

Abstract
Transcriptional dysregulation has long been recognized as central to the pathogenesis of Huntington's disease (HD). MicroRNAs (miRNAs) represent a major system of post-transcriptional regulation, by either preventing translational initiation or by targeting transcripts for storage or for degradation. Using next-generation miRNA sequencing in prefrontal cortex (Brodmann Area 9) of twelve HD and nine controls, we identified five miRNAs (miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p and miR-1247-5p) up-regulated in HD at genome-wide significance (FDR q-value<0.05). Three of these, miR-196a-5p, miR-196b-5p and miR-615-3p, were expressed at near zero levels in control brains. Expression was verified for all five miRNAs using reverse transcription quantitative PCR and all but miR-1247-5p were replicated in an independent sample (8HD/8C). Ectopic miR-10b-5p expression in PC12 HTT-Q73 cells increased survival by MTT assay and cell viability staining suggesting increased expression may be a protective response. All of the miRNAs but miR-1247-5p are located in intergenic regions of Hox clusters. Total mRNA sequencing in the same samples identified fifteen of 55 genes within the Hox cluster gene regions as differentially expressed in HD, and the Hox genes immediately adjacent to the four Hox cluster miRNAs as up-regulated. Pathway analysis of mRNA targets of these miRNAs implicated functions for neuronal differentiation, neurite outgrowth, cell death and survival. In regression models among the HD brains, huntingtin CAG repeat size, onset age and age at death were independently found to be inversely related to miR-10b-5p levels. CAG repeat size and onset age were independently inversely related to miR-196a-5p, onset age was inversely related to miR-196b-5p and age at death was inversely related to miR-615-3p expression. These results suggest these Hox-related miRNAs may be involved in neuroprotective response in HD. Recently, miRNAs have shown promise as biomarkers for human diseases and given their relationship to disease expression, these miRNAs are biomarker candidates in HD.
Author Summary
Huntington's disease (HD) is an inherited fatal neurological disorder that commonly affects people in midlife. Past studies have implicated abnormal patterns of gene expression as a candidate for causing the death of the brain cells affected in HD. MicroRNAs (miRNAs) are small molecules that regulate and target transcripts for either storage or destruction. We measured the levels of miRNAs, as well as the levels of gene expression (mRNAs) in twelve HD and nine control brain samples. We found five miRNAs that had greatly increased expression in the HD brains, including three that were not expressed in the normal samples. Four of these were related to important characteristics of the disease expression, including the age at disease onset, and the age at death of the individual. The genes that these miRNAs target for regulation were also altered in their expression with most being increased, suggesting they may have been targeted for storage. One of the miRNAs, miR-196a-5p was previously implicated in enhancing the survival of brain cells in HD. When we overexpressed miR-10b-5p in an HD cell model, the cells survived longer than untreated cells, suggesting these miRNAs may promote neuron survival and may hold new clues for treatments in HD.
Introduction
Huntington's disease (HD) (OMIM: 143100) is an inherited neurodegenerative disorder characterized by involuntary movement, dementia, and changes in personality. HD is transmitted as an autosomal dominant disorder, for which an expansion of a CAG trinucleotide repeat within the coding region of the huntingtin gene (HTT) is the disease causing mutation [1]. The CAG repeat codes for a polyglutamine domain in the Htt protein and results in neuronal cell death predominantly affecting the caudate nucleus and putamen although neuronal loss is widespread in the HD brain [2], [3]. While the biological processes leading to neurodegeneration in HD are poorly understood, transcriptional dysregulation has long been proposed as central to the pathogenesis of HD. Widespread alterations in gene expression have been reported [4] and several studies suggest that gene expression may be altered at one or more of the stages of RNA processing, translation, protein post-translational modification or trafficking [5], [6].
MicroRNAs (miRNAs) are small non-coding RNAs that function as translational regulators of mRNA expression. miRNAs may inhibit gene expression either by repressing translation, or by targeting mRNA for either storage or degradation [7]. Recently, dysregulation of miRNAs has been linked to neurological and neurodegenerative disorders [8] and several studies have explored the role of miRNAs in HD. Marti et al [9] performed miRNA-sequencing for two pooled HD samples and two pooled control samples and reported altered expression for a large number of miRNAs. Altered expression of miRNAs, quantified using microarray technology, has been reported in cellular models of HD [10]–[12] and in mouse models of HD [12]–[15] but a comprehensive study of miRNA and mRNA expression obtained through next-generation sequencing technology in human HD samples has not been performed.
In order to investigate (1) the presence of altered miRNA expression and (2) the potential role of miRNAs on the altered mRNA expression seen in HD, we performed both miRNA-sequencing and mRNA sequencing, using Illumina massively parallel sequencing in twelve HD and nine neurologically normal control brains. To our knowledge this is the first genome-wide quantification of miRNA expression comparing human HD and control brain, and the first to combine total miRNA expression with total mRNA expression obtained through massively parallel sequencing.
Results
Selection of prefrontal cortex and BA9
While the striatum is the region most heavily involved neuropathologically in HD [3], 80% to 90% of the neurons in that region will have degenerated by the time of death. These changes, together with the presence of reactive astrocytosis, alter the cellular composition of the striatum. In contrast, cortical involvement in HD is well defined [2], [16] and while it does not experience dramatic neuronal degeneration, cortical neurons are known to exhibit the effects of protein aggregation and nuclear inclusion bodies characteristic of the disease. Therefore, we selected the prefrontal cortex for these studies.
Five miRNAs are up-regulated in HD
After removing sample outliers using principal component analysis filtering, we identified five out of 1,417 detected mature miRNA species as differentially expressed between twelve HD and nine control prefrontal cortex samples using the R statistical package DESeq ( Tables 1 , 2 and 3 ; Figure 1 ). All five miRNAs were significantly up-regulated in HD. The largest effect between conditions was seen for miR-10b-5p, with a 28.41 fold increased expression in HD relative to control samples (mean control expression = 915.81; mean HD expression = 26,020.05, Figure 1). miR-1247-5p was expressed at moderate levels in both control (mean = 49.44) and HD brain (mean = 102.01). Three of the miRNAs, miR-196a-5p (mean control expression = 1.47; mean HD expression = 27.49), miR-196b-5p (mean control expression = 2.49; mean HD expression = 11.01) and miR-615-3p (mean control expression = 1.09, mean HD expression = 6.66), had near zero expression levels in all nine control samples.
Table 1. HD brain samples analyzed for mRNA-seq, miRNA-seq and RT-qPCR validation of miR-10b-5p.
| Sample ID | miRNA-seq | RT-qPCR | PMI (hr.) | RIN or RQN | Death age | Onset age | Duration (yr.) | CAG repeat size | Neuron Loss in Neocortical Gray Matter |
| HD-01 | Passed | Y | 37 | 7.1 | 55 | 44 | 11 | 45 | 1 |
| HD-02 | Passed | Y | 6 | 7.5 | 69 | 63 | 6 | 41 | 1 |
| HD-03 | Passed | Y | 21 | 7 | 71 | 52 | 19 | 43 | 1 |
| HD-05 | Passed | Y | 19 | 6.9 | 48 | 25 | 23 | 48 | 2 |
| HD-06 | Passed | Y | NA | 6.2 | 40 | 34 | 6 | 51 | 1 |
| HD-07 | Passed | Y | 8 | 8.5 | 72 | 55 | 17 | 41 | 1 |
| HD-08 | Passed | Y | 21 | 7.4 | 43 | NA | NA | 49 | 1 |
| HD-09 | Passed | Y | 4 | 7.8 | 68 | 45 | 23 | 42 | 1 |
| HD-10 | Passed | Y | 6 | 8.3 | 59 | 35 | 24 | 46 | 1 |
| HD-12 | Passed | Y | 13 | 6 | 68 | 52 | 16 | 42 | 0 |
| HD-13 | Passed | N | 25 | 6.1 | 57 | 40 | 17 | 49 | 1 |
| HD-14 | Passed | Y | 11 | 7.3 | 48 | 38 | 10 | 45 | 1 |
| Mean | - | - | 15.48 | 7.18 | 58.17 | 43.91 | 15.64 | 45.17 | 0.875 |
All of the HD samples passed mRNA-seq QC.
Scale of neuron loss: 0 = absent, 1 = mild, 2 = moderate.
Table 2. Control brain samples analyzed for mRNA-seq, miRNA-seq and RT-qPCR validation of miR-10b-5p.
| Sample ID | miRNA-seq | RT-qPCR | PMI (hr.) | RIN or RQN | Death age |
| C-14 | Passed | Y | 21 | 8 | 79 |
| C-21 | Passed | Y | 26 | 7.3 | 76 |
| C-29 | Passed | Y | 13 | 6.4 | 93 |
| C-31 | Passed | Y | 24 | 7.3 | 53 |
| C-32 | Passed | Y | 24 | 8.3 | 57 |
| C-33 | Passed | Y | 15 | 7.5 | 43 |
| C-35 | Failed PCA | N | 21 | 7.6 | 46 |
| C-36 | Passed | Y | 17 | 7.5 | 40 |
| C-37 | Failed PCA | N | 28 | 8.3 | 44 |
| C-38 | Passed | Y | 20 | 7.7 | 57 |
| C-39 | Passed | Y | 15 | 7.3 | 80 |
| Mean | - | - | 20.36 | 7.49 | 60.73 |
All the control samples passed mRNA-seq QC.
RIN = RNA Integrity Number, RQN = RNA Quality Number.
PMI = Postmortem Interval.
Table 3. Differentially expressed miRNAs from miRNA-seq.
| miRNA | Control expression | HD expression | Fold Change | p-value | q-value* |
| miR-196a-5p | 1.47 | 27.49 | 18.66 | 2.05E-10 | 2.91E-07 |
| miR-10b-5p | 915.81 | 26020.05 | 28.41 | 1.99E-08 | 1.41E-05 |
| miR-615-3p | 1.09 | 6.66 | 6.09 | 2.73E-05 | 1.29E-02 |
| miR-1247-5p | 49.44 | 102.01 | 2.06 | 7.67E-05 | 2.72E-02 |
| miR-196b-5p | 2.49 | 11.01 | 4.41 | 9.77E-05 | 2.77E-02 |
* FDR-adjusted q-value.
Figure 1. Differentially expressed miRNAs in Huntington's disease.

miR-10b-5p, miR-1247-5p, miR-196a-5p, miR-196b-5p, and miR-615-3p were identified as differentially expressed in Huntington's disease prefrontal cortex compared to non-neurological disease controls by Illumina miRNA-sequencing. Normalized expression values quantified from DESeq analysis are shown on the y-axis. miR-196a-5p, miR-196b-5p and miR-615-3p were essentially not expressed in control samples, while the mean HD expression was 27.49, 11.01 and 6.66 respectively. miR-1247-5p was expressed at moderate levels in both control (mean = 49.44) and HD brain (mean = 102.01). miR-10b-5p was expressed in control (mean = 915.81) and highly expressed in HD brain (mean = 26,020.05). For miRNA, *p<0.05 and ***p<0.001, as determined by DESeq, followed by the Benjamini-Hochberg multiple comparison correction. (HD = Huntington's disease).
Validation and replication of miRNA findings
miRNA expression differences were orthogonally validated using the Exiqon miRCURY LNA technology for reverse transcription quantitative PCR (RT-qPCR) in eleven of twelve sequenced HD samples and nine control samples originally studied for miRNA-seq. All five miRNAs were confirmed to be significantly up-regulated in HD (Table S1), consistent with our miRNA-sequencing findings.
To replicate our findings in an independent sample set, we performed RT-qPCR in an additional eight control and eight HD prefrontal cortical samples (Tables S2 and S3). Four out of five miRNA (miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p) were confirmed as significantly increased in expression in HD (Table S4).
Similar proportion of neurons in HD and control cortical brain homogenate samples
HD is characterized by progressive cortical atrophy, with recognizable neuropathologic abnormalities in the neocortical gray matter [2], [16]–[20] ( Table 1 ). To address whether miRNA expression changes in HD may be due to altered ratios in brain cell-type abundance, such as a change in the ratio of neurons to glial cells, we compared the number of neuronal and non-neuronal nuclei across conditions. Suspensions of cell nuclei of prefrontal cortex from 28 HD cases and 19 controls were immunocytochemically labeled with anti-NeuN, a neuron-specific nuclear antigen, followed by flow cytometric analysis. The mean and range of NeuN+ ratios for controls and cases were not significantly different (t = 1.67, p-value = 0.10; Figure S1), suggesting cortical neuron loss in the BA9 area in HD is relatively modest and does not account for the dramatic alterations in miRNA levels reported here.
Increased miR-10b-5p expression is not observed in Parkinson's disease (PD)
To establish whether miR-10b-5p change is a generalized response to neurodegeneration, we evaluated this miRNA in PD prefrontal cortex. While cortical neuronal loss is variable in PD, both PD and HD are neurodegenerative and caused by protein inclusions. We selected PD prefrontal cortex samples that exhibited reported neuron loss on their neuropathological evaluation (n = 6) and PD samples without reported cortical neuronal loss (n = 8). From total RNA, RT-qPCR was performed for miR-10b-5p (Table S5). No difference was seen in miR-10b-5p expression when stratifying PD based on the extent of neuron loss (t = 0.59, p-value = 0.58). Additionally, no significant difference in HD miR-10b-5p expression from qPCR was observed when stratifying HD cases based on a measure of cortical neuron loss (f = 0.28, p-value = 0.76).
Next, the relative expression of miR-10b-5p in PD was compared to all nineteen HD and eighteen control samples assayed. While no significant difference in miR-10b-5p expression was observed between control and PD samples (q = 0.05, p = 0.99), a significant difference was seen in HD compared to PD (q = 7.30, p<0.0001; Figure 2 ), suggesting increased miR-10b-5p expression, independent of neuron loss, is not a generalized response to neurodegeneration.
Figure 2. miR-10b-5p expression in control, Parkinson's disease and Huntington's disease prefrontal cortex.
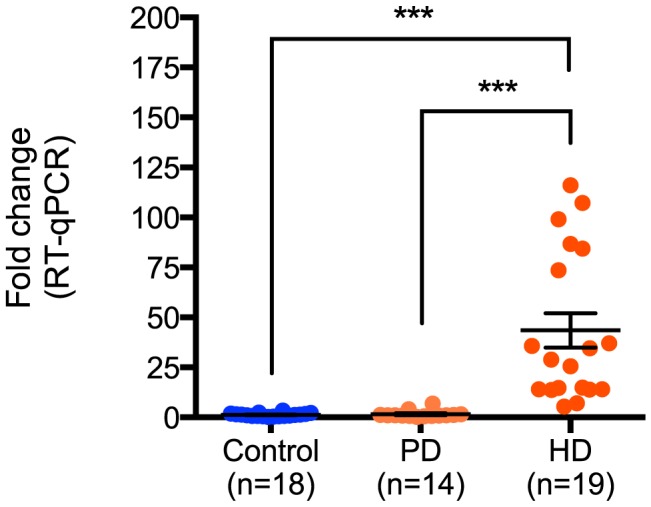
Up-regulation of miR-10b-5p was confirmed in HD by performing RT-qPCR, comparing nineteen Huntington's disease prefrontal cortex samples to eighteen non-neurological disease control samples (***p<0.001) or fourteen Parkinson's disease samples (***p<0.001). ΔΔCT values of miR-10b-5p in PD and HD as compared to controls are shown on the y-axis. The absence of up-regulation in PD frontal cortex suggests that up-regulation of miR-10b-5p may be HD specific. (CT = cycle threshold; RT-qPCR = reverse transcription quantitative PCR; PD = Parkinson's disease; HD = Huntington's disease).
Ectopic miR-10b-5p expression protects HD cell lines from polyglutamine-mediated cytotoxicity
To determine the functional importance of miR-10b-5p up-regulation in HD, we ectopically expressed miR-10b-5p in PC12 Q73 cells. These cell stably expressed huntingtin fragment derived from exon 1 (1–90), contain a pathogenic, 73 long polyglutamine repeat and a MYC epitope for protein identification. PC12 cells have been shown to terminally differentiate and form neural processes upon nerve growth factor (NGF) treatment [21], and HD models of these cells have been highly characterized, exhibiting phenotypic changes such as aggregate formation and polyglutamine-dependent cell death [22]–[26].
PC12 Q73 cells were transfected with miR-10b-5p mimic or a negative control mimic, cel-miR-67-3p, after 48 hours post-differentiation. Cell survival was quantified using a MTT cell viability assay 48 hours post-transfection. Increased survival, though modest (53.9% versus 48.2%), was statistically higher for cells transfected with miR-10b-5p compared to cells transfected with negative control miRNA (q = 4.58, p-value<0.0001; Figure 3 ). The enhanced survival via ectopic miR-10b-5p expression was further substantiated in experiments using viable fluorescent cell staining, where miR-10b-5p transfected cells showed increased cell viability over cells transfected with negative control miRNA (t = 2.381, p-value = 0.018).
Figure 3. miR-10b-5p overexpressing PC12 Q73 cells exhibit reduced cytotoxicity.

PC12 cells expressing huntingtin exon 1 with a polyglutamine expansion spanning 73 repeats were transfected with miR-10b-5p or cel-miR-67-3p as a negative control. On day 3 post-differentiation, a subset of cells were treated with 1 uM MG 132. A MTT assay was used to measure cell viability after four days post differentiation. On the Y-axis, the viability percentage was calculated from the initial cell count. Error bars represent SEM. (****p<0.0001; **p<0.001 *p<0.05).
Thus, miR-10b-5p may play a protective role in enhancing cell survival during stress. To model stress, we treated miRNA transfected cells with 1 uM MG 132, a potent proteasome inhibitor that increases huntingtin aggregation and cellular apoptosis in PC12 HD cell lines [27]. As expected, MG 132 treated cells had reduced cell viability as compared to untreated cells (cel-miR-67-3p, q = 6.52, adjusted p-value<0.0001; miR-10b-5p, q = 10.88, adjusted p-value<0.0001). However, MG 132 treated miR-10b-5p transfected PC12 Q73 cells exhibited improved survival over those transfected with negative control miRNA (q = 3.728, adjusted p-value = 0.045). No statistical difference was observed when comparing miR-10b-5p levels with MG 132 treatment to cel-miR-67-3p without treatment, (q = 2.95, adjusted p-value = 0.16), suggesting miR-10b-5p may enhance survival in times of cellular stress.
miRNA expression is related to clinical variables in HD
RNA sequence count data may be non-normally distributed [28], and tests of normality for miRNA expression levels in HD found that miR-10b-5p was negatively skewed (see Materials and Methods ). Therefore, to test the relationship of miRNA expression to clinical variables such as CAG repeat size, age at onset of motor symptoms, disease duration and age at death, as well as to the sample quality information for RIN/RQN (RNA integrity number/RNA quality number), we applied a step-wise backwards selection, negative binomial regression model.
Age at onset, duration and age at death are inter-dependent and could not be simultaneously included in the models. Furthermore, age at onset and age at death were strongly correlated with each other (Pearson r = 0.85, p-value = 5e-04) and both were correlated with CAG repeat size (r = −0.84, p-value = 6e-04, and r = −0.89, p-value = 1e-04 respectively) while duration was not correlated with age at onset, age at death or CAG repeat size in this sample. To determine which variables best modeled the relationship of the miRNAs to clinical variables, we compared the Akaike information criterion (AIC) for each variable (onset age, death age and duration) in regression analyses that adjusted for the effect of CAG repeat size. Of these three variables, duration was found to have the poorest fit with each of the five miRNAs and therefore we report analyses containing age at onset and age at death.
Among the HD brains, CAG repeat size, age at onset and age at death were all independently found to have a negative association with miR-10b-5p (CAG, β = −0.18, p-value = 2.7e-05; onset, β = −0.05, p-value = 1.9e-05; death, β = −0.07, p-value = 6.8e-07). CAG repeat size and age at onset were found to be independently, negatively related to miR-196a-5p (CAG, β = −0.15, p-value = 1.7e-02; onset, β = −0.07, p-value = 1.4e-03). Age at death was significantly related to miR-615-3p expression (β = −0.03, p-value = 0.0045) and age at onset was associated with miR-196b-5p (β = −0.04, p-value = 9e-04). No association to any clinical features was seen for miR-1247-5p. In order to fully evaluate whether there was any effect of disease duration on the observed relationships to the clinical features, duration was added back into final models. No substantial changes to the effect estimates were observed with the addition of duration to any of the models.
None of the miRNA levels was related to post-mortem interval in either control or HD case samples. The essentially null level of expression in controls prevented meaningful assessment of the relationship of miR-196a-5p, miR-196b-5p and miR-615-3p with clinical variables, in particular age at death, or sample variables, post-mortem interval (PMI), or RIN/RQN. Analysis of miR-10b-5p showed no association to age at death (β = −0.002, p-value = 0.60), or PMI (β = −0.014, p-value = 0.31), but did show association with RIN/RQN (β = 0.54, p-value = 7.2e-05) in controls. miR-1247-5p showed association with later age at death (β = −0.013, p-value = 0.024) in controls.
Expression of miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p are correlated
Among the twelve HD samples, the levels of four out of the five significantly differentially expressed miRNAs (miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p) were strongly correlated with each other, (Spearman r range 0.71–0.88; p range 0.0002–0.01). miR-1247-5p was not significantly correlated with these miRNAs (Spearman r range 0.13–0.51; p range 0.09–0.70). Because the values of miR-615-3p and miR-196a-5p were essentially zero in the control samples, correlations among the miRNAs were not performed for controls.
mRNA targets of miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p may have similar functions
Watson-Crick base-pairing between nucleotide position 2 through 8 on the mature miRNA, termed the ‘seed region,’ and the 3′ untranslated region (3′ UTR) of target mRNA determine the recognition, specificity and efficiency of miRNA silencing [29]. Seed sequences differ for miR-10b-5p (ACCCUGU), miR-615-3p (CCGAGCC) and miR-1247-5p (CCCGUCC) suggesting these miRNA have different targets, while miR-196a-5p and miR-196b-5p share a seed sequence (AGGUAGU) and only differ by a single base difference in mature miRNA sequence.
Targets of the five miRNAs were obtained from miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html), a repository of experimentally validated miRNA targets curated from literature and online resources [30]. miRWalk targets of miR-196a, miR-196b and miR-1247 were not strand specific. The miRWalk database contained 84 unique targets for miR-10b-5p, 80 for miR-196a, 40 for miR-196b, two for miR-1247 and twelve for miR-615-3p. Since miR-1247 had just two validated targets, it was removed from analysis.
Four target genes (DICER1, HOXA7, HOXB4, HOXD1) were shared across all four miRNAs. miR-10b-5p shared eleven targets with miR-196a-5p (HOXB8, COX8A, HOXA10, NPC1, FLT3, AKT1, NPM1, DROSHA, AGO2, NFYC, PAX7), and one with miR-615-3p (MAPK8). miR-196a and miR-196b shared 28 targets. In all, eleven of the 167 unique validated targets were Hox cluster genes (HOXA1, HOXA7, HOXA9, HOXA10, HOXB4, HOXB7, HOXB8, HOXC8, HOXD1, HOXD4, HOXD10).
To understand the influence these miRNAs may be having on shared biological processes, targets of each miRNA were analyzed using IPA Core Analysis. To find overlap in biological functions and canonical pathways of each miRNA and its targets, the IPA Core Comparison Analysis tool was used. After correcting for multiple comparisons, targets of miR-10b-5p, miR-196a, miR-196b and miR-615-3p shared significant overlap in 33 biological functions; the top three functional categories were “Cell Death and Survival,” (Benjamini-Hochberg adjusted p-value, range = 3.5e-07–1.5e-04), “Nervous System Development and Function” (range = 1.5e-07–1.5e-03) and “Cellular Assembly and Organization” (range = 2.5e-05–1.7e-03). Twelve pathways were shared among all four sets of miRNA targets, including “Huntington's Disease Pathway” (range = 7.6e-04–8.1e-03), (Gene set = AKT1, BAX, CAPSN1, CLTC, CREB1, EGFR, HDAC9, JUN, MAPK8).
mRNA targets of differentially expressed miRNAs are differentially expressed
Total mRNA-sequencing was performed in the same brain samples as miRNA-sequencing to examine whether gene expression was affected by miRNA up-regulation. Of the 169 unique gene targets for the five differentially expressed miRNAs, 167 were detected using mRNA-sequencing. 22 mRNA targets were significantly differentially expressed between the HD and control prefrontal cortex samples (False Discovery Rate (FDR) adjusted q-value = 0.05 after adjusting for 167 comparisons). Only one gene (keratin 5, KRT5) was down-regulated in HD ( Table 4 ), and four of these target genes were located in the Hox clusters (HOXD4, HOXA10, HOXB7 and HOXD10).
Table 4. 22 differential expressed targets of miR-10b-5p, miR-196a, miR-196b, miR-1247 and miR-615-3p.
| Target gene | miRNA | Location | Mean Control Expression (n = 9) | Mean HD Expression (n = 12) | Fold Change | p-value | q-value* |
| SERPINE1 | miR-10b-5p | 7q22.1 | 22.91 | 140.82 | 6.15 | 3.03E-11 | 5.06E-09 |
| CDKN1A | miR-196a | 6p21.2 | 336.73 | 841.75 | 2.5 | 1.58E-04 | 1.22E-02 |
| HOXD4 | miR-10b-5p | 2q31.1 | 1.74 | 18.33 | 10.51 | 2.38E-04 | 1.22E-02 |
| ANXA3 | miR-10b-5p | 4q21.21 | 259.93 | 553.71 | 2.13 | 2.92E-04 | 1.22E-02 |
| TWIST1 | miR-10b-5p | 7p21.2 | 43.43 | 105.16 | 2.42 | 5.63E-04 | 1.72E-02 |
| CD33 | miR-196a, miR-196b | 19q13.3 | 16.58 | 46.63 | 2.81 | 6.75E-04 | 1.72E-02 |
| DIO3 | miR-1247 | 14q32 | 10.89 | 41.93 | 3.85 | 7.29E-04 | 1.72E-02 |
| MMP2 | miR-10b-5p | 16q13-q21 | 58.67 | 137.83 | 2.35 | 8.26E-04 | 1.72E-02 |
| MMP9 | miR-10b-5p | 20q11.2-q13.1 | 5.32 | 17.33 | 3.26 | 9.33E-04 | 1.73E-02 |
| HOXA10 | miR-10b-5p, miR-196a, miR-196b | 7p15.2 | 1.06 | 17.06 | 16.12 | 1.21E-03 | 1.73E-02 |
| RHOD | miR-10b-5p | 11q14.3 | 12.71 | 37.96 | 2.99 | 1.23E-03 | 1.73E-02 |
| COL1A1 | miR-196a | 17q21.33 | 30.19 | 220.28 | 7.3 | 1.31E-03 | 1.73E-02 |
| HLA-E | miR-10b-5p | 6p21.3 | 3703.47 | 7769.76 | 2.1 | 1.34E-03 | 1.73E-02 |
| PPARA | miR-10b-5p | 22q13.31 | 444.7 | 865.02 | 1.95 | 1.53E-03 | 1.73E-02 |
| PAX6 | miR-196a | 11p13 | 693.52 | 1337.23 | 1.93 | 1.62E-03 | 1.73E-02 |
| EGFR | miR-10b-5p | 7p12 | 784.95 | 1762.88 | 2.25 | 1.66E-03 | 1.73E-02 |
| HOXB7 | miR-196a | 17q21.3 | 1.63 | 6.99 | 4.28 | 2.83E-03 | 2.78E-02 |
| PLAUR | miR-10b-5p | 19q13 | 56.15 | 119.67 | 2.13 | 3.65E-03 | 3.38E-02 |
| HOXD10 | miR-10b-5p | 2q31.1 | 1.25 | 9.33 | 7.45 | 4.73E-03 | 3.96E-02 |
| RUNX1 | miR-10b-5p | 21q22.3 | 87.69 | 224.88 | 2.56 | 4.74E-03 | 3.96E-02 |
| SOX2 | miR-10b-5p | 3q26.3-q27 | 1963.76 | 3492.72 | 1.78 | 5.32E-03 | 4.23E-02 |
| KRT5 | miR-196a | 12q13.13 | 113.74 | 51.99 | −2.19 | 6.00E-03 | 4.55E-02 |
* FDR-adjusted q-value for 167 targets of the five miRNAs.
miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p expression is related to Hox cluster gene expression
Four of the five up-regulated miRNAs are located intergenic to Hox gene clusters ( Figure 3 ). Because of gene duplication, miR-196a is derived from both the HOXB and HOXC clusters; miR-10b is located in the HOXD cluster and miR-615 is found in the HOXC cluster [31], [32]. A total of 55 genes (40 protein-coding genes, eleven antisense transcripts, three functional lncRNAs and one pseudogene) are located in the four Hox clusters [33], [34]. To evaluate evidence for a general regional up-regulation of Hox cluster genes, an expression analysis of the mRNA-sequence data was performed for all annotated genes within the Hox loci ( Table 5 ). Fifteen out of 55 genes within the Hox loci were differentially expressed in HD. Fourteen Hox genes were significantly up-regulated (FDR-adjust q-value<0.05, mean fold-change = 6.73, range 3.02 to 16.12) and a single Hox gene was down-regulated (HOXD1, FDR-adjust q-value = 3.92e-02, fold change = −2.45). The majority of differentially expressed Hox genes (13 out of 15) were essentially unexpressed in controls.
Table 5. Differential expression of Hox cluster genes in HD.
| Gene | Mean Control Expression (n = 9) | Mean HD Expression (n = 12) | Fold Change | p-value | q-value* |
| HOXA11 | 1.06 | 8.20 | 7.75 | 3.96e-05 | 2.07e-03 |
| HOXA5 | 1.06 | 7.63 | 7.21 | 1.03e-04 | 2.07e-03 |
| HOXD8 | 1.15 | 7.84 | 6.80 | 1.13e-04 | 2.07e-03 |
| HOXD4 | 1.74 | 18.33 | 10.51 | 2.38e-04 | 3.22e-03 |
| HOXB9 | 1.06 | 9.20 | 8.69 | 2.93e-04 | 3.22e-03 |
| HOXA10 | 1.06 | 17.06 | 16.12 | 1.21e-03 | 1.11e-02 |
| HOXC6 | 1.15 | 6.16 | 5.34 | 1.62e-03 | 1.27e-02 |
| HOXA11-AS | 1.25 | 7.39 | 5.90 | 2.49e-03 | 1.71e-02 |
| HOXB7 | 1.63 | 6.99 | 4.28 | 2.83e-03 | 1.73e-02 |
| HOXA13 | 1.45 | 8.74 | 6.03 | 4.07e-03 | 2.24e-02 |
| HOXD10 | 1.25 | 9.33 | 7.45 | 4.73e-03 | 2.36e-02 |
| HOXD1 | 55.91 | 22.80 | −2.45 | 8.55e-03 | 3.92e-02 |
| HOXC10 | 1.36 | 8.04 | 5.90 | 1.06e-02 | 4.14e-02 |
| HOXC4 | 3.57 | 10.77 | 3.02 | 1.12e-02 | 4.14e-02 |
| HOTAIRM1 | 3.52 | 12.52 | 3.56 | 1.13e-02 | 4.14e-02 |
* FDR-adjusted q-value for the 55 genes in the four Hox clusters.
The genes adjacent to the four differentially expressed miRNAs were highly expressed. Two genes immediately adjacent to miR-10b-5p were significantly up-regulated in HD (HOXD4, FDR-adjusted q = 3.22e-03; HOXD8, FDR-adjusted q = 2.07e-03), ( Figure 4 ). HOXB9 (FDR-adjusted q-value = 3.22e-03) immediately downstream of miR-196a-1 and HOXC10 (FDR-adjusted q-value = 4.14e-02) immediately upstream of miR-196a-2 were also up-regulated. Furthermore, all three Hox genes located upstream of miR-196b were significantly up-regulated in HD (HOXA10, FDR-adjusted q-value = 1.11e-02; HOXA11, FDR-adjusted q-value = 2.07e-03; HOXA13, FDR-adjusted q-value = 2.24e-02). HOXC6 (FDR-adjusted q-value = 1.27e-02) immediately upstream of miR-615 was also up-regulated.
Figure 4. Differentially expressed miRNAs in HD are located in Hox genes clusters.

Schematic representation of Hox clusters. Hox genes are represented as numbered boxes (labeled 1–13), miRNA are represented by triangles and other genes in the regions (functional lncRNA, PRAC) are represented by rectangles. Antisense transcripts and pseudogenes are not pictured. Nineteen genes within Hox cluster regions were found significantly differentially expressed in HD prefrontal cortex using mRNA-sequencing (FDR-adjusted p-value<0.05). Four miRNAs, one lncRNA, and fourteen Hox genes were significantly up-regulated in HD (indicated by red), many of which are adjacent to differentially expressed miRNAs. A single Hox gene (HOXD1) was down-regulated in HD (indicated by blue). (HD = Huntington's disease).
Discussion
Up-regulation of expression for five miRNAs in HD brain
We report a next-generation sequencing study of small RNAs, identifying 1,417 mature miRNA species in the prefrontal cortex (Brodmann Area 9) of twelve HD and nine control brains. Five of these, miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p and miR-1247-5p, were up-regulated in HD at genome-wide significance (FDR q-value<0.05), and three of these five, miR-196a-5p, miR-196b-5p and miR-615-3p, were expressed at near zero levels in the control brains. Up-regulation of miR-10b-5p was validated in the miRNA-sequencing samples and confirmed in an independent replication sample set. Several studies implicating a role for miRNAs in HD have been performed, although, to our knowledge this is the first genome-wide quantification of miRNA expression comparing individual human HD and control brain samples.
Packer et al. [11], studying an array of 365 mature miRNAs, had previously reported miR-196a-5p to be significantly increased by nearly six-fold in Brodmann Area 4 of HD grade 1 brains. Recently, a study by Cheng et al. [13] found increased miR-196a expression suppressed mutant HTT expression in both HD neuronal cell models and HD transgenic mouse models. These findings suggest increased expression of miR-196a may be an adaptive response, promoting neuronal survival and may have therapeutic implications for HD. Miyazaki et al. [35] studied miR-196a in spinal and bulbar muscular atrophy (SBMA), a neurodegenerative disease caused by a similar polyglutamine repeat expansion in the androgen receptor (AR) gene. They found increased miR-196a expression via adeno-associated virus vector-mediated delivery reduced AR mRNA levels leading to improved neurological function in transgenic SBMA mouse models. Together, these findings suggest a neuroprotective role for miR-196a and its targets and possible therapeutic implications across multiple polyglutamine-expansion neurodegenerative diseases. miR-196a-5p and miR-10b-5p were among the 56 miRNAs found to be elevated in response to mutant HTT over-expression in undifferentiated NT2 cells [36]. According to the miRNA search program “PubmiR,” [37] miR-196b-5p, miR-1247-5p and miR-615-3p have not been previously reported in HD miRNA studies.
A number of past studies have examined miRNA levels in HD, HD transgenic mice or cellular models; however, we did not replicate the results obtained in these studies. Gaughwin et al. [36] reported miR-34b elevated in plasma samples in HD, but we found neither miR-34b-3p nor miR-34b-5p to be altered in HD brain at genome-wide levels. We were not able to confirm any of the miRNAs reported in past microarray studies that examined targeted subsets of miRNAs, including the nine miRNAs reported as down-regulated in two mouse models of HD (YAC128 and R6/2) by Lee et al. [14] using a 567 miRNA microarray or the 38 miRNAs with altered expression in HD transgenic mice in a 382 miRNA microarray [15]. Johnson et al. [10]–[12] reported miR-29a and miR-330 to be significantly up-regulated in HD samples, neither of which was found to be altered in this study [10]. In a RT-qPCR study comparing 90 miRNAs in mouse Hdh (Q111/Q111) striatal cells to control mice [12], [38], none of the 27 reported differentially expressed miRNAs was different at genome-wide levels in our study. The most commonly reported altered miRNA in HD studies, miR-132, has been reported as both down-regulated [10], [14], [39] and up-regulated [11], but was not differentially expressed in our study.
While some of the lack of concordance may be a consequence of the differences between human and animal models of HD, it is also likely that some of the differences are a consequence of the different technologies employed by these studies. Microarrays may have different levels of detection for some miRNAs from that seen by miRNA sequencing. Finally, nearly all of the studies employ microarray methods. Microarrays that study only 365 (e.g. Packer et al. [11],) to 567 miRNAs (e.g. Lee et al. [14]) are not performing as many contrasts and thus do not adjust for as many contrasts as our genome wide analysis (e.g. 1,417 miRNAs detected) demands.
miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p implicate Hox cluster genes
Four (miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p) of the five differentially expressed miRNAs are related to Hox cluster genes as follows: (1) these four are located in intergenic regions of the Hox clusters, (2) eleven Hox genes are validated targets of these four miRNAs, (3) Hox genes adjoining differentially expressed miRNAs are differentially expressed and (4) multiple Hox cluster genes are differentially expressed in HD versus control brains ( Table 4 ).
Of the eleven Hox gene targets, eight did not differ in their expression across condition. A single target, HOXD1 was seen to be down-regulated in HD (FC = −2.45). HOXD1 is a reported target of four of the five miRNAs [40] which may explain its repression in HD.
Three Hox gene targets were up-regulated in HD (HOXB7, HOXD4 HOXD10). It is possible these up-regulated Hox genes share similar regulatory mechanisms, as the increased miRNA expression does not produce the expected miRNA-mediated gene silencing and suppress the observed up-regulation of the miRNA target genes. Coevolution of Hox genes and Hox-related miRNAs may further suggest that they share regulatory elements or mechanisms [41]. Furthermore, Hox genes and related miRNAs have been observed to have similar patterns of transcriptional activation and both are activated by retinoic acid [42]–[46]. Although miR-10b-5p has been validated as targeting HOXD4, they may exhibit patterns of co-expression. Specifically, Phua et al. [45] report miR-10b and HOXD4 are temporally co-expressed during neurodifferentiation. Here, we see a similar up-regulation and co-expression pattern in HD, where miR-10b and HOXD4 are both highly expressed.
Hox genes are a family of transcription factors that contribute to major morphological changes during embryonic development and are required for anterior-posterior body axis in bilaterally developing species [47]. They are highly involved in most aspects of early development, and are prominently expressed in the developing brain [48]. Hox-related miRNAs may also follow similar spatio-temporal patterns of expression during embryogenesis [49].
Hox genes are regulated by retinoic acid but also other factors, including basic fibroblast growth factor [50], steroid hormones [51], [52] and polycomb repressive complex group [53]. Polycomb group (PcG) proteins assemble into large silencing complexes and control histone-modifying activity. Hox genes are repressed by PcG complexes, specifically Polycomb Repressive Complex 2 (PRC2), which trimethylates histone H3 at lysine 27 (H3K27me3) [53].
Seong et al [54] observed knockout huntingtin mouse embryos lacked repression of HOXB1, HOXB2, and HOXB9 and showed diminished global H3K27me3, while a knock-in expanded repeat mouse exhibited increased H3K27me3 signal, suggesting mutant huntingtin may alter proper PRC2 activity. These findings raise the possibility that the increased expression of miRNAs and Hox genes reported here are related to enhanced H3K27me3 or impaired PcG repression. However, the role of Hox in the adult, HD brain is still unclear. Increased transcriptional activity of Hox may be compensatory, helping to preserve or re-establish cell polarity, or an indirect result of impaired epigenetic regulation.
miR-10b-5p response in HD may be protective
To functionally validate our miRNA-sequencing findings, we chose to assess miR-10b-5p. We believed this miRNA to be the most biologically active of the differentially expressed miRNAs. miR-10b-5p had the highest basal expression levels and the highest fold change between conditions. Additionally, miR-10b-5p levels were not increased in PD, a comparable protein aggregate, neurodegenerative disease, nor in PD samples with pathology in the prefrontal cortex equivalent to HD.
To determine whether miR-10b-5p had a protective or deleterious effect on neuron viability, we ectopically expressed miR-10b-5p in terminally differentiated PC12 Q73 cells. Since the levels the five differentially expressed miRNA were up-regulated, we felt overexpression of miR-10b-5p best represented the phenotype observed in HD brain.
We reported increased miR-10b-5p expression enhanced the survival of PC12 Q73 cells. Furthermore, we found that increased miR-10b-5p expression enhanced survival in the presence of apoptosis-inducing compound, MG 132. In this experiment, survival in cells with increased miR-10b-5p expression was comparable to that of unchallenged cells and significantly greater than untreated cells exposed to toxin. These finding provide support for the hypothesis that increased miR-10b-5p may be a neuroprotective response to the expanded polyglutamine repeat seen in HD and speaks to the role of this microRNA in the pathology of HD.
miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p have overlapping biological functions
Using pathway analysis, we showed that miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-3p targeted genes are predicted to be involved in apoptosis as well as nervous system development and function. In neuroblastoma SH-SY5Y cell lines, miR-10a, miR-10b and miR-615-5p expression levels significantly increased during all-trans-retinoic-acid (ATRA) treatment, indicating miR-10a/b and miR-615-5p may have a role in neurodifferentiation [44]. SH-SY5Y cells treated with antisense miR-10a or miR-10b had impaired neurite outgrowth and morphology but did not show changes in overall cell proliferation [44]. miR-10a and miR-10b were highly expressed in SK-N-BE, LAN5 and SH-SY5Y cell lines during ATRA treatment and ectopic expression of miR-10ab mirrored the phenotype of the ATRA treatment [42]. Taken together, these studies implicate these miRNAs in neuron differentiation, migration, and outgrowth.
In our past studies [16], we found increased neurite outgrowth in HD prefrontal cortex. Relative to controls, HD pyramidal neurons had a significantly increased number of primary dendritic segments, increased total dendritic length, and more dendritic branches than control neurons. Here, we report four miRNAs that have been observed in cell models to present a similar phenotype. It is possible that increased expression of these miRNAs and related targets represent an adaptive response of neurons stressed by a toxic expanded polyglutamine protein fragment.
miR-10b-5p, miR-196a-5p, miR-196b-5p and miR-615-5p are related to HD pathogenesis
Four of the five up-regulated miRNAs showed association to clinical features of HD (CAG repeat size, age of motor onset and age at death for miR-10b-5p; CAG repeat size and age at onset for miR-196a-5p, age at onset for miR-196b-5p and age at death for miR-615-3p). Due to the near zero level of expression in controls, it was not possible to assess the relationship of miR-196a-5p, mir-196b-5p and miR-615-3p to age at death, but miR-10b-5p was not correlated with age at death in controls. Thus, the increased expression of these miRNAs did not appear to be related to normal aging, but rather a component of gene regulation and transcription in the context of neurodegeneration. A growing body of literature points to the presence of toxic effects of the HD gene substantially before the onset of symptoms, perhaps from the time of conception [55]–[57].
Because age at death represents the lifetime exposure of the individual to the effects of the HD gene, we hypothesize that the association of miR-10b-5p and miR-615-3p with age at death may represent the lifetime exposure to the effects of the HD mutation. If the relationship of altered miRNA expression to age at death supports the view that the HD gene may have a life-long effect among expanded CAG-repeat carriers, this raises the possibility that the HD mutation may influence neuronal development in the developing brain through the action of one or more of these miRNAs and Hox cluster genes.
Target genes of over-expressed miRNAs show increased expression in HD
We report five miRNAs as being highly up-regulated in HD and though our expectation was to see the mRNA targets of these miRNAs as decreased, we observe increased expression of many of their shared mRNA targets. We believe these effects are not attributable to differences in cell populations studied, since flow cytometric analysis measuring neuron abundance found no significant difference across condition. Rather, we hypothesize positive miRNA-mRNA target relationships are a result of HD-specific alterations in mRNA processing.
Translation is a highly dynamic process. Cytoplasmic mRNA actively engaged in translation can cycle to a non-translated state and accumulate in stress granules or processing bodies (P-bodies). During cellular stress, mRNA can be sequestered to P-bodies or stress granules, to stall translation through translational repression machinery or miRNA silencing, until stress conditions have been resolved [7], [58]–[60]. P-bodies may also serve an important role in RNA transport. Because neurons are highly polarized, cytoplasmic transport of mRNA is essential for localized translation to discrete regions of the cell. During transport, it is believed that mRNAs are silenced by miRNA, upon rapid exchange at the synapse [60]–[62].
In HD cortical neurons, excitotoxicity, oxidative damage, aberrant gene expression and energetic defects lead to stress conditions and in response, cells may sequester mRNA to P-bodies and stress granules. Among the 55 Hox locus genes studied, only one of the fifteen significantly differentially expressed genes is down regulated ( Table 4 ). Thus, the increased levels of most of the validated gene targets of these four miRNAs may be reactionary, as they are sequestered to P-bodies for storage as part of a protective process to enhance cell viability [7].
To the best of our knowledge, no study has addressed the role of P-bodies or stress granules in HD. However, it was observed in live cortical neurons that wildtype huntingtin co-localized in P-bodies, specifically in neuronal RNA granules, along with Argonaute 2, the endonuclease required for RNA-mediated gene silencing by the RNA-induced silencing complex (RISC) [63], [64]. Therefore, it is reasonable to suggest mutant huntingtin may impair miRNA-mediated mRNA degradation and/or localized translation of specific mRNAs.
There is evidence that miRNA-mRNA regulatory mechanisms may be altered in other neurodegenerative diseases as well. In a joint examination of miRNA-mRNA expression in Alzheimer's disease (AD) and control prefrontal cortex, an overwhelming number of miRNA to mRNA targets were found to be positive correlated. Genomic variants in TDP-43 and FUS, genes that encode stress granule proteins, were found to cause familial Amyotrophic lateral sclerosis [65], [66] and several other stress granule proteins (TIA-1, G3BP) may also be pathogenic [67].
miRNAs as potential biomarkers in HD
These studies suggest potential relationships of these miRNAs to CAG repeat expansion, age at onset or age at death. If these findings hold up on further examination, these miRNAs may hold potential to provide insight into important biological and disease expression for HD. miRNA are extremely stable. The half-life of the majority of miRNAs has been predicted to be on average five days and plasma miRNAs have been found to be stable after being subjected to high heat, extreme pH, long-time storage at room temperature, or multiple freeze-thaw cycles [68]–[70]. If these miRNAs cross the blood-brain barrier and can be detected at reasonable levels in serum/plasma from mutant HD gene carriers, they may serve as biomarkers of disease expression.
Materials and Methods
Sample information
Frozen brain tissue from prefrontal cortex Brodmann Area 9 (BA9) was obtained from the Harvard Brain and Tissue Resource Center (HBTRC) McLean Hospital, Belmont MA. Twelve Huntington's disease (HD) samples and eleven neurologically-normal control samples were selected for the study ( Table 1 ). The HD subjects had no evidence of Alzheimer or Parkinson disease (PD) comorbidity based on neuropathology reports. For microscopic examination, 16 tissue blocks were systematically taken and histologically assessed as previously described [3]. All samples were male. HD samples and controls were not different for postmortem interval (PMI) (t = 1.07, p = 0.30), RNA integrity number (RIN) (t = 0.83, p = 0.41) or death age (t = 0.40, p = 0.69). CAG repeat size was known for all HD samples and onset age and disease duration was unknown for a single sample ( Table 1 ). Eight additional HD, nine control and fourteen PD cases were studied as part of validation and replication studies, and were obtained from the HBTRC and the Sun Health Research Institute Sun City, Arizona (Tables S2, S3 and S5).
RNA extraction
Total RNA, for all samples studied, was isolated using QIAzol Lysis Reagent and purified using miRNeasy MinElute Cleanup columns (Qiagen Sciences Inc, Germantown, MD). RNA quality for sequencing was assessed using either Agilent's BioAnalyzer 2100 system and RNA 6000 Nano Kits to find RNA Integrity Number (RIN) or Agilent 2200 TapeStation and DNA ScreenTape assay RNA Quality Number (RQN; Agilent, Foster City, CA). Both methods calculate the area under the peak for 18S and 28S RNA as a ratio of total RNA as well as the relative height of the 18S and 28S peaks to determine RNA quality [71]. The RIN/RQN values were similar for the twelve HD and eleven control specimens studied for miRNA and mRNA (t = 0.95, p = 0.36).
Illumina miRNA sequencing (miRNA-seq)
For each brain sample, 1 ug of RNA was used to construct sequencing libraries using Illumina's TruSeq Small RNA Sample Prep Kit, according to the manufacturer's protocol (Illumina, San Diego, CA). In brief, small RNA molecules were adapter-ligated, reverse transcribed, PCR amplified and gel purified to generate the library. Multiplexed samples were equimolarly pooled into sets of eight samples per flowcell lane and sequenced using 1×50 bp single-end reads on Illumina's HiSeq 2000 system at Tufts University sequencing core facility (http://tucf-genomics.tufts.edu/). Demultiplexing and FASTQ file generation (raw sequence read plus quality information in Phred format) were done using Illumina's Consensus Assessment of Sequence and Variation (CASAVA) pipeline.
Primary processing of Illumina miRNA-seq reads
Sequence read quality was evaluated using the FASTQ quality filter module from the FASTX-toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/), and only those reads with at least 80% of the base calls above Q20 (Phred score) were retained. The 3′ adapter sequence (5′-TGGAATTCTCGGGTGCCAAGG-3′) was removed from all reads using the FASTA/Q clipper module from the FASTX-toolkit. A minimum length threshold of 15 nucleotides was set for clipped reads because miRNAs of this length will contain the seed sequence. To avoid redundancy amongst identical read species, the reads were collapsed using the FASTA/Q collapser module from FASTX-toolkit to generate a FASTA file of only the unique read species.
Alignment and mapping of miRNA-seq reads
Quality-filtered, 3′ adapter-clipped reads were aligned to the UCSC human reference genome (build hg19) using Bowtie version 0.12.3 [72]. Alignment parameters were set to allow for no mismatch alignments and no limits on multiple mapping instances. Multiple-mapped identical sequences were summed for a single count for that annotated mature miRNA. The default settings were used for all other alignment options.
The miRNA aligned data are available on ArrayExpress (http://www.ebi.ac.uk/arrayexpress/), accession number E-MTAB-2206.
miRNA abundance estimation
Aligned reads that overlapped with the human miRNA annotation version 19 from miRBase (http://www.mirbase.org/ftp.shtml) were identified using default BEDTools' IntersectBed functionality [73]. To select for mature miRNA reads, sequences more than 27 bases in length were removed. Only those reads for which the aligned 5′ start-nucleotide matched exactly to the 5′ start-nucleotide of the annotated miRNA were retained for the analysis. After filtering, collapsed read counts were summed per annotated mature miRNA (Table S6).
miRNA differential expression
The R (http://www.R-project.org) package DESeq version 1.10.1 [28] was used to perform the differential expression analysis between HD and control samples using the read counts generated for each sample as described above. miRNAs with zero read counts across all case and control samples were removed from analysis. To accommodate the analysis of miRNAs with read counts of zero for some samples, a pseudo-count of one was added to all raw counts for every miRNA across all the samples, prior to performing DESeq's estimateSizeFactors and estimateDispersions functions with default options. DESeq assumes that count data follow a negative binominal distribution and factors in technical and biological variance when testing for differential gene expression between groups. DESeq's function, estimateSizeFactors, was used to obtain normalization factors for each sample and to normalize miRNA read counts.
The normalized counts were evaluated by principal component analysis (PCA) with the FactoMineR R package for all HD and control samples. The samples identified to be three or more standard deviations away from the mean on the first or second principal component were considered outliers and were removed from analysis. The first two principal components were used because they each explained more than 10% of the variance, while the remaining principal components explained less than 10% of the variance. Two control samples (C-35 and C-37) were identified as outliers based on PCA analysis.
miRNA differential expression analysis was performed with DESeq's nbinomTest function for the remaining nine control and twelve HD samples. All analyses were performed on DESeq normalized counts.
miRNA quantitative PCR
miRNA were assayed using Exiqon's miRCURY LNA Universal RT miRNA PCR following the manufacturer's protocol (Exiqon Inc, Denmark). In brief, reactions were incubated for 60 min at 42°C followed by heat-inactivation of reverse transcription for 5 min at 95°C and stored at 4°C. After cDNA synthesis, samples were diluted to 0.2 ng/ul in water. Brain samples were assayed using Exiqon ExiLENT SYBR Green master mix and LNA primer sets containing UniRT and miR-10b-5p, miR-196a-5p, miR-196b-5p, miR-615-3p or miR-1247. Reference primer hsa-SNORD48 PCR/UniRT was used for brain samples; U6 snRNA for cell lines. Samples were run in triplicate for each primer set in 384-well format (5 ul PCR Master mix, 1 ul PCR primer mix, 4 ul 0.2 ng cDNA). Reactions were cycled using Applied Biosystems 7900HT Fast Real-Time PCR System using manufacturer's instructions (Life Technologies, Carlsbad, CA). For analysis, threshold cycle (CT) was generated by ABi SDS v2.4 software. CT values for triplicate wells were normalized by average RNU48 value for brain or U6 for cells. miRNA fold change was calculated using the 2-ΔΔCT method [74].
Neuron abundance quantification
0.5–1.0 g of tissue in 5 ml of lysis buffer was homogenized using a dounce tissue grinder. Lysates were transferred to ultracentrifugation tubes, loaded on top of sucrose solution and centrifuged at 24,400 RPM for 2.5 hr at 4°C (Beckman Coulter, Pasadena, CA; L8-70 M with SW80 rotor). Nuclei pellets were resuspended in 500 ul PBS and incubated at 4°C in a staining solution containing 0.72% normal goat serum, 0.036% BSA, 1∶1200 anti-NeuN (Millipore, Germany), 1∶1400 Alexa488 goat anti-mouse secondary antibody (Life Technologies, Carlsbad, CA), for 45 min. Flow cytometry was performed at the Boston University Medical School Flow Cytometry Core Lab on a FACSVantage SE flow cytometer.
Illumina messenger RNA sequencing (mRNA-seq)
For each brain sample, 1 ug of RNA was used to construct sequencing libraries using Illumina's TruSeq RNA Sample Prep Kit according to the manufacturer's protocol. In brief, mRNA molecules were polyA selected, chemically fragmented, randomly primed with hexamers, synthesized into cDNA, 3′ end-repaired and adenylated, sequencing adapter ligated and PCR amplified. Each adapter-ligated library contained one of twelve TruSeq molecular barcodes. Multiplexed samples were equimolarly pooled into sets of three samples per flowcell lane and sequenced using 2×100 bp paired-end reads on Illumina's HiSeq 2000 system at Tufts University sequencing core facility (http://tucf-genomics.tufts.edu/). Demultiplexing and FASTQ file generation were accomplished using Illumina's CASAVA pipeline.
Primary processing of Illumina mRNA-seq reads
Forward and reverse sequencing reads were independently quality-filtered using the FASTQ quality filter module from the FASTX-toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/) with the same criteria as that applied for the processing of the miRNA-seq reads. Reads failing the quality threshold, as well as their corresponding mate reads, were removed.
Alignment and mapping of mRNA-seq reads
Quality-filtered paired-end reads were aligned to the UCSC human reference genome (build hg19) using TopHat version 2.0.4 [75], [76]. This version of TopHat incorporates the Bowtie version 2.0.0.7 algorithm to perform the alignment [72] as well as SAMtools version 0.1.18.0 for alignment file formatting [77]. For efficient read mapping, TopHat requires the designation of the mean and standard deviation of the distance between paired-end reads, the read inner-distance. To estimate the appropriate read inner-distance, we aligned a subset of 5 million reads from four HD and four control samples to the Ensembl human reference transcriptome (release 66) using Bowtie version 2.0.0.7. Using the CollectInsertSizeMetrics function from picardTools version 1.76 (http://sourceforge.net/projects/picard/files/picard-tools/), we estimated the average mean inner-distance per condition and subsequently applied these values for the TopHat alignment; 22 for HD samples 25 for controls respectively, (the current TopHat default setting is 20), (Table S7). To account for read variability, the standard deviation for inner-distance was set to 100. The number of allowed splice mismatches was set to 1. Default settings were used for all other alignment options.
mRNA gene abundance estimation
Gene expression quantification was performed using htseq-count version 0.5.3p9 (http://www-huber.embl.de/users/anders/HTSeq) and the GENCODE version 14 annotation gtf file as reference (http://www.gencodegenes.org/releases). Intersection non-empty mode and unstranded library type were specified as parameters for htseq-count. Default settings were used for all other options (Table S8).
mRNA differential expression analysis
The mRNA differential expression analysis between HD and control samples was performed using DESeq version 1.10.1 [28]; the workflow was the same as described for the miRNA differential expression analysis. No outliers were found based on the PCA of the DESeq-normalized count data. The nbinomTest function was run for eleven control samples and twelve HD samples to assess differentially expressed genes. Multiple comparison adjustment for multiple testing with the Benjamini-Hochberg correction was used to control for false discovery rate. For Hox gene differential expression analysis, 55 comparisons were used. Genes located within HOX-gene containing regions were queried through the Ensembl database (release 72), interfacing through the R package BiomaRt [78], [79]. Genes that were between HOXA1-HOXA13, HOXB1-HOXB13, HOXC4-HOXC13 and HOXD1-HOXD13 start sites were regarded as “Hox genes.” For miRNA target differential expression, 154 comparisons were used for Benjamini-Hochberg correction.
miRNA-mRNA target analysis
Information on experimentally validated miRNA targets of miR-10b-5p, miR-196a-5p and miR-615-3p were extracted from the miRWalk “Validated Targets” module [30]. Strand specificity was preserved. Targets for miR-196a-1 and miR-196a-2 were merged for analysis. IPA Core Analysis (analysis.ingenuity.com) was run as nervous system and CNS cell line specific across all species, using target gene lists imported from miRWalk output. “Bio Functions” and “Canonical Pathway” analyses were used. Right-tailed Fisher's Exact Tests were run through IPA software and p-values with FDR-adjusted q-values (p<0.05) were considered significant. Biological functions across the 3 significant miRNA were compared using the IPA Core Comparison Analysis tool. Benjamini-Hochberg Multiple Testing Correction p-values (p<0.05) were considered significant.
Linear modeling of miRNA relationship to clinical covariates
To account for the non-normality in the miRNA data, negative binominal general linear regressions were performed using Proc genmod in SAS. DESeq normalized counts were rounded to the nearest integer before running the model. To test the normality of gene expression data, Shapiro-Wilk tests were performed. Differentially expressed miRNA data trended as non-normally distributed in HD (miR-10b-5p, p = 0.04; miR-196a-5p, p = 0.05; miR-615-3p, p = 0.06), but not in controls (miR-10b-5p, p = 0.71; miR-196a-5p and miR-615-3p were essential zero).
Generation of transgenic cell lines
PC12 (rat adrenal gland phaeochromocytoma) cells were grown at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA) with 20% fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA), 100 units/ml penicillin and 100 units/ml streptomycin (Life Technologies, Carlsbad, CA). pcDNA3.1mycC expressing human huntingtin fragment (1–90) containing 73 polyglutamine repeats (Coriell Institute; CHDI-90000034) was used for stable transfection. Cells were seeded to 70% confluency and grown overnight. 15 µl of Attractene Transfection Reagent (Qiagen, Gaithersburg, MD) was added to 4 µg plasmid DNA diluted in 300 µl Opti-MEM (Life Technologies, Carlsbad, CA). Cells were grown in complete media and selected for four weeks using 500 mg/ml G418 (Life Technologies, Carlsbad, CA). To create monoclonal cultures, single colonies were isolated using dilution cloning, picked with filter paper, grown in a 6-well plate and screened for transgenic expression by Western blot analysis using mouse Anti- c-Myc (Novex, R950-25, Life Technologies, Carlsbad, CA).
Cell differentiation and miRNA overexpression
96-well culture plates were seeded with 10,000 cells per well. For differentiation, culture medium was replaced with medium composed of DMEM with 0.5% FBS, 100 mg/ml G418, 100 units/ml penicillin and 100 units/ml streptomycin and 100 ng/ml nerve growth factor (R&D Systems, Minneapolis, MN). After 48 hr, miRNA was transfected into HD cells using 0.25 ul Lipofectamine 2000 (Life Technologies, Carlsbad, CA) and 6.25 pmol miR-10b-5p or miRIDIAN microRNA Mimic Negative Control #1 (cel-miR-67-3p, Thermo Scientific, Waltham, MA) per well, following manufacturer's protocol. miR-10b-5p overexpression was verified using qPCR.
Cell viability assays
For MTT assays, 1 uM MG 132 (Tocris Bioscience, United Kingdom) was added to select wells containing 10,000 cells per well at 72 hr post-differentiation. Cell viability was assessed at 96 hr post-differentiation. Following manufacturer's protocol, CellTiter 96 Non-Radioactive Cell Proliferation Assay kit (Promega; Madison, WI) was used to determine cell number. Cells were incubated for 1.5 hr at 37°C and 5% CO2 with MTT dye solution. Undifferentiated HD cells were serially diluted across a 96-well plate to create a standard curve for cell number calculation. Absorbance was measured using Bio-Tek Synergy H1 spectrophotometer at 540 nm for miR-10b-5p transfected wells, with MG 132 (n = 44) and without MG 132 (n = 35) and cel-miR-67-3p transfected wells with MG 132 (n = 40) and without MG 132 (n = 40). One-way ANOVA way used for statistical analysis.
For cell viability staining, miR-10b-5p and negative control mimic were transfected after 48 hours of differentiation in 12-well culture plate with 4 replicates each, 250,000 cells per well. Molecular Probes Neurite Outgrowth Staining Kit (Life Technologies, Carlsbad, CA) was used according to manufacturer's protocol. Using Bio-Tek Synergy H1 microplate reader, fluorescent area scans were taken at 530 nm excitation/590 nm emission with a 5×5 matrix per well.
Supporting Information
Neuron counts from prefrontal cortical tissue homogenate. No significant difference is observed when comparing ratios of NeuN+ counts to total events quantified by flow cytometry.
(EPS)
miRNA RT-qPCR validation study results. RT-qPCR was used to validate the five differentially expressed miRNA in the same set of sample used for miRNA-sequence analysis. The table lists the difference and standard error of fold change between condition (2-ΔΔCt), as well as p-values from two-tailed Welch's t-tests, for ten control and eleven Huntington's disease (HD) samples.
(DOCX)
Sample information for eight Huntington's disease brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the eight Huntington's disease (HD) brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
Sample information for eight control brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the eight control brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
miRNA RT-qPCR replication study results. RT-qPCR was used to replicate the five differentially expressed miRNA in an independent sample set of Huntington's disease (HD) brains. The table lists the difference and standard error of fold change between condition (2-ΔΔCt), as well as p-values from one-tailed Welch's t-tests, for eight control and eight Huntington's disease samples.
(DOCX)
Sample information for fourteen Parkinson's disease brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the fourteen Parkinson's disease (PD) brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
Read statistics for miRNA-sequence analysis. Summary of Illumina miRNA-sequence read and quality control statistics generated from the FASTX-toolkit.
(DOCX)
Mean and standard deviation inner-distance estimates for TopHat2 alignment. Statistics used to estimate the distance between paired-end reads, generated from picardTools.
(DOCX)
Read statistics for mRNA-sequence analysis. Summary of Illumina mRNA-sequence read and quality control statistics generated from the FASTX-toolkit.
(DOCX)
Acknowledgments
We would like to acknowledge the Harvard Brain Tissue Resource Center (HBTRC) for providing all of the HD, PD and control samples used in these studies, Michael Nagle for his cell culture expertise and Tammy Gillis and Jayalakshmi Srinidhi Mysore for their assistance in CAG repeat size genotyping.
Funding Statement
The study was supported by funding from the US National Institutes of Health (R01NS073947 Epigenetic Markers in Huntington's disease Brain, R21NS076958 Dopaminergic Epigenomes From Human Brain, R01NS032765 Delineating the Huntington's Disease Mechanism by Manipulating the Mouse HD), and the Jerry McDonald HD Research Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. HDCRG (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Hadzi TC, Hendricks AE, Latourelle JC, Lunetta KL, Cupples LA, et al. (2012) Assessment of cortical and striatal involvement in 523 Huntington disease brains. Neurology 79: 1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, et al. (1985) Neuropathological classification of Huntington's disease. Journal of neuropathology and experimental neurology 44: 559–577. [DOI] [PubMed] [Google Scholar]
- 4. Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, et al. (2006) Regional and cellular gene expression changes in human Huntington's disease brain. Human molecular genetics 15: 965–977. [DOI] [PubMed] [Google Scholar]
- 5. Cha JH (2000) Transcriptional dysregulation in Huntington's disease. Trends in neurosciences 23: 387–392. [DOI] [PubMed] [Google Scholar]
- 6. Cha JH (2007) Transcriptional signatures in Huntington's disease. Progress in neurobiology 83: 228–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lavut A, Raveh D (2012) Sequestration of highly expressed mRNAs in cytoplasmic granules, P-bodies, and stress granules enhances cell viability. PLoS genetics 8: e1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Junn E, Mouradian MM (2012) MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacology & therapeutics 133: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marti E, Pantano L, Banez-Coronel M, Llorens F, Minones-Moyano E, et al. (2010) A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic acids research 38: 7219–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, et al. (2008) A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiology of disease 29: 438–445. [DOI] [PubMed] [Google Scholar]
- 11. Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL (2008) The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 28: 14341–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sinha M, Ghose J, Bhattarcharyya NP (2011) Micro RNA -214,-150,-146a and-125b target Huntingtin gene. RNA biology 8: 1005–1021. [DOI] [PubMed] [Google Scholar]
- 13. Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, et al. (2013) miR-196a Ameliorates Phenotypes of Huntington Disease in Cell, Transgenic Mouse, and Induced Pluripotent Stem Cell Models. American journal of human genetics 93 (2) 306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee ST, Chu K, Im WS, Yoon HJ, Im JY, et al. (2011) Altered microRNA regulation in Huntington's disease models. Experimental neurology 227: 172–179. [DOI] [PubMed] [Google Scholar]
- 15. Jin J, Cheng Y, Zhang Y, Wood W, Peng Q, et al. (2012) Interrogation of brain miRNA and mRNA expression profiles reveals a molecular regulatory network that is perturbed by mutant huntingtin. Journal of neurochemistry 123: 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sotrel A, Williams RS, Kaufmann WE, Myers RH (1993) Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology 43: 2088–2096. [DOI] [PubMed] [Google Scholar]
- 17. Cudkowicz M, Kowall NW (1990) Degeneration of pyramidal projection neurons in Huntington's disease cortex. Annals of neurology 27: 200–204. [DOI] [PubMed] [Google Scholar]
- 18. Gu X, Li C, Wei W, Lo V, Gong S, et al. (2005) Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46: 433–444. [DOI] [PubMed] [Google Scholar]
- 19. Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, et al. (2005) Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 65: 745–747. [DOI] [PubMed] [Google Scholar]
- 20. Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, et al. (2002) Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology 58: 695–701. [DOI] [PubMed] [Google Scholar]
- 21. Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America 73: 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kita H, Carmichael J, Swartz J, Muro S, Wyttenbach A, et al. (2002) Modulation of polyglutamine-induced cell death by genes identified by expression profiling. Human molecular genetics 11: 2279–2287. [DOI] [PubMed] [Google Scholar]
- 23. Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, et al. (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Human molecular genetics 10: 1829–1845. [DOI] [PubMed] [Google Scholar]
- 24. Igarashi S, Morita H, Bennett KM, Tanaka Y, Engelender S, et al. (2003) Inducible PC12 cell model of Huntington's disease shows toxicity and decreased histone acetylation. Neuroreport 14: 565–568. [DOI] [PubMed] [Google Scholar]
- 25. Apostol BL, Illes K, Pallos J, Bodai L, Wu J, et al. (2006) Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Human molecular genetics 15: 273–285. [DOI] [PubMed] [Google Scholar]
- 26. Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC (2004) Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. The Journal of biological chemistry 279: 4988–4999. [DOI] [PubMed] [Google Scholar]
- 27. Li X, Wang CE, Huang S, Xu X, Li XJ, et al. (2010) Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Human molecular genetics 19: 2445–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome biology 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dweep H, Sticht C, Kharkar A, Pandey P, Gretz N (2013) Parallel analysis of mRNA and microRNA microarray profiles to explore functional regulatory patterns in polycystic kidney disease: using PKD/Mhm rat model. PLoS One 8. 8 (1) e53780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Swalla BJ (2006) Building divergent body plans with similar genetic pathways. Heredity (Edinb) 97: 235–243. [DOI] [PubMed] [Google Scholar]
- 32. Yekta S, Tabin CJ, Bartel DP (2008) MicroRNAs in the Hox network: an apparent link to posterior prevalence. Nat Rev Genet 9: 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, et al. (2013) Ensembl 2013. Nucleic acids research 41: D48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, et al. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, et al. (2012) Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nature medicine 18: 1136–1141. [DOI] [PubMed] [Google Scholar]
- 36. Gaughwin PM, Ciesla M, Lahiri N, Tabrizi SJ, Brundin P, et al. (2011) Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington's disease. Human molecular genetics 20: 2225–2237. [DOI] [PubMed] [Google Scholar]
- 37.Windemuth AS, I; Pregibon, D; Marini. D. (2012) PubmiR: A Literature Search Tool for MicroRNA Research. Firefly BioWorks, Inc. [Google Scholar]
- 38. Sinha M, Ghose J, Das E, Bhattarcharyya NP (2010) Altered microRNAs in STHdh(Q111)/Hdh(Q111) cells: miR-146a targets TBP. Biochemical and biophysical research communications 396: 742–747. [DOI] [PubMed] [Google Scholar]
- 39. Soldati C, Bithell A, Johnston C, Wong K-Y, Stanton LW, et al. (2013) Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington's disease. J Neurochem 124: 418–430. [DOI] [PubMed] [Google Scholar]
- 40. Woltering JM, Durston AJ (2008) MiR-10 represses HoxB1a and HoxB3a in zebrafish. PLoS One 3: e1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tehler D, Hoyland-Kroghsbo NM, Lund AH (2011) The miR-10 microRNA precursor family. RNA Biol 8: 728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foley NH, Bray I, Watters KM, Das S, Bryan K, et al. (2011) MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell death and differentiation 18: 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang H, Xie C, Sun X, Ritchie RP, Zhang J, et al. (2010) miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J Biol Chem 285: 9383–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D (2011) MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J Biol Chem 286: 4150–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phua SL, Sivakamasundari V, Shao Y, Cai X, Zhang LF, et al. (2011) Nuclear accumulation of an uncapped RNA produced by Drosha cleavage of a transcript encoding miR-10b and HOXD4. PloS one 6: e25689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weiss FU, Marques IJ, Woltering JM, Vlecken DH, Aghdassi A, et al. (2009) Retinoic acid receptor antagonists inhibit miR-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology 137: 2136–2145 e2131–2137. [DOI] [PubMed] [Google Scholar]
- 47. Lemons D, McGinnis W (2006) Genomic evolution of Hox gene clusters. Science 313: 1918–1922. [DOI] [PubMed] [Google Scholar]
- 48. Pearson JC, Lemons D, McGinnis W (2005) Modulating Hox gene functions during animal body patterning. Nature reviews Genetics 6: 893–904. [DOI] [PubMed] [Google Scholar]
- 49. Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, et al. (2005) MicroRNA expression in zebrafish embryonic development. Science 309: 310–311. [DOI] [PubMed] [Google Scholar]
- 50. Diez del Corral R, Storey KG (2004) Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 26: 857–869. [DOI] [PubMed] [Google Scholar]
- 51. Svingen T, Tonissen KF (2006) Hox transcription factors and their elusive mammalian gene targets. Heredity (Edinb) 97: 88–96. [DOI] [PubMed] [Google Scholar]
- 52. Taylor HS, Arici A, Olive D, Igarashi P (1998) HOXA10 is expressed in response to sex steroids at the time of implantation in the human endometrium. J Clin Invest 101: 1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by polycomb and trithorax proteins. Cell 128: 735–745. [DOI] [PubMed] [Google Scholar]
- 54. Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, et al. (2010) Huntingtin facilitates polycomb repressive complex 2. Human molecular genetics 19: 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Humbert S (2010) Is Huntington disease a developmental disorder? EMBO reports 11: 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kirkwood SC, Siemers E, Hodes ME, Conneally PM, Christian JC, et al. (2000) Subtle changes among presymptomatic carriers of the Huntington's disease gene. Journal of neurology, neurosurgery, and psychiatry 69: 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, et al. (1991) Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J Neuropathol Exp Neurol 50: 729–742. [DOI] [PubMed] [Google Scholar]
- 58. Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R (2005) MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nature cell biology 7: 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Balagopal V, Parker R (2009) Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Current opinion in cell biology 21: 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W (2006) Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124. [DOI] [PubMed] [Google Scholar]
- 61. Cougot N, Bhattacharyya SN, Tapia-Arancibia L, Bordonne R, Filipowicz W, et al. (2008) Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. The Journal of neuroscience : the official journal of the Society for Neuroscience 28: 13793–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeitelhofer M, Karra D, Macchi P, Tolino M, Thomas S, et al. (2008) Dynamic interaction between P-bodies and transport ribonucleoprotein particles in dendrites of mature hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 28: 7555–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Savas JN, Ma B, Deinhardt K, Culver BP, Restituito S, et al. (2010) A role for huntington disease protein in dendritic RNA granules. The Journal of biological chemistry 285: 13142–13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Savas JN, Makusky A, Ottosen S, Baillat D, Then F, et al. (2008) Huntington's disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proceedings of the National Academy of Sciences of the United States of America 105: 10820–10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319: 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208. [DOI] [PubMed] [Google Scholar]
- 67. Wolozin B (2012) Regulated protein aggregation: stress granules and neurodegeneration. Molecular neurodegeneration 7: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, et al. (2011) Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic acids research 39: 5692–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, et al. (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proceedings of the National Academy of Sciences of the United States of America 105: 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen X, Ba Y, Ma L, Cai X, Yin Y, et al. (2008) Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell research 18: 997–1006. [DOI] [PubMed] [Google Scholar]
- 71. Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, et al. (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC molecular biology 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 75. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14 (4) R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durinck SH, W. (2013) biomaRt: Interface to BioMart databases (e.g. Ensembl, COSMIC, Wormbase and Gramene). R. 2.10.0 ed.
- 79. Kasprzyk A (2011) BioMart: driving a paradigm change in biological data management. Database (Oxford) 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Neuron counts from prefrontal cortical tissue homogenate. No significant difference is observed when comparing ratios of NeuN+ counts to total events quantified by flow cytometry.
(EPS)
miRNA RT-qPCR validation study results. RT-qPCR was used to validate the five differentially expressed miRNA in the same set of sample used for miRNA-sequence analysis. The table lists the difference and standard error of fold change between condition (2-ΔΔCt), as well as p-values from two-tailed Welch's t-tests, for ten control and eleven Huntington's disease (HD) samples.
(DOCX)
Sample information for eight Huntington's disease brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the eight Huntington's disease (HD) brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
Sample information for eight control brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the eight control brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
miRNA RT-qPCR replication study results. RT-qPCR was used to replicate the five differentially expressed miRNA in an independent sample set of Huntington's disease (HD) brains. The table lists the difference and standard error of fold change between condition (2-ΔΔCt), as well as p-values from one-tailed Welch's t-tests, for eight control and eight Huntington's disease samples.
(DOCX)
Sample information for fourteen Parkinson's disease brains used for RT-qPCR replication study. Post-mortem intervals (PMI), RNA integrity numbers (RIN) and ages at death for the fourteen Parkinson's disease (PD) brains used for RT-qPCR verification of the five differentially expressed miRNA.
(DOCX)
Read statistics for miRNA-sequence analysis. Summary of Illumina miRNA-sequence read and quality control statistics generated from the FASTX-toolkit.
(DOCX)
Mean and standard deviation inner-distance estimates for TopHat2 alignment. Statistics used to estimate the distance between paired-end reads, generated from picardTools.
(DOCX)
Read statistics for mRNA-sequence analysis. Summary of Illumina mRNA-sequence read and quality control statistics generated from the FASTX-toolkit.
(DOCX)