

Abstract
Background
Gastrointestinal nematodes of livestock have major socio-economic importance worldwide. In small ruminants, Chabertia spp. are responsible for economic losses to the livestock industries globally. Although much attention has given us insights into epidemiology, diagnosis, treatment and control of this parasite, over the years, only one species (C. ovina) has been accepted to infect small ruminants, and it is not clear whether C. erschowi is valid as a separate species.
Methods
The first and second internal transcribed spacers (ITS-1 and ITS-2) regions of nuclear ribosomal DNA (rDNA) and the complete mitochondrial (mt) genomes of C. ovina and C. erschowi were amplified and then sequenced. Phylogenetic re-construction of 15 Strongylida species (including C. erschowi) was carried out using Bayesian inference (BI) based on concatenated amino acid sequence datasets.
Results
The ITS rDNA sequences of C. ovina China isolates and C. erschowi samples were 852–854 bp and 862 -866 bp in length, respectively. The mt genome sequence of C. erschowi was 13,705 bp in length, which is 12 bp shorter than that of C. ovina China isolate. The sequence difference between the entire mt genome of C. ovina China isolate and that of C. erschowi was 15.33%. In addition, sequence comparison of the most conserved mt small subunit ribosomal (rrnS) and the least conserved nad2 genes among multiple individual nematodes revealed substantial nucleotide differences between these two species but limited sequence variation within each species.
Conclusions
The mtDNA and rDNA datasets provide robust genetic evidence that C. erschowi is a valid strongylid nematode species. The mtDNA and rDNA datasets presented in the present study provide useful novel markers for further studies of the taxonomy and systematics of the Chabertia species from different hosts and geographical regions.
Keywords: Chabertia spp, Nuclear ribosomal DNA, Internal transcribed spacer (ITS), Mitochondrial DNA, Phylogenetic analysis
Background
The phylum Nematoda includes many parasites that threaten the health of plants, animals and humans on a global scale. The soil-transmitted helminthes (including roundworms, whipworms and hookworms) are estimated to infect almost one sixth of all humans, and more than a billion people are infected with at least one species [1]. Chabertia spp. are common gastrointestinal nematodes, causing significant economic losses to the livestock industries worldwide, due to poor productivity, failure to thrive and control costs [2-6]. In spite of the high prevalence of Chabertia reported in small ruminants [7], it is not clear whether the small ruminants harbour one or more than one species. Based on morphological features (e.g., cervical groove and cephalic vesicle) of adult worms, various Chabertia species have been described in sheep and goats in China, including C. ovina, C. rishati, C. bovis, C. erschowi, C. gaohanensis sp. nov and C. shaanxiensis sp. nov [8-10]. However, to date, only Chabertia ovina is well recognized as taxonomically valid [11,12]. Obviously, the identification and distinction of Chabertia to species using morphological criteria alone is not reliable. Therefore, there is an urgent need for suitable molecular approaches to accurately identify and distinguish closely-related Chabertia species from different hosts and regions.
Molecular tools, using genetic markers in mitochondrial (mt) genomes and the internal transcribed spacer (ITS) regions of nuclear ribosomal DNA (rDNA), have been used effectively to identify and differentiate parasites of different groups [13-16]. For nematodes, recent studies showed that mt genomes are useful genetic markers for the identification and differentiation of closely-related species [17,18]. In addition, employing ITS rDNA sequences, recent studies also demonstrated that Haemonchus placei and H. contortus are distinct species [19]; Trichuris suis and T. trichiura are different nematode species [20,21].
Using a long-range PCR-coupled sequencing approach [22], the objectives of the present study were (i) to characterize the ITS rDNA and mt genomes of C. ovina and C. erschowi from goat and yak in China, (ii) to compare these ITS sequences and mt genome sequences, and (iii) to test the hypothesis that C. erschowi is a valid species in phylogenetic analyses of these sequence data.
Methods
Parasites and isolation of total genomic DNA
Adult specimens of C. ovina (n = 6, coded CHO1-CHO6) and C. erschowi (n = 9, coded CHE1-CHE9) were collected, post-mortem, from the large intestine of a goat and a yak in Shaanxi and Qinghai Provinces, China, respectively, and were washed in physiological saline, identified morphologically [8,10], fixed in 70% (v/v) ethanol and stored at -20°C until use. Total genomic DNA was isolated separately from 15 individual worms using an established method [23].
Long-range PCR-based sequencing of mt genome
To obtain some mt sequence data for primer design, we PCR-amplified regions of C. erschowi of cox1 gene by using a (relatively) conserved primer pair JB3-JB4.5 [24], rrnL gene was amplified using the designed primers rrnLF (forward; 5′-GAGCCTGTATTGGGTTCCAGTATGA-3′) and rrnLR (reverse; 5′-AACTTTTTTTGATTTTCCTTTCGTA-3′), nad1 gene was amplified using the designed primers nad1F (forward; 5′-GAGCGTCATTTGTTGGGAAG-3′) and nad1R (reverse; 5′-CCCCTTCAGCAAAATCAAAC-3′), cytb gene was amplified using the designed primers cytbF (forward; 5′-GGTACCTTTTTGGCTTTTTATTATA-3′) and cytbR (reverse; 5′-ATATGAACAGGGCTTATTATAGGAT-3′) based on sequences conserved between Oesophagostomum dentatum and C. ovina Australia isolate. The amplicons were sequenced in both directions using BigDye terminator v.3.1, ABI PRISM 3730. We then designed primers (Table 1) to regions within cox1, rrnL, nad1 and cytb and amplified from C. ovina (coded CHO1) in four overlapping fragments: cox1-rrnL, rrnL-nad1, nad1-cytb and cytb-cox1. Then we designed primers (Table 1) to regions within cox1, rrnL, nad5, nad1, nad2 and cytb and amplified from C. erschowi (coded CHE1) in six overlapping fragments: cox1- rrnL, rrnL-nad5, nad5-nad1, nad1-nad2, nad2-cytb and cytb-cox1. The cycling conditions used were 92°C for 2 min (initial denaturation), then 92°C/10 s (denaturation), 50 -58°C (C. erschowi) or 56 -65°C (C. ovina)/30 s (annealing), and 60°C/10 min (extension) for 10 cycles, followed by 92°C for 2 min, then 92°C/10 s, 50 -58°C (C. erschowi) or 56 -65°C (C. ovina)/30 s, and 60°C/10 min for 20 cycles, with a cycle elongation of 10 s for each cycle and a final extension at 60°C/10 min. Each amplicon, which represented a single band in a 1.0% (w/v) agarose gel, following electrophoresis and ethidium-bromide staining, was column-purified and then sequenced using a primer walking strategy [22].
Table 1.
Sequences of primers used to amplify mitochondrial DNA regions from Chabertia erschowi and Chabertia ovina from China
| Primer | Sequence (5′ to 3′) |
|---|---|
| For rrnS |
|
| CHOF |
TCGTTTAGTGGGTATGTGTGGTTCT (for C. ovina) |
| CHOR |
GCCTACTCCCTAACAAATGACGCTC (for C. ovina) |
| CHEF |
GTGGTTTTTAGGTTAGGGTTGAGTG (for C. erschowi) |
| CHER |
ACGCTCATACAAAGTAATAAACGCA (for C. erschowi) |
| For nad2 |
|
| CHOF |
TTTGTGG(C\T)TAAGAGTGTT(G\A)GCTATT (for C. ovina) |
| CHOR |
GAGCCGTAATCAAACATAGTAAATC (for C. ovina) |
| CHEF |
TTTGTGG(C\T)TAAGAGTGTT(G\A)GCTATT (for C. erschowi) |
| CHER |
ACCGTAATCAAACATAGTAAAATCT (for C. erschowi) |
| For C. ovina |
|
| COF |
TGGTTGTGTGGTTTGGGCTCAT |
| rrnLR |
ATGTCCTCACGCTAAGACTGCC |
| rrnLF |
AGTTTGCTTCTGCCCAGTGA |
| ND5R |
ACCGTAACCTCGCCCATCCTG |
| ND5F |
ACGGCGTTAGTGGAGGAGGA |
| ND1R |
CCACTAACCAACTCCCTTTCACCC |
| ND1F |
ATTGGTGCTTTGCGGGCCAGT |
| ND2R |
CCATAAACCTTTAAAACCTCCC |
| ND2F |
TTGTTGGTTGGGAGACTATG |
| CYR |
AAAGGGTCCTCAACCAAACA |
| CYF |
CCTGTTTGGGGACCTTCTATTG |
| COR |
CCGCAGTAAAATAAGCACGAGA |
| For C. erschowi |
|
| COF |
ACCGACGGCTTATGGAAT |
| rrnLR |
AGTGCAACCCAACATTATACCCT |
| rrnLF |
TAAAGTTTGCTTCTGCCCAGTGATA |
| ND1R |
ATAATAGCCAACAAAAGCACCGACA |
| ND1F |
CTTGTCGGTGCTTTGCG |
| CYR |
CCGCCTCAATAAACATCTC |
| CYF |
TGGTCCAGATTATTGAAGG |
| COR | TTACCCGTCAAATACAAAGT |
Sequencing of ITS rDNA and mt rrnS and nad2
The full ITS rDNA region including primer flanking 18S and 28S rDNA sequences was PCR-amplified from individual DNA samples using universal primers NC5 (forward; 5′-GTAGGTGAACCTGCGGAAGGATCATT-3′) and NC2 (reverse; 5′-TTAGTTTCTTTTCCTCCGCT-3′) described previously [25]. The primers rrnSF and rrnSR (Table 1) designed to conserved mt genome sequences within the rrnS gene were employed for PCR amplification and subsequent sequencing of this complete gene (~ 700 bp) from multiple individuals of Chabertia spp. The primers nad2F and nad2R (Table 1) designed to conserved mt genome sequences within the nad2 gene were employed for PCR amplification and subsequent sequencing of this complete gene (~ 900 bp) from multiple individuals of Chabertia spp..
Sequence analyses
Sequences were assembled manually and aligned against the complete mt genome sequences of C. ovina Australia isolate [26] using the computer program Clustal X 1.83 [27] to infer gene boundaries. Translation initiation and termination codons were identified based on comparison with that of C. ovina Australia isolate [26]. The secondary structures of 22 tRNA genes were predicted using tRNAscan-SE [28] and/or manual adjustment [29], and rRNA genes were identified by comparison with that of C. ovina Australia isolate [26].
Phylogenetic analyses
Amino acid sequences inferred from the 12 protein-coding genes of the two Chabertia spp. worms were concatenated into a single alignment, and then aligned with those of 14 other Strongylida nematodes (Angiostrongylus cantonensis, GenBank accession number NC_013065 [30]; Angiostrongylus costaricensis, NC_013067 [30]; Angiostrongylus vasorum, JX268542 [31]; Aelurostrongylus abstrusus, NC_019571 [32]; Chabertia ovina Australia isolate, NC_013831 [26]; Cylicocyclus insignis, NC_013808 [26]; Metastrongylus pudendotectus, NC_013813 [26]; Metastrongylus salmi, NC_013815 [26]; Oesophagostomum dentatum, FM161882 [17]; Oesophagostomum quadrispinulatum, NC_014181 [17]; Oesophagostomum asperum, KC715826 [33]; Oesophagostomum columbianum, KC715827 [33]; Strongylus vulgaris, NC_013818 [26]; Syngamus trachea, NC_013821 [26], using the Ancylostomatoidea nematode, Necator americanus, NC_003416 as the outgroup [29]. Any regions of ambiguous alignment were excluded using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) [34] with the default parameters (Gblocks removed 1.6% of the amino acid alignments) and then subjected to phylogenetic analysis using Bayesian Inference (BI) as described previously [35,36]. Phylograms were drawn using the program Tree View v.1.65 [37].
Results
Nuclear ribosomal DNA regions of the two Chabertia species
The rDNA region including ITS-1, 5.8S rDNA and ITS-2 were amplified and sequenced from C. ovina China isolates, and they were 852-854 bp (GenBank accession nos. KF913466-KF913471) in length, which contained 367-369 bp (ITS-1), 153 bp (5.8S rDNA) and 231-239 bp (ITS-2). These sequences were 862-866 bp in length for C. erschowi samples (GenBank accession nos. KF913448-KF913456), containing 375-378 bp (ITS-1), 153 bp (5.8S rDNA) and 239-245 bp (ITS-2).
Features of the mt genomes of the two Chabertia species
The complete mt genome sequence of C. ovina China isolate and C. erschowi were 13,717 bp and 13,705 bp in length, respectively (GenBank accession nos. KF660604 and KF660603, respectively). The two mt genomes contain 12 protein-coding genes (cox1-3, nad1-6, nad4L, cytb, atp6), 22 transfer RNA genes and two ribosomal RNA genes (rrnS and rrnL) (Table 2), but the atp8 gene is missing (Figure 1). The protein-coding genes are transcribed in the same directions, as reported for Oesophagostomum spp. [17,33]. Twenty-two tRNA genes were predicted from the mt genomes, which varied from 55 to 63 bp in size. The two ribosomal RNA genes (rrnL and rrnS) were inferred; rrnL is located between tRNA-His and nad3, and rrnS is located between tRNA-Glu and tRNA-Ser (UCN). Three AT-rich non-coding regions (NCRs) were inferred in the mt genomes (Table 2). For these genomes, the longest NCR (designated NC2; 250 bp for C. ovina China isolate and 240 bp for C. erschowi in length) is located between the tRNA-Ala and tRNA-Pro (Figure 1), have an A + T content of 83.75% and 84%, respectively.
Table 2.
Mitochondrial genome organization of Chabertia erschowi (CE) and Chabertia ovina China isolate (COC) and Australia isolate (COA)
| Gene and region |
Positions and nt sequence lengths (bp) |
Initiation/termination codons |
||||
|---|---|---|---|---|---|---|
| CE | COC | COA | CE | COC | COA | |
|
cox1 |
2-1579 (1578) |
2-1579 (1578) |
2-1579 (1578) |
ATT/TAA |
ATT/TAA |
ATT/TAA |
| tRN A-Cys (C) |
1583-1637 (55) |
1583-1639 (57) |
1583-1639 (57) |
|
|
|
| tRNA-Met (M) |
1639-1697 (59) |
1640-1699 (60) |
1640-1699 (60) |
|
|
|
| tRNA-Asp (D) |
1699-1758 (60) |
1700-1758 (59) |
1699-1759 (61) |
|
|
|
| tRNA-Gly (G) |
1760-1816 (57) |
1759-1814 (56) |
1757-1814 (58) |
|
|
|
|
cox2 |
1817-2512 (696) |
1815-2510 (696) |
1814-2509 (696) |
ATT/TAA |
ATA/TAA |
ATA/TAA |
| tRNA-His (H) |
2512-2566 (55) |
2512-2568 (57) |
2511-2567 (57) |
|
|
|
|
rrnL |
2573-3542 (970) |
2572-3533 (962) |
2570-3531 (962) |
|
|
|
|
nad3 |
3543-3881 (339) |
3534-3869 (327) |
3532-3867 (336) |
ATT/TAA |
ATT/TAA |
ATT/TAA |
| Non-coding region (NC1) |
3882-3965 (84) |
3870-3949 (80) |
3868-3947 (80) |
|
|
|
|
nad5 |
3967-5548 (1582) |
3950-5531 (1582) |
3948-5529 (1582) |
ATT/ TAA |
ATT/T |
ATT/TAT |
| tRNA-Ala (A) |
5549-5603 (55) |
5532-5588 (57) |
5530-5586 (57) |
|
|
|
| Non-coding region (NC2) |
5604-5853 (250) |
5589-5828 (240) |
5587-5825 (239) |
|
|
|
| tRNA-Pro (P) |
5854-5909 (56) |
5829-5882 (54) |
5826-5880 (55) |
|
|
|
| tRNA-Val (V) |
5927-5982 (56) |
5930-5984 (55) |
5914-5970 (57) |
|
|
|
|
nad6 |
5983-6417 (435) |
5985-6416 (432) |
5970-6401 (432) |
ATA/ TAA |
TTG/TAA |
TTG/TAA |
|
nad4L |
6420-6653 (234) |
6418-6651 (234) |
6402-6635 (234) |
ATT/ TAA |
ATT/TAG |
ATT/TAG |
| tRNA-Trp (W) |
6681-6736 (56) |
6655-6712 (58) |
6639-6697 (59) |
|
|
|
| tRNA-Glu (E) |
6739-6794 (56) |
6740-6797 (58) |
6725-6784 (60) |
|
|
|
|
rrnS |
6797-7492 (696) |
6798-7493 (696) |
6780-7479 (700) |
|
|
|
| tRNA-Ser UCN (S2) |
7493-7547 (55) |
7494-7548 (55) |
7480-7536 (57) |
|
|
|
| tRNA-Asn (N) |
7547-7603 (57) |
7548-7605 (58) |
7535-7593 (59) |
|
|
|
| tRNA-Tyr (Y) |
7610-7666 (57) |
7608-7664 (57) |
7595-7652 (58) |
|
|
|
|
nad1 |
7667-8539 (873) |
7665-8537 (873) |
7652-8524 (873) |
ATT/TAA |
ATT/TAA |
ATT/TAA |
|
atp6 |
8539-9138 (600) |
8538-9137 (600) |
8525-9121 (597) |
ATT/TAA |
ATT/TAA |
ATT/TAG |
| tRNA-Lys (K) |
9150-9211 (62) |
9144-9206 (63) |
9128-9191 (64) |
|
|
|
| tRNA-LeuUUR (L2) |
9222-9276 (55) |
9215-9269 (55) |
9197-9252 (56) |
|
|
|
| tRNA-Ser AGN (S1) |
9277-9335 (59) |
9270-9327 (58) |
9252-9304 (53) |
|
|
|
|
nad2 |
9336-10175 (840) |
9328-10167 (840) |
9308-10147 (840) |
GTT/TAA |
ATT/TAA |
ATA/TAA |
| tRNA-Ile (I) |
10176-10234 (59) |
10175-10235 (61) |
10151-10211 (61) |
|
|
|
| tRNA-Arg (R) |
10235-10289 (55) |
10240-10294 (55) |
10215-10270 (56) |
|
|
|
| tRNA-Gln (Q) |
10290-10345 (56) |
10299-10353 (55) |
10271-10326 (56) |
|
|
|
| tRNA-Phe (F) |
10346-10403 (58) |
10354-10412 (59) |
10326-10385 (60) |
|
|
|
|
cytb |
10404- 11516 (1113) |
10413-11525 (1113) |
10385-11497 (1113) |
ATT/TAG |
ATT/TAA |
ATT/TAA |
| tRNA-Leu CUN (L1) |
11517-11572 (56) |
11529-11584 (56) |
11501-11562 (62) |
|
|
|
|
cox3 |
11573-12338 (766) |
11585-12350 (766) |
11557-12327 (771) |
ATT/T |
ATT/T |
ATT/TAA |
| tRNA-Thr (T) |
12339-12397 (59) |
12351-12404 (54) |
12323-12377 (55) |
|
|
|
|
nad4 |
12398-13627 (1230) |
12405-13634 (1230) |
12376-13608 (1233) |
TTG/TAA |
TTG/TAA |
ATT/TAA |
| Non-coding region (NC3) | 13628 – 1 (75) | 13635-1 (84) | 13609-1 (75) | |||
Figure 1.

Structure of the mitochondrial genomes for Chabertia. Genes are designated according to standard nomenclature, except for the 22 tRNA genes, which are designated using one-letter amino acid codes, with numerals differentiating each of the two leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGN and UCN, respectively). “NCR-1, NCR-2 and NCR-3” refer to three non-coding regions.
Comparative analyses between C. ovina and C. erschowi
The mt genome sequence of C. erschowi was 13,705 bp in length, 12 bp shorter than that of C. ovina China isolate, and 23 bp longer than that of C. ovina Australia isolate. The arrangement of the mt genes (i.e., 13 protein genes, 2 rrn genes and 22 tRNA genes) and NCRs were the same. A comparison of the nucleotide sequences of each mt gene as well as the amino acid sequences conceptually translated from individual protein-coding genes of the two Chabertia are given in Table 3. The greatest nucleotide variation between the C. ovina China isolate and C. erschowi was in the nad2 gene (19.4% and 17.92%), whereas least differences (7.33%) were detected in the rrnS gene, respectively (Table 3). The nucleotide sequence difference between the entire mt genome of C. ovina China isolate and that of C. erschowi was 15.33%. Sequence difference between the entire mt genome of C. ovina Australia isolate and that of C. erschowi was 15.48%. Sequence difference between the entire mt genome of C. ovina China isolate and that of C. ovina Australia isolate was 4.28%.
Table 3.
Nucleotide and/or predicted amino acid (aa) sequence differences for mt protein-coding and ribosomal RNA genes among Chabertia erschowi (CE) and Chabertia ovina China isolate (COC) and Australia isolate (COA)
| Gene |
Nucleotide length (bp) |
Nucleotide difference (%) |
Number of aa |
aa difference (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CE | COC | COA | CE/COC | CE/COA | COC/COA | CE | COC | COA | CE/COC | CE/COA | COC/COA | |
|
atp6 |
600 |
600 |
597 |
14.33 |
14.83 |
5.00 |
199 |
199 |
198 |
11.06 |
14.57 |
7.54 |
|
nad1 |
873 |
873 |
873 |
13.63 |
13.97 |
4.35 |
290 |
290 |
290 |
5.17 |
8.62 |
3.79 |
|
nad2 |
840 |
840 |
840 |
19.40 |
20.12 |
3.93 |
279 |
279 |
279 |
17.92 |
17.20 |
2.15 |
|
nad3 |
339 |
336 |
336 |
17.40 |
17.70 |
6.55 |
112 |
112 |
112 |
16.96 |
16.07 |
7.14 |
|
nad4 |
1230 |
1230 |
1233 |
17.64 |
18.82 |
5.43 |
409 |
409 |
410 |
14.67 |
17.80 |
4.88 |
|
nad4L |
234 |
234 |
234 |
11.97 |
12.39 |
4.70 |
77 |
77 |
77 |
7.79 |
7.79 |
0 |
|
nad5 |
1582 |
1582 |
1582 |
17.51 |
17.32 |
4.87 |
527 |
527 |
527 |
14.42 |
13.66 |
2.47 |
|
nad6 |
435 |
432 |
432 |
19.08 |
19.31 |
5.56 |
144 |
143 |
143 |
15.97 |
17.36 |
1.40 |
|
cox1 |
1578 |
1578 |
1578 |
11.98 |
12.86 |
4.06 |
525 |
525 |
525 |
0.57 |
0.57 |
0 |
|
cox2 |
696 |
696 |
696 |
13.36 |
13.65 |
4.89 |
231 |
231 |
231 |
0.87 |
0.87 |
0 |
|
cox3 |
766 |
766 |
771 |
14.75 |
14.01 |
5.06 |
255 |
255 |
256 |
3.14 |
2.35 |
1.17 |
|
cytb |
1113 |
1113 |
1113 |
16.80 |
16.89 |
4.67 |
370 |
370 |
370 |
9.73 |
9.73 |
1.08 |
|
rrnS |
696 |
696 |
700 |
7.33 |
7.71 |
1.86 |
- |
- |
- |
- |
- |
- |
| rrnL | 970 | 962 | 962 | 13.61 | 13.92 | 3.20 | - | - | - | - | - | - |
The difference in the concatenated amino acid sequences of the 12 protein-coding genes of the C. ovina China isolate and those of C. erschowi was 9.36%, 10% between those of the C. ovina Australia isolate and those of C. erschowi, and 2.37% between those of the C. ovina China isolate and those of C. ovina Australia isolate. The amino acid sequence differences between each of the 12 protein-coding genes of the C. ovina Australia isolate and the corresponding homologues of C. erschowi ranged from 0.57-17.92%, with COX1 being the most conserved and NAD2 the least conserved proteins (Table 3). Phylogenetic analyses of concatenated amino acid sequence data sets, using N. americanus as the outgroup, revealed that the Chabertia and Oesophagostomum were clustered together, with absolute support (posterior probability (pp) = 1.00) support (Figure 2).
Figure 2.
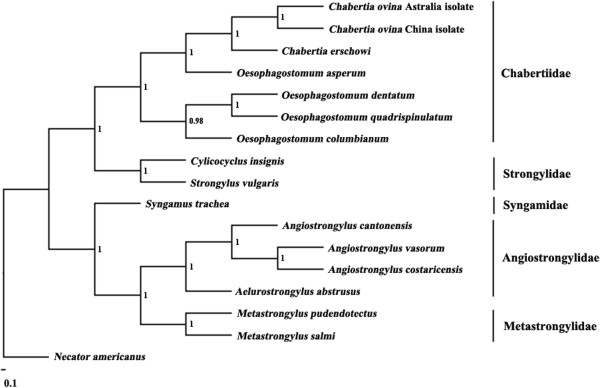
Inferred phylogenetic position of Chabertia within Strongylida nematodes. Analysis of the concatenated amino acid sequence data representing 12 protein-coding genes by Bayesian inference (BI), using Necator americanus (NC_003416) as the outgroup.
Sequence variation in complete nad2 gene was assessed among 15 individuals of Chabertia from goats and yaks. Sequences of the six C. ovina China isolate individuals were the same in length (840 bp) (GenBank accession nos. KF913472-KF913477). Nucleotide variation among the six C. ovina China isolate individuals was detected at 18 sites (18/840; 2.1%). Sequences of the nine C. erschowi individuals were the same in length (840 bp) (GenBank accession nos. KF913484-KF913492). Nucleotide variation also occurred at 23 sites (23/840; 2.7%). All 15 alignments of the nad2 sequences revealed that all individuals of Chabertia differed at 182 nucleotide positions (182/840; 21.7%). Phylogenetic analysis of the nad2 sequence data revealed strong support for the separation of C. ovina and C. erschowi individuals into two distinct clades (Figure 3A).
Figure 3.
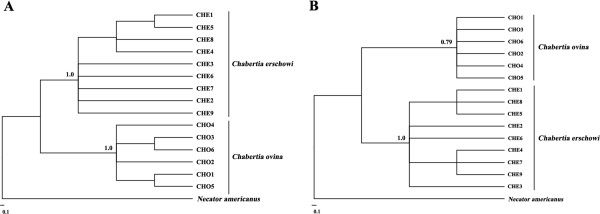
Inferred genetic relationships of 15 individual Chabertia specimens. The analyses were carried out by Bayesian inference (BI) based on mitochondrial rrnS (A) and nad2 (B) sequence data, using Necator americanus as the outgroup.
Sequence variation in complete rrnS gene was assessed among 15 individuals of Chabertia from goat and yak. Sequences of the rrnS gene from the six C. ovina China isolate individuals were the same in length (696 bp) (GenBank accession nos. KF913478-KF913483). Nucleotide variation among the six C. ovina China isolate individuals was detected at seven sites (7/696; 1.0%). Sequences of the rrnS gene from the nine C. erschowi individuals were the same in length (696 bp) (GenBank accession nos. KF913457-KF913465). Nucleotide variation also occurred at 6 sites (6/696; 0.9%). All 15 alignments of the rrnS sequences revealed that all individuals of Chabertia differed at 56 nucleotide positions (56/696; 8.05%). Phylogenetic analysis of the rrnS sequence data revealed strong support for the separation of C. ovina and C. erschowi individuals into two distinct clades (Figure 3B).
The ITS-1 and ITS-2 sequences from 10 individual adults of C. ovina China isolate were compared with that of 6 individual adults of C. erschowi. Sequence variations were 0–2.9% (ITS-1) and 0–2.7% (ITS-2) within the two Chabertia species, respectively. However, the sequence differences were 6.3-8.2% (ITS-1) and 10.4-13.6% (ITS-2) between the C. ovina China isolate and C. erschowi.
Discussion
Chabertia spp. is responsible for economic losses to the livestock industries globally. Although several Chabertia species have been described from various hosts based on the microscopic features of the adult worms (e.g. cervical groove and cephalic vesicle), it is not clear whether C. erschowi is valid as a separate species due to unreliable morphological criteria. For this reason, we employed a molecular approach, so that comparative genetic analyses could be conducted.
In the present study, substantial levels of nucleotide differences (15.33%) were detected in the complete mt genome between C. ovina China isolate and C. erschowi, and 15.48% between C. ovina Australia isolate and C. erschowi. These mtDNA data provide strong support that C. erschowi represents a single species because a previous comparative study has clearly indicated that variation in mtDNA sequences between closely-related species were typically 10%-20% [13].
The difference in amino acid sequences of the concatenated 12 proteins encoded by the complete mt genome between C. ovina China isolate and C. erschowi is 9.36%, and 10% between the C. ovina Australia isolate and C. erschowi. This level of amino acid variation is higher than those of other nematodes. Previous studies of other congener nematodes have detected low level differences in 12 protein sequences. For example, differences in amino acid sequences between A. duodenale and A. caninum is 4.1% [29,38], and between Toxocara malaysiensis and Toxocara cati is 5.6% [39], and between O. dentatum and O. quadrispinulatum is 3.22% [17]. In addition, substantial levels of nucleotide differences (6.3%-8.2% in ITS-1 and 10.4-13.6% in ITS-2) were also detected between C. ovina China isolate and C. erschowi. These results also indicate that C. erschowi is a separate species from C. ovina. This proposal was further supported by phylogenetic analysis based on mtDNA sequences (Figure 3), although, to date, only small numbers of adult worms have been studied molecularly. Clearly, larger population genetic and molecular epidemiological studies should be conducted using the mt and nuclear markers defined in this study to further test this proposal/hypothesis.
Conclusion
The findings of this study provide robust genetic evidence that C. erschowi is a separate and valid species from C. ovina. The mtDNA and rDNA datasets reported in the present study should provide useful novel markers for further studies of the taxonomy and systematics of Chabertia spp. from different hosts and geographical regions.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ and GHL conceived and designed the study, and critically revised the manuscript. GHL, LZ and HQS performed the experiments, analyzed the data and drafted the manuscript. GHZ, JZC and QZ helped in study design, study implementation and manuscript revision. All authors read and approved the final manuscript.
Contributor Information
Guo-Hua Liu, Email: liuguohua5202008@163.com.
Lei Zhao, Email: xinyao-2007@163.com.
Hui-Qun Song, Email: songhuiqun@caas.cn.
Guang-Hui Zhao, Email: zgh083@163.com.
Jin-Zhong Cai, Email: caijinz@vip.sina.com.
Quan Zhao, Email: zhaoquan0825@163.com.
Xing-Quan Zhu, Email: xingquanzhu1@hotmail.com.
Acknowledgements
This work was supported in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037) and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006).
References
- Hotez PJ, Fenwick A, Savioli L, Molyneux DH. Rescuing the bottom billion through control of neglected tropical diseases. Lancet. 2009;373:1570–1575. doi: 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- Broughan JM, Wall R. Faecal soiling and gastrointestinal helminth infection in lambs. Int J Parasitol. 2007;37:1255–1268. doi: 10.1016/j.ijpara.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Sweeny JP, Robertson ID, Ryan UM, Jacobson C, Woodgate RG. Impacts of naturally acquired protozoa and strongylid nematode infections on growth and faecal attributes in lambs. Vet Parasitol. 2012;184:298–308. doi: 10.1016/j.vetpar.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Sweeny JP, Ryan UM, Robertson ID, Jacobsen C. Molecular identification of naturally acquired strongylid infections in lambs-An investigation into how lamb age influences diagnostic sensitivity. Vet Parasitol. 2012;187:227–236. doi: 10.1016/j.vetpar.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Sweeny JP, Ryan UM, Robertson ID, Niemeyer D, Hunt PW. Development of a modified molecular diagnostic procedure for the identification and quantification of naturally occurring strongylid larvae on pastures. Vet Parasitol. 2012;190:467–481. doi: 10.1016/j.vetpar.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Roeber F, Jex AR, Campbell AJ, Nielsen R, Anderson GA, Stanley KK, Gasser RB. Establishment of a robotic, high-throughput platform for the specific diagnosis of gastrointestinal nematode infections in sheep. Int J Parasitol. 2012;42:1151–1158. doi: 10.1016/j.ijpara.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Tariq KA, Chishti MZ, Ahmad F, Shawl AS. Epidemiology of gastrointestinal nematodes of sheep managed under traditional husbandry system in Kashmir valley. Vet Parasitol. 2008;158:138–143. doi: 10.1016/j.vetpar.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Xiong DS, Kong FY. Chabertia erschowi.n.sp.-A new parasitic nematode of sheep and goat in China. J Beijing Agr Univ. 1956;2:115–122. (in Chinese) [Google Scholar]
- Zhang JL. Chabertia shaanxiensis sp. nov (Nematode: Strongylidae) from the cattle. J Anim Vet Adv. 1985;16:137–141. [Google Scholar]
- Zhang LP, Li JB, Liu SG, An RY. Comparison of the head structure of two Chabertia species by scanning electron microscope. J Chin Electron Micro Soc. 1998;17:345–346. (in Chinese) [Google Scholar]
- Khalafalla RE, Elseify MA, Elbahy NM. Seasonal prevalence of gastrointestinal nematode parasites of sheep in Northern region of Nile Delta, Egypt. Parasitol Res. 2011;108:337–340. doi: 10.1007/s00436-010-2066-9. [DOI] [PubMed] [Google Scholar]
- Sissay MM, Uggla A, Waller PJ. Prevalence and seasonal incidence of nematode parasites and fluke infections of sheep and goats in eastern Ethiopia. Trop Anim Health Prod. 2007;39:521–531. doi: 10.1007/s11250-007-9035-z. [DOI] [PubMed] [Google Scholar]
- Blouin MS. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527–531. doi: 10.1016/S0020-7519(01)00357-5. [DOI] [PubMed] [Google Scholar]
- Liu GH, Chen F, Chen YZ, Song HQ, Lin RQ, Zhou DH, Zhu XQ. Complete mitochondrial genome sequence data provides genetic evidence that the brown dog tick Rhipicephalus sanguineus (Acari: Ixodidae) represents a species complex. Int J Biol Sci. 2013;9:361–369. doi: 10.7150/ijbs.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbar A, Mohandas N, Jex AR, Gasser RB. The mitochondrial genome of Protostrongylus rufescens – implications for population and systematic studies. Parasit Vectors. 2013;6:263. doi: 10.1186/1756-3305-6-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Li C, Li JY, Zhou DH, Xiong RC, Lin RQ, Zou FC, Zhu XQ. Characterization of the complete mitochondrial genome sequence of Spirometra erinaceieuropaei (Cestoda: Diphyllobothriidae) from China. Int J Biol Sci. 2012;8:640–649. doi: 10.7150/ijbs.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RQ, Liu GH, Hu M, Song HQ, Wu XY, Li MW, Zhang Y, Zou FC, Zhu XQ. Oesophagostomum dentatum and Oesophagostomum quadrispinulatum: characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp Parasitol. 2012;131:1–7. doi: 10.1016/j.exppara.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012;6:e1539. doi: 10.1371/journal.pntd.0001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson LA, Chilton NB, Gasser RB. Differentiation of Haemonchus placei from H. contortus (Nematoda: Trichostrongylidae) by the ribosomal DNA second internal transcribed spacer. Int J Parasitol. 1995;25:483–488. doi: 10.1016/0020-7519(94)00156-I. [DOI] [PubMed] [Google Scholar]
- Cutillas C, Callejón R, de Rojas M, Tewes B, Ubeda JM, Ariza C, Guevara DC. Trichuris suis and Trichuris trichiura are different nematode species. Acta Trop. 2009;111:299–307. doi: 10.1016/j.actatropica.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Liu GH, Zhou W, Nisbet AJ, Xu MJ, Zhou DH, Zhao GH, Wang SK, Song HQ, Lin RQ, Zhu XQ. Characterization of Trichuris trichiura from humans and T. suis from pigs in China using internal transcribed spacers of nuclear ribosomal DNA. J Helminthol. 2014;88:64–68. doi: 10.1017/S0022149X12000740. [DOI] [PubMed] [Google Scholar]
- Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2:2339–2344. doi: 10.1038/nprot.2007.358. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, Cafarchia C, Beveridge I, Zhu X. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1:3121–3128. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- Bowles J, Blair D, Mcmanus DP. Genetic variants within the genus Echinococcus identified by mitochondrial DNA sequencing. Mol Biochem Parasitol. 1992;54:165–174. doi: 10.1016/0166-6851(92)90109-W. [DOI] [PubMed] [Google Scholar]
- Zhu X, Chilton NB, Jacobs DE, Boes J, Gasser RB. Characterisation of Ascaris from human and pig hosts by nuclear ribosomal DNA sequences. Int J Parasitol. 1999;29:469–478. doi: 10.1016/S0020-7519(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Jex AR, Hall RS, Littlewood DT, Gasser RB. An integrated pipeline for next generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522–533. doi: 10.1093/nar/gkp883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE. A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea) Int J Parasitol. 2002;32:145–158. doi: 10.1016/S0020-7519(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Lv S, Zhang Y, Zhang L, Liu Q, Liu HX, Hu L, Wei FR, Graeff-Teixeira C, Zhou XN, Utzinger J. The complete mitochondrial genome of the rodent intra-arterial nematodes Angiostrongylus cantonensis and Angiostrongylus costaricensis. Parasitol Res. 2012;111:115–123. doi: 10.1007/s00436-011-2807-4. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Jabbar A, Mohandas N, Schnyder M, Deplazes P, Littlewood DT, Jex AR. Mitochondrial genome of Angiostronglus vasorum: comparison with congeners and implications for studying the population genetics and epidemiology of this parasite. Infect Genet Evol. 2012;12:1884–1891. doi: 10.1016/j.meegid.2012.07.022. [DOI] [PubMed] [Google Scholar]
- Jabbar A, Jex AR, Mohandas N, Hall RS, Littlewood DT, Gasser RB. The mitochondrial genome of Aelurostrongylus abstrusus-diagnostic, epidemiological and systematic implications. Gene. 2013;516:294–300. doi: 10.1016/j.gene.2012.10.072. [DOI] [PubMed] [Google Scholar]
- Zhao GH, Hu B, Cheng WY, Jia YQ, Li HM, Yu SK, Liu GH. The complete mitochondrial genomes of Oesophagostomum asperum and Oesophagostomum columbianum in small ruminants. Infect Genet Evol. 2013;19:205–211. doi: 10.1016/j.meegid.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Liu GH, Wang Y, Xu MJ, Zhou DH, Ye YG, Li JY, Song HQ, Lin RQ, Zhu XQ. Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae) Infect Genet Evol. 2012;12:1635–1641. doi: 10.1016/j.meegid.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Page RD. TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Jex AR, Waeschenbach A, Hu M, van Wyk JA, Beveridge I, Littlewood DT, Gasser RB. The mitochondrial genomes of Ancylostoma caninum and Bunostomum phlebotomum--two hookworms of animal health and zoonotic importance. BMC Genomics. 2009;10:79. doi: 10.1186/1471-2164-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MW, Lin RQ, Song HQ, Wu XY, Zhu XQ. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics. 2008;9:224. doi: 10.1186/1471-2164-9-224. [DOI] [PMC free article] [PubMed] [Google Scholar]