

Abstract
Cisplatin, carboplatin, and oxaliplatin are widely used anticancer drugs. Their efficacy is strongly reduced by development of cell resistance, a phenomenon not entirely understood, with contribution of drug detoxification, defective accumulation, and efflux from the cell. Down-regulation of CTR1, responsible for Cu uptake by the cell, and up-regulation of the Cu-ATPases, ATP7A and ATP7B, which accept Cu from the cytosolic chaperone Atox1 and transfer the metal ion into the secretory pathway where it is incorporated into cuproenzymes, have been associated to augmented drug resistance. To gain information on translocation of Pt drugs by human Cu-ATPases, we performed electrical measurements on COS-1 cell microsomal fraction, enriched with recombinant ATP7A, ATP7B, and selected mutants, adsorbed on a solid supported membrane (SSM). The experimental results demonstrate that Pt drugs activate Cu-ATPases and undergo ATP-dependent translocation with a mechanism identical to that of Cu. We then used NMR spectroscopy and ESI-MS to determine the binding mode of these drugs to the first N-terminal metal binding domain of ATP7A (Mnk1).
Keywords: platinum drugs, copper ATPases, NMR spectroscopy, charge measurements
ATP7A and ATP7B share 54% sequence identity and are associated, respectively, with Menkes and Wilson’s diseases. Both ATPases are 160–170 kDa proteins with eight transmembrane (TM) domains and six cytosolic N-terminal metal binding domains (NMBD). N- and P-domains are involved in ATP binding and hydrolysis. The actuator (A)-domain interacts with the ATP-binding domain and is required for conformational changes during ATP hydrolysis (Figure 1).[1] For many P-type ATPases the ion transport requires passing through the membrane portion of the protein.[2] It has been demonstrated that, similarly to Cu, Pt binds to the CXXC motifs of the NMBDs of ATP7B and that such interaction mediates cancer cell resistance to cisplatin.[3,4]
Figure 1. Structures of platinum complexes (top), topology of P-type Cu-ATPases ATP7A/ATP7B (middle), and schematic diagram of a microsome containing ATP7A/ATP7B adsorbed on a SSM (bottom).
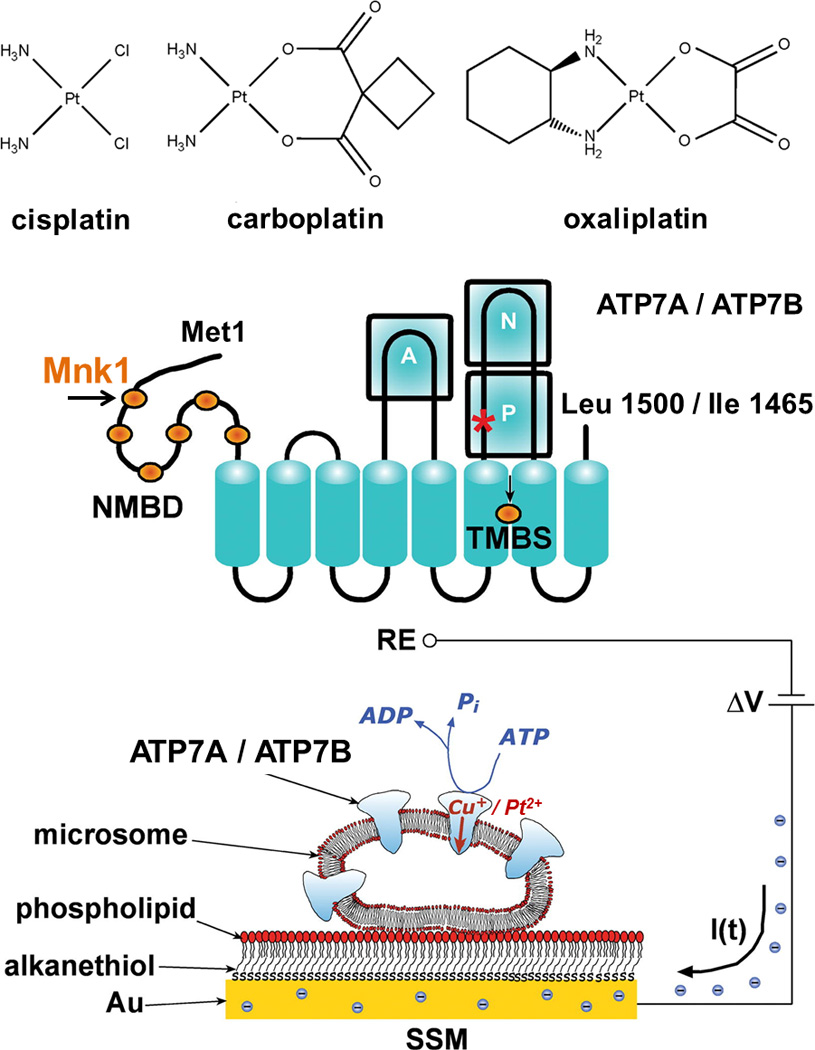
When charge displacement occurs, a compensating current I(t) flows along the external circuit (the blue spheres are electrons) to keep constant the potential difference (ΔV) applied across the whole system. RE is the reference electrode.
Recently, the SSM technique was employed to investigate charge movements (Cu+) in recombinant human Cu-ATPases.[5–7] Addition of ATP to microsomes enriched with recombinant ATP7A/B and adsorbed on a SSM is followed by an electrogenic event recorded as a current transient (see Figure 1 and Supporting Methods). The current transient is due to flow of electrons along the external circuit toward the electrode surface, and is required to compensate for the potential difference across the vesicular membrane produced by displacement of positive charges (attributed to vectorial translocation of bound Cu+) into the microsome upon ATP utilization. The current recorded by the SSM method is a measure of the rate of change of the transmembrane potential, and is not sensitive to stationary currents. Therefore, only electrogenic steps within the first catalytic cycle are measured, while steady-state events after the first cycle are not detected.
In the present work, we first examined the effect of Zeise’s salt (K[PtCl3(C2H4)]), characterized by very labile chloride ligands due to the strong labilizing effect of ethylene, and then extended the investigation to the aquated species of cisplatin and oxaliplatin (cis-[Pt(NH3)2(H2O)(SO4)] and cis-[Pt(R,R-DACH)(H2O)(SO4)], DACH = 1,2-diaminocyclohexane) pre-incubated for 30 min in 300 mM KCl. In the presence of chloride cis-[Pt(NH3)2(H2O)xCl2-x] and cis-[Pt(R,R-DACH)(H2O)xCl2-x] species are formed, however, for the time of the experiment (<90 min),the monoaqua and diaqua species are still present in relevant amount (~50% of the total) and thus they may represent the active form of the drugs in vivo.
Charge translocation in the presence of copper and/or platinum species
The SSM setup was initially tested for Cu translocation. Microsomes containing ATP7A/B adsorbed on the SSM were incubated in a buffer solution containing 5 µM CuCl2 for ~30 min (in the presence of excess DTT Cu2+ is reduced to Cu+). Then the solution was exchanged with an identical buffer solution containing 5 µM CuCl2 and, in addition, 100 µM ATP and the current transient measured (Figure 2, black lines and columns). After this preliminary experiment the system was flushed with a buffer solution containing 5 µM Pt complex and then incubated for ½ hour. Then the solution was exchanged with an identical buffer solution containing 5 µM Pt complex and, in addition, 100 µM ATP and the current transient detected (Figure 2, red lines and columns). A similar overall charge was translocated in all cases (Figure 2, black and red columns), but the current transient detected in the case of Zeise’s salt (Figures 2A and 2B) exhibits a slower decay than those observed in the cases of CuCl2, cisplatin (Figure 2C), and oxaliplatin (Figure 2D). Therefore, current measurements point to a comparable movement of Cu or Pt-related charge through ATP7A/B upon ATP utilization. Importantly, no current transient was detected if the 100 µM ATP jump was performed with buffer solution not containing CuCl2 or Pt complex.
Figure 2. Current measurements on ATP7A (A, C, and D) and ATP7B (B) in the presence of Zeise’s salt (A and B), aquated cisplatin (C) and oxaliplatin (D).

Representative current transients induced by 100 µM ATP concentration jumps in the presence of 5 µM CuCl2 (black lines), 5 µM Pt complex (red lines), 5 µM CuCl2 and 5 µM Pt complex (green lines), and 5 µM CuCl2, 5 µM Pt complex and 1 mM BCS (blue lines). The insets show the charges related to ATP-induced current transients obtained under the various experimental conditions (the same color code is used for lines and columns). Error bars: SD of six measurements on the same SSM.
To check if there was interference between the two metallic species, we carried out an ATP concentration jump in the presence of both CuCl2 and Pt complex at 5 µM concentration each: no current transient was detected (Figure 2, green lines and columns). A cross check experiment was performed by addition of 1 mM bathocuproine disulfonate (BCS) to the buffer solution containing CuCl2 and Pt complex. Following an ATP jump, a current transient was recovered (to ~60–70% of the charge measured in the presence of 5 µM Pt complex alone) (Figure 2, blue lines and columns). Thus, chelating Cu+ with BCS allows Pt-related charge translocation by ATP7A/B to be partially restored.
To further investigate Pt-related charge movements upon ATP utilization, analogous experiments were performed using specific mutants of ATP7A and ATP7B.
First, the effect of Asp1044 mutation in ATP7A was analyzed. Asp1044 is the residue receiving the ATP γ-phosphate at the catalytic site in the cytosolic P-domain of ATP7A, to form the aspartyl phosphorylated intermediate. Its mutation produces catalytic inactivation of the ATPase. We found that the D1044A mutant yields no current transient upon ATP jump either in the presence of 5 µM CuCl2 or in the presence of 5 µM Pt drug (Figure S1A). This result indicates that the ATP-induced movement of Cu or Pt-related charge through the ATPase is directly correlated to the formation of the acyl-phosphate intermediate by ATP consumption.
We then considered the effects of site-directed mutations C575A/C578A of the CXXC motif in the 6th NMBD of ATP7B, which was shown to be critical for Cu-related charge displacement.[5] The C575A/C578A double mutant was found to be catalytically inactive,[5] therefore no current transient was detected following an ATP jump in the presence of 5 µM Pt drug, as well as in the presence of 5 µM CuCl2 (Figure S1B). Thus, it is concluded that C575A/C578A mutation in the 6th NMBD interferes with both Cu and Pt binding, thereby preventing ATP-dependent charge movements.
Platinum binding to apoMnk1
Having found that the 6th NMBD of ATP7B was essential for charge movements, we focused our investigation on the interaction of one of these domains with Pt substrates. All NMBDs share similar characteristics and we chose the first domain of ATP7A, Mnk1.
Fully 15N-labeled and selectively 13C-cysteine-labeled Mnk1 (15N,13C-Cys Mnk1 hereafter) was bacterially expressed with a recognition site for the restriction protease Factor Xa and a (His)6 tag at the C-terminal end, crucial for its purification by metal affinity chromatography. After cleavage of the tag, four amino acids (IEGR) of the restriction site were left at the C-terminus of the protein. The N-terminal methionine was partially processed in E. coli, yielding a 76-amino acid protein (numbering the first codon (Met) as residue 1). Therefore, the ESI-MS spectrum of apoMnk1 is formed by two series of multiply charged states corresponding to the protein with and without the first Met residue (Figure S2).
When cisplatin is added to apoMnk1 (100 µM) in equimolar amount under anaerobic conditions, ESI-MS spectra of the reaction mixture show a new set of signals, whose intensity increases over time, indicating the formation of adducts between Mnk1 and the Pt complex. The mass difference between corresponding charged states indicates binding of a {Pt(NH3)2Cl}+ and a {Pt(NH3)2}2+ fragment to the protein. In the early stage of the reaction the monodentate adduct {Pt(NH3)2Cl}+-Mnk1 is by far the major species. After 24 h incubation, the signals corresponding to the chelate adduct {Pt(NH3)2}2+-Mnk1 become the most intense (Figures 3A and 3B).
Figure 3. ESI-MS spectra of 15N,13C-Cys Mnk1 treated with 1 mol equiv of cisplatin (A and B) or oxaliplatin (C and D) after 3 (A and C) and 24 h (B and D) of incubation.
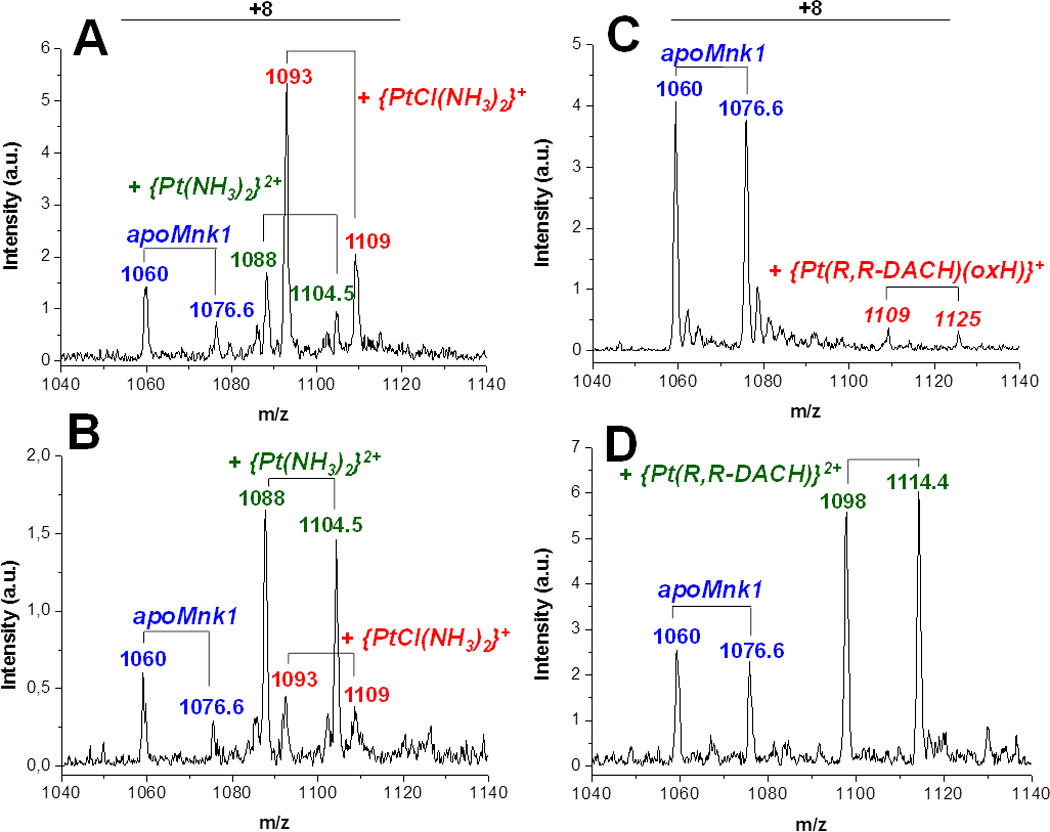
Peaks corresponding to +8 charged state are shown. Each species comprises two pairs of signals corresponding to Mnk1 with and without Met1 as first residue.
A similar behavior was observed in the reaction of apoMnk1 with oxaliplatin, however the reaction appears to be slower: after 3 h incubation only weak signals relative to the monodentate adduct {Pt(R,R-DACH)(oxalateH)}+-Mnk1 are present together with those of apoMnk1; after 24 h, the apoMnk1 signals decrease while intense signals relative to the chelate adduct {Pt(R,R-DACH)}2+-Mnk1 appear (Figures 3C and 3D).
The starting 1H,15N-HSQC spectrum of 15N-labeled cisplatin incubated with apoMnk1 shows signals of the dichlorido species (cis-[PtCl2(15NH3)2], δ 15N = −67 ppm) and monoaqua species (cis-[PtCl(15NH3)2(H2O)]+, δ 15N = −66 and −83 ppm for 15NH3 trans to Cl and to H2O, respectively).[8] After 3 h incubation, the intensity of these cross-peaks decreases simultaneously with the appearance of two cross-peaks relative to the intermediate species characterized by one 15NH3 trans to Cl and one 15NH3 trans to S (δ 15N ~ −66 and −38 ppm, respectively). After 24 h incubation, only two cross-peaks are present in the spectrum that are characteristic of 15NH3 trans to S atoms (δ 15N ~ −43 and −36 ppm, respectively), indicating complete loss of chloride ions from the Pt complex (Figures 4A–C).
Figure 4. 2D 1H,15N-HSQC spectra of cis-PtCl2(15NH3)2 incubated at pH 7.0 with 1 mol equiv of apoMnk1 (A–C) and Cu+-Mnk1 (D–F) at 0 (A and D, blue contours), 3 (B and E, red contours) and 24 h (C and F, green contours) after mixing.
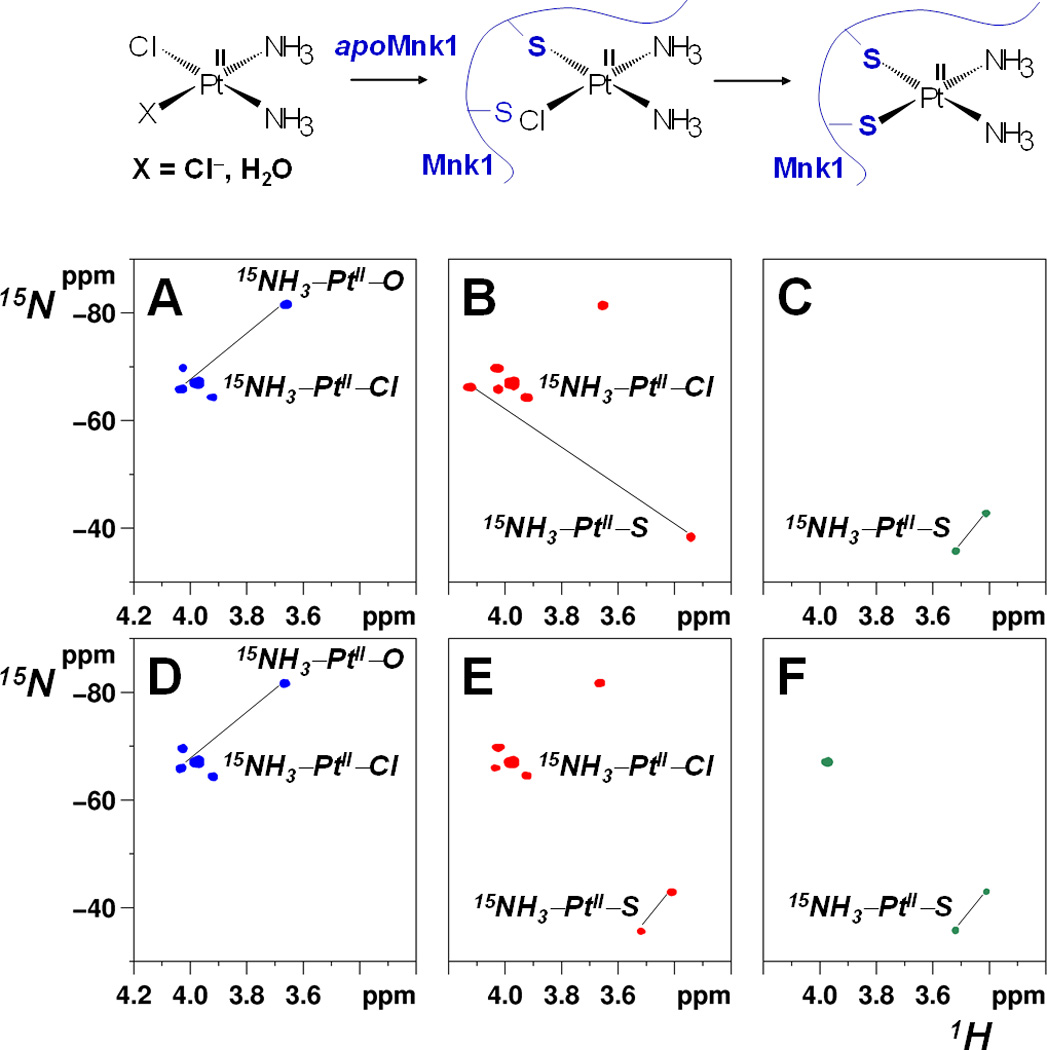
Cross-peaks are assigned to 15NH3 trans to O, Cl, or S donor atoms, respectively. Cross-peaks belonging to the same species are connected by a straight line. The reaction scheme of cisplatin with apoMnk1 is shown at the top.
To prove that cysteine residues of the Cu+-binding motif, CXXC, are involved in Pt coordination, 1H,13C-HSQC spectra were recorded on 15N,13C-Cys Mnk1 incubated with one equivalent of cisplatin (Figures S3A–C). After 3h incubation, original cross-peaks of apoMnk1 nearly disappear with concomitant appearance of new cross-peaks for α-CH and β-CH2 groups of Cys15 and Cys18, that correspond to monodentate adduct with the Pt drug (S3B). A different set of Cys signals is observed after 24 h corresponding to formation of the chelate adduct (Figure S3C). The 1H,13C-HSQC spectra also display natural-abundance ε-CH2 cross-peaks of lysines, which all resonate in the same frequency region. These latter signals do not experience any relevant change upon reaction with cisplatin (Figure S4).
To explore the exchange behavior of the chelate adduct, after 24 h incubation of 15N,13C-Cys Mnk1 with one equivalent of unlabeled cisplatin, a second equivalent of 15N-labeled cisplatin was added. After additional 24 h (and up to several days of incubation) there is no evidence of 15NH3 signals trans to S atoms and the 1H,13C-HSQC spectrum of the protein is not perturbed further, indicating that no exchange occurs between the second equivalent of cisplatin and Pt already chelated by the two Cys residues of Mnk1 in the chelate adduct ({Pt(NH3)2}2+-Mnk1).
Since in the cytosol there is a relevant concentration of glutathione (GSH, ~5 mM), we wanted to explore if the reaction between cisplatin and Mnk1 would take place also in the presence of excess GSH (10-fold). Results showed that Mnk1 competes successfully with GSH for complexation to platinum (Figure S5).
Platinum binding to Cu+-Mnk1
One equivalent of 15N-labeled cisplatin was added to preformed Cu+-Mnk1 adduct. Both ESI-MS (Figure S6) and NMR measurements (Figures 4D–F) indicate that the Pt drug can bind to Cu+-Mnk1 as found for the apoprotein, but in the case of the holoprotein there is no evidence of monodentate adduct formation. Instead, the chelate adduct (both 15NH3 trans to S atoms) is formed already after 3 h incubation, thus indicating that Cu+ can catalyze Pt drug chelation by two Cys residues with complete loss of chlorido ligands. Notably, the chemical shifts of the ammine ligands of cisplatin are the same as those observed in the former experiment with apoMnk1 after 24 h incubation. Also the 1H,13C-HSQC spectrum of 15N,13C-Cys Cu+-Mnk1, after 3 h incubation with cisplatin, shows a great decrease in intensity of the signals of the holoprotein while signals of {Pt(NH3)2}2+-Mnk1 appear (compare Figure S3E with Figures S3D and S3C), clearly indicating that cisplatin can displace Cu+ bound to CXXC motifs, thus potentially interfering with cellular Cu homeostasis. Furthermore, after 24 h incubation, a remarkable reduction in intensity of 15NH3 and 15N,13C-Cys Mnk1 signals (Figures 4F and S3F) is observed, accompanied by coalescence of amide signals in the middle of the 1H,15N-HSQC spectrum, a typical sign of protein unfolding (Figure S7). Notably, the {Pt(NH3)2}2+-Mnk1 chelate adduct alone unfolds at a much slower rate (Figures S3C and S7C), thus indicating that the Cu+ is displaced by cisplatin to promote protein unfolding.
Taken together, our measurements clearly prove ATP-induced Pt-related charge translocation in ATP7A/B. The charge translocation occurs with rates analogous to that of Cu in the case of aquated cisplatin or oxaliplatin. Numerical integration of the ATP-induced current transients in the presence of CuCl2 or Pt complex yields similar values for the translocated charge (Figure 2, black and red columns), thus indicating that an equal amount of positive charge is displaced by ATP7A/B upon ATP utilization within a single catalytic cycle. Analysis of bacterial Cu-ATPase, i.e. CopA from A. fulgidus[9] and L. pneumophila[10], suggested that the transport stoichiometry could probably be two Cu+ ions per hydrolyzed ATP. Thus, assuming the same stoichiometry (2Cu+ per ATP) in the case of human ATP7A/B, our measurements indicate that one Pt species bearing two positive charges, or two Pt species bearing a single positive charge, is(are) translocated following utilization of one ATP molecule. Based on the stability of the ammine groups which are not trans-labilized by cysteines, we propose that the complex that translocates bears the ammine groups.
The lack of charge translocation following mutation of the catalytic Asp1044 of ATP7A or the CXXC motif in the 6th NMBD of ATP7B clearly demonstrates that the Pt-related charge movement is dependent on formation of a phosphorylated intermediate as well as conformational adjustments analogous to those required for Cu-related charge translocation.
It is of interest that the presence of both Cu and Pt in the buffer solution abolishes charge movements, indicating that simultaneous occupancy of activating sites by the two metal ions prevents catalytic activation, as previously observed.[11] Interestingly, the use of a Cu chelator (BCS) partially restores charge movement, demonstrating direct involvement of Pt drug.
Copper binding to CXXC motifs of NMBDs triggers protein conformational changes that weaken the interactions between the N-terminus and the ATP-binding domain and favor catalytic phosphorylation.[12] Based on the NMR experiments performed with 15N-labeled cisplatin, we propose the mechanism depicted on the top of Figure 4 for reaction of cisplatin with one of such NMBDs. The drug initially forms a monodentate adduct ({Pt(NH3)2Cl}+-Mnk1) with the protein. This is an early adduct which may be relevant for Pt-related charge translocation. On a longer timescale, Pt releases the second chloride while keeping the two ammines, thus forming the chelate adduct ({Pt(NH3)2}2+-Mnk1). Once the chelate adduct is formed no Pt exchange can occur at the CXXC motif at a detectable rate.
At variance with what occurs with MXXM motifs of CTR1,[13] the S atoms of cysteines are not able to labilize the ammine ligands of the Pt drug that are trans to them. This surprising difference was already noticed in a previous paper from our group reporting the interaction of Pt drugs with Atox1 which is structurally similar to Mnk1.[14] It is possible that the protein exerts a protective effect, as witnessed by the strong shielding in the 1H dimension of the cross peaks of the cisplatin-Mnk1 adducts.
Cisplatin is able to displace readily Cu+ bound to the CXXC motif and coordinates directly in a chelate mode, however the displaced Cu ion destabilizes the Pt-Mnk1 structure. An analogous unfolding behavior has previously been observed for the structurally similar Atox1.[15]
In conclusion, our studies show that cisplatin and oxaliplatin can activate the catalytic cycle of ATP7A/B, leading to charge movement. Cisplatin can react with the ATPase N-terminal extension, initially forming quite long-lasting monodentate adducts, that then evolve to stable and unreactive chelate adducts. It is likely that translocation by ATPases (at short incubation time) and sequestration in the N-terminal extension (at long incubation time) may contribute to cisplatin resistance in vivo, with predominance of either phenomenon depending on dosage and time of administration. Resistance would be certainly higher under conditions of ATPase protein overexpression.
Supplementary Material
Footnotes
We thank the Universities of Bari and Florence, the Ente Cassa di Risparmio di Firenze, the Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici (CIRCMSB), the Italian Ministero dell’Università e della Ricerca (PON 01078, PRIN 2010M2JARJ, PON01_00937, PRIN 20083YM37E), and the European Commission (COST Actions CM0902 and CM1105) for support. Expression of recombinant ATP7A, ATP7B and respective mutants was supported by USA National Institutes of Health Grant RO301-69830 from the NHLBI to GI.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Francesco Tadini-Buoninsegni, Department of Chemistry “Ugo Schiff”, University of Florence, via della Lastruccia 3, 50019 Sesto Fiorentino (Italy).
Gianluca Bartolommei, Department of Chemistry “Ugo Schiff”, University of Florence, via della Lastruccia 3, 50019 Sesto Fiorentino (Italy).
Maria Rosa Moncelli, Department of Chemistry “Ugo Schiff”, University of Florence, via della Lastruccia 3, 50019 Sesto Fiorentino (Italy).
Giuseppe Inesi, California Pacific Medical Center Research Institute, San Francisco, CA 94107, USA.
Angela Galliani, Department of Chemistry, University of Bari “Aldo Moro”, via E. Orabona 4, 70125 Bari (Italy).
Marilù Sinisi, Department of Chemistry, University of Bari “Aldo Moro”, via E. Orabona 4, 70125 Bari (Italy).
Maurizio Losacco, Department of Chemistry, University of Bari “Aldo Moro”, via E. Orabona 4, 70125 Bari (Italy).
Giovanni Natile, Department of Chemistry, University of Bari “Aldo Moro”, via E. Orabona 4, 70125 Bari (Italy).
Fabio Arnesano, Department of Chemistry, University of Bari “Aldo Moro”, via E. Orabona 4, 70125 Bari (Italy).
References
- 1.Gourdon P, Sitsel O, Lykkegaard KJ, Birk ML, Nissen P. Biol. Chem. 2012;393:205–216. doi: 10.1515/hsz-2011-0249. [DOI] [PubMed] [Google Scholar]
- 2.Bublitz M, Morth JP, Nissen P. J. Cell Sci. 2011;124:2515–2519. doi: 10.1242/jcs.088716. [DOI] [PubMed] [Google Scholar]
- 3.Dolgova NV, Olson D, Lutsenko S, Dmitriev OY. Biochem. J. 2009;419:51–56. doi: 10.1042/BJ20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Safaei R, Adams PL, Maktabi MH, Mathews RA, Howell SB. J. Inorg. Biochem. 2012;110:8–17. doi: 10.1016/j.jinorgbio.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, Pilankatta R, Lewis D, Inesi G. FEBS Lett. 2010;584:4619–4622. doi: 10.1016/j.febslet.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis D, Pilankatta R, Inesi G, Bartolommei G, Moncelli MR, Tadini-Buoninsegni F. J. Biol. Chem. 2012;287:32717–32727. doi: 10.1074/jbc.M112.373472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, Fendler K. Arch. Biochem. Biophys. 2008;476:75–86. doi: 10.1016/j.abb.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 8.Berners-Price SJ, Ronconi L, Sadler PJ. Prog. Nucl. Magn. Reson. Spectrosc. 2006;49:65–98. [Google Scholar]
- 9.Gonzalez-Guerrero M, Eren E, Rawat S, Stemmler TL, Arguello JM. J. Biol. Chem. 2008;283:29753–29759. doi: 10.1074/jbc.M803248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP, Nissen P. Nature. 2011;475:59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 11.Safaei R, Otani S, Larson BJ, Rasmussen ML, Howell SB. Mol. Pharmacol. 2008;73:461–468. doi: 10.1124/mol.107.040980. [DOI] [PubMed] [Google Scholar]
- 12.Huster D, Lutsenko S. J. Biol. Chem. 2003;2789:32212–32218. doi: 10.1074/jbc.M305408200. [DOI] [PubMed] [Google Scholar]
- 13.Arnesano F, Scintilla S, Natile G. Angew. Chem. 2007;119:9220–9222. doi: 10.1002/anie.200703271. Angew. Chem. Int. Ed. Engl.2007, 46, 9062–9064. [DOI] [PubMed] [Google Scholar]
- 14.Arnesano F, Banci L, Bertini I, Felli IC, Losacco M, Natile G. J. Am. Chem. Soc. 2011;133:18361–18369. doi: 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- 15.Palm ME, Weise CF, Lundin C, Wingsle G, Nygren Y, Bjorn E, Naredi P, Wolf-Watz M, Wittung-Stafshede P. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6951–6956. doi: 10.1073/pnas.1012899108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.