits the hedgehog pathway by inducing loss of the cilium (Jung et al., 2011). HPI-4 caused complete deciliation in the tubular cells, along with a large increase in cytosolic Ac-tub staining (Figure 9A). Of interest, HPI-4 abolished TGFβ plus LCM–induced SMA expression (Figure 9B). Similar morphological and functional observations were made in fibroblasts, in which HPI-4 prevented the TGFβ-induced increase in SMA expression (Figure 9, C and D). Because the exact mechanism of action of HPI-4 is not known, we used an alternative, nonpharmacological and specific means to interfere with the cilium by downregulating Kif3a, a kinesin critical for cilium integrity and function (Lin et al., 2003). Knockdown of Kif3a in the epithelium resulted in the truncation of the cilia, manifesting as bulky circular patches of Ac-tub staining (Figure 9E). Kif3a silencing substantially inhibited LCM plus TGFβ–induced SMA expression (Figure 9F). These data are in complete agreement with a recent article showing impaired TGFβ signaling in Tg737 Oakridge polycystic kidney mouse fibroblasts, which possess stunted primary cilia due to an Ift88 mutation (Clement et al., 2013). Of interest, the same cilium defect was associated with increased shear stress–induced endothelial–mesenchymal transition (Egorova et al., 2011). This scenario raised the possibility that although the initial presence of the cilium is necessary for efficient TGFβ-induced EMyT, the ensuing loss itself might facilitate the full-blown transition. We attempted to address this possibility by varying the time of Kif3a siRNA transfection before stimulation. We surmised that the cilium should be intact at the time of TGFβ plus LCM stimulation, but acceleration of its disruption could result in greater SMA expression. Because the TGFβ plus LCM treatment itself induces deciliation after ∼24 h, we had a very narrow time frame in which to interfere with the cilium (i.e., to keep it at the time of the stimulus but then facilitate its loss). We transfected the cells with Kif3a siRNA at 6, 12, and 24 h before TGFβ plus LCM stimulation for 72 h. Intriguingly, compared with the corresponding nonrelated siRNA control, Kif3a siRNA transfection consistently increased the ensuing SMA expression when it was applied 6 h before the stimulation, whereas it markedly suppressed SMA expression if it preceded the stimulation by 24 h (Figure 9G) or more (Figure 9F). The intermediate transfection time (12 h) had only a slight inhibitory effect. As verified in parallel experiments, Kif3a siRNA did not yet cause a significant change in Kif3a protein expression (Figure 9H) or cilium structure (Supplemental Figure S3) after 6 h of transfection, whereas it induced marked Kif3a decrease and cilium disruption at 24 h. A plausible interpretation of these results is that Kif3a siRNA applied 6 h before stimulation did not interfere with the cilium at the time of stimulation but accelerated its loss thereafter, which was accompanied by heightened SMA expression. Taken together, the results show that elimination or disruption of the cilium before TGFβ addition mitigates the transition of various precursors into myofibroblast, but once the critical signaling has been initiated, ciliary loss might facilitate the transition.
FIGURE 9:

Efficient induction of EMyT requires the primary cilium. (A) LLC-PK1 cells were incubated for 24 h with DMSO or 30 μM HPI-4 and then stained for Ac-tub. (B) Confluent LLC-PK1 were incubated with DMSO or 30 μM HPI-4 for 24 h in serum-free medium and then left untreated or exposed to LCM plus TGFβ supplemented with DMSO or HPI-4. Western blotting was then performed for the indicated proteins. (C) Human skin fibroblasts were grown to confluence, incubated as in A, and then fixed and stained for Ac-tub. (D) Confluent human skin fibroblasts were incubated as in B and then either left in serum-free medium or treated with TGFβ for 72 h. Subsequently Western blotting was performed for the indicated proteins. (E) LLC-PK1 cells were transfected with nonrelated (NR) or Kif3a siRNA at 40% confluence, grown to confluence, serum starved for 24 h, and stained for Ac-tub. (F) LLC-PK1 cells were transfected as in E, treated as indicated for 72 h, and subjected to Western blotting for the indicated proteins. (G, H) The process of deciliation may facilitate SMA expression. (G) LLC-PK1 cells were transfected with NR or Kif3a siRNA at varying times (6, 12, and 24 h) before treatment (labeled as -6, -12, and -24 h, respectively) with serum-free medium or LCM plus TGFβ for an additional 72 h. Subsequently, Western blotting was performed for SMA and GAPDH as loading control. Note the reversal (early stimulation and late inhibition) of the SMA response. (H) To check for Kif3a expression at the time of stimulation, cells were transfected as shown and after the indicated time processed for Western blotting for Kif3A and GAPDH.
DISCUSSON
Changes in the primary cilium during myofibroblast generation
This study characterizes changes in the primary cilium during myofibroblast generation and allows insight into the underlying mechanisms. One of our key findings is that the emergence of the myofibroblast phenotype, irrespective of whether the precursors are mesenchymal or epithelial cells, is associated with loss of the primary cilium. In epithelial cells, in which double-hit conditions (LCM plus TGFβ or wounding plus TGFβ) are necessary for myofibroblast transition, the same combined conditions are required for loss of the cilium. In contrast, in mesenchymal cells, in which TGFβ can provoke myofibroblast transition, it is sufficient for deciliation. These results suggest that transition to the myofibroblast state, rather than a particular stimulus or pathway, is the critical factor for deciliation. On the basis of these findings, we propose that the myofibroblast (or at least the nascent myofibroblast) is a unique cilium-less entity.
Although the cilium is ultimately lost, the initial phase of EMyT is associated with ciliary growth. Because early EMT/EMyT is characterized by the loss of adherens junctions (AJs; Kalluri and Weinberg, 2009), we considered that the absence of cell contacts might be linked to cilium growth. Indeed, we found that LCM-induced junction uncoupling (which results in E-cadherin endocytosis and subsequent degradation; Ivanov et al., 2004; Masszi et al., 2004) or specific siRNA-mediated E-cadherin knockdown caused substantial cilium growth, suggesting that AJ integrity is a critical regulator of cilium homeostasis. Consistent with this intriguing possibility, a study showed that scratch wounding of the cornea results in reassembly and elongation of cilia in corneal endothelial cells, which under normal conditions contain no or very short cilia (Blitzer et al., 2011). Furthermore, regeneration after acute tubular necrosis was associated with cilium elongation in tubular cells (Verghese et al., 2009). Longer cilia have been proposed to exhibit increased sensitivity to flow (Schwartz et al., 1997), which may maintain sufficient ciliary signaling, necessary for optimal healing (Verghese et al., 2009). In any case, cilium elongation is usually coincident with normal tissue repair. However, our studies suggest that this regenerative phase may signify a vulnerable, “sensitized” state, and TGFβ exposure in this context can result in cilium loss, a condition associated with dysregulated regeneration, fibrosis, or carcinogenesis (Seeley and Nachury, 2010; Seeger-Nukpezah and Golemis, 2012). Regulation of cilium length is a complex and poorly understood process (Miyoshi et al., 2011; Broekhuis et al., 2013). Although the mechanism by which intercellular junctions modulate cilium length was not the focus of this work, it is worth mentioning a few possibilities. Considering only potential direct links, E-cadherin has been reported to associate with polycystin-1 at the cell junctions (Huan and van Adelsberg, 1999), and the level of WT polycystin-1 was proposed to regulate cilium length in a dose-dependent manner (Hopp et al., 2012). It remains to be established whether E-cadherin depletion affects polycystin-1 stability or the levels available in the cilium and whether this has an effect on cilium length. E-cadherin was also found to associate with TRPV4 (Sokabe and Tominaga, 2010), a mechanosensitive cation (Ca2+) channel that forms a functional complex with polycystin-2 (Kottgen et al., 2008). An increase in intracellular Ca2+ is a negative regulator of cilium length (Miyoshi et al., 2011). Future studies should test whether loss of E-cadherin affects basal Ca2+ levels in a TRPV4-dependent manner.
Critical signaling mechanisms leading to cilium loss during myofibroblast transition: key role for myosin
Because after a transient elongation the cilium is lost during myofibroblast transition, we sought to understand the signaling events leading to this end state. One of the main features of the myofibroblast phenotype is increased myosin-based contractility (Hinz, 2010), and our data suggest that this is also the key determinant underlying loss of the cilium. This conclusion is supported by our findings that 1) inducing increased myosin activity by various means, including the expression of active RhoA, Rac1, or diphosphomimetic MLC, triggers deciliation; and 2) suppressing myosin expression or activation by various means, including down-regulation of the myosin heavy chain, MRTF, Smad3, or Rac1, mitigates the loss of the cilium. Moreover, we established that sustained MLC phosphorylation is needed for the loss of the cilium—that is, transient increases in contractility can occur without altered cilium status. The EMyT-associated changes in MLC phosphorylation and cilium loss can be integrated into a coherent picture (Figure 10). TGFβ activates Smad3, which is necessary for the two-hit-induced cilium loss. Moreover, the absence of contacts potentiates early Smad3 signaling, presumably due to nuclear translocation of TAZ, a Smad3 retention factor (Varelas et al., 2008, 2010). This is one reason why contact-deprived cells are topically susceptible to TGFβ. Contact disassembly also leads to activation of small GTPases RhoA and Rac1, which in turn induce actin polymerization, nuclear translocation of MRTF, and MLC phosphorylation (Fan et al., 2007; Busche et al., 2008). However, two remarks must be made here. First, contact disruption alone causes only transient changes in MRTF translocation and MLC phosphorylation, but these responses are prolonged by TGFβ. Second, although RhoA activation is involved in the early responses, RhoA silencing did not affect sustained MLC expression/phosphorylation or cilium loss. This is congruent with our finding that RhoA is down-regulated during EMyT, consistent with its reported TGFβ-induced, Smurf1-mediated degradation in EMT (Ozdamar et al., 2005). However, the role of other Rho proteins cannot be excluded, as TGFβ was shown to induce RhoB (Vasilaki et al., 2010), and various Rho isoforms have partly overlapping function during EMT (Hutchison et al., 2009). Whereas RhoA was dispensable, Rac1 was necessary for sustained MLC phosphorylation and cilium loss. These results are in good agreement with the emerging role of Rac as a key mediator of EMT (Radisky et al., 2005; Lee et al., 2012). Moreover, this allowed us to integrate Rac1- and Smad3-dependent events into a unifying mechanism, since both are necessary for enhanced ROS production (Rac as NADPH oxidase component and Smad3 as inducer of Nox4), which is essential for myofibroblast transition and EMT (Hecker et al., 2009; Bondi et al., 2010; Boudreau et al., 2012). Accordingly, our double-hit conditions induced a robust expression of Nox4 that was substantially mitigated by Smad3 knockdown, which also reduced MLC phosphorylation. ROS can promote myosin phosphorylation predominantly by inhibiting MLC phosphatase (Jernigan et al., 2008; Tsai and Jiang, 2010). Taking the results together, we propose that Rac1- and Smad3-dependent, Nox4-mediated ROS production is an important contributor to EMyT-associated MLC phosphorylation and deciliation. In further support of this mechanism, the NADPH oxidase inhibitor/ROS scavenger apocynin inhibited both responses.
FIGURE 10:
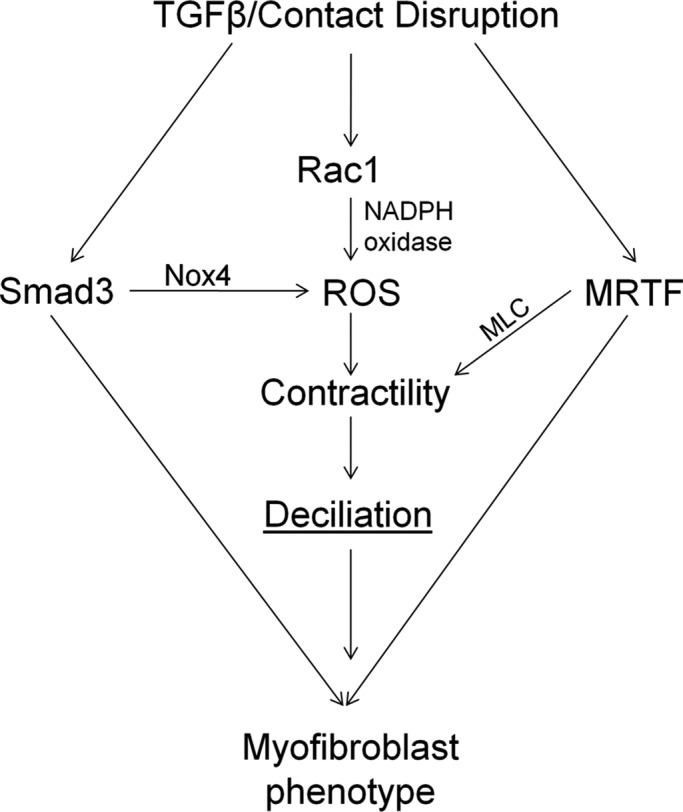
The major processes and their mediators underlying EMyT-associated loss of the cilium. For further explanation see the Discussion.
Potential mechanisms underlying myosin-dependent cilium loss
What is the mechanism by which myofibroblast transition and the associated myosin phosphorylation lead to cilium loss? In principle, the cilium could be lost by shedding (autotomy; Overgaard et al., 2009) or disassembly (resorption; Santos and Reiter, 2008). A study indicated that enhanced contractility interferes with ciliogenesis (Pitaval et al., 2010). In our system, fully developed cilia were eliminated from the cells—that is, EMyT interfered with ciliary maintenance rather than “ciliogenesis”—although the underlying mechanisms might be overlapping. We cannot exclude that both autotomy and resorption (disassembly) could contribute. Nonetheless we favor disassembly as a dominant process for the following reasons. First, using sucrose gradient centrifugation of cellular supernatants after EMyT, we were unable to detect increased cilium shedding, although we could verify this process after acute chemical deciliation (Supplemental Figure S4). Second, the remaining cilia seen in a few percent of cells after EMyT were shorter after 48-h treatment compared with 12 h, which is consistent with resorption (Supplemental Figure S5). Nonetheless, since deciliation seems to be a stochastic process that occurs 12–48 h after EMyT induction, the detection of shedding will depend on the integrity of the shed cilium and the stability of Ac-tub in this time frame. Long-term imaging with live cilium markers may address this question in the future. With regard to potential molecular mechanisms, myosin-II–mediated membrane scission has been described in the Golgi apparatus (Miserey-Lenkei et al., 2010). A similar process might operate at the cilium, or dysregulated Golgi fission might perturb the Golgi-to-cilium traffic, a regulator of cilium maintenance (Deretic, 2013). However, myosin-mediated Golgi fragmentation required Rab6 (Miserey-Lenkei et al., 2010), and our preliminary data show no obvious effect of dominant-negative Rab6 on EMyT-induced deciliation. Alternatively, myosin activity may regulate cilium maintenance by influencing the ciliary barrier at the base of the organelle. Intriguingly, myosin was found to directly bind to septin 2 (Joo et al., 2007), which is a critical component of the ciliary diffusion barrier, necessary for cilium homeostasis (Hu et al., 2010). It is thus conceivable that myosin might interact with septins at the cilium, and this might alter septin function. Early and sporadic reports suggested the presence of myosin II at the base of motile cilia (Gordon et al., 1980; Sandoz et al., 1982; Klotz et al., 1986). However, it remains to be tested whether this is so in the primary cilium and whether local or global contractility is involved in deciliation.
Our data showing that myosin II as a prime regulator of the cilium during myofibroblast transition extend the emerging concept that the microfilament cytoskeleton plays a key role in cilium homeostasis. A recent siRNA screen identified a set of actin skeleton components as positive or negative regulators of ciliogenesis. For example, the actin polymerization inducers Arp3 (Kim et al., 2010) and cortactin (Bershteyn et al., 2010) were shown to inhibit ciliogenesis and/or stimulate cilium disassembly. It is noteworthy that cortactin was detected in the basal body and can also bind to a cytosolic component of the NADPH oxidase (Touyz et al., 2005) and myosin light chain kinase (Dudek et al., 2002; Usatyuk et al., 2012). Thus cortactin might be an adaptor that interacts with or localizes key components of the myosin-dependent deciliation machinery.
Role of the primary cilium during EMyT and myofibroblast generation
Finally we consider the pathophysiologic role of the cilium in myofibroblast transition. Initially, the cilium supports EMyT, since deciliation before the process inhibits the emergence of the myofibroblast phenotype. This finding is fully consistent with the ciliary localization of various signaling complexes—for example, the Hh pathway—with well-established roles in fibrogenesis. Further, while the present work was being submitted, a study from the Christensen lab was published (Clement et al., 2013) showing that TGFβ signaling is impaired in the classic mouse model of polycystic kidney disease. The role of the cilium, however, seems to be more complex, as a previous study revealed that endothelial cells from the same mouse model are predisposed to shear stress–induced mesenchymal transition (Egorova et al., 2011). It remains to be tested whether the cilium has a differential effect on TGFβ-induced and mechanically provoked EMT. In any case, our data are consistent with both an early and critical role of the cilium in the TGFβ-triggered myofibroblast transition and the possibility that the ensuing cilium loss may accelerate this process. Nonetheless, the cilium is lost during the transition, and this suggests that the myofibroblast is characterized by profoundly reprogrammed ciliary signaling. Indeed, we found strongly suppressed responsiveness to PDGF-AA or Shh. However, these changes may represent dysregulated rather than overall inhibited fibrogenic signaling. Consistent with such interpretation, loss of the cilium has also been associated with enhanced Wnt/β-catenin and mTOR signaling, both involved in matrix deposition and fibrosis (Bell et al., 2011; Lancaster et al., 2011). During early phases of experimental kidney fibrosis, Shh stimulates pericyte proliferation, whereas PDGF-AA induces pericyte migration from the vessel wall to the interstitium (Chen et al., 2011; Fabian et al., 2012). If the cilium is then lost, the transforming pericytes may “remain put” in the interstitium, laying down an excess of extracellular matrix. Such “acquired ciliopathy” may also contribute to the disease. On the other hand, activation of myofibroblasts is part of normal wound healing as well. Thus cessation of certain ciliary pathways may serve regeneration by limiting proliferative and migratory responses. The role of acquired cilium loss in fibrotic or regenerative responses may be context dependent. In summary, our study shows for the first time that myofibroblast transition is associated with myosin-dependent cilium loss, and this unique feature may have important physiologic and pathological implications.
MATERIALS AND METHODS
Reagents
Hedgehog pathway inhibitor 4 (HPI-4), apocynin, and EGF were purchased from Sigma Aldrich (St. Louis, MO). TGFβ and recombinant mouse Shh ligand were from R&D Systems (Minneapolis, MN), and the PDGF-AA ligand was from BioVision (Farmingdale, NY). Commercially available antibodies were obtained from the following sources: Cell Signaling Technologies, Danvers, MA (Smad3, phospho–myosin light chain II [Thr-18/Ser-19], myosin light chain II, Rac1/2/3, and RhoA); Abcam, Cambridge, MA (acetylated tubulin, Kif3a, and Ki67); Sigma-Aldrich (tubulin, SMA [1A4] and nonmuscle myosin heavy chain II); Santa Cruz Biotechnology, Santa Cruz, CA (phospho–extracellular signal-regulated kinase 1/2 [ERK1/2; K23], ERK1 [E4], glyceraldehyde-3-phosphate dehydrogenase [GAPDH; 0411]); BD Transduction Laboratories, Mississauga, ON, Canada (E-cadherin); and Novus Biologicals, Littleton, CO (Nox4). Rabbit polyclonal anti-MRTF (BSAC) was described previously (Sasazuki et al., 2002). Rabbit polyclonal anti–polycystin-2 was a kind gift from the Johns Hopkins Center for Polycystic Kidney Disease (Baltimore, MD). All secondary antibodies were from Jackson ImmunoResearch Laboratories (Westgrove, PA).
Cell culture
LLC-PK1 (Cl 4) cells, a porcine proximal tubular epithelial cell line (a kind gift from R. C. Harris, Vanderbilt University School of Medicine, Nashville, TN), and C3H-10T1/2 cells, a mouse embryonic mesenchymal cell line (American Type Culture Collection, Manassas, VA), were cultured in low-glucose DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% streptomycin/penicillin solution (Invitrogen). Primary human skin fibroblasts (a gift from B. Hinz, University of Toronto, Toronto, Canada) were cultured in high-glucose DMEM and seeded onto collagen I–coated plates with similar supplements. To induce contact disruption, cells were washed three times with phosphate-buffered saline (PBS; Invitrogen) and then cultured in nominally calcium-free DMEM (LCM; Invitrogen). Scratch wounding was induced by running a pipette tip over a fully confluent monolayer. Where indicated, cells were treated with 4 ng/ml TGFβ, 8 ng/ml Shh, 10 ng/ml EGF, or 100 ng/ml PDGF-AA. For siRNA transfections, LLC-PK1 cells were cultured in antibiotic-free medium.
Plasmids and transfection
The 9xGli1-luciferase construct (Genentech, South San Francisco, CA; described by Barnes et al., 2005) harbors nine copies of the Gli1-binding site of mouse hepatocyte factor-3β enhancer upstream of herpes simplex virus thymidine kinase promoter and the luciferase gene. The Smad3-responsive reporter construct SBE4-Luc was provided by A. B. Roberts (National Institutes of Health, Bethesda, MD). Thymidine kinase promoter–driven Renilla plasmid (pRL-TK) was purchased from Promega (Madison, WI). Expression plasmids for the WT (TβRI(WT)), kinase-dead (TβRI(KR)), and constitutively active (TβRI(TD)) TGFβRI have been described (Feng et al., 1995; Choy and Derynck, 1998). Vectors encoding constitutively active RhoA (Q63L RhoA) and Rac1 (Q61L Rac1) have been described (Zhang et al., 1995; Masszi et al., 2003). DD-MLC (described in Di Ciano-Oliveira et al., 2003) was generated by replacing Thr-18 and Ser-19 of WT-MLC with aspartic acid residues. Cells were transfected with 0.5–1 μg of DNA/sample in 2 ml of medium, using FuGENE 6 (Roche Applied Science, Branford, CA) at 2.5 μl/μg DNA (for the TGFβ receptor constructs) or jetPRIME (Polyplus Transfection SA, New York, NY) at 2 μl/μg DNA (for the other constructs), according to manufacturer's recommendation.
RNA interference
The following porcine-specific sequences were used to target the indicated proteins by RNA interference: E-cadherin, 5′-CUCUGCUGGUGUUUGAUUA-3′; Smad3, 5′-AAGAGUUCACUCCACAUUCUC-3′; MYH9, 5′-AAGAAGGCGAAGGCGAACAAG-3′; Rac1/2, 5′-AAAUACCUGGAGUGCUCGGCG-3′; MRTF A, 5′-AACCAAGGAGCUGAAGCCAAA-3′; MRTF B, 5′-AACGACAAACACCGTAGCAAA-3′; Kif3a, 5′-AAGCAGAGAACUUGAGGAAAA-3′; and RhoA, 5′-AAAGCAGGTAGAGTTGGCTTT-3′. Oligonucleotides were synthesized by Applied Biosystems (Burlington ON, Canada) or Thermo Scientific (Waltham, MA). The control (nonrelated) siRNA was purchased from Applied Biosystems (Silencer Negative Control siRNA #2). Transfections were performed using Lipofectamine RNAiMAX (Invitrogen), applying a 2 or 3 μl/sample for 50 or 100 nM siRNA, respectively.
Luciferase reporter assays
Luciferase reporter assays were completed similar to our previous studies (Masszi et al., 2010; Charbonney et al., 2011). Transfection with the luciferase construct (0.25 μg/well) along with the pRL-TK Renilla (0.025 μg/well) was performed using FuGENE6. At 24 h later cells were serum starved for 2 h, followed by the indicated treatment. Cells were lysed using 1× Passive Lysis Buffer (Promega), and luciferase activity was measured using the Dual Luciferase Assay System kit (Promega).
Western blotting
After the indicated treatments, cells were washed once with ice-cold PBS and then lysed using Triton lysis buffer (30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 100 mM NaCl, 1 mM ethylene glycol tetraacetic acid, 20 mM sodium fluoride, and 1% Triton X-100), which was supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, and Complete Mini Protease Inhibitor (Roche). For experiments analyzing relative phospho–myosin II levels, RIPA buffer was used (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, pH 7.4) supplemented as before. SDS–PAGE and subsequent Western blotting were performed on samples with equal protein concentrations as determined using BCA Protein Assay reagents (Thermo Scientific). Where indicated, densitometry was performed using Quantity One software and a GS800 densitometer (Bio-Rad, Hercules, CA).
Immunofluorescence microscopy
Cells to be imaged were plated onto glass coverslips (in the case of human skin fibroblasts, glass coverslips were first coated with collagen I) and then fixed using 4% paraformaldehyde (PFA; Canemco & Marivac, Lakefield, QC, Canada) for 20 min after the indicated treatments. PFA was quenched using 100 mM glycine in PBS for 10 min, followed by cell permeabilization with PBS containing 0.1% Triton X-100. Fixed samples were blocked using 3% bovine serum albumin in PBS (1 h to overnight), followed by primary antibody incubation for 1 h. Cells were then washed with PBS and incubated with fluorescently labeled secondary antibodies for 1 h with the addition of 4′,6-diamidino-2-phenylindole (Lonza, Basel, Switzerland) for nuclear labeling. For imaging of acetylated tubulin or E-cadherin, cells were fixed using ice-cold methanol for 6 min, followed by blocking. Coverslips were mounted using Dako (Glostrup, Denmark) fluorescence mounting medium (Glostrup). Fixed samples were analyzed using an LSM 700 confocal microscope (Zeiss, Oberkochen, Germany), Plan-Apochromat 63×/numerical aperture 1.4 oil objective (Zeiss), and Zen 2010 software (Zeiss). All acetylated tubulin images were generated by collecting Z-sections (0.2-μm step size) and then collapsing into a maximum-intensity projection. Modifications were restricted to contrast and brightness for all images. Unless otherwise indicated, when quantification was performed to assess the percentage of cells having a primary cilium or the length of the cilium, 10–12 frames of ∼30 cells were counted per individual experiment. Cilium length measurements were conducted using MetaMorph software (Molecular Devices, Sunnyvale, CA).
Scanning electron microscopy
Fixation and imaging of samples were performed as described (Prashar et al., 2012).
ROS measurement
ROS production was determined by a quantitative nitro blue tetrazolium (NBT) assay as described previously (Serrander et al., 2007), with minor modifications. Briefly, cells grown in six-well plates were exposed to serum-free medium or LCM plus TGFβ treatment for 48 h and, after washing, incubated for 45 min in the corresponding medium supplemented with 1.67 mg/ml NBT. Cells were then fixed with ice-cold methanol and photographed with a Nikon Coolpix camera attached to Nikon eclipse TS100 microscope (40× objective), using the Nikon digital sight system and SPOT software. NBT was then extracted and solubilized using a mix of 560 μl of 2 M KOH and 480 μl of dimethyl sulfoxide. Absorbance of reduced NBT was measured at 630 nm. Data were normalized to protein levels determined in parallel samples.
Screening of cellular growth medium for primary cilia shedding
To detect the presence of shed primary cilia, the growth medium was collected after the indicated treatments. As a positive control, cells were cultured for 48 h in serum-free medium, followed by replacement of growth medium with high-Ca2+ deciliation solution (112 mM NaCl, 3.4 mM KCl, 10 mM CaCl2, 2.4 mM NaHCO3, 20 mM HEPES, pH 7.0) (Raychowdhury et al., 2005). Cells were then shaken for 4 min (360 rpm at 37°C; Mitchell et al., 2004), and the deciliation solution containing the primary cilium was collected. The cellular debris was sedimented by centrifugation at 1000 × g for 5 min at 4°C. The primary cilia in the resulting supernatant were enriched on a 60% (wt/vol) sucrose cushion by two rounds of centrifugation at 6000 × g for 10 min at 4°C. The enriched cilia supernatant was overlaid on 8 ml of a 20–60% discontinuous sucrose gradient in a 13-ml ultracentrifuge tube and subjected to equilibrium sedimentation at 150,000 × g for 5 h at 4°C (L8-80M ultracentrifuge; Beckman, Brea, CA). After centrifugation, 450-μl fractions were collected from the top of the tube. The resulting fractions were analyzed by Western blot for the presence of acetylated tubulin. The primary cilium fraction was found close to the 20%–60% sucrose interphase.
Statistical analysis
Data are presented as representative blots or images from at least three similar experiments or as means ± SEM for the number of experiments indicated. Statistical significance was determined by two-tailed Student's t test or one-way analysis of variance (ANOVA; Tukey or Dunn post hoc testing for parametric and nonparametric ANOVA, as appropriate), using Prism and Instat software. p < 0.05 was accepted as significant. *p < 0.05, **p < 0.001, ***p < 0.0001.
Supplementary Material
Acknowledgments
This work was supported by grants from the Canadian Institute of Health Research (MOP-86535 and MOP-106625) and the Kidney Foundation of Canada to A.K. We are indebted to K. Szászi for suggestions and critical reading of the manuscript and M. Terebiznik, A. Prashar, M. Ailenberg, and B. Calvieri for kind help with the scanning electron microscopy studies.
Abbreviations used:
- Ac-tub
acetylated tubulin
- EGF
epidermal growth factor
- EMT
epithelial–mesenchymal transition
- EMyT
epithelial–myofibroblast transition
- ERK
extracellular signal-regulated kinase
- Hh
hedgehog
- LCM
low-calcium medium
- MLC
myosin light chain
- MRTF
myocardin-related transcription factor
- PDGF
platelet-derived growth factor
- pMLC
phospho–myosin light chain
- ROS
reactive oxygen species
- SBE4
Smad binding element 4
- Shh
Sonic hedgehog
- siRNA
small interfering RNA
- SMA
smooth muscle actin
- TGFβ
transforming growth factor-β1
- TGFβRI
transforming growth factor-β1 receptor I
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-07-0429) on January 8, 2014.
REFERENCES
- Barnes EA, Heidtman KJ, Donoghue DJ. Constitutive activation of the shh-ptc1 pathway by a patched1 mutation identified in BCC. Oncogene. 2005;24:902–915. doi: 10.1038/sj.onc.1208240. [DOI] [PubMed] [Google Scholar]
- Bell PD, et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J Am Soc Nephrol. 2011;22:839–848. doi: 10.1681/ASN.2010050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M, Atwood SX, Woo WM, Li M, Oro AE. Mim and cortactin antagonism regulates ciliogenesis and hedgehog signaling. Dev Cell. 2010;19:270–283. doi: 10.1016/j.devcel.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer AL, Panagis L, Gusella GL, Danias J, Mlodzik M, Iomini C. Primary cilia dynamics instruct tissue patterning and repair of corneal endothelium. Proc Natl Acad Sci USA. 2011;108:2819–2824. doi: 10.1073/pnas.1016702108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, Barnes JL. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau HE, Casterline BW, Rada B, Korzeniowska A, Leto TL. Nox4 involvement in TGF-beta and Smad3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radical Biol Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekhuis JR, Leong WY, Jansen G. Regulation of cilium length and intraflagellar transport. Int Rev Cell Mol Biol. 2013;303:101–138. doi: 10.1016/B978-0-12-407697-6.00003-9. [DOI] [PubMed] [Google Scholar]
- Busche S, Descot A, Julien S, Genth H, Posern G. Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-MAL signalling. J Cell Sci. 2008;121:1025–1035. doi: 10.1242/jcs.014456. [DOI] [PubMed] [Google Scholar]
- Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–435. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- Charbonney E, Speight P, Masszi A, Nakano H, Kapus A. β-Catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol Biol Cell 22, 4472–4485. 2011. [DOI] [PMC free article] [PubMed]
- Chen YT, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- Chew TL, Masaracchia RA, Goeckeler ZM, Wysolmerski RB. Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (gamma-PAK) J Muscle Res Cell Motil. 1998;19:839–854. doi: 10.1023/a:1005417926585. [DOI] [PubMed] [Google Scholar]
- Choy L, Derynck R. The type II transforming growth factor (TGF)-beta receptor-interacting protein Trip-1 acts as a modulator of the TGF-beta response. J Biol Chem. 1998;273:31455–31462. doi: 10.1074/jbc.273.47.31455. [DOI] [PubMed] [Google Scholar]
- Clement CA, et al. TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013;3:1806–1814. doi: 10.1016/j.celrep.2013.05.020. [DOI] [PubMed] [Google Scholar]
- Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- Deretic D. Crosstalk of Arf and Rab GTPases en route to cilia. Small GTPases. 2013;4, 70–77 doi: 10.4161/sgtp.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciano-Oliveira C, Sirokmany G, Szaszi K, Arthur WT, Masszi A, Peterson M, Rotstein OD, Kapus A. Hyperosmotic stress activates Rho: differential involvement in Rho kinase-dependent MLC phosphorylation and NKCC activation. Am J Physiol Cell Physiol. 2003;285:C555–C566. doi: 10.1152/ajpcell.00086.2003. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Birukov KG, Zhan X, Garcia JG. Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun. 2002;298:511–519. doi: 10.1016/s0006-291x(02)02492-0. [DOI] [PubMed] [Google Scholar]
- Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, Poelmann RE, Hierck BP. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res. 2011;108:1093–1101. doi: 10.1161/CIRCRESAHA.110.231860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian SL, Penchev RR, St-Jacques B, Rao AN, Sipila P, West KA, McMahon AP, Humphreys BD. Hedgehog-GLI pathway activation during kidney fibrosis. Am J Pathol. 2012;180:1441–1453. doi: 10.1016/j.ajpath.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the Rho-Rho kinase-phospho-myosin pathway. Mol Biol Cell. 2007;18:1083–1097. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Filvaroff EH, Derynck R. Transforming growth factor-beta (TGF-beta)-induced down-regulation of cyclin a expression requires a functional TGF-beta receptor complex. Characterization of chimeric and truncated type I and type II receptors. J Biol Chem. 1995;270:24237–24245. doi: 10.1074/jbc.270.41.24237. [DOI] [PubMed] [Google Scholar]
- Gordon RE, Lane BP, Miller F. Identification of contractile proteins in basal bodies of ciliated tracheal epithelial cells. J Histochem Cytochem. 1980;28:1189–1197. doi: 10.1177/28.11.7000888. [DOI] [PubMed] [Google Scholar]
- Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NAPDH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, Harris PC. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257–4273. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan Y, van Adelsberg J. Polycystin-1, the pkd1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest. 1999;104:1459–1468. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison N, Fligny C, Duffield JS. Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. 2013;1832:962–971. doi: 10.1016/j.bbadis.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison N, Hendry BM, Sharpe CC. Rho isoforms have distinct and specific functions in the process of epithelial to mesenchymal transition in renal proximal tubular cells. Cell Signal. 2009;21:1522–1531. doi: 10.1016/j.cellsig.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–188. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L515–L529. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo E, Surka MC, Trimble WS. Mammalian Sept2 is required for scaffolding nonmuscle myosin II and its kinases. Dev Cell. 2007;13:677–690. doi: 10.1016/j.devcel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Jung IH, Jung DE, Park YN, Song SY, Park SW. Aberrant hedgehog ligands induce progressive pancreatic fibrosis by paracrine activation of myofibroblasts and ductular cells in transgenic zebrafish. PLoS One. 2011;6:e27941. doi: 10.1371/journal.pone.0027941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009a;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Tsiokas L. Cilia and cell cycle re-entry: more than a coincidence. Cell Cycle. 2011;10:2683–2690. doi: 10.4161/cc.10.16.17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y, Choi ME. Transforming growth factor-beta (TGF-beta1) activates Tak1 via Tab1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem. 2009b;284:22285–22296. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz C, Bordes N, Laine MC, Sandoz D, Bornens M. Myosin at the apical pole of ciliated epithelial cells as revealed by a monoclonal antibody. J Cell Biol. 1986;103:613–619. doi: 10.1083/jcb.103.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottgen M, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol. 2011;13:700–707. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Moon HJ, Lee JM, Joo CK. Smad3 regulates Rho signaling via Net1 in the transforming growth factor-beta-induced epithelial-mesenchymal transition of human retinal pigment epithelial cells. J Biol Chem. 2010;285:26618–26627. doi: 10.1074/jbc.M109.073155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Chen QK, Lui C, Cichon MA, Radisky DC, Nelson CM. Matrix compliance regulates Rac1b localization, NAPDH oxidase assembly, and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:4097–4108. doi: 10.1091/mbc.E12-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the Kif3a subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am J Pathol. 2004;165:1955–1967. doi: 10.1016/s0002-9440(10)63247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Speight P, Charbonney E, Lodyga M, Nakano H, Szaszi K, Kapus A. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol. 2010;188:383–399. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miserey-Lenkei S, Chalancon G, Bardin S, Formstecher E, Goud B, Echard A. Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat Cell Biol. 2010;12:645–654. doi: 10.1038/ncb2067. [DOI] [PubMed] [Google Scholar]
- Mitchell KA, Gallagher BC, Szabo G, Otero Ade S. NDP kinase moves into developing primary cilia. Cell Motil Cytoskeleton. 2004;59:62–73. doi: 10.1002/cm.20025. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Kasahara K, Miyazaki I, Asanuma M. Factors that influence primary cilium length. Acta Med Okayama. 2011;65:279–285. doi: 10.18926/AMO/47009. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin Cancer Biol. 2012;22:446–454. doi: 10.1016/j.semcancer.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Ng YY, Huang TP, Yang WC, Chen ZP, Yang AH, Mu W, Nikolic-Paterson DJ, Atkins RC, Lan HY. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998;54:864–876. doi: 10.1046/j.1523-1755.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- Okada H, Ban S, Nagao S, Takahashi H, Suzuki H, Neilson EG. Progressive renal fibrosis in murine polycystic kidney disease: an immunohistochemical observation. Kidney Int. 2000;58:587–597. doi: 10.1046/j.1523-1755.2000.00205.x. [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev. Mol Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn-Heaford HL, Ryan AJ, Murthy S, Racila AM, He C, Sieren JC, Spitz DR, Carter AB. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. 2012;287:3301–3312. doi: 10.1074/jbc.M111.308387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard CE, Sanzone KM, Spiczka KS, Sheff DR, Sandra A, Yeaman C. Deciliation is associated with dramatic remodeling of epithelial cell junctions and surface domains. Mol Biol Cell. 2009;20:102–113. doi: 10.1091/mbc.E08-07-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Papadimitriou E, Vasilaki E, Vorvis C, Iliopoulos D, Moustakas A, Kardassis D, Stournaras C. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-beta and miR-24: role in epithelial-to-mesenchymal transition. Oncogene. 2012;31:2862–2875. doi: 10.1038/onc.2011.457. [DOI] [PubMed] [Google Scholar]
- Petronio MS, Zeraik ML, Fonseca LM, Ximenes VF. Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules. 2013;18:2821–2839. doi: 10.3390/molecules18032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitaval A, Tseng Q, Bornens M, Thery M. Cell shape and contractility regulate ciliogenesis in cell cycle-arrested cells. J Cell Biol. 2010;191:303–312. doi: 10.1083/jcb.201004003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prashar A, Bhatia S, Tabatabaeiyazdi Z, Duncan C, Garduno RA, Tang P, Low DE, Guyard C, Terebiznik MR. Mechanism of invasion of lung epithelial cells by filamentous Legionella pneumophila. Cell Microbiol. 2012;14:1632–1655. doi: 10.1111/j.1462-5822.2012.01828.x. [DOI] [PubMed] [Google Scholar]
- Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80:41–50. doi: 10.1038/ki.2011.77. [DOI] [PubMed] [Google Scholar]
- Radisky DC, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychowdhury MK, McLaughlin M, Ramos AJ, Montalbetti N, Bouley R, Ausiello DA, Cantiello HF. Characterization of single channel currents from primary cilia of renal epithelial cells. J Biol Chem. 2005;280:34718–34722. doi: 10.1074/jbc.M507793200. [DOI] [PubMed] [Google Scholar]
- Raymond K, Cagnet S, Kreft M, Janssen H, Sonnenberg A, Glukhova MA. Control of mammary myoepithelial cell contractile function by alpha3beta1 integrin signalling. EMBO J. 2011;30:1896–1906. doi: 10.1038/emboj.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Samarin SN, Ivanov AI, Flatau G, Parkos CA, Nusrat A. Rho/Rock-II signaling mediates disassembly of epithelial apical junctions. Mol Biol Cell. 2007;18:3429–3439. doi: 10.1091/mbc.E07-04-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoz D, Gounon P, Karsenti E, Sauron ME. Immunocytochemical localization of tubulin, actin, and myosin in axonemes of ciliated cells from quail oviduct. Proc Natl Acad Sci USA. 1982;79:3198–3202. doi: 10.1073/pnas.79.10.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos N, Reiter JF. Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn. 2008;237:1972–1981. doi: 10.1002/dvdy.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasazuki T, et al. Identification of a novel transcriptional activator, BSAC, by a functional cloning to inhibit tumor necrosis factor-induced cell death. J Biol Chem. 2002;277:28853–28860. doi: 10.1074/jbc.M203190200. [DOI] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schwartz EA, Leonard ML, Bizios R, Bowser SS. Analysis and modeling of the primary cilium bending response to fluid shear. Am J Physiol. 1997;272:F132–F138. doi: 10.1152/ajprenal.1997.272.1.F132. [DOI] [PubMed] [Google Scholar]
- Sebe A, Masszi A, Zulys M, Yeung T, Speight P, Rotstein OD, Nakano H, Mucsi I, Szaszi K, Kapus A. Rac, Pak and p38 regulate cell contact-dependent nuclear translocation of myocardin-related transcription factor. FEBS Lett. 2008;582:291–298. doi: 10.1016/j.febslet.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger-Nukpezah T, Golemis EA. The extracellular matrix and ciliary signaling. Curr Opin Cell Biol. 2012;24:652–661. doi: 10.1016/j.ceb.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley ES, Nachury MV. The perennial organelle: assembly and disassembly of the primary cilium. J Cell Sci. 2010;123:511–518. doi: 10.1242/jcs.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. Nox4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major alpha-tubulin k40 acetyltransferase alphatat1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci USA. 2010;107:21517–21522. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe T, Tominaga M. The TRPV4 cation channel: a molecule linking skin temperature and barrier function. Commun Integr Biol. 2010;3:619–621. doi: 10.4161/cib.3.6.13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. The type I TGF-beta receptor engages Traf6 to activate Tak1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- Speight P, Nakano H, Kelley TJ, Hinz B, Kapus A. Differential topical susceptibility to TGFb in intact and injured regions of the epithelium: key role in myofibroblast transition. Mol Biol Cell. 2013;24:3326–3336. doi: 10.1091/mbc.E13-04-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa H, et al. Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011;300:F511–F520. doi: 10.1152/ajprenal.00038.2010. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. P47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- Tsai MH, Jiang MJ. Reactive oxygen species are involved in regulating alpha1-adrenoceptor-activated vascular smooth muscle contraction. J Biomed Sci. 2010;17:67. doi: 10.1186/1423-0127-17-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usatyuk PV, et al. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. J Biol Chem. 2012;287:9360–9375. doi: 10.1074/jbc.M111.294546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, Wrana JL. Taz controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. The crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-Smad pathway. Dev Cell. 2010;19:831–844. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Vasilaki E, Papadimitriou E, Tajadura V, Ridley AJ, Stournaras C, Kardassis D. Transcriptional regulation of the small GTPase RhoB gene by TGFb-induced signaling pathways. FASEB J. 2010;24:891–905. doi: 10.1096/fj.09-134742. [DOI] [PubMed] [Google Scholar]
- Verghese E, Ricardo SD, Weidenfeld R, Zhuang J, Hill PA, Langham RG, Deane JA. Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol. 2009;20:2147–2153. doi: 10.1681/ASN.2008101105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol. 2010;21:1247–1253. doi: 10.1681/ASN.2010060616. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- Zheng G, et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol. 2009;175:580–591. doi: 10.2353/ajpath.2009.080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.