

PERK is phosphorylated at Y561 in the juxtamembrane domain, and the adaptor protein Nck1, by directly interacting with phospho-Y561 PERK, negatively regulates PERK activity. Strong evidence is given supporting the biological relevance of Nck1 regulation of PERK function in modulating pancreatic β-cell proinsulin content.
Abstract
PERK, the PKR-like endoplasmic reticulum (ER) kinase, is an ER transmembrane serine/threonine protein kinase activated during ER stress. In this study, we provide evidence that the Src-homology domain–containing adaptor Nck1 negatively regulates PERK. We show that Nck directly binds to phosphorylated Y561 in the PERK juxtamembrane domain through its SH2 domain. We demonstrate that mutation of Y561 to a nonphosphorylatable residue (Y561F) promotes PERK activity, suggesting that PERK phosphorylation at Y561 (pY561PERK) negatively regulates PERK. In agreement, we show that pY561PERK delays PERK activation and signaling during ER stress. Compatible with a role for PERK in pancreatic β-cells, we provide strong evidence that Nck1 contributes to PERK regulation of pancreatic β-cell proteostasis. In fact, we demonstrated that down-regulation of Nck1 in mouse insulinoma MIN6 cells results in faster dephosphorylation of pY561PERK, which correlates with enhanced PERK activation, increased insulin biosynthesis, and PERK-dependent increase in proinsulin content. Furthermore, we report that pancreatic islets in whole-body Nck1-knockout mice contain more insulin than control littermates. Together our data strongly suggest that Nck1 negatively regulates PERK by interacting with PERK and protecting PERK from being dephosphorylated at its inhibitory site pY561 and in this way affects pancreatic β-cell proinsulin biogenesis.
INTRODUCTION
The endoplasmic reticulum (ER) is a subcellular organelle responsible for the synthesis, processing, and quality control of transmembrane and secretory proteins. Perturbation of ER homeostasis leads to accumulation of misfolded proteins and causes ER stress. This triggers the activation of three ER transmembrane proteins: inositol-requiring enzyme 1α (IRE1α), PKR‑like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), which together initiate a cytoplasmic signaling network defined as the unfolded protein response (UPR; Ron and Walter, 2007). The UPR encompasses a translational and transcriptional program that helps cells alleviate ER stress. However, chronic or irreversible ER stress initiates a UPR-mediated apoptotic pathway that induces cell death (Szegezdi et al., 2006).
PERK, a type I ER transmembrane Ser/Thr protein kinase, is activated after dimerization and autophosphorylation induced by ER stress (Shi et al., 1998; Harding et al., 1999). Activated PERK phosphorylates the α-subunit of the eukaryotic initiation factor 2 (eIF2α) at Ser-51, which results in translation attenuation but paradoxically promotes translation of the activating transcription factor 4 (ATF4), which controls an important part of the UPR transcriptional program (Harding et al., 2000b; Vattem and Wek, 2004). Of interest, a role for PERK in pancreatic β-cells was highlighted after the discovery of the Wolcott–Rallison syndrome (WRS), a neonatal/early infancy form of diabetes caused by mutations in the human PERK gene resulting in PERK loss of function (Delepine et al., 2000). Furthermore, PERK−/− mice, which phenocopy WRS dysfunctions, also display postnatal hyperglycemia, which was initially attributed to the loss of pancreatic β-cell mass associated with a dramatic increase in β‑cell apoptosis (Harding et al., 2001; Zhang et al., 2002). However, it was later demonstrated that the loss of β-cell mass in PERK−/− mice is not due to apoptosis. It instead results from reduction in β-cell proliferation and differentiation during the neonatal period, which impedes postnatal gain of pancreatic β-cell mass (Zhang et al., 2006). Finally, conditional deletion of PERK in young adult or mature mice significantly increased β-cell death, revealing that PERK also contributes to the maintenance of β‑cell function in adults. However, β-cell proliferation was dramatically increased, highlighting that PERK regulates β-cell function in adults through a different mechanism than that during early postnatal development (Gao et al., 2012).
A direct role for PERK in β‑cell proliferation and proinsulin/insulin content was further supported by the fact that acute inhibition of PERK generated by adenoviral vector–mediated expression of a dominant-negative Perk mutant lacking the kinase domain in INS 832/13 rat insulin–secreting β‑cells (AdDN-Perk INS 832/13 β-cells) led to reduced proliferation and insulin content (Feng et al., 2009). Unexpectedly, this study also showed that although insulin content was reduced, proinsulin was abnormally retained in the ER, suggesting an additional role for PERK in trafficking and maturation of secretory proteins. In agreement, shPerk-expressing INS 832/13 β-cells in which PERK protein level was reduced by 56–66% decreased insulin synthesis, and defective ER–Golgi anterograde proinsulin trafficking was also observed (Gupta et al., 2010). In contrast, acute inhibition of PERK using a specific PERK pharmacological inhibitor (PERKi) in mouse insulinoma MIN6 cells increased proinsulin synthesis (Harding et al., 2012). However, PERKi still induced rapid buildup of insoluble proinsulin associated with a subtle defect in proinsulin maturation, further supporting the concept that PERK is required for proper ER quality control of proinsulin folding and trafficking. The discrepancy regarding the effects of PERK inhibition on insulin synthesis might be explained by the different degree of PERK inhibition reached using short hairpin RNA (shRNA) or pharmacological inhibitor.
We previously reported that Nck regulates PERK-mediated eIF2αS51 phosphorylation (Kebache et al., 2004; Latreille and Larose, 2006). Nck is a Src-homology (SH) domain–containing adaptor protein, consisting of three N-terminal SH3 domains and one C‑terminal SH2 domain (Chen et al., 1998). Nck is well known to couple activated receptor tyrosine kinases (RTKs) through its SH2 domain to SH3 domain–bound effectors involved in actin cytoskeleton remodeling (Rivera et al., 2006; Abella et al., 2010). In mammals, two different genes encode highly identical Nck isoforms, Nck1 and Nck2, which are believed to be functionally redundant (Vorobieva et al., 1995; Chen et al., 1998; Braverman and Quilliam, 1999). Recently PERK was shown to be phosphorylated at Tyr-615 in its kinase domain, and this modification was found to be essential for maximal PERK activation upon ER stress (Su et al., 2008). In this study, we report that PERK phosphorylation at Tyr-561 (Y561) in the juxtamembrane creates a specific binding site for Nck and limits PERK activation. Furthermore, we provide strong evidence that this interaction regulates the ability of pancreatic β-cells to synthesize proinsulin.
RESULTS
Nck regulates PERK
We previously showed that mouse embryonic fibroblasts (MEFs) lacking Nck (Nck1−/−;Nck2−/−) display increased basal levels of eIF2α phosphorylated at Ser-51 (peIF2αS51), concomitant with up-regulated expression of UPR markers, including ATF4, Gadd34, Grp94, and BiP (Latreille and Larose, 2006). However, the effect of Nck deficiency on PERK activity was not addressed. To investigate PERK activation in Nck1−/−;Nck2−/− MEFs, we measured the levels of PERK phosphorylated at Thr-980. MEFs lacking Nck spontaneously displayed significantly increased levels of PERK phosphorylated at Thr-980 (pThr-980) compared with wild type (Figure 1A). In addition, upon thapsigargin (Tg)-induced ER stress, Nck1−/−;Nck2−/− MEFs also displayed increased levels of pThr-980 PERK compared with wild type (Figure 1B). In unstressed cells, PERK migrates as a doublet of 130–150 kDa that is proposed to represent differential species of phosphorylation states of the protein (Harding et al., 1999). Conversely, ER stress results in PERK migration toward 170 kDa, representing hyperphosphorylation of the protein. The shift in PERK migration toward 170 kDa was more pronounced in Nck1−/−;Nck2−/− MEFs compared with controls (Figure 1B, PERK blot), supporting PERK hyperactivation in cells depleted of Nck. In agreement, ER stress–induced peIF2αS51 levels also significantly increased in MEFs lacking Nck regardless of an apparent increase in eIF2α compared with controls (Figure 1B). These results reveal that Nck negatively controls PERK activation in unstressed and ER stress conditions.
FIGURE 1:
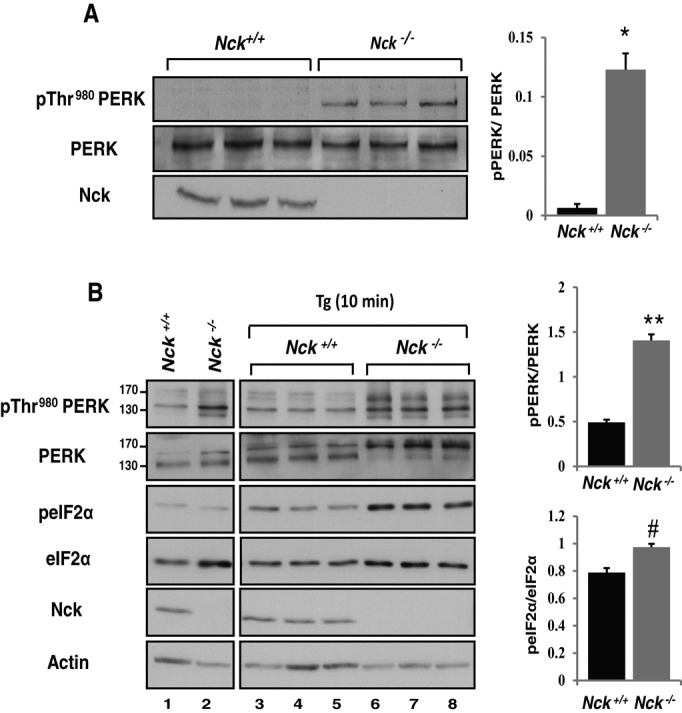
Lack of Nck enhances PERK activity. (A) PERK activity (pThr-980) in Nck+/+ and Nck−/− MEFs. Equivalent amount of proteins (50 μg) from total cell lysates prepared from three independent culture dishes for each genotype were subjected to immunoblotting with indicated antibodies. (B) Nck+/+ and Nck−/− MEFs were left untreated (lanes 1 and 2) or treated with 1 μM Tg for 10 min (lanes 3–8). Cell lysates (50 μg of protein) from independent culture dishes were analyzed by immunoblotting with the indicated antibodies. Bar charts show ratio of pThr-980 PERK to total PERK in unstressed cells (A) and pThr-980 PERK to total PERK and peIF2αS51 to total eIF2α upon Tg treatment as determined by densitometry (B). Data are means ± SEM (n = 3; *p = 0.004, **p < 0.001, #p = 0.01).
Nck and PERK interact in vitro
To determine whether Nck interacts with PERK, we performed in vitro binding assays using glutathione S-transferase (GST) chimera of the cytoplasmic segment of PERK (GST-cPERK) and recombinant Nck proteins. Using this approach, we observed that both Nck1 and Nck2 directly interact with GST-cPERK but not with GST alone (Figure 2A). In this setting, the GST moiety of GST‑cPERK acts as a dimerizing module causing activation of the PERK kinase activity (Harding et al., 1999). In agreement, GST‑cPERK wild type (WT) migrates slowly on SDS–PAGE compared with the GST-cPERK catalytically inactive K618A mutant (Figures 2B). Using Cos-1 cell lysate, we demonstrated that GST‑cPERK WT interacts with endogenous Nck, whereas GST‑cPERK K618A fails to bind Nck (Figure 2B). These results indicate that the interaction of Nck with PERK is direct and dependent on PERK kinase activity.
FIGURE 2:
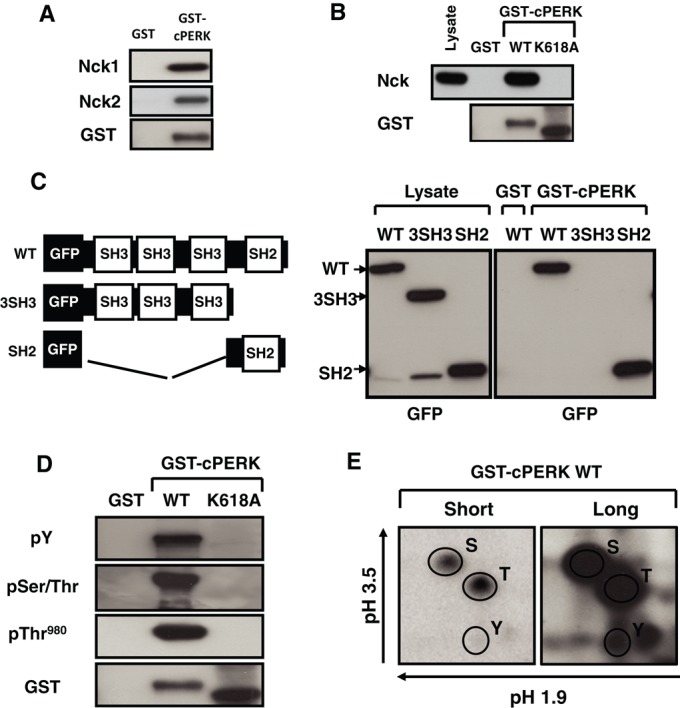
Nck and PERK interaction. (A) In vitro binding of recombinant GST-cPERK WT and Nck. Nck1 or Nck2 bound to GST-cPERK was revealed by immunoblotting using a panNck antibody, and GST-cPERK was revealed using a GST antibody. (B) Pull-down assays using GST and GST-cPERK WT or catalytically inactive (K618A) incubated with Cos-1 cell lysates. Bound endogenous Nck was revealed by immunoblotting using a panNck antibody, and a GST antibody was used to show GST proteins. (C) Schematic representation of GFP-Nck1 full length and deletion mutants (left). Pull-down assays using GST and GST-cPERK WT incubated with Cos-1 cell lysates expressing indicated GFP-Nck1 constructs. GFP-Nck1–related proteins in cell lysates and pull-down assays were revealed by immunoblotting using a GFP antibody. (D) Phosphorylation of GST-cPERK WT and K618A (100 ng) was monitored using indicated antibodies. GST-proteins were revealed by immunoblotting using a GST antibody. (E) Two-dimensional phosphoamino acid analysis of GST-cPERK WT subjected to in vitro autophosphorylation in the presence of [γ‑32P]ATP. Phosphoserine, phosphothreonine, and phosphotyrosine are indicated. Data are typical of three independent experiments.
Nck contains three SH3 domains known to recognize proline-rich sequences, followed by one SH2 domain engaging phosphotyrosine residues in target proteins (Figure 2C; Chen et al., 1998; Braverman and Quilliam, 1999). To define the molecular determinant(s) mediating Nck and PERK interaction, we expressed GFP-Nck1 WT and deletion mutants in Cos-1 cells and performed pull-down assays using GST‑cPERK. Deletion of the Nck1 SH2 domain (3SH3) abrogated binding to PERK, whereas deletion of the three SH3 domains (SH2) did not impair this interaction (Figure 2C). Therefore we conclude that the SH2 domain of Nck1 mediates the interaction with PERK.
PERK is highly phosphorylated on serine and threonine residues upon activation, but studies established that PERK is also phosphorylated on tyrosine, which is essential for optimal PERK activation (Su et al., 2008). Our finding that Nck interacts with PERK through its SH2 domain strongly suggests that GST‑cPERK is indeed phosphorylated on tyrosine. Therefore we examined phosphorylation of GST-cPERK by immunoblotting using appropriate antibodies. In contrast to the catalytically inactive GST-cPERK K618A, we found that GST-cPERK WT is phosphorylated on serine and threonine (pSer/Thr), including Thr-980, and on tyrosine, as reported previously (Figure 2D; Su et al., 2008). Furthermore, phosphoamino acid analysis of GST-cPERK revealed the presence of phosphoserine, phosphothreonine, and phosphotyrosine residues (Figure 2E). These results demonstrate that PERK activity correlates with PERK autophosphorylation on serine/threonine and tyrosine residues in vitro and strongly suggest that PERK tyrosine phosphorylation might mediate direct interaction with Nck.
Identification of pY561 in the PERK juxtamembrane domain as the Nck-binding site
We screened PERK cytoplasmic amino acid sequence and found that mouse PERK Y561 (human Y565) perfectly matches the Nck SH2 domain consensus binding motif pYDXV(S/A/T/Y)X(D/E) (Figure 3A; Frese et al., 2006). This motif is highly conserved among mammalian PERK orthologues but is absent in Caenorhabditis elegans and Drosophila melanogaster (unpublished data). In addition, the PERK Y561 surrounding amino acid sequence revealed a high degree of homology with Nck SH2 domain–binding sequences identified in enteropathogenic Escherichia coli Tir, nephrin, and Git-1 proteins (Gruenheid et al., 2001; Frese et al., 2006), suggesting that phosphorylation of PERK at Y561 could be a potential binding site for Nck. To address this, we generated a PERK mutant in which Y561 is replaced by a nonphosphorylatable phenylalanine (Y561F) and tested its ability to interact with Nck. GST-cPERK Y561F failed to bind Nck in pull-down experiments (Figure 3B). However, GST‑cPERK Y561F migrates like the active GST‑cPERK WT on SDS–PAGE (Figure 3B, Coomassie staining), suggesting that it is catalytically active. This was confirmed by detection of pThr-980 on GST‑cPERK Y561F, although GST‑cPERK Y561F displayed a significant decrease in overall tyrosine phosphorylation compared with WT (Figure 3C). Together these results demonstrate that pY561, which markedly contributes to PERK global tyrosine autophosphorylation, mediates PERK interaction with Nck.
FIGURE 3:
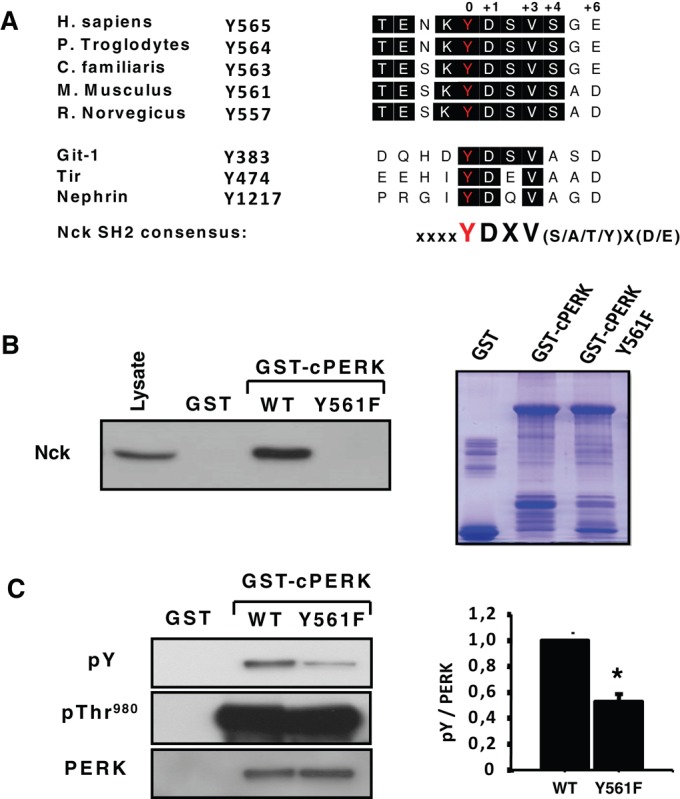
PERK juxtamembrane pY561 is the Nck-binding site. (A) Alignment of amino acids in the PERK orthologues juxtamembrane domain showing a conserved tyrosine residue (mouse Y561) matching the Nck SH2 domain consensus binding motif (Frese et al., 2006) found in enteropathogenic E. coli Tir, Git-1, and nephrin proteins (Gruenheid et al., 2001; Frese et al., 2006). (B) Left, pull-down assay using GST and indicated GST-cPERK proteins incubated with Cos-1 cell lysates. Bound endogenous Nck was revealed by immunoblotting using a panNck antibody (n = 3). Right, purified, bacterially expressed GST-cPERK proteins (1 μg) were resolved on SDS–PAGE and stained with Coomassie blue. (C) Global phosphorylation on tyrosine and at Thr-980 of GST-cPERK WT and Y561F mutant (100 ng) shown by immunoblotting with the indicated antibodies. Bar chart represents the ratio of global tyrosine phosphorylation of respective GST-cPERK over total GST-cPERK determined by densitometry. Data are mean ± SEM (n = 3; *p < 0.001).
Phosphorylation of PERK at Y561 modulates PERK activity
To address whether phosphorylation of PERK at Y561 affects PERK activity, we carried out a comparative analysis of GST-cPERK WT and Y561F ability to phosphorylate recombinant His-eIF2α in vitro in the presence of [γ‑32P]ATP. We observed that incorporation of radioactivity into His-eIF2α was significantly increased in reactions performed with GST-cPERK Y561F compared with WT (Figure 4A), suggesting that phosphorylation at Y561 negatively regulates PERK activity. This was further confirmed in PERK−/− MEFs transiently overexpressing full-length PERK WT, Y561F, or K618A, in which we assessed peIF2αS51 in response to ER stress. In agreement with our in vitro observations, we found that in response to dithiothreitol (DTT) treatment, peIF2αS51 levels were increased in PERK−/− MEFs expressing PERK Y561F compared with WT (Figure 4B and Supplemental Figure S1). In contrast, PERK−/− MEFs expressing the catalytically inactive PERK K618A mutant failed to induce peIF2αS51 upon DTT treatment, demonstrating that DTT-induced peIF2αS51 is specific to PERK in these conditions. These results demonstrate that PERK Y561F displays enhanced ER stress–induced peIF2αS51, further supporting a negative regulatory role for PERK Y561 phosphorylation in regulating PERK activation and signaling.
FIGURE 4:
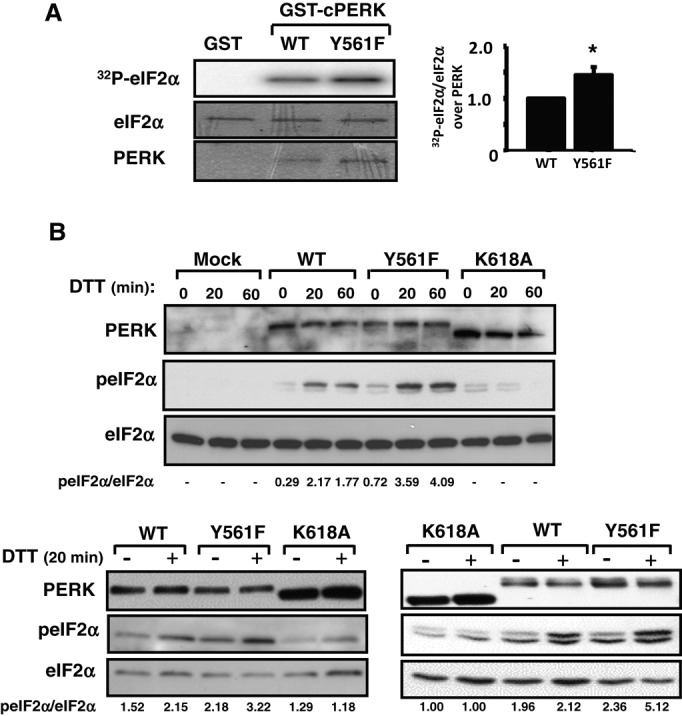
PERK Y561F mutation enhances PERK catalytic activity. (A) Phosphorylation of recombinant His-eIF2α by GST-cPERK WT and Y561F mutant in in vitro kinase assay. Bar chart shows quantification by densitometry of His-eIF2α phosphorylation normalized for the amounts of eIF2α and GST-cPERK present in the reactions. Data are mean ± SEM (n = 3; *p < 0.01). (B) PERK−/− MEFs transiently transfected with empty vector (Mock), full-length PERK WT, Y561F, or K618A cDNAs were treated 48 h later with 1 mM DTT for 0, 20, or 60 min. Cell lysates normalized for protein content (50 μg of protein) were subjected to immunoblotting with indicated antibodies. Shown are three independent experiments, with peIF2αS51/total eIF2α ratio, as determined by densitometry, reported under the blots.
PERK is phosphorylated at Y561 in MIN6 cells
To detect PERK phosphorylation at Y561, we generated a specific antibody against a synthetic phosphopeptide encompassing the amino acid sequence surrounding mouse PERK Y561. We hypothesized that phosphorylation of PERK at Y561 (pY561) would occur upon ER stress, given that previous studies showed that PERK phosphorylation on specific tyrosine residues was detected in this condition (Su et al., 2008; Krishnan et al., 2011; Bettaieb et al., 2012). However, we could not detect pY561 PERK in control or Tg-treated MIN6 cells (unpublished data). The difficulty in detecting pY561 PERK could be due to its low levels or fast cycling between phosphorylated and unphosphorylated states in vivo. Therefore, to stabilize tyrosine phosphorylation of PERK, we treated MIN6 cells with pervanadate (PV), a nonspecific tyrosine phosphatase inhibitor, and subsequently assessed pY561 PERK by immunoblotting. Of interest, we detected high levels of pY561 PERK and a new PERK species with distinct mobility in PV-treated cells (Figure 5A, left). It is noteworthy that PV treatment did not activate PERK, as neither pThr-980 PERK nor peIF2αS51 levels were increased in this condition. Treating MIN6 cells with bpVPhen, another pY phosphatase inhibitor (Posner et al., 1994), also led to PERK phosphorylation at Y561 without altering PERK activation and signaling (Figure 5A, right). To demonstrate that the immunoreactive band detected with the pY561 PERK antibody is specific, we transfected MIN6 cells with PERK WT, Y561F, or K618A and treated them with PV. As expected, immunoblotting of total cell lysates with pY561 PERK antibody showed a positive signal only in cells overexpressing PERK WT (Figure 5B). Collectively these data provide strong evidence that PERK is phosphorylated at Y561 in unstressed cells.
FIGURE 5:
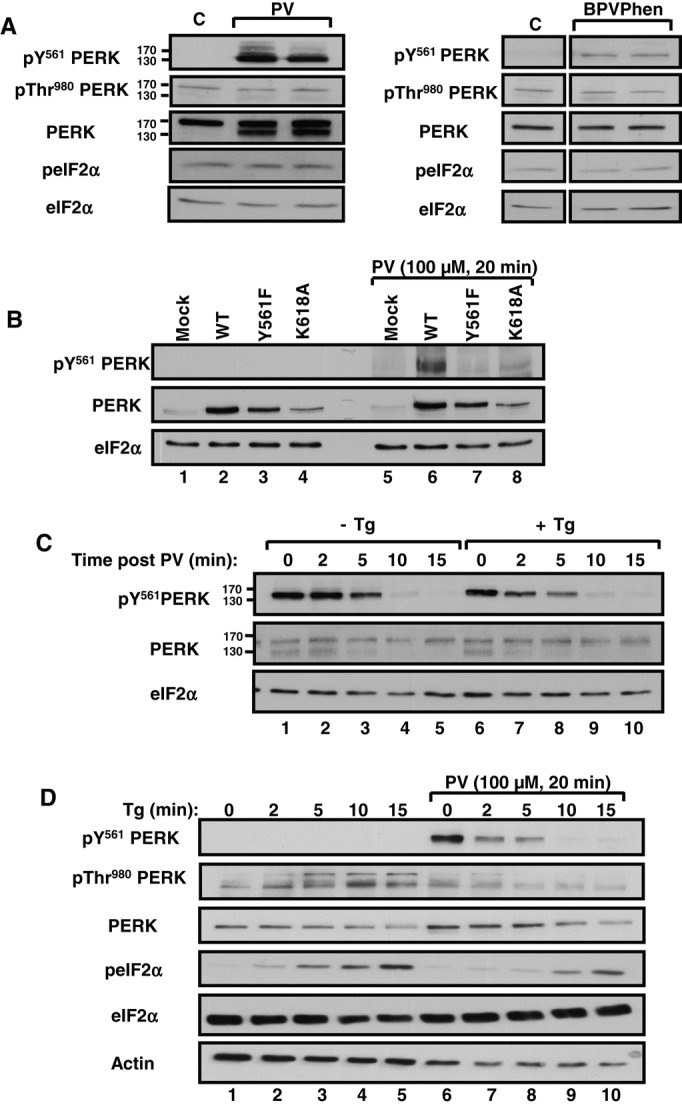
PERK phosphorylation at Y561 delays PERK activation and signaling in MIN6 cells. (A) Left, MIN6 cells were left untreated (lane 1) or treated with 100 μM PV for 20 min (lanes 2 and 3). Cell lysates (50 μg of protein) were subjected to immunoblotting with indicated antibodies. Right, MIN6 cells were left untreated (–) or treated (+) with bpVPhen (200 μM, 10 min). Total cell lysates (50 μg of protein) from independent culture dishes were simultaneously subjected to immunoblotting using indicated antibodies (n = 3). (B) Specificity of the pY561 PERK antibody. MIN6 cells transiently transfected with empty vector (Mock), PERK WT, Y561F, and K618A cDNA-containing vectors were, 24 h later, left untreated (lanes 1–4) or treated with 100 μM PV for 20 min (lanes 5–8). Total cell lysates (50 μg of protein) were subjected to immunoblotting with indicated antibodies (n = 3). (C) MIN6 cells were pretreated with 100 μM PV for 20 min (lanes 1–10) and then washed with PBS and either left untreated (lanes 1–5) or treated with 1 μM Tg (lanes 6–10) for indicated times. Cell lysates (50 μg of protein) were subjected to immunoblotting using indicated antibodies. (D) MIN6 cells were left untreated (lanes 1–5) or pretreated with 100 μM PV for 20 min (lanes 6–10). Then cells were washed with PBS and treated with Tg at 1 μM for indicated times. Cell lysates (50 μg of protein) were subjected to immunoblotting using indicated antibodies. Data are representative of three independent experiments.
Supporting the labile nature of pY561 PERK, we observed rapid loss of PERK phosphorylation at Y561 upon washing out PV (Figure 5C, lanes 1–5). Of interest, treating cells with Tg upon PV removal greatly accelerated dephosphorylation of PERK at Y561 (Figure 5C, lanes 6–10). We obtained similar findings using DTT to induce ER stress (Supplemental Figure S2A). In agreement, the PERK species that migrate faster in PV‑pretreated MIN6 cells disappeared concomitantly with the loss of pY561 PERK upon PV removal, and this was accelerated by ER stress (Figure 5C and Supplemental Figure S2A). Together these results demonstrate that ER stress promotes pY561 PERK dephosphorylation.
To determine whether PERK phosphorylation at Y561 modulates ER stress–induced PERK activation and signaling, we followed pThr-980 PERK and peIF2αS51 in ER-stressed MIN6 cells pretreated or not with PV (Figure 5D). As expected, pThr-980 PERK increased upon ER stress, in agreement with PERK activation in cells not pretreated with PV (Figure 5D, lanes 1–5). Conversely, pY561 PERK decreased upon Tg treatment in PV-pretreated MIN6 cells and inversely correlated with increased levels of peIF2αS51 (Figure 5D, lanes 6–10). Of interest, PV pretreatment resulted in lower levels of Tg-induced pThr-980 PERK and peIF2αS51, demonstrating that phosphorylation of PERK at Y561 correlates with reduced PERK activation and signaling. Similar results were observed in PV‑pretreated MIN6 cells exposed to DTT (Supplemental Figure S2B) and bpVPhen-pretreated Cos-1 cells exposed to Tg or DTT (Supplemental Figure S2C). Collectively these data suggest that pY561 PERK impairs ER stress–induced PERK activation and signaling.
Nck interacts with pY561 PERK in MIN6 cells
Given that PERK is phosphorylated at Y561 in PV-treated MIN6 cells (Figure 5) and that Nck interacts with pY561 PERK in vitro (Figure 3B), we assessed whether Nck and pY561 PERK interact in vivo by performing classic coimmunoprecipitation assays using MIN6 cells pretreated or not with PV. For this, Nck immunoprecipitates were probed for pY561 PERK. As shown in Figure 6, pY561 PERK was detected in Nck immunoprecipitates from PV-pretreated MIN6 cells, and this interaction decreased over time upon Tg exposure (Figure 6 and Supplemental Figure S3A). These results show that the interaction of Nck with PERK occurs in unstressed cells and is rapidly lost when levels of pY561 PERK decrease upon ER stress.
FIGURE 6:
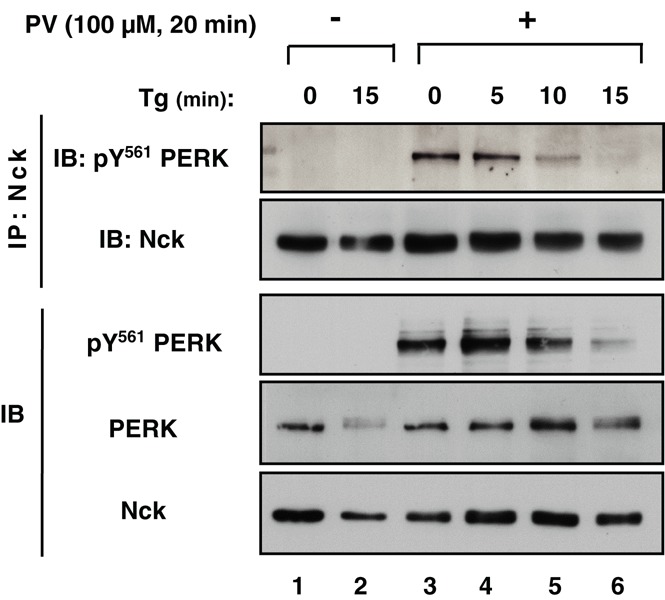
Nck interacts with PERK phosphorylated at Y561 in MIN6 cells. MIN6 cells were left untreated (lanes 1 and 2) or pretreated with 100 μM PV for 20 min (lanes 3–6) and then washed with PBS and treated with Tg (1 μM) for indicated times (lanes 3–6). Cell lysates (500 μg of protein) were subjected to Nck immunoprecipitation (IP: Nck), and Nck immunoprecipitates were subjected to immunoblotting with anti–phospho-Y561 PERK antibody. Total cell lysates (50 μg of protein) were subjected to immunoblotting with indicated antibodies (IB).
Nck1 modulates PERK and proinsulin levels in pancreatic β-cells
To assess the importance of Nck1 in regulating PERK, we generated MIN6 cells stably expressing a specific shRNA against Nck1 (shNck1), which show >80% down-regulation of Nck1 (Figure 7A). We then demonstrated that down-regulation of Nck1 in MIN6 cells significantly increased PERK activation (pThr-980) and signaling (peIF2αS51) in unstressed conditions (Figure 7A). Moreover, we observed that down-regulation of Nck1 in PV-pretreated cells increased the rate of Tg-induced pY561 PERK dephosphorylation and correlated with increased levels of peIF2αS51 (Figure 7B and Supplemental Figure S3B). These results suggest that Nck1 protects pY561 PERK from being dephosphorylated during ER stress and thereby limits PERK activation and signaling.
FIGURE 7:
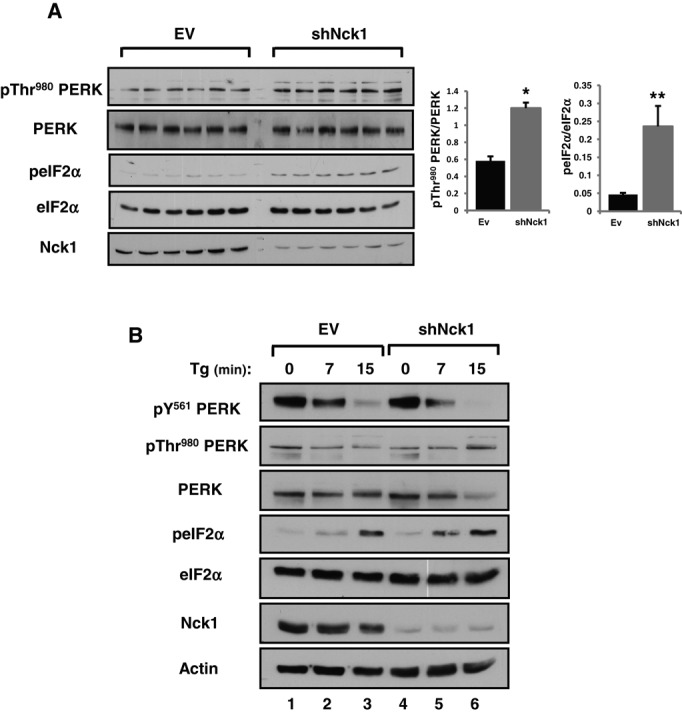
Nck1 modulates PERK activation and phosphorylation at Y561 in MIN6 cells. (A) LMP-EV and LMP-shNck1 MIN6 cell lysates (50 μg of protein) from six independent culture dishes were subjected to immunoblotting with indicated antibodies. Bar charts show the ratio of pThr-980 PERK to total PERK and peIF2αS51 to total eIF2α determined by densitometry. Data are mean ± SEM (n = 3; *P < 0.001, **P = 0.002, Mann–Whitney). (B) LMP-EV and LMP-shNck1 MIN6 cells were pretreated with 100 μM PV for 20 min and then washed with PBS and further treated with 1 μM Tg for 0, 7, and 15 min. Cell lysates (50 μg of protein) were subjected to immunoblotting with indicated antibodies (n = 3).
Given that PERK physiological activity controls proinsulin synthesis and processing in pancreatic β-cells (Gupta et al., 2010; Harding et al., 2012), we assessed proinsulin levels in shNck1 MIN6 cells that display increased physiological PERK activity. Of interest, we found that together with increased PERK activity (Figure 7A), proinsulin levels were significantly up-regulated in shNck1 MIN6 cells (Figure 8A). Increased proinsulin appears to be specific since the levels of GLUT2 and RasGAP were not changed in shNck1 MIN6 cells. In addition, we demonstrated that increased proinsulin levels correlate with increased proinsulin synthesis, as supported by increased [35S]Met/Cyst incorporation into insulin in shNck1 MIN6 cells compared with control cells (Figure 8B). In agreement with PERK-mediated modulation of proinsulin levels, we demonstrated that overexpression of PERK at low levels (transfection of 0.5–2.0 μg) in MIN6 cells slightly enhanced PERK activation and proinsulin content (Supplemental Figure S4). However, consistent with PERK activation inducing attenuation of general translation, we found that higher levels of PERK overexpression (transfection of 5 and 10 μg) led to marked PERK activation and reduced proinsulin content. To demonstrate that PERK mediates the effect of downregulating Nck1 on proinsulin content, we overexpressed the catalytically inactive PERK K618A in shNck1 MIN6 cells. We expect that overexpression of PERK K618A will counteract PERK physiological activity by dimerizing with endogenous PERK. Accordingly, we demonstrated that PERK K618A significantly reverses the effect of downregulating Nck1 on proinsulin levels in MIN6 cells (Figure 8C). We extended the significance of these findings by measuring insulin levels in isolated pancreatic islets of Nck1−/− mice. We observed that Nck1−/− islets displayed increased insulin content compared with Nck1+/+ islets (Figure 8D). Collectively our results provide strong evidence that in absence of ER stress, Nck1 binding to PERK protects PERK from being dephosphorylated at its negative regulatory site pY561 and thereby limits PERK activation and signaling. Furthermore, our findings indicate that Nck1 is a negative regulator of PERK activation and PERK‑mediated proinsulin biosynthesis in pancreatic β-cells.
FIGURE 8:
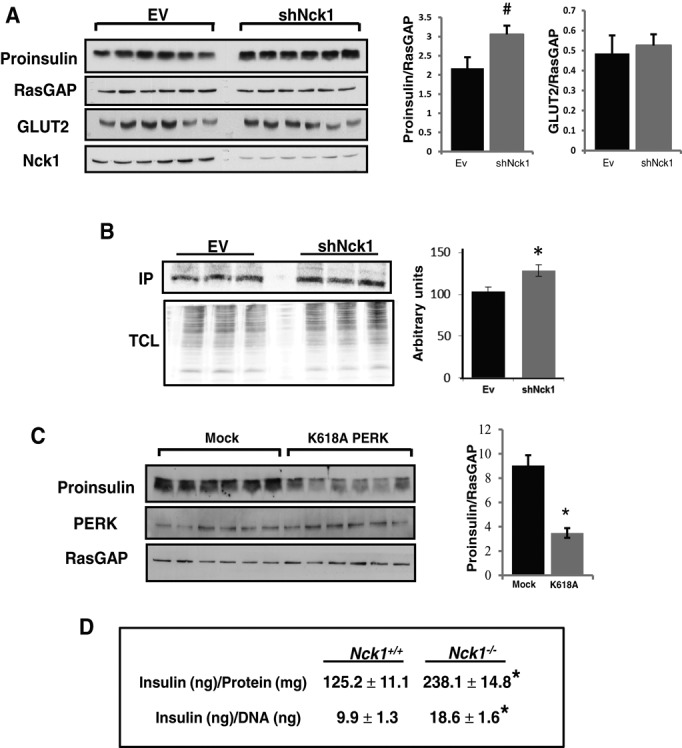
Nck1 modulates proinsulin levels and insulin biosynthesis in MIN6 cells and insulin content in isolated pancreatic islets. (A) LMP-EV and LMP-shNck1 MIN6 cell lysates (50 μg of protein) from independent culture dishes were subjected to immunoblotting with indicated antibodies. Bar charts show quantification by densitometry of the indicated protein levels. Data are mean ± SEM (n = 3; #p = 0.036). (B) [35S]Met/Cyst incorporation into insulin immunoprecipitates (IP; top) and total cell lysate proteins (TCL) from LMP-EV and LMP-shNck1 MIN6 cells. Bar chart represents quantification in arbitrary units of [35S]Met/Cyst incorporation into insulin IPs. Data are mean ± SEM (n = 4; *p = 0.047). (C) Total cell lysates (50 μg of protein) prepared from independent culture dishes of shNck1 MIN6 cells transiently transfected with pcDNA3.1 (Mock) or pcDNA3.1-PERK K618A (K618A) were subjected to immunoblotting using the indicated antibodies. Bar chart shows the ratio of proinsulin to RasGAP as determined by densitometry. Data are mean ± SEM (n = 3; *p < 0.001). (D) Insulin levels in Nck1+/+ and Nck1−/− isolated pancreatic islets. Data are mean ± SEM (n = 6; *p < 0.05).
DISCUSSION
PERK, a member of the UPR, is activated in response to stress impairing ER homeostasis. In these conditions, it induces phosphorylation of eIF2α at Ser-51, which attenuates mRNA translation initiation, thereby decreasing general protein synthesis (Harding et al., 2000b). In the present study, we provide strong evidence that the SH2/SH3 domain–containing adaptor protein Nck1 is a negative regulator of PERK, which affects β-cell proinsulin synthesis in physiological conditions. Indeed, we show that stable depletion of Nck1 in MIN6 cells results in enhanced physiological PERK activity, accompanied by increased proinsulin synthesis. As reported by others (Gupta et al., 2010), these results confirm that physiological PERK activity positively regulates proinsulin biosynthesis, as opposed to its well-established role as a negative regulator of general protein synthesis through phosphorylation of eIF2αSer-51 under ER stress conditions (Harding et al., 2000a). However, whereas partial PERK depletion in INS832/13 cells was reported to specifically decrease proinsulin biosynthesis (Gupta et al., 2010), acute pharmacological inhibition of PERK in MIN6 cells increased proinsulin biosynthesis (Harding et al., 2012). This discrepancy on the role of PERK on proinsulin biosynthesis can be due to the degree of PERK inhibition achieved by the different approaches used to modulate PERK activity and its regulation of general protein synthesis. Indeed, partial decrease in PERK activity by shPerk down-regulates proinsulin biosynthesis without affecting β cell general protein synthesis (Gupta et al., 2010), whereas the robustness of the effect of acute and complete inhibition of PERK activity by small molecules (PERKi) could increase proinsulin biosynthesis through nonspecific increase of global protein synthesis (Harding et al., 2012).
Nck directly interacts with and negatively regulates PERK
Nck couples activated receptor tyrosine kinases (RTKs) to effectors that promote RTK intracellular signaling (Chen et al., 1998). Of interest, our study demonstrates an alternative role for Nck in regulating PERK signal transduction. In fact, as for RTKs, Nck, through its SH2 domain, directly interacts with tyrosine-phosphorylated PERK, but in contrast to mediating RTK signaling, it dampens PERK activation and signaling. Supporting this, we showed that Nck directly interacts with PERK phosphorylated at Y561 and that PERK mutation Y561F, which abolished Nck binding, promotes PERK activation and signaling. Furthermore, we showed that in response to ER stress, Nck1 down-regulation resulted in faster PERK pY561 dephosphorylation and activation. These results suggest that phosphorylation of PERK at Y561 regulates Nck1 and PERK interaction, whereas Nck1 binding to PERK delays PERK pY561 dephosphorylation and activation upon ER stress. Taken together, our results also suggest that the interaction between Nck1 and PERK is dynamic because it requires PERK phosphorylation at Y561, which is dependent on PERK kinase activity, as demonstrated by the inability of the PERK-inactive mutant K618A to be tyrosine phosphorylated at Y561 and to interact with Nck1. However, it remains to be determined whether Nck1 participates in a feedback mechanism to restore PERK inactivation when ER stress is resolved or inactivating PERK after reaching a threshold level of PERK activation during ER stress.
Previous reports identified several proteins regulating PERK activity through direct interaction with PERK luminal or cytoplasmic domains. Among these, GRP78/BiP, an ER chaperone that binds to PERK luminal domain, prevents PERK dimerization (Bertolotti et al., 2000; Hendershot, 2004). Protein kinase inhibitor of 58 kDa (p58IPK), which acts as a cochaperone for BiP in the ER (Rutkowski et al., 2007), is also part of a negative feedback loop through its direct interaction with PERK luminal domain, leading to inhibition of PERK kinase activity during late phases of the UPR (Yan et al., 2002; van Huizen et al., 2003). In addition, calcineurin, a cytoplasmic Ca2+-dependent phosphatase that is upregulated and activated upon ER stress, interacts with the cytoplasmic domain of preactivated PERK to further promote PERK activation (Bollo et al., 2010). Finally, PARP16, an ER-anchored protein member of the poly(ADP-ribose) polymerase family facing the cytoplasm, interacts with PERK and is required for PERK activation upon ER stress, as well as being sufficient for PERK activation in the absence of ER stress (Jwa and Chang, 2012). We demonstrate for the first time that Nck1, which is detected at the ER (Latreille and Larose, 2006), directly interacts with PERK cytoplasmic domain through the phosphorylation of Y561 in the PERK juxtamembrane region and that this interaction is PERK kinase and Nck1 SH2 dependent. We provide evidence that PERK is phosphorylated at Y561 in vivo and demonstrate that substitution of Y561 with a phenylalanine enhances PERK catalytic kinase activity, as monitored by phosphorylation of eIF2αS51. We show that phosphorylation of PERK at Y561 delays PERK activation and signaling upon ER stress and that PERK phosphorylation at Y561 is rapidly lost in response to ER stress. Collectively these novel findings strongly suggest a negative regulatory role for PERK phosphorylation at Y561 on PERK activation. Previous studies reported that PERK is phosphorylated on tyrosine residues (Ma et al., 2001; Su et al., 2008). In particular, PERK tyrosine phosphorylation in the kinase domain at Y615 was associated with optimal activation of PERK under ER stress (Su et al., 2008), but our findings reveal that PERK phosphorylation at Y561 negatively regulates PERK activation. Of interest, Nck, which directly binds to pY561PERK, dissociates from PERK upon ER stress, correlating with PERK pY561 dephosphorylation and activation. In addition, we show that Nck1 protects pY561PERK from dephosphorylation. Together these data support a role for Nck1 in negatively regulating PERK activation by interacting with and protecting pY561PERK from being dephosphorylated.
Nck1 could negatively regulate PERK through various mechanisms. In the absence of stress, Nck1 bound to PERK juxtamembrane domain prevents spontaneous PERK dimerization (i.e., through an allosteric or steric mechanism). During ER stress, PERK reaches a threshold level of functional activity (i.e., PERK Thr-980, eIF2αS51), whereas Nck1 reassociation to the juxtamembrane domain of PERK contributes to restoring inactive PERK monomeric states. Alternatively, Nck1 binding to PERK juxtamembrane domain allosterically regulates BiP interaction with PERK to control both PERK basal activity and activation upon ER stress. On the other hand, binding of Nck1 to the PERK juxtamembrane domain induces changes in PERK conformation (e.g., allosteric modulation or sterical hindrance), which disfavors its cytoplasmic domain interaction with activators such as calcineurin during the early phase of UPR activation.
Association of Nck1 with PERK phosphorylated at Y561 could hinder recognition by tyrosine phosphatase(s) dephosphorylating PERK at pY561, thereby limiting its activation. Conceptually, the latter mechanism is supported by our findings that PERK mutation at Y561 for a phenylalanine promotes PERK activation and down-regulation of Nck1 in MIN6 cells accelerates dephosphorylation of pY561PERK and PERK activation upon ER stress. Protein tyrosine phosphatase 1B (PTP1B) and T-cell protein tyrosine phosphatase (TCPTP) are highly related ER tyrosine phosphatases (Andersen et al., 2001) known to modulate PERK activation and signaling in MIN6 cells (Bettaieb et al., 2011, 2012). Although down-regulation of PTP1B results in enhanced PERK activation and signaling, TCPTP down-regulation has the opposite effect (Bettaieb et al., 2011). Because increased PERK phosphorylation at Y561 correlates with decreased PERK activation during ER stress, we suggest that TCPTP, as opposed to PTP1B, can mediate dephosphorylation of PERK at Y561 to facilitate PERK activation. On the other hand, PTP1B deficiency enhances PERK activation and signaling in brown adipose tissue, possibly through increased PERK phosphorylation at Y615 (Bettaieb et al., 2012). Given that Nck interacts with PTP1B through its SH3 domains (Clemens et al., 1996), Nck bound to PERK can recruit PTP1B to dephosphorylate PERK at Y615, thereby promoting PERK inactivation. Whether TCPTP dephosphorylates pY561PERK or Nck hampers dephosphorylation of pY561PERK by TCPTP or recruits PTP1B and PERK in a common complex requires further investigation.
Finally, as an adaptor protein, Nck1 bound to PERK through its SH2 domain could couple PERK to PERK negative regulator(s) bound to its SH3 domains. In fact, we previously showed that Nck1 overexpression decreases both basal and ER stress-induced eIF2αS51 phosphorylation, whereas MEFs lacking Nck spontaneously display high levels of peIF2αS51 and PERK downstream signaling markers (Kebache et al., 2004; Latreille and Larose, 2006). In addition, we provided evidence that Nck1 assembles a holophosphatase complex containing the Ser/Thr phosphatase PP1c and eIF2, which could be responsible for the decrease of peIF2αS51 in cells overexpressing Nck1 (Kebache et al., 2004; Latreille and Larose, 2006). Here we show that MEFs lacking Nck and MIN6 cells with low levels of Nck1 display enhanced PERK activation, revealing that Nck1 contributes to negative regulation of PERK. Therefore consistent with our previous findings (Kebache et al., 2004; Latreille and Larose, 2006), it is possible that Nck1, while bound to PERK, brings PP1c in close proximity, maintaining PERK dephosphorylated on critical Ser/Thr residues involved in PERK activation.
Nck1 modulates proinsulin biosynthesis and content in pancreatic β-cells by regulating PERK
We discovered that proinsulin levels are significantly increased in Nck1-depleted MIN6 cells as well as insulin levels in pancreatic isolated islets of Nck1−/−mice. We provide strong evidence that Nck1, by regulating physiological PERK activity, affects proinsulin biosynthesis. However, how PERK, known to attenuate translation during ER stress, promotes proinsulin biosynthesis in physiological conditions needs further investigation. Our study provides new insights into the dynamic regulation of PERK and further supports an important physiological function for PERK in pancreatic β-cells. Overall it relies on original findings that provide new perspectives on understanding β-cell function and dysfunction.
MATERIALS AND METHODS
Cells
Cos-1 cells and MEFs were cultured in high-glucose DMEM (Invitrogen, Burlington, ON, Canada) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 0.75 mg/ml penicillin, and 0.1 mg/ml streptomycin and kept at 37°C in a 95% O2–5% CO2 environment. Mouse insulinoma (MIN6) cells were cultured in high-glucose DMEM as reported but with 0.55 μM β-mercaptoethanol and 15% FBS.
Cell treatments
Thapsigargin (Sigma, St. Louis, MO), DTT (Roche, Mississauga, ON, Canada), and protein tyrosine phosphatase inhibitors (peroxide derivative of Na3VO4 [Sigma] and bpVPhen) were used as indicated in figure legends. Cos-1, MIN6, and PERK−/− MEF cells transfected with indicated plasmids using Lipofectamine Plus (Invitrogen) were lysed 48 h posttransfection in lysis buffer (Latreille and Larose, 2006) and total cell lysates subjected to either immunoblotting or immunoprecipitation.
Antibodies
PERK polyclonal antiserum was obtained after rabbit immunization with a GST chimera of the PERK cytoplasmic segment (amino acids [aa] 537–1114; Harding et al., 1999). PERK phosphospecific Y561 (pY561 PERK) polyclonal antibody was generated by GenScript (Genscript USA, Piscataway, NJ) using a synthetic phosphopeptide derived from PERK juxtamembrane domain (QTESKpYDSVSADVS). Pan Nck (Nck) antibody recognizes both Nck isoforms (Lussier and Larose, 1997). Nck1 polyclonal antibody was generated as previously reported (Latreille et al., 2011). Human peIF2αSer52 (mouse peIF2αSer-51) antibody was from BioSource International (44-728G; Medicorp, Montreal, QC, Canada). pThr981 PERK (Sc-32577), eIF2α (Fl-315), pTyr (PY99), GST (B-14), GFP (B-2), RasGAP, insulin (H-86), and GLUT2 (H-67) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). pSer/Thr antibodies were from BD Transduction Laboratories (Lexington, KY), and β-actin antibody (AC-74) was from Sigma. All other chemicals were from standard commercial sources.
Constructs
Plasmids encoding GST chimera of the PERK cytoplasmic segment (GST‑cPERK, aa 537–1114) WT, kinase dead (K618A), Myc-PERK full-length WT, and K618A were provided by David Ron (University of Cambridge, Cambridge, United Kingdom). GST‑cPERK mutated in Y561F was generated by overlapping PCR using the primers 5′-ACTGACATCGGCACTCACGGAGTCGAATTTACTTTCAGTCTGGCACTG-3′ and 5′-GACTCCGTGAGTGCCGATGTCAGT-3′. Myc-PERK full-length Y561F mutant was obtained by site-directed mutagenesis (Mutagenex). Recombinant Nck proteins were produced by thrombin cleavage of purified bacterially produced GST-Nck. GFP-Nck1 full length, the three SH3 domains (3SH3, aa 1–267), and the SH2 domain (SH2, aa 268–377) were generated by subcloning corresponding cDNAs into pEGFP-C1 (Clontech, Mountain View, CA). All constructs were confirmed by DNA sequencing.
In vitro binding assays
GST and GST-cPERK WT or K618A (100 ng) bound on glutathione beads were incubated with recombinant Nck1 or Nck2 (100 ng) for 3 h at 4°C in the binding buffer (Latreille and Larose, 2006). Beads were washed with binding buffer, and proteins were recovered in Laemmli buffer and subjected to SDS–PAGE. Nck was revealed by immunoblot using Nck antibody and enhanced chemiluminescence as recommended by the manufacturer (Amersham GE Healthcare, Piscataway, NJ). Transfected Cos-1 cells were washed with phosphate-buffered saline (PBS) and lysed in binding buffer. GST‑cPERK (5–10 μg) was used in pull-down experiments performed at 4°C for 3 h. Bound proteins were resolved by SDS–PAGE and subjected to immunoblotting.
In vitro kinase assays
In vitro eIF2α phosphorylation assays were performed by incubating recombinant His-eIF2α (300 ng) and GST-cPERK (200 ng) in kinase buffer (10 mM Tris-HCl, pH 7.4, 50 mM KCl, 2 mM MgCl2, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 4 μg/ml aprotinin). Reactions were initiated by adding [γ‑32P]ATP (10 μCi) in a final volume of 25 μl, incubated for 30 min at 30°C, and then stopped with Laemmli buffer. Samples were subjected to SDS–PAGE and dried gels exposed for autoradiography.
Phosphoamino acid analysis
In vitro 32P-labeled GST-cPERK WT (200 ng) produced as described was subjected to SDS–PAGE and transferred onto polyvinylidene fluoride (PVDF) membrane, and upon autoradiography the corresponding PERK band was excised and washed with H2O. Hydrolysis was performed for 1 h at 100°C in 6 N HCl. Lyophilized supernatants were resuspended in buffer at pH 1.9 containing 1 mg/ml phosphoserine, phosphothreonine, and phosphotyrosine as internal standards. Phosphoamino acids were separated by two-dimensional electrophoresis (pH 1.9 and 3.5) on TLC plates (EM Sciences), visualized by autoradiography, and identified according to ninhydrin-stained standards.
shNck1 MIN6 cells
A mouse shRNA Nck1 was designed according to RNA interference oligoRetriever using the sequence of a mouse small interfering RNA (siRNA; Nck1 (Integrated DNA Technologies, Coralville, IA) efficient in decreasing Nck1 expression in transient transfection. This shNck1 was subcloned into MSCV‑LMP retroviral vector (Open Biosystems, Fisher Scientific, Ottawa, ON, Canada) to obtain mouse shNck1 flanked by microRNA-30 sequence and confirmed by sequencing. Retroviral productions of the empty LMP and the shNck1-containing LMP were achieved according to classic procedures. MIN6 cells infections were established by adding the appropriate virus preparation and medium at a ratio of 1:1 in the presence of Polybrene (8 μg/ml). Stable pools of control and shNck1 MIN6 cells were selected in medium containing puromycin.
Isolated pancreatic islets
Isolated islets (110–200) obtained as described (Peyot et al., 2004) were subjected to acid-ethanol extraction (0.2 M HCl in 75% ethanol) to determine insulin content using radioimmunoassay (Linco Research, St. Charles, MO). In parallel, isolated islets were lysed in Tris-EDTA containing 0.5% Triton X-100 to quantify protein using Bio-Rad assay (Bio-Rad, Mississauga, ON, Canada) and DNA using SYBR green.
Insulin biosynthesis
Control and shNck1 MIN6 cells were incubated for 1 h in MIN6 cell medium lacking methionine and cysteine and supplemented with 15% dialyzed FBS. Cells were then labeled using [35S]Met/Cyst labeling protein mix (Perkin Elmer, Woodbridge, ON, Canada) at 100 μCi/ml for 30 min. Cells were then washed with ice-cold PBS and lysed, and 400 μg of protein from cell lysates was used to immunoprecipitate insulin. Samples were run on a 6 M urea/16% tricine gel and transferred onto a PVDF membrane before being dried and exposed for detection of 35S label incorporated into insulin using a phosphoimager (Typhoon FLA 9500; GE Canada, Mississauga, ON, Canada).
Statistics
Statistical significance was determined using Student's t test with p ≤ 0.05. In all tests, two groups with only one changed parameter were compared. Mann–Whitney test was used for the indicated experiments.
Supplementary Material
Acknowledgments
We thank David Ron for providing PERK-related reagents. Nck+/+ and Nck−/− MEFs and Nck1+/+ and Nck1−/− mice were from Tony Pawson (Mount Sinai Hospital Research Institute, Toronto, Canada). We thank Genevieve Bourret (McGill University, Montreal, Canada) for help in generating the PERK antibody. We thank Peter Siegel (McGill University) for providing the MSCV‑LMP vector and Jason Northey (McGill University) for help and advice on retroviral production and stable infection of MIN6 cells. This work was supported by grants to L.L. from the Canadian Diabetes Association in honor of the late Lilian I. Dale and the Canadian Institutes of Health (MOP115045). L.Y. is supported by a doctoral award from the Canadian Diabetes Association.
Abbreviations used:
- ATF4
activating transcription factor 4
- BiP
binding immunoglobulin protein
- DTT
dl-dithiothreitol
- eIF2
eukaryotic initiation factor 2
- ER
endoplasmic reticulum
- Gadd34
growth arrest and DNA damage-inducible protein 34
- Grp 94
glucose-related protein 94
- Nck
noncatalytic region of tyrosine kinase adaptor protein
- PERK
PKR-like endoplasmic reticulum protein kinase
- PV
pervanadate
- Tg
thapsigargin
- UPR
unfolded protein response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-09-0511) on December 26, 2013.
*Present address: Institute of Molecular Health Sciences, ETH Zurich, 8093 Zurich, Switzerland.
REFERENCES
- Abella JV, Vaillancourt R, Frigault MM, Ponzo MG, Zuo D, Sangwan V, Larose L, Park M. The Gab1 scaffold regulates RTK-dependent dorsal ruffle formation through the adaptor Nck. J Cell Sci. 2010;123:1306–1319. doi: 10.1242/jcs.062570. [DOI] [PubMed] [Google Scholar]
- Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bettaieb A, et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;286:9225–9235. doi: 10.1074/jbc.M110.186148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A, Matsuo K, Matsuo I, Wang S, Melhem R, Koromilas AE, Haj FG. Protein tyrosine phosphatase 1B deficiency potentiates PERK/eIF2alpha signaling in brown adipocytes. PLoS One. 2012;7:e34412. doi: 10.1371/journal.pone.0034412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollo M, Paredes RM, Holstein D, Zheleznova N, Camacho P, Lechleiter JD. Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PLoS One. 2010;5:e11925. doi: 10.1371/journal.pone.0011925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman LE, Quilliam LA. Identification of Grb4/Nckbeta, a src homology 2 and 3 domain-containing adapter protein having similar binding and biological properties to Nck. J Biol Chem. 1999;274:5542–5549. doi: 10.1074/jbc.274.9.5542. [DOI] [PubMed] [Google Scholar]
- Chen M, She H, Davis EM, Spicer CM, Kim L, Ren R, Le Beau MM, Li W. Identification of Nck family genes, chromosomal localization, expression, and signaling specificity. J Biol Chem. 1998;273:25171–25178. doi: 10.1074/jbc.273.39.25171. [DOI] [PubMed] [Google Scholar]
- Clemens JC, Ursuliak Z, Clemens KK, Price JV, Dixon JE. A Drosophila protein-tyrosine phosphatase associates with an adapter protein required for axonal guidance. J Biol Chem. 1996;271:17002–17005. doi: 10.1074/jbc.271.29.17002. [DOI] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Feng D, Wei J, Gupta S, McGrath BC, Cavener DR. Acute ablation of PERK results in ER dysfunctions followed by reduced insulin secretion and cell proliferation. BMC Cell Biol. 2009;10:61. doi: 10.1186/1471-2121-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese S, Schubert WD, Findeis AC, Marquardt T, Roske YS, Stradal TE, Heinz DW. The phosphotyrosine peptide binding specificity of Nck1 and Nck2 Src homology 2 domains. J Biol Chem. 2006;281:18236–18245. doi: 10.1074/jbc.M512917200. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, Diehl JA. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol. 2012;32:5129–5139. doi: 10.1128/MCB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, Finlay BB. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol. 2001;3:856–859. doi: 10.1038/ncb0901-856. [DOI] [PubMed] [Google Scholar]
- Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59:1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000a;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000b;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem. 2012;287:44338–44344. doi: 10.1074/jbc.M112.428987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- Jwa M, Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nat Cell Biol. 2012;14:1223–1230. doi: 10.1038/ncb2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebache S, Cardin E, Nguyen DT, Chevet E, Larose L. Nck-1 antagonizes the endoplasmic reticulum stress-induced inhibition of translation. J Biol Chem. 2004;279:9662–9671. doi: 10.1074/jbc.M310535200. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latreille M, Laberge MK, Bourret G, Yamani L, Larose L. Deletion of Nck1 attenuates hepatic ER stress signaling and improves glucose tolerance and insulin signaling in liver of obese mice. Am J Physiol Endocrinol Metab. 2011;300:E423–E434. doi: 10.1152/ajpendo.00088.2010. [DOI] [PubMed] [Google Scholar]
- Latreille M, Larose L. Nck in a complex containing the catalytic subunit of protein phosphatase 1 regulates eukaryotic initiation factor 2alpha signaling and cell survival to endoplasmic reticulum stress. J Biol Chem. 2006;281:26633–26644. doi: 10.1074/jbc.M513556200. [DOI] [PubMed] [Google Scholar]
- Lussier G, Larose L. A casein kinase I activity is constitutively associated with Nck. J Biol Chem. 1997;272:2688–2694. doi: 10.1074/jbc.272.5.2688. [DOI] [PubMed] [Google Scholar]
- Ma Y, Lu Y, Zeng H, Ron D, Mo W, Neubert TA. Characterization of phosphopeptides from protein digests using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and nanoelectrospray quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:1693–1700. doi: 10.1002/rcm.426. [DOI] [PubMed] [Google Scholar]
- Peyot ML, Nolan CJ, Soni K, Joly E, Lussier R, Corkey BE, Wang SP, Mitchell GA, Prentki M. Hormone-sensitive lipase has a role in lipid signaling for insulin secretion but is nonessential for the incretin action of glucagon-like peptide 1. Diabetes. 2004;53:1733–1742. doi: 10.2337/diabetes.53.7.1733. [DOI] [PubMed] [Google Scholar]
- Posner BI, et al. Peroxovanadium compounds. A new class of potent phosphotyrosine phosphatase inhibitors which are insulin mimetics. J Biol Chem. 1994;269:4596–4604. [PubMed] [Google Scholar]
- Rivera GM, Antoku S, Gelkop S, Shin NY, Hanks SK, Pawson T, Mayer BJ. Requirement of Nck adaptors for actin dynamics and cell migration stimulated by platelet-derived growth factor B. Proc Natl Acad Sci USA. 2006;103:9536–9541. doi: 10.1073/pnas.0603786103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, Hegde RS. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell. 2007;18:3681–3691. doi: 10.1091/mbc.E07-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J Biol Chem. 2008;283:469–475. doi: 10.1074/jbc.M704612200. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem. 2003;278:15558–15564. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobieva N, Protopopov A, Protopopova M, Kashuba V, Allikmets RL, Modi W, Zabarovsky ER, Klein G, Kisselev L, Graphodatsky A. Localization of human ARF2 and NCK genes and 13 other NotI-linking clones to chromosome 3 by fluorescence in situ hybridization. Cytogenet Cell Genet. 1995;68:91–94. doi: 10.1159/000133898. [DOI] [PubMed] [Google Scholar]
- Yan W, Gale MJ, Jr, Tan SL, Katze MG. Inactivation of the PKR protein kinase and stimulation of mRNA translation by the cellular co-chaperone P58(IPK) does not require J domain function. Biochemistry. 2002;41:4938–4945. doi: 10.1021/bi0121499. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.