

Abstract
Despite of remarkable improvement of postoperative 5-FU–based adjuvant chemotherapy, the relapse rate of gastric cancer patients who undergo curative resection followed by the adjuvant chemotherapy remains substantial. Therefore, it is important to identify prediction markers for the chemotherapeutic efficacy of 5-FU. We recently identified NF-κB as a candidate relapse prediction biomarker in gastric cancer. To evaluate the biological significance of NF-κB in the context of 5-FU–based chemotherapy, we analyzed the NF-κB-dependent biological response upon 5-FU treatment in gastric cancer cell lines. Seven genes induced by 5-FU treatment in an NF-κB-dependent manner were identified, five of which are known p53 targets. Knockdown of RELA, which encodes the p65 subunit of NF-κB, decreased both p53 and p53 target protein levels. In contrast, NF-κB was not affected by TP53 knockdown. We also demonstrated that cell lines bearing Pro/Pro homozygosity in codon72 of p53 exon4, which is important for NF-κB binding to p53, are more resistant to 5-FU than those with Arg/Arg homozygosity. We conclude that NF-κB plays an important role in the response to 5-FU treatment in gastric cancer cell lines, with a possible compensatory function of p53. These results suggest that NF-κB is a potential 5-FU-chemosensitivity prediction marker that may reflect 5-FU-induced stress-response pathways, including p53.
Introduction
The majority of gastric cancer in the world is diagnosed in East Asia [1], where the standard therapy for advanced gastric cancers remains surgery and chemotherapy. Recently developed adjuvant chemotherapeutic regimens after curative gastrectomy for advanced gastric cancer have made remarkable progress in terms of controlling relapse and disease-free survival, particularly in the Japanese population [2], [3]. However, 30–40% of patients still experience relapse despite receiving chemotherapy after curative gastrectomy [3], suggesting that patient selection based on molecular information could potentially be very effective for increasing chemotherapy-mediated non-relapse and survival rates.
To select for gastric cancer patients who might benefit from chemotherapy, it is important to understand individual sensitivities before chemotherapy [4]. Post-operative adjuvant chemotherapy of gastric cancer provides an opportunity to test patient-derived tumors before they receive chemotherapy. In an attempt to identify potential biomarkers in this setting at the protein level, we previously reported a cell line panel screening system using quantitative protein expression profiling with Reverse-Phase Protein Arrays (RPPAs) [5], [6] combined with a cell-based growth assay system based on the concept of NCI-60 cell line screening panel [7], [8]. Candidate biomarkers were isolated based on correlation coefficients from protein expression and drug sensitivity matrix and then further validated using surgically-removed specimens [9]. Based on this approach we identified two biomarkers at the protein level, including NF-κB and JNK, whose levels had good correlation with chemotherapeutic response. The higher expression of NF-κB seemed to correlate with a poorer prognosis, while JNK showed an inverse correlation. These markers were also validated at the molecular level using gastrointestinal cancer cell lines. It has been shown that siRNA-mediated knockdown of p65 almost exclusively affects 5-FU sensitivity among currently-used chemotherapeutic drugs; but this is not the case for JNK knockdown [9]. Therefore, we concluded that NF-κB plays a dominant role in 5-FU treatment and JNK may be an indicator of chronic inflammation of the gastric background mucosae [10]. As an extension of this validation study, we sought to explore these proteins functionally and clarify the role of NF-κB as a stress-inducible transcription factor during 5-FU treatment. We also evaluated the role of p53 after 5-FU-mediated transactivation of NF-κB [10], [11] because it is well known that p53 is activated in response to this genotoxic agent [12]. In this study we report a potential compensatory role of NF-κB for p53 through analysis of a p53-NF-κB binding polymorphic site, codon 72 of p53. Together, these findings suggest that NF-κB/p53-codon72 could be a robust biomarker for 5-FU sensitivity.
Materials and Methods
Cell Lines
Nine human gastric cancer cell lines, including Kato-III, KE39, MKN74, MKN7, NUGC4, GSS, GCIY, and MKN45 were obtained from the RIKEN BioResource Center Cell Bank. IWT-1 was a de novo cell line that established in our laboratory from a Japanese male gastric cancer patient who had relapsed peritonitis carcinomatosa. The use of IWT-1 cell line has been approved by the Iwate Medical University Institutional Review Board (H25-116, and HG H25-15) and the family of donor patient who had died at the time of establishment of the cell line with a written informed consent with respect to taking the samples and making the cell line. Cells were grown to 70–80% confluency in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) at 37°C in the presence of 5% CO2.
Preparation of Cell Lysate
Cells were harvested by centrifugation and cell pellets were lysed using Pink Buffer containing 9 M urea (Sigma-Aldrich, St. Louis, MI, USA), 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesul-fonate(CHAPS;Calbiochem, Merck Millipore, Darmstadt, Germany), 2% pH 8.0–10.5 pharma-lyte (GE Healthcare Japan, Tokyo, Japan), and 65 mM DTT (GE Healthcare Japan, Tokyo, Japan) as previously described [5], [13].
Western Blot
SDS-PAGE was performed using NuPAGE 4–12% Bis-TrisGel electrophoresis (Invitrogen, Carlsbad, CA, USA), XCell Sure Lock Mini-cell (Invitrogen, Carlsbad, CA, USA), and Power PAC HC (BIO-RAD, Hercules, CA, USA). The resolved proteins on the gel were transferred to a nitrocellulose membrane using iBlot Dry Blotting System (Invitrogen, Carlsbad, CA, USA). The resulting membranes were blocked with 5% iBlot (Applied Biosystems, Foster City, CA, USA) and 0.1% Tween-20 (Bio-Rad, Hercules, CA, USA) in TBS (TBST) for 1 h. Membranes were then incubated with the indicated primary antibodies, including pan-actin, p53 (Thermo Scientific, Kalamazoo, MI, USA); p21, TIGAR, and PUMAα/β (Santa Cruz Biotechnology, Dallas, TX, USA); and NF-κB, α-tubulin, and PCNA (Cell Signaling Technology Japan, Tokyo, Japan). Next, the membranes were washed twice for 5 min with TBST, incubated with an HRP-conjugated secondary antibody for 1 h, and then washed twice for 5 min in TBST. Chemiluminescence detection reagents were incubated with the membranes for 1–5 min and then images were acquired using Image Quant LAS500 (GE Healthcare Japan, Tokyo, Japan). To evaluate protein induction by 5-FU, western blots were quantified using ImageJ (http://rsbweb.nih.gov/ij/).
Immunocytochemistry
Cells were grown to 70–80% confluency in RPMI-1640 supplemented with 10% FBS in 4-chamber polystyrene vessel tissue culture-treated glass slides and then treated with 5-FU as indicated in each experiment. After the cells were exposed to 50 µM of 5-FU for 4 h to see an early transcriptional response, they were fixed in 4% paraformaldehyde, permeabilized using 0.2% Triton X-100 in PBS, and stained with DAPI (0.6 µM DAPI, 50 µl RNase, and 5 ml PBS) at room temperature for 12 min. Cells were then incubated with the following primary antibodies: anti-NF-κB p65, phospho-NF-κB p65 (Ser536), and phospho-p53 (Ser15) (Cell Signaling Technology Japan, Tokyo, Japan), and p53 (Thermo Scientific, Kalamazoo, MI, USA). Finally, the cells were incubated with either Alexa Fluor488- or 568-conjugated secondary antibody (Life Technologies Japan, Tokyo, Japan). A BX43 fluorescent microscope (Olympus, Tokyo, Japan) was used for image acquisition.
Gene Expression Profiling
MKN45 cells were harvested after treatment with or without 50 µM of 5-FU for 4 h. RNA was then extracted from the harvested cells and gene expression profiling was performed according to the manufacturer’s instructions (Sure Print G3 Human GE8×60 K, Agilent Technologies Japan, Tokyo, Japan). Raw data were first normalized by dividing each probe signal by the 75th percentile of the entire signal. Each microarray experiment was performed in duplicate resulting in two control and two 5-FU treatment microarray data sets. To identify genes that were differentially expressed in response to 5-FU, each control data set was compared separately to each 5-FU treatment set (4 comparisons). Differentially expressed genes were those that had a change in expression >2-fold in each comparison. We identified the final set of 10 differentially expressed genes based on their frequency in the 4 comparisons [14]. To confirm the reproducibility of these expression changes, quantitative real-time RT-PCR of 5-FU treated samples at 0, 4, 8, 12, and 24 h was performed for each gene. Primer sequences are listed in Table S1. For genes induced by 5-FU, analysis of promoter binding sites was performed using JASPAR algorithm (JASPAR, http://jaspar.genereg.net/). A 1000 bp promotor sequence specific to respective genes was obtained from Transcriptional Regulatory Element Database (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home). Binding sites were predicted by scanning promoter sequences with the consensus sequences of NF-κB and p53 with 70% of profile score threshold.
RELA and TP53 Gene Knockdown
Cells were grown to 70–80% confluency in RPMI-1640 supplemented with 10% FBS in 6-well cell culture plates and then treated with NF-κB p65 or p53 siRNA (Cell Signaling Technology Japan, Tokyo, Japan) for 48 h. Briefly, knockdown was performed using Trans IT-TKO (Mirus Bio Corporation, Madison, WI, USA) at a concentration of 3% for 10 min at room temperature. Appropriate concentrations of siRNAs for each cell line was mixed with the Trans IT-TKO solutions followed by a 20 min incubation at room temperature. The siRNA concentrations used were as follows: 100 nM p65 siRNA for MKN45, and MKN74 cells, and 150 nM for GSS and Kato-III cells; and 100 nM p53 siRNA for MKN45, and GSS cells, and 50 nM for MKN74 cells. After 48 h, cells were harvested and protein levels were examined by Western blot. Two siRNA constructs possessing different sequences to the same target gene was used for each gene to confirm knockdown specificity. Cell cycle distribution was assessed using the Tali Image-based Cytometer (Life Technologies, Carlsbad, CA, USA). To see the maximum effect of siRNA on the 5-FU response, the drug was added 48 h after siRNA was transfected, and the respective cell cycle was measured after 24 h incubation with 5-FU. All experiments were repeated at least three times.
TP53 Status and Codon72 Variant
DNA was extracted from gastric cancer cell lines using QIAmp DNA Mini Kit (Qiagen Japan, Tokyo, Japan). PCR amplification for the p53 exon4 codon72 variant (Table S1) and the p53 exon5–9 mutation was performed as previously described [15], [16]. Each PCR product was sequenced using the ABI PRISM 3030xl genetic analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. Sequencing results were analyzed using FinchTV (PerkinElmer Japan, Tokyo, Japan) and MEGA 5.1 Beta 3 [17].
Growth Suppression Assay
Ten thousand cells per well were seeded in a 96-well microplate. Twenty-four hours later cells were treated with 5-FU for 24 h. After 5-FU treatment, the fraction of living cells was measured using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan) and a TriStar LB 941 microplate reader (Berthold Technologies, Bad Wildbad, Germany). Fifty percent growth inhibition concentration (GI50) was calculated using Prism software (Graph Pad Software, La Jolla, CA, USA). The GI50 values were used to determine correlations between 5-FU efficacy and protein levels based on the Pearson’s product-moment correlation coefficient (r).
Results
Fluorouracil Induces NF-κB
To confirm typical NF-κB behavior in response to 5-FU treatment, we traced the subcellular localization of NF-κB in the MKN45 (p53 wild type), MKN74 (p53 mutant), GSS (p53 mutant), and Kato-III (p53 homozygously deleted) cell lines. Western blot analysis with nuclear and cytoplasmic fractions demonstrated that 5-FU induced NF-κB in both compartments in MKN45 but was not observed in p53 mutant cell lines (Fig. 1A–D). We also examined the effect of 5-FU on NF-κB localization in the cells by immunocytochemistry (Fig. 1E–H). NF-κB localized to the cytoplasm in untreated MKN45, whereas 5-FU treatment caused an increase in NF-κB nuclear localization (Fig. 1E); however, no increase was observed in the p53 mutant cell lines (Fig. 1F–H). We also observed a drastic increase in phosphorylated NF-κB (p65 Ser536) in the nucleus of MKN45 treated with 5-FU, indicating NF-κB was transactivated by 5-FU (Fig. 1E). Constitutive nuclear localization and occasional phosphorylation of p53 was observed in MKN74 but did not seem to be induced by 5-FU (Fig. 1G). Nuclear localization of p53 was induced by 5-FU in GSS, but the activated signal was faint (Fig. 1H).
Figure 1. Induction and localization of NF-κB to the nucleus after 5-FU treatment in MKN45, Kato-III, MKN74, and GSS cells.

A–D, Western blot analysis of the induction of NF-κB in the nucleus and the cytoplasm by 50 µM of 5-FU for 4 h for the indicated cell lines. PCNA and α-tubulin were used as the nuclear and cytoplasmic loading controls, respectively. E–H, Immunocytochemistry of p65 and p53 induction and localization by 5-FU treatment for the indicated cell lines. p65 (red), phosphor-p65 (Ser536; red), p53 (green), phospho-p53 (Ser15; red), and DAPI (blue) staining of the nucleus without (control) and with 5-FU treatment. I, Gene expression with quantitative real-time RT-PCR in a time course of 5-FU treatment. TIGAR, PUMA, CDKN1A, and BTG2 were identified as 5-FU induced genes. The quantitative values were relative to β-actin expression.
p53 Targets are Induced upon 5-FU Treatment
Since NF-κB is a transcription factor (TF), its nuclear localization upon 5-FU treatment strongly suggests transactivation. We identified the top 7 transcripts among over 60,000 that were induced after 4 h of 5-FU treatment in MKN45 using gene expression profiling (Table 1). Interestingly, five of the 7 transcripts, namely BBC3 (which encodes p53 up-regulated modulator of apoptosis, PUMA), BTG2, C12orf5 (which encodes probable fructose-2,6-bisphosphatase TIGAR), CDKN1A, and GPR87, are known p53 targets [18]–[22]. The presence of promoter binding sites were predicted by scanning promoter sequences with the consensus sequences of NF-κB and p53 using JASPAR algorithm (Table 1) [23].
Table 1. Genes induced by 5-FU.
| Promoter binding site | |||||
| Genes (Alias) | p53 inducible(reference number) | Refseq | Promoter ID | NF-κB | p53 |
| BBC3(PUMA) | Yes [20] | NM_014417.4 | 21819 | Yes | Yes |
| BTG2 | Yes [21] | NM_006763.2 | 1764 | Yes | Yes |
| C12orf5(TIGAR) | Yes [19] | NM_020375.2 | 8600 | Yes | No |
| CDKN1A (p21/Cip1) | Yes [18] | NM_000389.4 | 35406 | Yes | Yes |
| EDN2 | – | NM_001956.3 | 3557 | Yes | No |
| GPR87 | Yes [22] | NM_023915.3 | 30365 | Yes | No |
| GRIN2C | – | NM_000835.3 | 18146 | Yes | No |
Promoter ID was obtained from http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home.
We found that p53 levels were induced in the nucleus similar to those of NF-κB in response to 5-FU treatment (Fig. 1E). Treatment of 5-FU also increased the levels of p53 phosphorylation at Ser15, suggesting its transactivation (Fig. 1E, ref. [24]). In fact, the majority of total p53 induced by 5-FU seemed to be phosphorylated. We also observed a time-dependent induction of genes, including C12orf5(TIGAR), BBC3(PUMA), CDKN1A(p21), and BTG2 by 5-FU using RT-PCR (Fig. 1I). Taken together, these results suggest that the cellular response to 5-FU treatment may involve both NF-κB and p53 for transcriptional activation in this context, in MKN45.
RELA Knockdown has a Greater Effect on p53 Target Proteins than TP53 Gene Knockdown
To evaluate the regulatory effect of NF-κB and p53 in response to 5-FU treatment, we analyzed the protein levels of p65, p53, as well as known p53 targets, p21, TIGAR, and PUMA, by western blotting following RELA and TP53 knockdown in MKN45, MKN74, GSS, and Kato-III. RELA knockdown caused a marked decrease in p53 levels in all cell lines. As expected, the levels of p21, TIGAR, and PUMA were also decreased (Fig. 2A). Conversely, while TP53 knockdown decreased p53 levels, it did not affect p65 levels. As expected, the levels of p53 target proteins were decreased by TP53 knockdown; however, this reduction was less than the reduction caused by RELA knockdown (Fig. 2B). In the cell cycle analysis, the MKN74, GSS, and Kato-III cell lines showed a slight increase in the S or G2 phase fraction whereas MKN45 exhibited an increase in the G1 fraction after 24 h exposure to 5-FU in the p65 and p53 knockdown similar to the corresponding scrambles (Fig. 2C). These results may indicate the robustness of the cellular stress response machinery that maintains the cell cycle distribution despite the knockdown of p65 and p53.
Figure 2. RELA and TP53 knockdown.
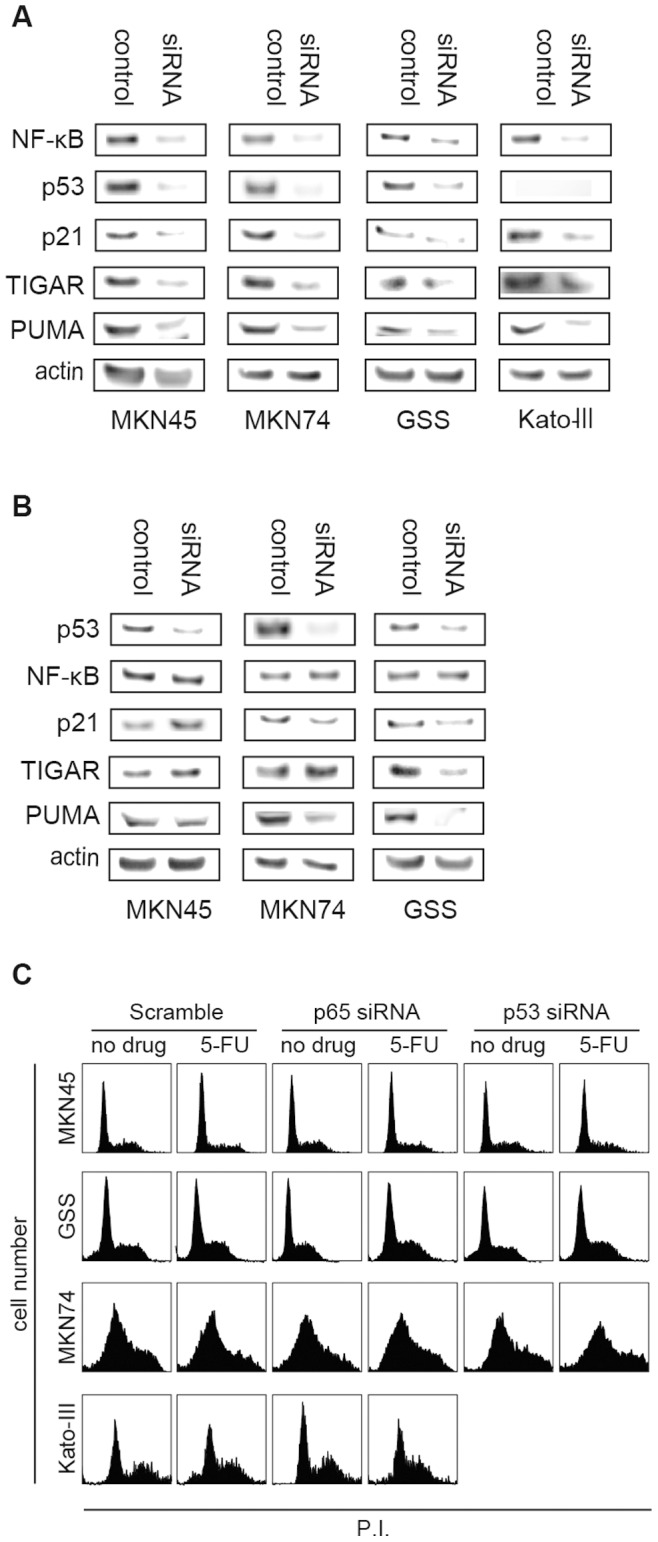
A, Western blot analysis of RELA knockdown in four gastric cancer cell lines. In addition to p53 and p65 proteins, p53 targets, including p21, TIGAR, and PUMA, were evaluated. Actin was used as a loading control. Results from cells incubated with RELA siRNA (siRNA) and without target siRNA (control) are shown. B, Western blot analysis of TP53 knockdown in three gastric cancer cell lines. p53 allele of Kato-III is homozygously deleted so TP53 knockdown was not performed. C, Drug-induced cell cycle analysis. The horizontal axis, the strength of propidium iodine (P.I.), and the vertical axis indicate the cell numbers of each cell line.
TP53 Codon72 Pro Variant Exhibits Low 5-FU Sensitivity and High NF-κB Levels
Gene knockdown experimental results indicated that the interaction between NF-κB and p53 proteins might be important in the context of 5-FU treatment. To investigate the possibility that the TP53 codon72 variant may affect cellular responses to 5-FU treatment, we sequenced TP53 codon72 as well as mutations in the DNA binding domain coding regions (i.e., exons 5–8) of 9 gastric cancer cell lines (Table 2). The status of the codon72 variation and TP53 mutation did not demonstrate clear associations.
Table 2. TP53 status in gastric cancer cell lines.
| base change, position | Effect | codon72 | |
| Kato-III | NA | NA | NA |
| KE39 | G to T (GTG to TTG) | Val to Leu | Arg/Arg |
| exon8, codon272 | |||
| MKN74 | A to C (ATC to CTC) | Ile to Leu | Arg/Arg |
| exon7, codon251 | |||
| MKN7 | wt | wt | Arg/Arg |
| NUGC4 | wt | wt | Arg/Pro |
| IWT-1 | C deletion (TCC to TCT) | Frameshift | Arg/Arg |
| exon7, codon241 | |||
| GSS | T to G (CTT to CGT) | Leu to Arg | Pro/Pro |
| exon6, codon194 | |||
| GCIY | T to G (CAT to CAG) | His to Gln | Pro/Pro |
| exon5, codon179 | |||
| MKN45 | wt | wt | Pro/Pro |
NA, Not applicable; wt, wild type.
We next investigated the association between 5-FU sensitivity and TP53 status (Fig. 3A) as well as the endogenous levels of NF-κB and p53 (Fig. 3B). GSS, GCIY, and MKN45 lines, which possess the Pro homozygous variant, exhibited low sensitivity to 5-FU, whereas the KE39, MKN74, MKN7, NUGC4, and IWT1 lines possessing Arg allele exhibited relatively high sensitivity. Kato-III, which has a large TP53 deletion [25], was the most sensitive to 5-FU. NF-κB protein levels were particularly correlated with 5-FU sensitivity (r = 0.68; p = 0.04; Fig. 3C); however, there was no clear correlation between p53 levels and 5-FU sensitivity (r = −0.04; p = 0.95; Fig. 3D). These results suggest that Pro homozygosity is associated with 5-FU resistance, while neither p53 mutation nor endogenous p53 levels directly affects the 5-FU sensitivity.
Figure 3. 5-FU sensitivity of gastric cancer cell lines and their endogenous NF-κB and p53 levels.

A, Growth suppression assay in 5-FU treatment in gastric cancer cell lines. Horizontal axes show GI50 value of 5-FU. B, Endogenous protein levels of NF-κB, p53, and actin by Western blot in gastric cancer cell lines. Actin is used as a loading control. C, Scattergram of the distribution of protein levels of NF-κB relative to actin and GI50 of 5-FU in each cell line. D, Scattergram of the distribution of p53 protein levels and GI50 of 5-FU in each cell line. Pearson’s product-moment correlation coefficient (r) as well as the p value is indicated in each scattergram.
Discussion
We have previously identified NF-κB as a potential prediction marker for post-operative 5-FU-based chemosensitivity for advanced gastric cancer [9]. NF-κB is an inducible transcription factor comprised of p65 (RelA), c-Rel, Rel-B, p50/NF-κB1, and p52/NF-κB2 [26], and plays a central role in immune responses and inflammatory cytokine regulation [27]–[29]. Chang et al previously demonstrated that NF-κB induced up or down regulation of differentially expressed genes in 5-FU-induced intestinal mucositis by inducing proinflammatory cytokines, such as IL-6, TNF-α, and IL-1β [10]. These 5-FU-induced inflammatory responses have been considered to be part of stress-avoiding processes that may lead to desensitization of 5-FU efficacy in gastric mucosa [30]. Although it has been suggested that activation of NF-κB is not directly associated with tumor development and progression [31], NF-κB has been considered to be a major biomarker and therapeutic target [32]. The direct evidence of reduced chemoresistance to 5-FU by siRNA for RELA together with the high discriminatory power of NF-κB nuclear staining in therapeutic outcomes of surgically removed tissues prompted us to perform further validation of NF-κB from a biological viewpoint.
Our transcriptional profiling results revealed that several p53 downstream genes were up-regulated in response to 5-FU, in which transactivation of NF-κB also occurred. These two major transcription factors have previously been shown to be co-regulated in response to genotoxic agents [33]–[35] and TNF-α [36], [37]. In addition, it has been proposed that co-activation of p53 and NF-κB in tumors treated with genotoxic agents could lead to therapeutic failure due to NF-κB-mediated survival signaling [38].
Individual knockdown of these transcription factors revealed genes downstream of p53 were affected by p65 knockdown, whereas the effect was limited by p53 knockdown. Previous reports have suggested a cooperative relationship between p53 and NF-κB in the context of autophagy, apoptosis, and S-phase checkpoint activation [34], [39]–[41]. Our results also indicated that NF-κB may compensate for the transcriptional activity of p53 when the intact function is lost in response to 5-FU treatment. In fact, a majority of gastric cancers bear mutations in the p53 DNA binding domain, thus rendering it transcriptionally inactive [42]. A recent study by Frank et al. reported that codon72 polymorphism of TP53 substantially affects the ability of p53 to cooperate with NF-κB for gene transactivation, particularly in the induction of apoptosis through caspase 4/11 [40]. Together with the present finding that some transcriptional activity of p53 require NF-κB in response to 5-FU, p53 codon 72 polymorphism for its NF-κB binding may be more influential than mutational status of TP53 or protein expression status of p53.
The impact of the codon72 polymorphism on spontaneous cancer risk has been previously investigated but not conclusively established due to limited human population and animal models [40], [43]–[45]. However, the codon72 polymorphism may play a role in maintaining established cancer cells than triggering cellular malignant transformation. In fact, previous studies have reported that the Pro/Pro allele is associated with resistance to chemotherapy and poor prognosis in the oral cavity [46] as well as colorectal [47], breast [48] and gastric [49] cancers and neuroblastomas [43]. A series of in vitro studies also support this hypothesis showing that the Arg allele is a more potent apoptosis inducer than the Pro allele [40], [50], [51]. Apoptosis is one of the major mechanisms induced by 5-FU and thus it is reasonable to hypothesize that the efficacy of 5-FU-based chemotherapy is associated with specific p53 polymorphisms [49]. Our in vitro findings support these epidemiological and experimental data and suggest a possible mechanism for the 5-FU-mediated p53-NF-κB interaction at the p53-codon72 binding site.
As expected, our study demonstrated that cell lines with the Pro allele were more resistant to 5-FU than those with the Arg allele. The growth suppression profile of 9 gastric cancer cell lines showed a good correlation to NF-κB protein levels. These results suggest that the Arg/Arg genotype has a stronger induction of apoptosis than the Pro/Pro genotype in the presence of 5-FU. Among the cell lines (all derived from Japanese gastric cancer patients), the ratio of Arg/Arg:Arg/Pro:Pro/Pro was 4∶1∶3, while that in healthy Japanese patients was 4.5∶4.4∶1 [52]. This may reflect a selection process that occurs during tumor development and the establishment as a cell line. Previous meta-analyses in cancer risk and p53-codon72 polymorphisms suggest that the Pro/Pro genotype has a higher cancer risk (lower for the Arg/Arg genotype) in Asian populations [45], [53]. However, the significance of “cancer risk” for cancer malignancy or treatment response remains to be elucidated, because it is generally difficult to conduct a clinical study dominated by genetic polymorphisms and assess the true genetic effects of treatment. To date, most reports describing an association between the p53 codon72 polymorphism and chemotherapeutic responses have demonstrated that the Arg/Arg genotype has a favorable response in a wide range of cancers treated with conventional genotoxic drugs [49], [54], [55]. We propose a putative mechanism for response to 5-FU via NF-κB and p53 protein binding associated with the p53 polymorphism, and thus a combinational diagnosis of NF-κB protein expression and codon72 may be a useful indicator for post-operative adjuvant chemotherapy. Aside from a few large scale datasets [52], [56], the extent of demographical and ethnic distributions of the polymorphism remains unclear. Accumulating data on ethnic differences for the polymorphism may explain the differences in cancer risk or chemotherapeutic response rate in patient population.
In summary, our findings indicate that NF-κB regulates p53 transcriptional activity in response to 5-FU, which may be associated with a polymorphic site of p53 at codon72. Further clinical and epidemiological studies should assess the utility of concomitant pathological/genetic evaluation of NF-κB/p53-codon72 in surgically-removed gastric cancer specimens in order to predict the efficacy of post-operative 5-FU-based adjuvant chemotherapy.
Supporting Information
Primer Sequences.
(DOCX)
Funding Statement
This work was supported by MIAST (Medical Innovation by Advanced Science and Technology) project of Grant-in-Aid for Strategic Medical Science Research Center from the Ministry of Education, Culture, Science and Technology of Japan, 2010–2014 (S.S.N., K.Ku., G.W.); and Grant-in-Aid for Scientific Research (C) (11863286) (S.S.N.), and (12877914) (K.Ko.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893–2917. [DOI] [PubMed] [Google Scholar]
- 2. Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, et al. (2007) Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 357: 1810–1820. [DOI] [PubMed] [Google Scholar]
- 3. Sasako M, Sakuramoto S, Katai H, Kinoshita T, Furukawa H, et al. (2011) Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 29: 4387–4393. [DOI] [PubMed] [Google Scholar]
- 4. Matsuo T, Nishizuka SS, Ishida K, Endo F, Katagiri H, et al. (2013) Evaluation of chemosensitivity prediction using quantitative dose-response curve classification for highly advanced/relapsed gastric cancer. World J Surg Oncol 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishizuka S, Charboneau L, Young L, Major S, Reinhold WC, et al. (2003) Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A 100: 14229–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spurrier B, Ramalingam S, Nishizuka S (2008) Reverse-phase protein lysate microarrays for cell signaling analysis. Nat Protoc 3: 1796–1808. [DOI] [PubMed] [Google Scholar]
- 7. Nishizuka S, Chen ST, Gwadry FG, Alexander J, Major SM, et al. (2003) Diagnostic markers that distinguish colon and ovarian adenocarcinomas: identification by genomic, proteomic, and tissue array profiling. Cancer Res 63: 5243–5250. [PubMed] [Google Scholar]
- 8. Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ Jr, et al. (1997) An information-intensive approach to the molecular pharmacology of cancer. Science 275: 343–349. [DOI] [PubMed] [Google Scholar]
- 9. Ishida K, Nishizuka SS, Chiba T, Ikeda M, Kume K, et al. (2012) Molecular marker identification for relapse prediction in 5-FU-based adjuvant chemotherapy in gastric and colorectal cancers. PLoS One 7: e43236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang CT, Ho TY, Lin H, Liang JA, Huang HC, et al. (2012) 5-Fluorouracil induced intestinal mucositis via nuclear factor-kappaB activation by transcriptomic analysis and in vivo bioluminescence imaging. PLoS One 7: e31808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang W, Cassidy J, O’Brien V, Ryan KM, Collie-Duguid E (2004) Mechanistic and predictive profiling of 5-Fluorouracil resistance in human cancer cells. Cancer Res 64: 8167–8176. [DOI] [PubMed] [Google Scholar]
- 12. Vilgelm AE, Washington MK, Wei J, Chen H, Prassolov VS, et al. (2010) Interactions of the p53 protein family in cellular stress response in gastrointestinal tumors. Mol Cancer Ther 9: 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishizuka S, Ramalingam S, Spurrier B, Washburn FL, Krishna R, et al. (2008) Quantitative protein network monitoring in response to DNA damage. J Proteome Res 7: 803–808. [DOI] [PubMed] [Google Scholar]
- 14. Nishizuka S, Winokur ST, Simon M, Martin J, Tsujimoto H, et al. (2001) Oligonucleotide microarray expression analysis of genes whose expression is correlated with tumorigenic and non-tumorigenic phenotype of HeLa x human fibroblast hybrid cells. Cancer Lett 165: 201–209. [DOI] [PubMed] [Google Scholar]
- 15. Kimura Y, Oda S, Egashira A, Kakeji Y, Baba H, et al. (2004) A variant form of hMTH1, a human homologue of the E coli mutT gene, correlates with somatic mutation in the p53 tumour suppressor gene in gastric cancer patients. J Med Genet 41: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kusser WC, Levin DB, Glickman BW (1993) Sensitive two-stage PCR of p53 genomic DNA exons 5–9. PCR Methods Appl 2: 250–252. [DOI] [PubMed] [Google Scholar]
- 17. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aliouat-Denis CM, Dendouga N, Van den Wyngaert I, Goehlmann H, Steller U, et al. (2005) p53-independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol Cancer Res 3: 627–634. [DOI] [PubMed] [Google Scholar]
- 19. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, et al. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126: 107–120. [DOI] [PubMed] [Google Scholar]
- 20. Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694. [DOI] [PubMed] [Google Scholar]
- 21. Rouault JP, Prevot D, Berthet C, Birot AM, Billaud M, et al. (1998) Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcriptional regulatory complex. J Biol Chem 273: 22563–22569. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y, Qian Y, Lu W, Chen X (2009) The G protein-coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res 69: 6049–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, et al. (2010) JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38: D105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ju J, Schmitz JC, Song B, Kudo K, Chu E (2007) Regulation of p53 expression in response to 5-fluorouracil in human cancer RKO cells. Clin Cancer Res 13: 4245–4251. [DOI] [PubMed] [Google Scholar]
- 25. Yamada Y, Yoshida T, Hayashi K, Sekiya T, Yokota J, et al. (1991) p53 gene mutations in gastric cancer metastases and in gastric cancer cell lines derived from metastases. Cancer Res 51: 5800–5805. [PubMed] [Google Scholar]
- 26. Perkins ND (2012) The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer 12: 121–132. [DOI] [PubMed] [Google Scholar]
- 27. Baldwin AS (2012) Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol Rev 246: 327–345. [DOI] [PubMed] [Google Scholar]
- 28. Goetz CA, Baldwin AS (2008) NF-κB pathways in the immune system: control of the germinal center reaction. Immunol Res 41: 233–247. [DOI] [PubMed] [Google Scholar]
- 29. Karin M, Greten FR (2005) NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5: 749–759. [DOI] [PubMed] [Google Scholar]
- 30. Nakanishi C, Toi M (2005) Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5: 297–309. [DOI] [PubMed] [Google Scholar]
- 31. DiDonato JA, Mercurio F, Karin M (2012) NF-κB and the link between inflammation and cancer. Immunol Rev 246: 379–400. [DOI] [PubMed] [Google Scholar]
- 32. Uetsuka H, Haisa M, Kimura M, Gunduz M, Kaneda Y, et al. (2003) Inhibition of inducible NF-κB activity reduces chemoresistance to 5-fluorouracil in human stomach cancer cell line. Exp Cell Res 289: 27–35. [DOI] [PubMed] [Google Scholar]
- 33. Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC (2004) p53 induces NF-κB activation by an IκB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem 279: 26115–26125. [DOI] [PubMed] [Google Scholar]
- 34. Ryan KM, Ernst MK, Rice NR, Vousden KH (2000) Role of NF-κB in p53-mediated programmed cell death. Nature 404: 892–897. [DOI] [PubMed] [Google Scholar]
- 35. Schumm K, Rocha S, Caamano J, Perkins ND (2006) Regulation of p53 tumour suppressor target gene expression by the p52 NF-κB subunit. EMBO J 25: 4820–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, et al. (2007) Mutant p53 enhances nuclear factor κB activation by tumor necrosis factor alpha in cancer cells. Cancer Res 67: 2396–2401. [DOI] [PubMed] [Google Scholar]
- 37. Wu H, Lozano G (1994) NF-κB activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem 269: 20067–20074. [PubMed] [Google Scholar]
- 38. Schneider G, Kramer OH (2011) NFκB/p53 crosstalk-a promising new therapeutic target. Biochim Biophys Acta 1815: 90–103. [DOI] [PubMed] [Google Scholar]
- 39. Barre B, Perkins ND (2010) The Skp2 promoter integrates signaling through the NF-κB, p53, and Akt/GSK3beta pathways to regulate autophagy and apoptosis. Mol Cell 38: 524–538. [DOI] [PubMed] [Google Scholar]
- 40. Frank AK, Leu JI, Zhou Y, Devarajan K, Nedelko T, et al. (2011) The codon 72 polymorphism of p53 regulates interaction with NF-κB and transactivation of genes involved in immunity and inflammation. Mol Cell Biol 31: 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schneider G, Henrich A, Greiner G, Wolf V, Lovas A, et al. (2010) Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 29: 2795–2806. [DOI] [PubMed] [Google Scholar]
- 42. Fenoglio-Preiser CM, Wang J, Stemmermann GN, Noffsinger A (2003) TP53 and gastric carcinoma: a review. Hum Mutat 21: 258–270. [DOI] [PubMed] [Google Scholar]
- 43. Cattelani S, Ferrari-Amorotti G, Galavotti S, Defferrari R, Tanno B, et al. (2012) The p53 codon 72 Pro/Pro genotype identifies poor-prognosis neuroblastoma patients: correlation with reduced apoptosis and enhanced senescence by the p53–72P isoform. Neoplasia 14: 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whibley C, Pharoah PD, Hollstein M (2009) p53 polymorphisms: cancer implications. Nat Rev Cancer 9: 95–107. [DOI] [PubMed] [Google Scholar]
- 45. Zhou Y, Li N, Zhuang W, Liu GJ, Wu TX, et al. (2007) P53 codon 72 polymorphism and gastric cancer: a meta-analysis of the literature. Int J Cancer 121: 1481–1486. [DOI] [PubMed] [Google Scholar]
- 46. Kuroda Y, Nakao H, Ikemura K, Katoh T (2007) Association between the TP53 codon72 polymorphism and oral cancer risk and prognosis. Oral Oncol 43: 1043–1048. [DOI] [PubMed] [Google Scholar]
- 47. Godai TI, Suda T, Sugano N, Tsuchida K, Shiozawa M, et al. (2009) Identification of colorectal cancer patients with tumors carrying the TP53 mutation on the codon 72 proline allele that benefited most from 5-fluorouracil (5-FU) based postoperative chemotherapy. BMC Cancer 9: 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Proestling K, Hebar A, Pruckner N, Marton E, Vinatzer U, et al. (2012) The Pro allele of the p53 codon 72 polymorphism is associated with decreased intratumoral expression of BAX and p21, and increased breast cancer risk. PLoS One 7: e47325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang ZH, Hua D, Li LH, Zhu JD (2008) Prognostic role of p53 codon 72 polymorphism in gastric cancer patients treated with fluorouracil-based adjuvant chemotherapy. J Cancer Res Clin Oncol 134: 1129–1134. [DOI] [PubMed] [Google Scholar]
- 50. Dumont P, Leu JI, Della Pietra AC 3rd, George DL, Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33: 357–365. [DOI] [PubMed] [Google Scholar]
- 51. Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, et al. (2007) The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat Struct Mol Biol 14: 912–920. [DOI] [PubMed] [Google Scholar]
- 52. Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, et al. (2005) Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer 114: 730–737. [DOI] [PubMed] [Google Scholar]
- 53. Francisco G, Menezes PR, Eluf-Neto J, Chammas R (2011) Arg72Pro TP53 polymorphism and cancer susceptibility: a comprehensive meta-analysis of 302 case-control studies. Int J Cancer 129: 920–930. [DOI] [PubMed] [Google Scholar]
- 54. Santos AM, Sousa H, Portela C, Pereira D, Pinto D, et al. (2006) TP53 and P21 polymorphisms: response to cisplatinum/paclitaxel-based chemotherapy in ovarian cancer. Biochem Biophys Res Commun 340: 256–262. [DOI] [PubMed] [Google Scholar]
- 55. Xu Y, Yao L, Ouyang T, Li J, Wang T, et al. (2005) p53 Codon 72 polymorphism predicts the pathologic response to neoadjuvant chemotherapy in patients with breast cancer. Clin Cancer Res 11: 7328–7333. [DOI] [PubMed] [Google Scholar]
- 56. Tommiska J, Eerola H, Heinonen M, Salonen L, Kaare M, et al. (2005) Breast cancer patients with p53 Pro72 homozygous genotype have a poorer survival. Clin Cancer Res 11: 5098–5103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer Sequences.
(DOCX)