

Abstract
Apolipoprotein E (apoE), an anti-atherogenic apolipoprotein, plays a significant role in the metabolism of lipoproteins. It lowers plasma lipid levels by acting as a ligand for low-density lipoprotein receptor (LDLr) family of proteins, in addition to playing a role in promoting macrophage cholesterol efflux in atherosclerotic lesions. The objective of this study is to examine the effect of acrolein modification on the structure and function of rat apoE and to determine sites and nature of modification by mass spectrometry. Acrolein is a highly reactive aldehyde, which is generated endogenously as one of the products of lipid peroxidation and is present in the environment in pollutants such as tobacco smoke and heated oils. In initial studies, acrolein-modified apoE was identified by immunoprecipitation using an acrolein-lysine specific antibody, in the plasma of ten-week old male rats that were exposed to filtered air (FA) or low doses of environmental tobacco smoke (ETS). While both groups displayed acrolein-modified apoE in the lipoprotein fraction, the ETS group had higher levels in lipid-free fraction compared to the FA group. This observation provided the rationale to further investigate the effect of acrolein modification on rat apoE at a molecular level. Treatment of recombinant rat apoE with a 10-fold molar excess of acrolein resulted in: (i) a significant decrease in lipid-binding and cholesterol efflux abilities, (ii) impairment in the LDLr- and heparin-binding capabilities, and (iii) significant alterations in the overall stability of the protein. The disruption in the functional abilities is attributed directly or indirectly to acrolein modification yielding: an aldimine adduct at K149 and K155 (+38); a propanal adduct at K135 and K138 (+56); an Nε-(3-methylpyridinium)lysine (MP-lysine) at K64, K67 and K254 (+76), and Nε-(3-formyl-3,4-dehydropiperidino)lysine (FDP-lysine) derivative at position K68 (+94), as determined by Matrix-Assisted Laser Desorption/Ionization-Time of Flight/Time of Flight Mass Spectrometry (MALDI-TOF/TOF MS). The loss of function may also be attributed to alterations in the overall fold of the protein as noted by changes in the guanidine HCl-induced unfolding pattern and to protein cross-linking. Overall, disruption of the structural and functional integrity of apoE by oxidative modification of essential lysine residues by acrolein is expected to affect its role in maintaining plasma cholesterol homeostasis and lead to lipid dysregulation.
Keywords: Mass spectrometry, Cardiovascular disease, rat apolipoprotein E, acrolein, lipid binding, LDL receptor binding, cholesterol efflux, environmental tobacco smoke
Apolipoprotein E (apoE) is a 34-kDa anti-atherogenic protein that plays a crucial role in cardiovascular disease by regulating plasma cholesterol levels and lipoprotein metabolism.1, 2 By assisting in the transportation of very low-density lipoproteins (VLDL) and chylomicron remnants, apoE is able to remove excess cholesterol and triglyceride from the blood and into the liver for further processing. It mediates this role by acting as a ligand for cell surface localized lipoprotein receptors such as the low density lipoprotein receptor (LDLr) and heparan sulfate proteoglycans (HSPG).2, 3 ApoE has the ability to exist in lipid-free and lipid- (lipoprotein)-bound states, and undergoes a dramatic conformational change when going from one state to the other. Its ability to act as a ligand for the LDLr is elicited only in the lipid-bound state; 4, 5 thus, its lipid-binding ability is a key functional prerequisite for apoE. ApoE also plays a role in promoting ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux from macrophages in atherosclerosis, a function that is typically mediated by apoAI under normal physiological conditions.6
The importance of apoE in lipoprotein metabolism is exemplified by studies with apoE-null mice, which display massive accumulation of cholesterol and triglyceride-rich lipoproteins in plasma, and develop early and severe atherosclerotic lesions.7, 8 On the other hand, targeted over-expression of apoE in this mouse model lead to a marked resistance to diet-induced hypercholesterolemia and decreased plasma cholesterol levels.9 In humans, apoE deficiency leads to type III hyperlipoproteinemia and premature development of atherosclerosis characterized by elevated plasma cholesterol levels and accumulation of VLDL.10-12 These early studies established the key role of apoE in regulating plasma cholesterol and triglyceride levels, elevated levels of which are recognized as two of several risk factors for heart disease. The broad objective of this study is to investigate the role of oxidative stress modification of apoE and the potential molecular and physiological implications of the process.
Oxidative stress is recognized as a major factor in the onset of atherosclerosis and cardiovascular disease in humans 13, with aging and life style (diet, smoking, exercise) playing significant roles.14 Several lines of evidence implicate increased susceptibility of LDL to oxidative modification as potential factors.15-17 Oxidative modification of LDL leads to their uptake by scavenger receptors located on macrophages, which eventually are converted to foam cells and deposited as fatty streaks in the vasculature, a signature feature of atherosclerotic plaques.18 Both oxidized lipids 19-22 and oxidized proteins are believed to play crucial roles in atherogenesis, with oxidative damage to proteins such as apoB-100 likely increasing the atherogenicity of LDL.23-26 However, little is known about the potential oxidative damage to apoE, a key protein component of chylomicrons, VLDL and a sub-type of HDL, and the contribution of oxidatively modified apoE to atherogenesis.
In this study we follow modification of apoE by acrolein, one of the end products of lipid peroxidation generated endogenously in a process triggered by oxidative stress.27 Acrolein is an α,β-unsaturated aldehyde (2-propenal, CH2=CH-CHO) that is also present in the environment in pollutants such as tobacco smoke and heated oils. It is the strongest electrophile among all α,β-unsaturated aldehydes. Acrolein has been shown to play a significant role in oxidative modification of apoB-100 on LDL.28 In this study, we note that endogenous generation of acrolein modifies apoE in rats; exposure of rats to environmental tobacco smoke (ETS) results in increased presence of acrolein-modified apoE in the lipoprotein-dissociated state. Further, direct exposure of purified recombinant rat apoE to acrolein caused impairment in LDLr-and HSPG-binding abilities, and in lipid-binding and cholesterol efflux capabilities. We attributed the dysfunction of apoE to significant protein side chain modification of essential lysine residues mediated by acrolein and to overall alterations in the global protein fold.
Experimental Procedures
Animal exposures
The animal studies were carried out in accordance with the declaration of Helsinki and with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. They were in conformity with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by the University of California Davis, Institutional Review Board. Standardized research cigarettes were purchased from the University of Kentucky (Louisville, KY). Male Sprague-Dawley rats (10 weeks old) were exposed to aged and diluted side stream cigarette smoke (total suspended particulates (TSP) 0.97 ± 0.05 mg/m3) as a surrogate to ETS. Rats (12 per group) were exposed for 6 h/ day for 3 consecutive days (body weight, 278 ± 9 g, mean ± S.D.); animals in the control group received filtered air (FA) under sham conditions (body weight, 276 ± 10 g, mean ± S.D.). Research cigarettes (3R4F) obtained from the University of Kentucky were maintained under humified and temperature-controlled conditions in a dessicator prior to use. Cigarettes were combusted using a TE10 smoking machine (Teague Enterprises, Woodland, CA) using a smoke puff volume setting of 35 ml of 2 seconds duration once per minute over a period of 8 minutes prior to the automatic replacement with a new cigarette in the smoking machine. Between smoke puffs, side stream cigarette smoke was collected and mixed with the mainstream puff volume, then passed through an aging and dilution chamber prior to further dilution and introduction into a whole body animal exposure system. Tobacco exposure conditions to aged and diluted smoke for the exposure period were: relative humidity, 65 ± 1.7%; temperature ,73 ± 1.2 °F; TSP, 0.97 ± 0.05 mg/m3, nicotine, 0.25 ± 0.06 mg/m3; and, carbon monoxide, 3.6 ± 0.3 ppm. The conditions of exposure to ETS are highly representative of concentrations encountered in the home or other places where smoking occurs. It is known for people who smoke, that they create a personal cloud of TSP approximating 2 mg/m3. Blood was withdrawn on the 3rd smoke exposure day, centrifuged at 1000 × g for 15 min to obtain plasma.
Plasma lipoprotein isolation
Total lipoprotein fraction (d <1.21 g/ml) was isolated from plasma by density gradient ultracentrifugation. Prior to use, the lipoprotein and lipid-free fractions (top and bottom fractions, respectively) were dialyzed against three changes of degassed 10 mM sodium phosphate, pH 7.4 containing 150 mM NaCl (phosphate buffered saline, PBS). Plasma apoE levels were determined by ELISA using mAb3H1. Plasma triglyceride and cholesterol levels were measured by enzymatic-endpoint reagent kits, according to the manufacturer's instructions, on a Gilford Impact500E auto analyzer (Ciba-Corning Diagnostics Corp., Oberlin, OH).
Immunoprecipitation
Protein G-Sepharose (GE Healthcare, Pittsburgh, PA) was incubated with 0.1μg mAb5F6 (an antibody specific for acrolein-lysine adducts, obtained from Japan Institute for the Control of Aging, Fukuroi, Shizuoka, 437-0122 Japan), in PBS containing 0.1% Tween 20 for 1 h at 4 °C and washed four times with this buffer to remove unbound antibody. The total lipoprotein or lipid-free fraction (25 μg protein) isolated from plasma of rats exposed to FA or ETS was then incubated with conjugated Protein G-Sepharose for 1 h at 24 °C, followed by 4 washes as above to remove unbound proteins. The samples were treated with non-reducing SDS sample treatment buffer for Western blot analysis using horseradish peroxidase (HRP)-conjugated goat anti-human apoE antibody (BIODESIGN International (Saco, ME).
Acrolein modification of rat apoE
Recombinant rat apoE bearing a hexa-His tag was overexpressed, isolated and purified using a HiTrap nickel-affinity column (HisTrap™ HP, GE Healthcare, Uppsala, Sweden) as described recently29. In initial experiments, purified apoE (5 mg) was incubated with acrolein (Alfa Aesar, Ward Hill, MA) (1:2.5 to 2000:1 molar ratio, acrolein:apoE) in PBS for 4 h at 37 °C. In control reactions, apoE was incubated as such, with no additives. Excess un-reacted acrolein was removed by extensive dialysis against PBS for 48 h with 3 changes. Unmodified and acrolein-modified apoE were visualized by SDS-PAGE on a 4-20% acrylamide gradient gel (Invitrogen, Carlsbad, CA). In subsequent experiments, we focused on apoE modified with acrolein at 10:1 molar ratio (acrolein:apoE).
Western Blot
Western blot analysis of unmodified and acrolein-modified apoE (∼0.5 μg) was carried out using anti-apoE-HRP antibody (1:1000 dilution), or, mAb5F6 (1:500 dilution) 30 followed by HRP-conjugated anti-mouse IgG (1:10,000 dilution) (Chemicon) for detecting acrolein-lysine modifications (Nε -(3-formyl-3,4-dehydropiperidino)lysine (FDP-lysine), using the enhanced chemiluminescence (ECL) detection system (GE Healthcare, Uppsala, Sweden).
Circular dichroism (CD) spectroscopy
The secondary structure of rat apoE was examined by CD spectroscopy on a Jasco J-810-150S spectropolarimeter at 24 °C.31 Far-UV CD scans were recorded between 185 nm and 260 nm in 10 mM sodium phosphate buffer, pH 7.4 using protein concentrations of 0.2 mg/ml in a 0.1 cm path length cuvette. Far-UV CD profiles were the average of four independent scans recorded with a response time of 1s and bandwidth of 1 nm. The molar ellipticity ([θ]) in deg. cm2dmol-1 at 222 nm was obtained using the equation:
| (1) |
where MRW is the mean residue weight (obtained by dividing molecular weight by the number of residues) calculated to be 115.64, θ is the measured ellipticity in degrees at 222 nm, l is the cuvette path-length (in cm) and c is the protein concentration (in g/ml). The percent α-helix content was calculated as described by others32:
| (2) |
GdnHCl-induced unfolding
Unfolding of rat apoE was assessed by following changes in molar ellipticity at 222 nm as a function of increasing concentration of GdnHCl as described earlier.33 The samples (0.2 mg/ml) were treated with 0-6 M GdnHCl in 10mM sodium phosphate buffer, pH 7.4 for 18 h at 24 °C. The % maximal change was calculated from the ellipticity values at 222 nm as:
| (3) |
Free energy of stabilization of unmodified and acrolein-modified apoE was calculated using the relationship:
| (4) |
where ΔGD is the free energy change between 0 and 6 M GdnHCl, ΔGDH2O is the free energy of denaturation in the absence of denaturant, Δn is the difference in the number of GdnHCl binding sites between the native and denatured states of protein, R is the gas constant, T is the temperature, k is the GdnHCl binding constant (0.8), and a is the molarity of GdnHCl. The free energy change ΔGD was calculated as described previously.34,35
Fluorescence spectroscopy
Steady state fluorescence analyses were performed on a Perkin-Elmer LS55B fluorometer at 24 °C. Fluorescence emission spectra of unmodified or acrolein-modified apoE (0.02 mg/ml) were recorded in PBS between 290 and 490 nm following excitation at 280 nm, at a scan speed 50 nm/min (excitation and emission slit widths); an average of 10 scans were recorded.
Lipid binding assay
The ability of unmodified and acrolein-modified apoE to bind lipids was determined as described previously.29, 36 The assay was performed in a Perkin Elmer UV/VIS spectrophotometer equipped with a Peltier controlled (PTP-6) cell holder. 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti Polar Lipids, Alabaster, AL) MLV (500 μg) was pre-incubated at 24 °C in PBS in the cuvette followed by addition of 100 μg apoE. The decrease in absorbance was measured at 325 nm. Data were normalized to initial absorbance at 325 nm prior to addition of protein. The time required for initial absorbance to decrease by 50% (T1/2) and the rate constant (K), (reciprocal of T1/2), were determined.
Preparation of DMPC/apoE complex
DMPC/apoE complexes were prepared as described earlier 37 using rat apoE or human apoE3(1-191). ApoE was incubated with DMPC vesicles (2.5:1 weight ratio or 125:1 molar ratio) at 24 °C for 16 h; lipid-bound protein was separated from the unbound protein by density gradient ultracentrifugation using a KBr gradient. About 10 fractions were collected and protein assay carried out on each fraction by the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL), and phospholipid assay using the phospholipid assay kit (Wako Chemicals USA, Inc., Richmond, VA). Fractions containing both protein and lipid were pooled and concentrated. The lipoprotein complexes were characterized in terms of particle size and diameter, lipid and protein composition as described earlier.38, 39 The protein and lipid contents of the complexes were estimated using the DC™ protein (BioRad Laboratories, Hercules, CA) and phospholipid assay. Non-denaturing PAGE of the isolated lipoprotein complexes was carried out to evaluate the molecular mass and average particle size using Amersham High Molecular Weight Standard (GE Healthcare, Uppsala, Sweden) on a 4–20% gradient gel for 24 h at 110 V and stained with Amido Black.
LDLr binding assay
To examine the ability of acrolein-modified apoE to bind to the LDLr, a co-immunoprecipitation (co-IP) assay was performed as described previously.40 A construct bearing the LDLr ligand binding domains LA3-LA6 with a c-Myc epitope was employed. This construct represents the essential ligand binding elements of the extra cellular soluble portion of the mature LDLr and is represented as sLDLr. DMPC/unmodified apoE or DMPC/acrolein-modified apoE (0, 1 and 10 μg protein) was incubated with 10 μg of sLDLr in the presence of 2 mM Ca+2 in PBS for 1 h at 4 °C. This was followed by co-IP with an anti-c-Myc antibody-linked agarose (Sigma-Aldrich, St. Louis, MO) to capture the DMPC/apoE/sLDLr complexes. ApoE was detected by Western blot analysis using HRP-conjugated polyclonal apoE antibody. A replica experiment was conducted wherein an anti-c-Myc antibody (9E10) was utilized to identify the presence of LDLr in each reaction.
Heparin binding assay
A typical functional feature of apoE is its ability to interact with cell surface-localized HSPG in the blood vessels. In vitro, this is followed using the HiTrap heparin-Sepharose column (GE Healthcare, Uppsala, Sweden) appended to ÄKTA FPLC system (Amersham Pharmacia Biotech, Uppsala, Sweden) as described by us previously.41, 42 Unmodified or acrolein-modified apoE was dialyzed against 20 mM sodium phosphate buffer, pH 7.4, and injected onto the column at a flow rate of 1.0 ml/min. The bound protein was eluted with a salt gradient (0 to 1.0 M NaCl) and monitored at 280 nm. The salt concentration in the eluate was monitored by conductivity measurements.
Cholesterol efflux activity
The ability of unmodified or acrolein-modified apoE to stimulate cholesterol efflux was assessed in J774 mouse macrophages (which do not synthesize apoE) as described previously.43-45 Cells were plated onto 24-well culture plates and labeled with [3H]cholesterol (1 μCi/mL) in RPMI-1640 with 1% fetal bovine serum (FBS) for 48 h. A cAMP analogue (cpt-cAMP) was added (18 h) to up-regulate ABCA1 expression. Cells were rinsed extensively, and then exposed to unmodified or acrolein-modified apoE in serum-free RPMI-1640 medium at the indicated concentrations. In control experiments, unmodified or acrolein-modified human plasma-derived apoAI was used under identical conditions. The amount of [3H]cholesterol appearing in the medium was expressed as a percentage of the radioactivity initially present in cells at time zero. Background release of [3H]cholesterol to serum-free medium was subtracted from values obtained with added proteins.
Mass Spectrometric Analysis
Purified recombinant rat apoE was incubated with acrolein (10-fold molar excess over apoE) at 37 °C for 4 h. For identification of acrolein-modified sites, all sample proteolytic digestions were performed with sequencing-grade trypsin (Promega, Madison, WI), AspN (from a mutant of Pseudomonas fragi) (Sigma-Aldrich, St. Louis, MO) and GluC (from Staphylococcus aureus V8) (Sigma-Aldrich, St. Louis, MO) or AspN + GluC in solution. Samples were incubated with a 50:1 ratio (protein:protease w/w) of GluC for 40 h at 37 °C in 100 mM ammonium bicarbonate, pH 8.0. Approximately 18 h prior to termination of GluC digestion, a 100:1 ratio (protein:protease w/w) of AspN was added to the mixture and the incubation continued to completion. The proteolytic digest was cleaned using C18 ZipTip® Pipette Tips (Millipore Corporation, Billerica, MA) as per manufacturer's instructions. The digests of unmodified or acrolein-modified apoE were then subjected to matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry (MALDI TOF/TOF MS). The samples were directly spotted on MALDI target plates, mixed with alphacyano-4-hydroxy cinnamic acid (CHCA) matrix (Protea Co, Morgantown, WV) in 50% acetonitrile (Millipore Corporation, Billerica, MA), 0.1% trifluoroacetic acid (Fisher Scientific, Fair Lawn, NJ) and then analyzed on an AB 4800 mass analyzer (Applied Biosystems, (South San Francisco, CA). Mass spectra were collected from 1000 laser shots per spot and tandem mass spectrometric (MS/MS) data were collected from 3000 laser shots in 4800 mass analyzer. The peptides with signal-to-noise ratio above 15 at the MS mode were selected as the strongest peaks first for MS/MS experiment; a maximum of 20 MS/MS was allowed per spot. Mass calibration in MALDI TOF MS and MALDI TOF/TOF MS/MS was achieved through internal calibration (TOF/TOF Calibration Mixture, AB SCIEX). The spectra of digested peptides were acquired in the positive reflectron mode using an accelerating voltage of 20kV. Raw data files obtained from the AB 4800 mass analyzer were processed using GPS Explorer ™ version 3.6 (Applied Biosystems, South San Francisco, CA) and then searched against Swiss-Prot protein sequence database downloaded from UniProt under the genus restriction of Rattus using the in-house licensed Mascot searching program (version 2.1.03). Note that the residue numbering matches that of the native sequence: the His-tag introduced for the purpose of isolation and purification is not numbered. The following parameters were specified: (i) enzyme, AspN+GluC; (ii) missed cleavage, 2; (iii) variable modification, methionine oxidation, Acrolein 112(K), Acrolein38 (K), Acrolein56 (C), Acrolein56 (H), Acrolein56 (K), Acrolein76 (K) and Acrolein94 (K); (iv) peptide tolerance, 300 ppm; and (v) MS/MS tolerance of 0.8 Da. The peptides containing acrolein-modified amino acids with p<0.05 and, peptide ion score and ion score confidence were further confirmed by manually checking the MS/MS spectra. Quantification of acrolein labeling was carried out using peak area-based label free quantitation method.46 MS/MS spectra were further processed by Data Explorer software (Version 4.9) (Applied Biosystems) with the noise removal method to reduce background noise and improve the signal to noise ratio.
Results
Rat apoE is a 34 kDa, 294 amino acid protein with 73.5 % sequence similarity with human apoE3, Figure 1. It is rich in basic residues bearing 32 Arg and 11 Lys, many of which play a critical role in interacting with the LDLr family of proteins to facilitate receptor-mediated endocytosis, and in maintaining the structural integrity of the protein. Lysine residues are considered particularly susceptible to oxidative modification; in initial studies, we determined if plasma apoE is modified by acrolein in vivo as a result of natural cellular oxidation or due to environmental oxidative stress by exposing 10 -week old male rats to FA or low dose of ETS. ELISA revealed that the total plasma apoE levels were slightly lower in ETS group compared to the FA group (0.034 ± 0.016 versus 0.049 ± 0.009 mg/ml for ETS and FA groups, respectively) (P < 0.2); when the lipid-free fraction alone was examined, the apoE level was found to be marginally higher in the ETS than in the FA group (0.025 ± 0.001 versus 0.019 ± 0.006 mg/ml for ETS and FA groups, respectively) (P < 0.2). The lipoprotein and lipid-free fractions of the ETS and FA-exposed groups were then evaluated for acrolein modification. IP analysis was performed in lipoprotein fractions pooled from 3 rats, Figure 2A. Acrolein modification of apoE was noted in both groups, with no significant differences in the extent of modification between the two. On the other hand, in the lipid-free fractions, Figure 2B, the ETS group displayed more acrolein-modified apoE compared to the FA group. An exception was one pooled fraction in the FA group (lane 2) that showed an intense acrolein-modified apoE band (the reason for this outlier is not known; it is possibly due to variations in endogenous lipid peroxidation, as discussed later). Examining individual rat plasma samples, we identified the possible contribution of one individual outlier that displays the intense acrolein modification in the FA group (not shown).
Figure 1.

Comparison of structural features of human apoE3 and rat apoE. The amino acid sequence of human apoE3 (Top) and rat apoE (Bottom) are shown (the His-tag sequence is omitted for convenience). For both, the residues predicted to adopt an amphipathic α-helix or random coil (based on http://bioinf.cs.ucl.ac.uk/psipred) are indicated as “H” and “C”, respectively, below the sequence. In addition, for human apoE3 the α-helical segments (derived from the NMR structure PDB ID# 2L7B) are shown as grey cylinders for validation of the prediction. All Lys residues are shown in bold, the major LDLr binding sites in human apoE3 in red, the predicted LDLr binding sites in rat apoE based on sequence alignment in blue. The major HSPG binding sites in the NT domain are underlined for both (those for rat apoE are based on sequence alignment). Residues modified by acrolein in rat apoE under the conditions employed in this study as identified by MALDI TOF/TOF are indicated with a star (*)below the sequence.
Figure 2. Effect of FA or ETS exposure on oxidative status of plasma apoE: Detection of acrolein-modified apoE.
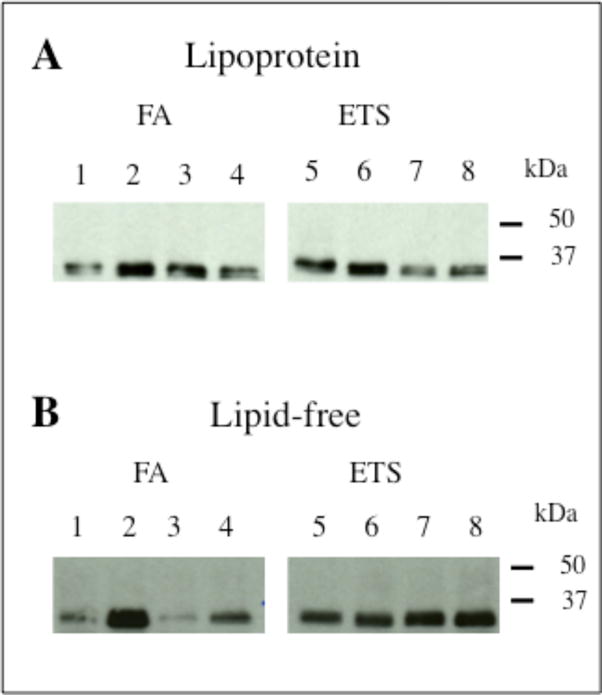
The lipoprotein (A) and lipid-free fractions (B) (25 μg protein) of rats exposed to FA or ETS were incubated with 5F6 conjugated to Protein G-Sepharose to capture all acrolein-modified proteins, followed by Western blot using anti-apoE-HRP.
In terms of the lipid peroxidation status of plasma (i.e., free and protein-bound Schiff base conjugates), there was no significant difference in the total MDA levels between the two groups (FA group = 16.5 ± 1.3 and ETS = 15.7 ± 1.6 μM). In addition, the susceptibility of LDL Cu2+-induced lipid peroxidation was evaluated. Lipid peroxidation was initiated by addition of 50 μM freshly prepared CuSO4 to LDL from FA or ETS group maintained at 37 °C. For the two groups, the total diene concentration (169.7 ± 46.2 and 168.3 ± 57.8 nmol/mg LDL protein for FA and ETS groups, respectively) and the propagation rates, 2.8 ± 0.8 and 2.8 ± 0.9 nmol dienes formed/min/mg LDL protein, respectively (data not shown) were similar; however, the lag period for the ETS group was shorter (∼12 min) than that for the FA group (∼20 min). Further, there was no difference in the total cholesterol levels between the two groups (FA, 74.4 ± 3.7 mg/dL; ETS, 72.1 ± 1.2 mg/dL); the triglyceride levels were ∼25% higher in the ETS compared to the FA group (115 ± 9.5 and 92 ± 17.2 mg/dL, respectively; n=12, P < 0.3). Nonetheless, the observation that plasma apoE can be oxidatively modified provided the rationale for us to propose that acrolein modification disrupts key functional features of apoE.
To test the molecular basis of this hypothesis, purified recombinant rat apoE was treated with PBS or increasing amounts acrolein in PBS (1:2.5 to 2000:1 molar ratio, acrolein:apoE), followed by extensive dialysis to remove unbound acrolein. SDS-PAGE analysis under reducing conditions, Figure 3A, indicated a variety of species depending on the molar excess of acrolein used: at 1:2.5, 1:1 and 2:1 ratios (lanes 2, 3 and 4, respectively), the major species were monomeric with minimal inter-molecular cross-linking. At 20:1 ratio (lane 5) there was evidence of significantly modified monomeric species as seen by the slight shift in the 34 kDa band to lower mobility; in addition covalently cross-linked dimeric and some oligomeric species were noted (indicated by arrows). Between 200:1 and 2000:1 ratios (lanes 6-9), there was a progression towards formation of higher molecular weight species. In subsequent experiments we used 10:1 ratio, the rationale being that there are 11 lysines in rat apoE and there would be ∼1 acrolein for each lysine. At this ratio, there is evidence of cross-linked dimer, as seen in the immunoblot using anti-apoE-HRP, Figure 3B. Acrolein modification was verified by Western blot using mAb5F6, Figure 3C, lane 2; the results confirm that acrolein modification of recombinant apoE generates epitopes that are recognized by 5F6, indicative of lysine modification. In addition, the presence of higher-molecular weight bands is indicative of acrolein-facilitated inter-molecular cross-linking via lysines (indicated with arrows). The presence of monomeric band in the immunoblot is indicative of lysine modification without inter-molecular cross-linking. As expected, the 5F6 antibody does not recognize unmodified recombinant apoE as noted in control experiments, Figure 3C, lane 1.
Figure 3. Acrolein modification of recombinant rat apoE.
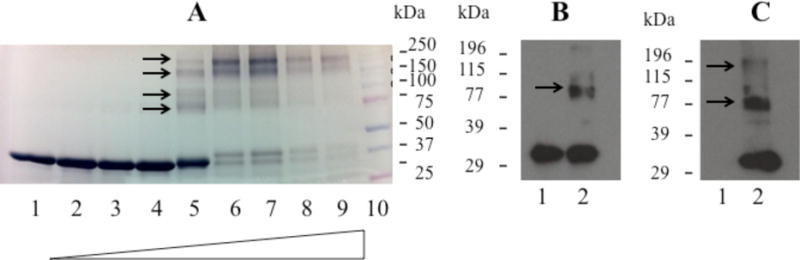
About 10 μg of apoE was treated with PBS (Lane 1) or increasing amounts of acrolein at the ratios indicated below, electrophoresced on a 4-20% acrylamide gradient gel and stained with Amido Black (Panel A). The lane assignments for the various acrolein: apoE molar ratios were as follows: Lane 2, 1:2.5; Lane 3, 1:1; Lane 4, 2:1; Lane 5, 20:1; Lane 6, 200:1; Lane 7, 400:1; Lane 8, 1000:1; Lane 9, 2000:1. Lane 10 shows the low molecular mass standards. Arrows draw attention to cross-linked species. Immunoblot analyses of 0.5 μg unmodified (Lane 1) and acrolein-modified (Lane 2) apoE was carried out with anti-apoE-HRP (Panel B) and 5F6 (Panel C) antibody for apoE modified at 10:1 acrolein: apoE ratio.
Far-UV CD spectra of acrolein-modified apoE shows typical features of a highly helical protein, characterized by troughs at 208 and 222 nm, Figure 4A. The α-helical content was calculated from the molar ellipticity values at 222 nm by eqn. 2 to be 46 ± 6 %, which is very similar to that noted for unmodified apoE, 45 ± 5%, as reported recently.29 GdnHCl-induced unfolding of acrolein-modified apoE was performed by following changes in the molar ellipticity at 222 nm as a function of varying GdnHCl concentration, Figure 4B. Unmodified apoE undergoes a two-phase unfolding process with a distinct plateau between the two phases, corresponding to unfolding of the C-terminal and N-terminal domains (midpoints of denaturation around 0.8 and 2.2 M GdnHCl, respectively). Each phase of the biphasic denaturation profile was treated as individual two-state denaturation equilibrium as described earlier.34 The corresponding ΔGDH2O values using eqn. 4 are 4.7 ± 1.7 and 9.9 ± 1.6 kcal/mol, respectively. In contrast, acrolein-modified apoE displayed a single-phase denaturation profile, with a midpoint of denaturation corresponding to 2.06 ± 0.3 M GdnHCl and a ΔGDH2O of 1.5 ± 0.2 kcal/mol, suggesting the possibility of domain-domain interaction and a change in the overall tertiary fold. This is supported by a 25% decrease in intrinsic fluorescence emission at 350 nm (predominantly due to Trp), upon excitation at 280 nm, Figure 4C. Together these results are indicative of alterations in the tertiary fold, including intra-molecular crosslinking between the N and C-terminal domains.
Figure 4. Effect of acrolein modification on secondary structure and tertiary fold of apoE.
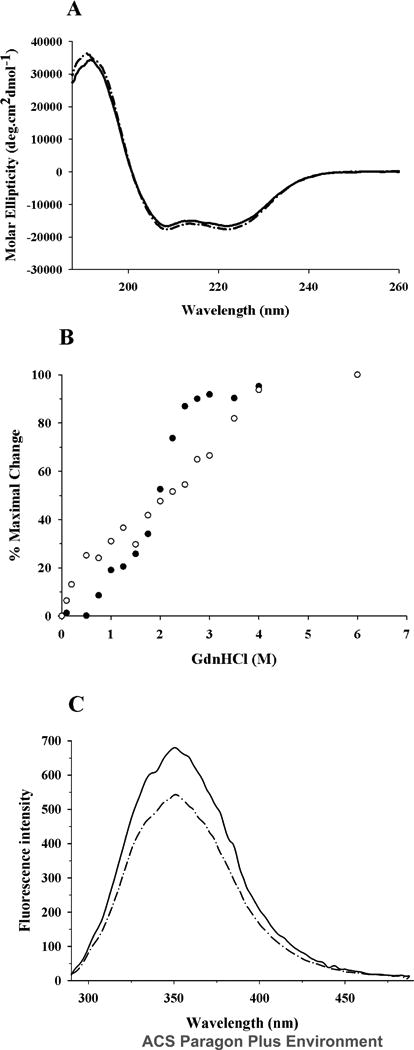
A. Far-UV CD spectra. The spectra of unmodified (solid line) and acrolein-modified apoE (dashed line) (0.2 mg/ml) were recorded in 20 mM sodium phosphate buffer, pH 7.4. The scans were obtained from 185 to 260 nm using a 0.1 cm path length cuvette, scan speed of 20 nm per minute, and response time of 1 second (average of 3 scans shown). B. GdnHCl-induced unfolding. The % maximal change in ellipticity of 0.2 mg/ml unmodified (filled circles) and acrolein-modified (open circles) apoE at 222 nm was plotted as a function of increasing GdnHCl concentration. The % maximal change was calculated using eqn. 3. C. Fluorescence emission spectra. Fluorescence emission spectra of unmodified (solid line) and acrolein-modified apoE (dashed line) (0.02 mg/ml) were recorded in PBS. Emission scans were recorded between 290 and 490 nm following excitation at 280 nm (5 nm excitation and emission slit widths).
To determine if acrolein modification alters the functional abilities of apoE, the following assays were performed: (i) the LDLr binding ability of acrolein-modified apoE was followed using sLDLr/LA3-LA6/Myc. Following incubation of DMPC/apoE (unmodified or acrolein-modified) with sLDLr/LA3-LA6/Myc, the receptor-bound complexes were captured by co-IP with anti-c-Myc-agarose and detected by anti-apoE-HRP antibody, Figure 5 (Top panel) or anti-Myc antibody (Bottom panel). Lipid-free apoE (0.5 μg) and sLDLr/LA3-LA6/Myc (10 μg) were loaded in lane 1, Top and Bottom panels respectively, as immunoblot controls. DMPC/unmodified apoE elicits the ability to bind the sLDLr/LA3-LA6/Myc in a concentration-dependent manner in the presence of Ca2+ (0, 1 and 10 μg apoE, lanes 2, 3 and 4, respectively). The binding ability was abolished when Ca2+ was omitted and 2 mM EDTA included to chelate residual Ca2+ in the incubation mixture (data not shown), indicative of the specificity of interaction of apoE with sLDLr, and the requirement of Ca2+ for maintaining the structural and functional integrity of the latter. In contrast, DMPC/acrolein-modified rat apoE was unable to interact with the sLDLr (0,1 and 10 μg apoE, lanes 5, 6 and 7, respectively). The lipid-bound form of human apoE3/(1-191) (10 μg) bearing the LDLr binding segment was used as a positive control, lane 8.
Figure 5. Effect of acrolein modification on LDLr binding ability of apoE.
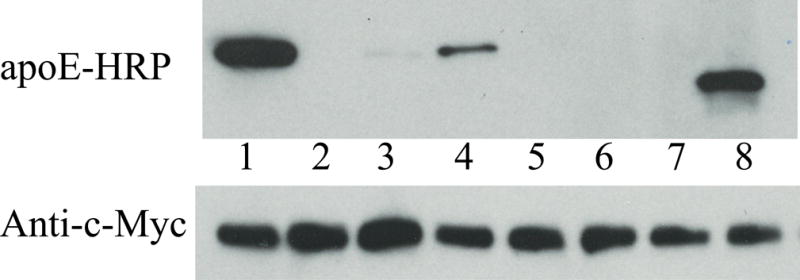
DMPC/apoE complexes were incubated with 10 μg of sLDLr in PBS and 2 mM CaCl2 for 1 h at 4 °C, followed by co-IP with anti-c-Myc antibody-linked agarose to capture the DMPC/apoE/sLDLr complexes. LDLr-bound apoE was detected by Western blot using anti-apoE-HRP antibody (Top panel). A duplicate analysis was carried out in parallel with anti-c-Myc antibody (Bottom panel) to confirm the presence of similar levels of LDLr in each reaction mixture. The lane assignments were as follows: Lane 1: Western blot control in both panels (0.5 μg apoE in Top panel and 10 μg sLDLr/LA3-LA6/Myc in Bottom panel); lanes 2, 3 and 4: 0, 1 and 10 μg protein, respectively, of DMPC/unmodified apoE ; lanes 5, 6 and 7: 0, 1 and 10 μg protein, respectively, of DMPC/acrolein-modified apoE. Lane 8 contains DMPC/human apoE3/(1-191) (10 μg), which served as an additional control to support the robustness of the assay.
(ii) The heparin binding ability of acrolein-modified apoE was assessed in comparison with that of unmodified protein, Figure 6. Unmodified apoE binds to heparin-Sepharose, requiring 0.49 ± 0.01 M NaCl to elute the bound protein. Acrolein-modified apoE also binds to heparin-Sepharose; however, its binding is relatively weaker, requiring 0.39 ± 0.04 M to be eluted.
Figure 6. Heparin binding ability of unmodified and acrolein-modified apoE.
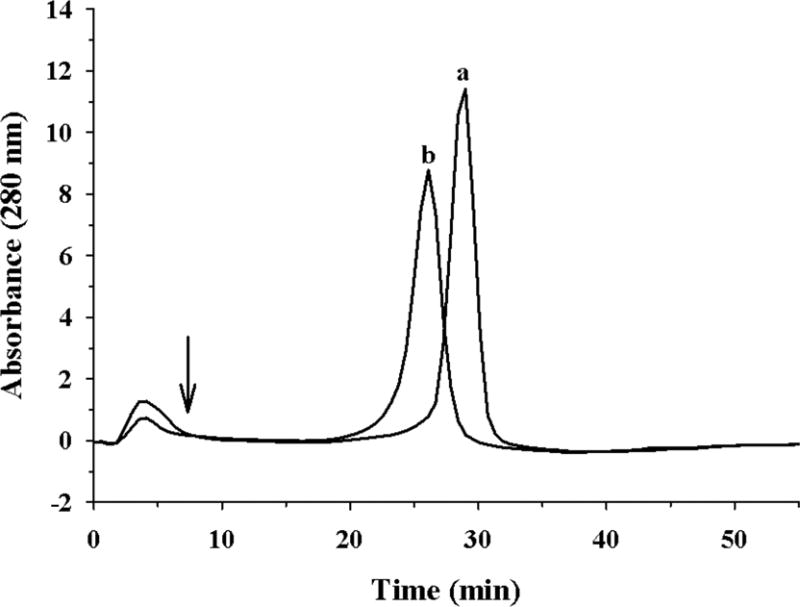
Unmodified (a) or acrolein-modified (b) apoE (100 μg) was loaded onto a heparin-Sepharose column in 20 mM sodium phosphate pH 7.4 in an ÄKTA FPLC system. The flow rate was maintained at 1 ml/min. A salt gradient of 0 – 1.0 M NaCl was used to elute the bound protein; the elution of the protein was monitored at 280 nm. The arrow represents the start of the gradient.
(iii) The ability of acrolein-modified apoE to interact with lipids was next evaluated in comparison with that of unmodified apoE. The rationale was to see if the increase in modified apoE in the lipid-free bottom fraction from plasma of ETS-exposed rats was due to decreased lipid-binding ability. Typically, lipid binding of an apolipoprotein is assessed by determining its ability to transform DMPC MLV to discoidal bilayer complexes at 24 °C, the gel-crystalline transition temperature of DMPC. Conversion of the large vesicles (∼200 nm diameter) to the smaller lipid/protein complexes (diameter ∼ 20 nm) is accompanied by a decrease in turbidity that can be followed as changes in absorbance at 325 nm, especially in the initial stages of the assay. Figure 7A shows changes in absorbance obtained when DMPC MLV (500 μg lipid) was treated with unmodified (curve a), acrolein-modified apoE (curve b) (100 μg protein) or PBS (curve c). The absorbance of DMPC vesicles remained mostly unchanged in absence of protein. It decreased rapidly upon addition of unmodified apoE (T1/2 = 8.8 ± 1.0 min and rate constant, K = 0.12 ± 0.01 min-1), as reported earlier. On the other hand, the T1/2 and K were significantly altered in acrolein-modified apoE, 22 ± 1.7 min and 0.045 ± 0.003 min-1 respectively. Upon prolonged incubation, DMPC/acrolein-modified apoE complex formation occurs eventually. The lipoprotein complexes formed after overnight incubation were isolated by density gradient ultracentrifugation revealing more heterogeneous particles that are slightly larger in size, ∼ 17 nm diameter, and, mass, ∼670 kDa, compared to that noted for unmodified apoE (∼ 15 nm and ∼ 630 kDa, respectively) as seen by non-denaturing PAGE, Figure 7B.
Figure 7. Lipid-binding ability of unmodified and acrolein-modified apoE.
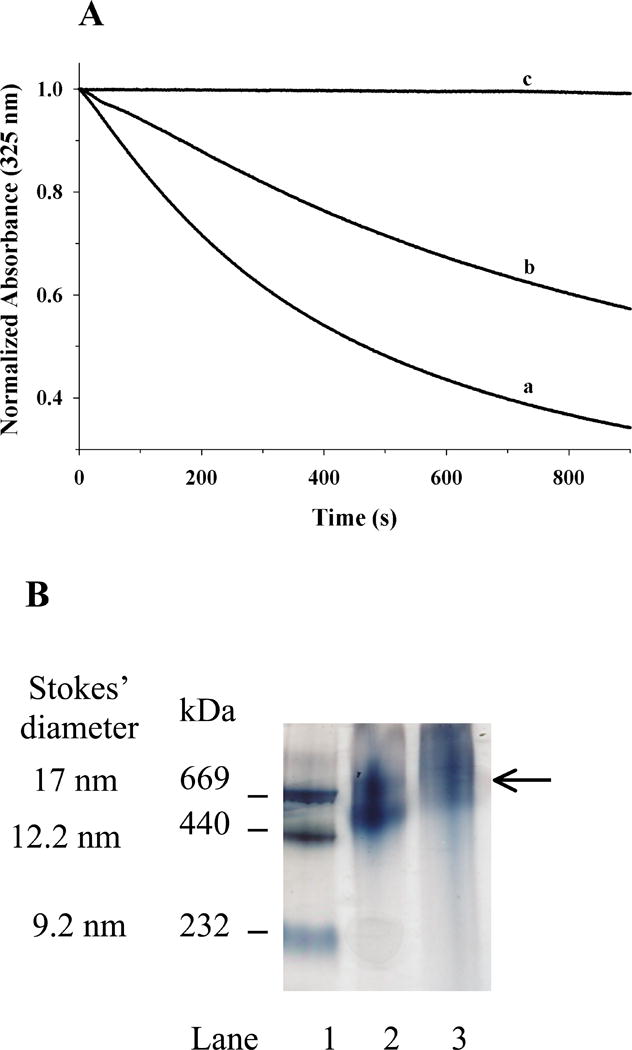
A. Transformation of DMPC MLV to smaller lipid/protein complexes by apoE. About 500 μg DMPC MLV was treated with 100 μg of unmodified- (curve a) or acrolein-modified (curve b) apoE or PBS (curve c), and the change in absorbance at 325 nm was followed as a function of time at 24 °C. The data are normalized to absorbance at 0 min. Representative curves from 3 different experiments are shown. B. Non-denaturing PAGE analysis of DMPC/apoE complexes prepared with unmodified or acrolein-modified apoE. About 50 μg of DMPC/apoE complexes were loaded on a 4-20% acrylamide gradient gel and electrophoresced in 10 mM Tris-glycine, pH 8.4 for 24 h at 110V and stained with Amido Black. Lane 1, high molecular mass standard; lane 2, DMPC/unmodified-apoE; lane 3, DMPC/acrolein-modified apoE complex (arrow draws attention to the major band). The molecular mass and average particle sizes were calculated from a calibration curve using the following standards and their corresponding molecular masses and Stokes diameters: thyroglobulin (669 kDa, 17 nm); ferritin (440 kDa, 12.2 nm); and, catalase (232 kDa, 9.2 nm).
(iv) Subsequently, we determined if acrolein modification alters the ability of apoE to promote cholesterol and phospholipid efflux from macrophages in comparison with unmodified and acrolein-modified apoAI, Figure 8, Panel A. Unmodified rat apoE (10 μg/ml) stimulates a robust cholesterol efflux from macrophages (∼80% compared to that by human apoAI) in the presence of cAMP, and lower levels in the absence of cAMP. Upon acrolein modification (10:1 and 50:1 acrolein:apoE m/m), its ability to promote efflux decreased significantly to ∼ 5%. This trend was similar to that noted for apoAI, with acrolein modification leading to a dramatic decrease in cholesterol efflux ability, an observation also noted by others.47 The ability to promote efflux was dose dependent in the range studied (0.1 to 30 μg/ml) for unmodified apoAI and apoE, with maximal effect noted at ∼10 μg/ml, Figure 8, Panel B; throughout the concentration range studied, acrolein-modified apoE (and apoAI) was still not effective, displaying only 5- 10% efficiency as unmodified protein. The decreased cholesterol efflux capability of acrolein-modified apoE appears to correlate with the extent of modification, Figure 8, Panel C with 10:1 and 50:1 ratios (acrolein:apoE m/m ratio) eliciting 67% and 36%, respectively, of unmodified apoE.
Figure 8. Effect of acrolein modification on cholesterol efflux capability of apoE.
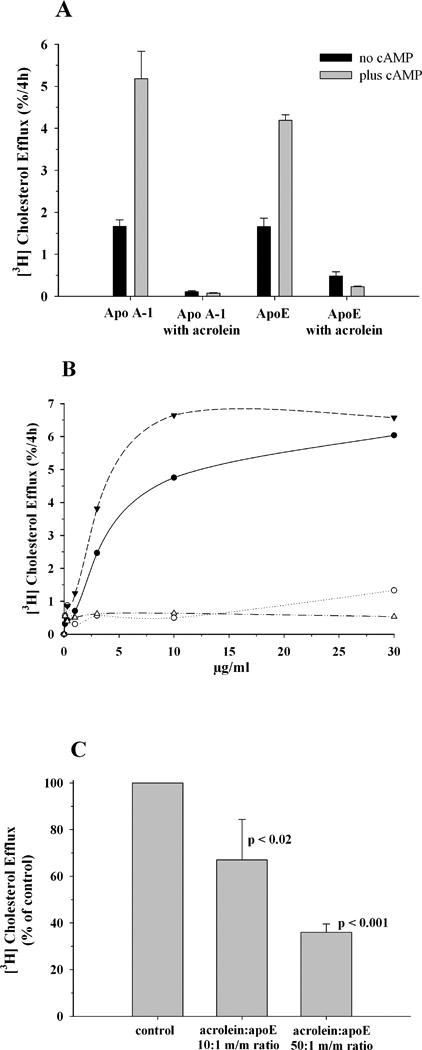
J774 macrophages were labeled with [3H]cholesterol (1 μCi/mL) in RPMI-1640 with 1% FBS for 48 h. A cAMP analogue was added to up-regulate ABCA1 expression for a period of 18 h, followed by exposure to 10 μg/ml (Panel A) unmodified or acrolein modified human apoAI or rat apoE (50:1 acrolein:protein m/m ratio) in serum-free RPMI-1640 medium. The amount of [3H]cholesterol appearing in the medium after 4 h was expressed as a percentage of the radioactivity initially present in cells at time zero. A parallel experiment with no added cAMP was conducted to assess the efflux not mediated by ABCA1. Background release of [3H]cholesterol to serum-free medium was subtracted from values obtained with added proteins. Panel B shows dose-dependent (0.1 -30 μg/ml) efflux for unmodified (filled circles) or acrolein-modified rat apoE (open circles), and unmodified (filled triangles) or acrolein-modified (open triangles) human apoAI. All other conditions are as described above. Panel C. Correlation between extent of acrolein modification and efflux capability of apoE. Under conditions as described under Panel A, efflux was measured with 3 μg/ml unmodified or acrolein-modified (10:1 and 50:1, acrolein:apoE m/m ratio) apoE. Values are expressed as % of control unmodified apoE, mean ± SD.
To determine if the loss of function is due to modification of specific sites by acrolein, we carried out mass spectrometric analysis. Previous studies indicate that acrolein reacts predominantly with the amino group of lysine, in addition to the sulfhydryl group of cysteine, and the imidazole group of histidine.48 Rat apoE lacks Cys residues; therefore we designed our studies to determine if Lys or His are modified. Initially, unmodifed apoE was subjected to trypsin, AspN, GluC or AspN + GluC cleavage to determine optimal cleavage conditions. The cleaved samples were cleaned using C18 ZipTip® Pipette Tips and analyzed by MALDI TOF. The predicted number of cleavages and cleavage sites obtained using the Peptide Cutter software (web.expasy.org/peptide_cutter/) are shown in Figure 9A. Tryptic digest of rat apoE is expected to yield 43 peptides following complete cleavage of peptide bonds at the carbonyl side of Lys and Arg; most of them are too small to be detected in the MALDI TOF scanning range, Figure 9B, Panel (i). Further, there is a possibility that trypsin may poorly recognize modified Lys.47, 49 On the other hand, AspN and GluC, which cleave peptide bonds at the amino side of Asp, and carbonyl side of Asp and Glu, respectively, could individually generate fewer but more detectable peptides than that obtained from trypsin digestion, Figure 9B, Panels (ii) and (iii), respectively. However, we found that a double digest (AspN + GluC), Figure 9B, Panel (iv), was the most optimal under our conditions, yielding more detectable peptide peaks in the scanning range. In subsequent studies, we therefore used AspN + GluC double enzyme digestion to definitively assign modification sites in acrolein-modified apoE.
Figure 9. Proteolytic cleavage of rat apoE.
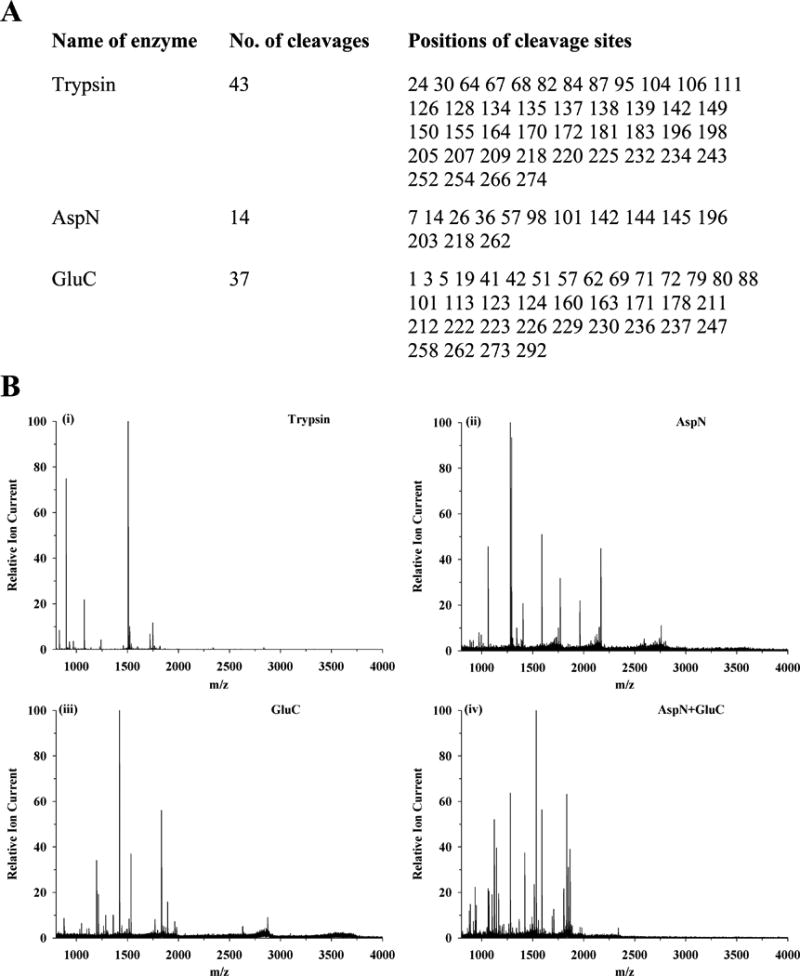
(Panel A) Predicted cleavage sites in rat apoE using trypsin, AspN or GluC based on PeptideCutter (web.expasy.org/peptide_cutter/). (Panel B) MALDI TOF MS pattern of enzyme-digested apoE peptides as described under EXPERIMENTAL PROCEDURES: Panel (i) Trypsin, Panel (ii) AspN, Panel (iii) GluC, and, Panel (iv) AspN + GluC.
MALDI-TOF/TOF analysis of AspN + GluC digested acrolein-modified apoE was carried out in comparison with unmodified apoE. It yielded ∼50% and 45% sequence coverage for unmodified and acrolein-modified apoE, respectively; 5 major peptides showed acrolein-modification of Lys residues (Table 1), based on increase in mass from the MS/MS spectra. These five peptides contain 8 (K64, K67, K68, K135, K138, K149, K155, K254) of 11 Lys in rat apoE. The MS/MS spectrum of one of the peptides (peptide 248-262: IFQARIKGWFEPLVE) is shown in the unmodified and modified states, Figure 10, Panels A and B, respectively. The spectra of the four other peptides 63-69: VKAYKKE, 58-69: DTMTEVKAYKKE, 125-146: LRSRLSTHLRKMRKRLMRDADD, and 145-160: DDLQKRLAVYKAGAQE are shown in Supporting Information Figure S1, Panels A, B, C and D, respectively. The MS/MS spectrum for peptide 248-262, Figure 10A and 10B shows that b7, b8, b11 and b14 and y10 and y12 gained 76 mass increases, indicating that Lys254 has been modified. Interestingly, Lys254 appeared to be modified even in the absence of added acrolein, suggesting air oxidation during sample handling; nevertheless, the modification at this site was ∼36-fold higher upon exogenous addition of acrolein as inferred from label-free quantification method. Modification at the other seven Lys was noted only in acrolein-exposed samples. Lastly, peptide 58-69 revealed an increase in mass units by 16 at Met60 that is attributed to oxidative modification of the –S-CH3 group to methionine sulfoxide.
Table 1. MALDI TOF/TOF analysis of peptides obtained from AspN + GluC digestion of acrolein modified rat apoE.
| Position | Sequence | Predicted (m/z) |
Observed (m/z) |
Modification |
|---|---|---|---|---|
| 63-69 | VKAYK67KE | 865.5 | 941.9 | K67+76 |
| 58-69 | DTM60TEVK64AYK67K68E | 1442.7 | 1704.5 | M60+16, K64+76, K67+76, K68+94 |
| 125-146 | LRSRLSTHLRK135MRK138RLMRDADD | 2754.2 | 2866.2 | K135+56, K138+56 |
| 145-160 | DDLQK149RLAVYK155AGAQE | 1804.9 | 1880.8 | K149+38, K155+38 |
| 248-262 | IFQARIK254GWFEPLVEa | 1833.0 | 1908.7 | K254+76 |
Unmodified or acrolein modified apoE (20 μg) was treated with AspN + GluC, and subjected to MALDI TOF/TOF analysis following cleanup using C18 ZipTip® Pipette Tips as described under EXPERIMENTAL PROCEDURES. The 5 major peptides with modified residues are shown. Data are shown only for acrolein-modified apoE. The modified residues are underlined and shown in bold, their positions indicated in superscript. The observed m/z values were obtained by processing the raw data using the GPS Explorer ™ version 3.6 (Applied Biosystems, South San Francisco, CA) and then searched against Swiss-Prot protein sequence database. The mass increase at defined sites is shown in the last column.
Peptide 248-262 showed modification (+76 Da) even in absence of added acrolein, possibly due to air oxidation of sample handling. However, the modification at this site was ∼36-fold higher upon exogenous addition of acrolein as inferred from quantification based on peak area.
Figure 10. MALDI TOF/TOF identification of Lys254 in the AspN + GluC digestion of unmodified and acrolein-modified apoE.
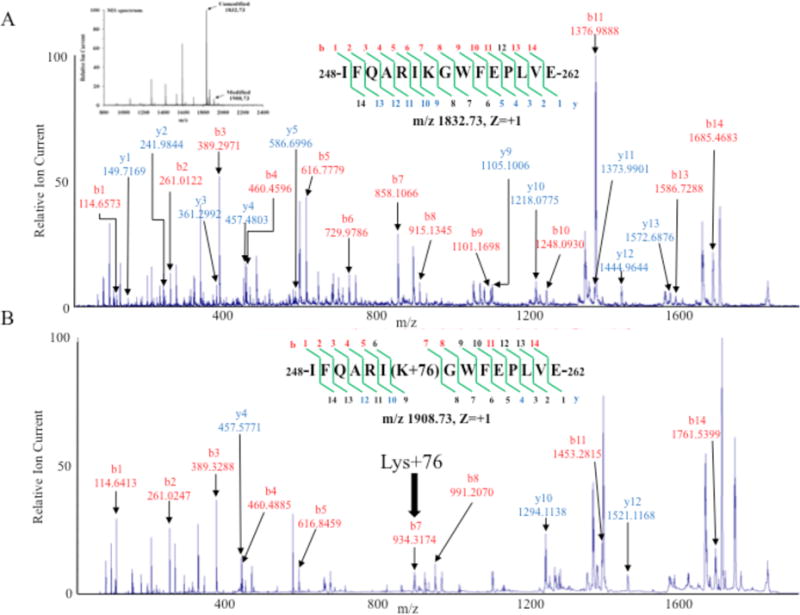
MS/MS analysis of peptide 248-262 obtained from AspN + GluC digestion of unmodified and acrolein-modified. Panel A: [IFQARIK248GWFEPLVE]+ (m/z 1833.0, z = +1). Panel B: [IFQARIK248+76GWFEPLVE]+ (m/z 1908.73, z = +1). Bold arrow draws attention to b7 ion from acrolein-modified apoE with 76 Da mass increase. Inset in Panel A shows the mass spectrum for each parent ion for unmodified and acrolein-modified apoE peptide.
Overall, there were four different types of acrolein-modified lysines: an aldimine adduct (+38) at Lys149 and K155; a propanal adduct (+56) at Lys135 and Lys138; Nε-(3-methylpyridinium)lysine (MP-lysine) (+76) at Lys64, Lys67 and Lys254, and FDP-lysine (+94) at Lys68, Figure 11 and Table 1. The mass increase of 38 Da is believed to correspond to a Schiff base adduct between the aldehyde group of acrolein and the ε-amino group of Lys; this, in turn, reacts with a second acrolein via a Michael addition eventually cyclizing to MP-Lys, a stable product registering a mass increase of 76 Da, via an imine intermediate.50 The mass increase of 56 Da arises when acrolein undergoes nucleophilic addition at the double bond leading to the Michael addition-type adduct; the increase of 94 Da possibly arises from reaction of a Lys with two acrolein molecules via Michael addition followed by condensation and dehydration reactions.28, 30, 51, 52
Figure 11. Chemical structures of acrolein adducts identified in acrolein-modified rat apoE by MALDI TOF.
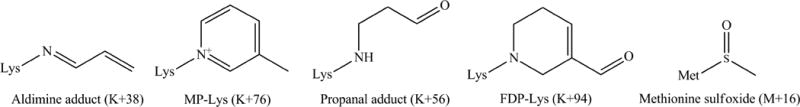
The numbers in parenthesis indicate the observed increases in mass units under the conditions employed in this study.
Discussion
Several lines of evidence indicate a correlation between lipoprotein oxidation and atherogenicity of a lipoprotein particle, with oxidized LDL (containing apoB-100 as the major protein and generally lacking apoE) playing a significant role in the etiology of atherosclerosis. 53, 54 We propose that oxidative modification of apoE located on other lipoproteins may be an additional mechanism for predisposing subjects towards an unfavorable plasma lipid (pro-atherogenic) profile. Acrolein is one of the major pro-oxidants generated in situ, and is also one of the predominant environmental pollutants. Lysine and histidines are likely targets for acrolein modification.30, 51, 52 Our aim was to determine if plasma apoE is subjected to modification by acrolein from either of these sources. To address this issue, initial studies were carried out to determine the effect of exposure of rats to FA or ETS on plasma apoE.
Despite the marginal differences in the triglyceride levels and the LDL susceptibility to oxidation, the FA and ETS exposure studies offered three major findings: (i) sufficient acrolein is generated in situ to cause modification of plasma proteins as noted in acrolein modification of apoE in rats exposed to FA, (ii) ETS exposure leads to further acrolein modification of apoE as noted in its increased presence in the lipid-free fraction during ultracentrifugation, and, (iii) acrolein modification appears to alter the lipoprotein binding property of apoE. These observations warranted further investigations to determine the molecular basis of the effect of acrolein modification on apoE at the structural and functional level.
As an exchangeable apolipoprotein, apoE can exist in a lipid-free or lipid (lipoprotein)-associated state. In the lipid-associated, but not in lipid-free state, apoE serves as a ligand for the LDLr. Rat apoE bears an overall sequence identity of 73.5% with human apoE3 and 73.9% with apoE4, and is expected to adopt a similar fold and three-dimensional structure as human apoE, based on the biphasic GdnHCl-induced unfolding pattern that we observed recently.29 It is a highly helical protein (55% α-helix) that displays two mid-points of denaturation corresponding to 0.8 and 2.2 M GdnHCl, suggesting the presence of two independently folded domains as noted by others for human apoE3.5, 32 In apoE3, they correspond to unfolding of the CT and NT domains, respectively. Human apoE3 is composed of a series of amphipathic α-helices that are organized as an N-terminal (NT) domain (residues 1-191) and a C-terminal (CT) domain (201-299). The NT domain is a four-helix bundle bearing the LDLr binding sites, while the CT domain bears high-affinity lipid-binding sites. The CT domain also mediates apoE self-association leading to tetramers by helix-helix interactions in lipid-free state. In humans, apoE is polymorphic, and basic residues in the 3 major isoforms (apoE2, apoE3 and apoE4) appear to be critical in determining the predisposition of individuals towards hyperlipidemia and Alzheimer's disease: in apoE3 (allelic frequency ∼78%), considered the normal anti-atherogenic isoform, positions 112 and 158 have a Cys and Arg, respectively; in apoE2 (allelic frequency ∼7%) these positions bear a Cys. Although rat apoE bears an Arg at position 112 like human apoE4, it likely resembles apoE3 from functional standpoint as it bears Thr61 as in mouse and other mammalian apoE.5, 57 From its sequence similarity and secondary structural predictions, it is expected to adopt a fold similar to apoE3. The Lys residues that are considered essential for LDLr binding are conserved in rat apoE. Of the 11 Lys residues, 8 are located towards the N-terminal part of rat apoE between residues 64 and 155 and 3 towards the C-terminal part. Thus, oxidative modification of lysine residues can be expected to have a significant impact on the ability of apoE to regulate plasma lipid homeostasis.
Acrolein modification generated epitopes on rat apoE that are recognized by mAb5F6, the anti-acrolein-lysine antibody. SDS-PAGE analysis reveals the presence of monomers, cross-linked dimers and oligomeric species in acrolein-modified apoE. Increasing the amount of acrolein resulted in increased extent of modification, with covalently cross-linked species appearing when there is at least one acrolein per lysine in apoE, Figure 3. The 34-kDa band in the immunoblot could also represent intra-molecular cross-linking in apoE, as suggested by GdnHCl-induced unfolding pattern of acrolein-modified apoE. The distinct biphasic pattern of unfolding that represents unfolding of the two domains seen in unmodified apoE is lost upon acrolein modification, Figure 4B. Acrolein-mediated covalent cross-linking between lysines in the two domains would prevent independent unfolding, and potentially alter the overall tertiary fold as seen in decreased intrinsic fluorescence emission, Figure 4C, without any significant change in the secondary structure, Figure 4A.
Two major physiological functions of apoE are LDLr- and HSPG-binding activities, 58, 59 both involving basic residues. Both functions appear to be impaired with acrolein modification. The LDLr is a multi-domain receptor that plays a crucial role in removing atherogenic neutral-lipid containing lipoproteins form the circulation. The ligand binding domain of the LDLr is accommodated within the 7 adjacent modular LDL-A repeats LA1-LA7, 60 of which LA5 is required for high-affinity binding of apoE containing lipoprotein particles.40, 61A single Ca2+ ion contributes to the structural integrity of each module by coordinating with 4 conserved acidic residues. In the case of the high affinity LDLr interaction with human apoE3, basic residues located between 130 and 150 in helix 4 of the NT domain and Arg172 have been shown to be essential for binding with the LDLr ligand binding modules.62, 63 Pioneering studies by Weisgraber and colleagues have demonstrated that amino acid substitution or in vitro chemical modification of the lysine and arginine residues in apoE leads to a dramatic loss in LDLr binding ability.5, 64 The binding involves numerous electrostatic interactions that govern specificity between the basic residues on apoE and the acidic residues surrounding the Ca2+ in the receptor; in addition, the interface also involves a Trp and a stacking of His, 65, 66 while the helix curvature that is conferred by the lipid or lipoprotein surface, also likely plays an essential role in the binding interaction. Together these and other factors such as creation of a multivalent ligand induced by lipid binding40, 67 contribute to the high affinity binding of lipid-associated apoE predominantly via residues TEELRVRLASHLRKLRKRLLR. The sequence identity in this segment between rat and human apoE is ∼81% (compared to overall identity of 73.5%), Figure 1, with all the basic residues being conserved (TEELRSRLSTHLRKMRKRLMR), signifying that these residues play a critical role in the binding interaction in rat apoE as well. Loss of LDLr binding ability upon acrolein modification, Figure 5, is indicative of direct modification of one or more of these Lys residues and/or changes in the overall fold of the protein. Mass spectral data provide direct evidence for modification of Lys135 and Lys138, Table 1, and Supporting Information Figure S1, Panel C, leading to a propanal adduct (+56 Da) in both these lysines, Figure 11.
In the case of heparin interaction, acrolein modification of Lys 135 and 138 appears to be responsible for a significant decrease in the binding ability of rat apoE. Interaction with cell surface HSPG is a major factor in the physiological role of apoE in the hepatic clearance of lipoprotein remnants. This is mediated through the LDLr and LDLr-related protein (LRP) coupled to HSPG pathway. Together, these uptake pathways play a significant role in clearing fasting and post-prandial remnant clearance.68 ApoE initially interacts with HSPG, followed by the transfer of the lipoprotein particle to LRP. Defective binding to HSPG can therefore potentially lead to accumulation of the pro-atherogenic remnant particles.68 The details of the interaction between human apoE and heparin have emerged from site-directed mutagenesis 69, NMR and surface plasmon resonance studies 69, 70 and by monoclonal antibodies.71 ApoE/heparin interaction possibly occurs as a two-step process, the first step involving a fast association with electrostatic interactions, and the second step involving hydrophobic interactions.69 In these studies, heparin, a highly sulfated version of HSPG that is isolated from bovine intestinal mucosa, was used as an in vitro model for studying cell surface HSPG. They point to the importance of Arg142, Lys143, Arg145, Lys146 and Arg147 in the NT domain of human apoE isoforms in high-affinity binding.70 They are available for interaction in both lipid-free and lipoprotein-associated apoE. These residues are directly involved in ionic interaction with the negatively charged sulfated groups of heparin. In human apoE3, Lys143 and Lys146 appear to have unusually low pKa (9.5 and 9.2, respectively), which was attributed to the strong positive electrostatic potential in their microenvironment.72, 73 Based on sequence alignment, the corresponding heparin-binding sites in rat apoE are Lys 135 and Lys 138, respectively, in the NT domain, 29 both of which are modified by acrolein. It has been suggested that the position of Lys in an amphipathic α-helix within salt-bridge forming distance such as an (E/D)XXK, KXX(E/D), (E/D)XXXK or KXXX(E/D) motif may alter its nucleophilicity and thereby enhance reactivity with acrolein as noted for apoAI and an HDL peptide mimetic.47, 74 However, in contrast to apoAI where most of the Lys is in this configuration, only 3 out of 11 Lys are within salt-bridge forming distance in rat apoE, making the enhanced nucleophilicity a less likely explanation for the altered reactivity to acrolein. Regardless of the nature of the chemical interaction, the modification significantly weakens the overall ionic interaction of rat apoE with the sulfated moieties of heparin.
It is possible that modified rat apoE retains its ability to bind heparin via sites in the CT domain. In human form, Lys233 appears to bear high affinity for heparin only in isolated CT domain.69 The corresponding site in rat apoE is an Arg (Arg225), which does not appear to bear any modification. Thus Arg 225 in the CT domain is a likely site for interaction of acrolein-modified rat apoE with heparin, which may explain the weak interaction. Interestingly, we observed in a prior study that acrolein modification of isolated NT domain of human apoE3 practically abolished its heparin binding ability.42 Nevertheless, the physiological implication of weak HSPG binding of acrolein-modified apoE is poor clearance of remnant lipoprotein particles in oxidative stress conditions such as aging and exposure to tobacco smoke and other environment pollution. It can be envisaged that acrolein can cause significant oxidative modification of the lysine residues as a result of ETS exposure, thereby disrupting its interaction with HSPG and the LDLr.
Acrolein also significantly impairs the lipid-binding ability of apoE as seen in the 60% decrease in the rate of conversion of DMPC vesicles to discoidal particles. This has important physiological implications, as lipid binding is an essential prerequisite that confers increased LDLr binding affinity as noted above. This observation derives support from in vivo studies, which revealed an increased presence of acrolein-modified apoE in the lipid-free fraction in the ETS group compared to the FA group. It is likely that oxidative modification of apoE weakened its lipid association and triggered its dissociation from the lipoprotein particle. Previous studies from our lab indicate that acrolein modification of the isolated N-terminal domain of human apoE3 also displayed a significant loss in its lipid binding ability.42 The alternative possibility that apoE was oxidatively modified after its dissociation from the lipoprotein particle, or that oxidative modification of lipids weakened the lipid-binding affinity of apoE cannot be excluded at this point.
Prior investigations established the critical role of human apoE isoforms in reverse cholesterol transport and in promoting ABCA1-mediated cholesterol efflux from cholesterol-laden macrophages, particularly in atherosclerotic lesions.6, 75-77 The ability of rat apoE to promote ABCA1-dependent cholesterol efflux noted in the present study is similar to that reported human apoE343 with J774 mouse macrophages and with HeLa cells transfected with ABCA1 cDNA. This is consistent with previous reports showing that apolipoproteins are good acceptors of lipids from cells expressing ABCA1.80-83 Although the molecular details of the process are not clear, the CT domain of apoAI and apoE appears to be critical for mediating efficient cholesterol efflux. Structural features of the α-helix such as hydrophobicity and amphipathic nature, rather than specific residues, are important determinants that govern cellular cholesterol efflux abilities of apolipoproteins.
In the present study, we show that acrolein modification significantly impairs the ability of rat apoE to promote cholesterol efflux. The site modified in the CT domain is Lys254, which may be a contributory factor towards decreasing ABCA1-mediated cholesterol efflux. Although there were no significant changes in the α-helical content following acrolein modification, it is possible that the Lys modifications disrupt intra-helical ionic interactions with Glu residues at i, i+3 or i+4 position and destabilize the overall helix stability. GdnHCl-induced denaturation data indicate that the biphasic mode of unfolding is lost in acrolein-modified apoE, suggesting inter domain (intra- or inter-molecular) cross-linking. This would alter the helix curvature, and explain the impairment of cholesterol efflux. Other studies show that interaction of apoAI with acrolein resulted in modification of Lys226 located in helix 10 in the C-terminal domain, which disrupted its ability to promote ABCA1-mediated cholesterol efflux in baby hamster kidney cells.47
Taken together with our in vitro biochemical analysis and other researchers' reports, we predict that apoE is a potential in vivo target for interaction with acrolein in tobacco smoke. Oxidation of protein appears to represent the final stage of LDL oxidation process85, 86, when the aldehydes react with side chain amino groups. It has been shown that acrolein preferentially modified lysine residues of LDL in vitro. Immunohistochemical studies from other labs show the presence of acrolein-modified proteins in atherosclerotic lesions from human aorta.30, 87 In the present study, we demonstrate that acrolein-modified apoE is found to a similar extent in the total lipoprotein fractions in both groups, while the ETS group had a greater extent of modified apoE in the lipid-free fraction. It is possible that acrolein present in the tobacco smoke either directly modified apoE or that other prooxidants triggered formation of acrolein in lipoprotein particles, eventually modifying apoE.
It is anticipated that other apolipoproteins may be potentially oxidized by ETS exposure as well, an aspect that requires further attention. The implication of our observations is that in addition to smokers, second hand smokers and individuals routinely exposed to environmental pollutants may be susceptible to developing heart disease due to the oxidative damage to apoE and other apolipoproteins. Further studies are required to determine the global effects of age- and ETS-induced oxidative stress on the plasma lipoproteome.
Supplementary Material
Acknowledgments
We thank Dr. Ronald Krauss and his group at the Children's Hospital Oakland Research Institute for help with determination of triglyceride and cholesterol levels, Ken Irvine for initial set up of acrolein modification experiment and Dale Uyeminami for oversight of animal care and the operation of the smoke inhalation facilities at University of California, Davis.
Funding sources: This work was funded by the Tobacco Related Disease Research Program (TRDRP 17RT-0165), NIH-GM105561 and HL096365, the Drake Family Trust (VN), American Heart Association (STK), NSF/CSU-LSAMP (HRD-0802628), Women & Philanthropy Scholarship and the CSULB Ronald E. McNair Post-Baccalaureate Achievement Program (TNT).
Abbreviations
- ApoE
Apolipoprotein E
- DMPC
1,2-Dimyristoyl-sn-glycero-3-phosphocholine
- ETS
Environmental tobacco smoke
- FA
Filtered air
- FDP-lysine
Nε -(3-formyl-3,4-dehydropiperidino)-lysine
- HSPG
heparan sulfate proteoglycan
- LDL
Low density lipoprotein
- LDLr
Low density lipoprotein receptor
- MALDI TOF
Matrix-Assisted Laser Desorption/Ionization - Time Of Flight
- MLV
Multilamellar vesicles
- MP-lysine
Nε-(3-methylpyridinium)lysine
- PBS
phosphate buffered saline, 10 mM sodium phosphate, pH 7.4, 150 mM sodium chloride
- VLDL
Very low density lipoprotein
Footnotes
Supporting Information Available: MALDI TOF/TOF identification of M60, K64, K67, K68, K135, K138, K149 and K155 in AspN + GluC digested acrolein-modified apoE (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: From lipid transport to neurobiology. Prog Lipid Res. 2010;50:62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Narayanaswami V, Maiorano JN, Dhanasekaran P, Ryan RO, Phillips MC, Lund-Katz S, Davidson WS. Helix orientation of the functional domains in apolipoprotein e in discoidal high density lipoprotein particles. J Biol Chem. 2004;279:14273–14279. doi: 10.1074/jbc.M313318200. [DOI] [PubMed] [Google Scholar]
- 5.Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 6.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2001;21:13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]
- 7.Shimano H, Ohsuga J, Shimada M, Namba Y, Gotoda T, Harada K, Katsuki M, Yazaki Y, Yamada N. Inhibition of diet-induced atheroma formation in transgenic mice expressing apolipoprotein E in the arterial wall. J Clin Invest. 1995;95:469–476. doi: 10.1172/JCI117687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 9.Kashyap VS, Santamarina-Fojo S, Brown DR, Parrott CL, Applebaum-Bowden D, Meyn S, Talley G, Paigen B, Maeda N, Brewer HB., Jr Apolipoprotein E deficiency in mice: gene replacement and prevention of atherosclerosis using adenovirus vectors. J Clin Invest. 1995;96:1612–1620. doi: 10.1172/JCI118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghiselli G, Schaefer EJ, Gascon P, Breser HB., Jr Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science. 1981;214:1239–1241. doi: 10.1126/science.6795720. [DOI] [PubMed] [Google Scholar]
- 11.Kurosaka D, Teramoto T, Matsushima T, Yokoyama T, Yamada A, Aikawa T, Miyamoto Y, Kurokawa K. Apolipoprotein E deficiency with a depressed mRNA of normal size. Atherosclerosis. 1991;88:15–20. doi: 10.1016/0021-9150(91)90252-x. [DOI] [PubMed] [Google Scholar]
- 12.Mabuchi H, Koizumi J, Shimizu M, Takeda R. Development of coronary heart disease in familial hypercholesterolemia. Circulation. 1989;79:225–232. doi: 10.1161/01.cir.79.2.225. [DOI] [PubMed] [Google Scholar]
- 13.Gueraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, Jouanin I, Siems W, Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- 14.Grassi D, Desideri G, Ferri L, Aggio A, Tiberti S, Ferri C. Oxidative stress and endothelial dysfunction: say no to cigarette smoking! Curr Pharm Des. 2010;16:2539–2550. doi: 10.2174/138161210792062867. [DOI] [PubMed] [Google Scholar]
- 15.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50(Suppl):S376–381. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsimikas S. In vivo markers of oxidative stress and therapeutic interventions. Am J Cardiol. 2008;101:34D–42D. doi: 10.1016/j.amjcard.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Fraley AE, Tsimikas S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr Opin Lipidol. 2006;17:502–509. doi: 10.1097/01.mol.0000245255.40634.b5. [DOI] [PubMed] [Google Scholar]
- 18.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 19.Sevanian A, Bittolo-Bon G, Cazzolato G, Hodis H, Hwang J, Zamburlini A, Maiorino M, Ursini F. LDL- is a lipid hydroperoxide-enriched circulating lipoprotein. J Lipid Res. 1997;38:419–428. [PubMed] [Google Scholar]
- 20.Cazzolato G, Avogaro P, Bittolo-Bon G. Characterization of a more electronegatively charged LDL subfraction by ion exchange HPLC. Free Radic Biol Med. 1991;11:247–253. doi: 10.1016/0891-5849(91)90120-r. [DOI] [PubMed] [Google Scholar]
- 21.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 22.Subbanagounder G, Watson AD, Berliner JA. Bioactive products of phospholipid oxidation: isolation, identification, measurement and activities. Free Radic Biol Med. 2000;28:1751–1761. doi: 10.1016/s0891-5849(00)00233-1. [DOI] [PubMed] [Google Scholar]
- 23.Ziouzenkova O, Asatryan L, Akmal M, Tetta C, Wratten ML, Loseto-Wich G, Jurgens G, Heinecke J, Sevanian A. Oxidative cross-linking of ApoB100 and hemoglobin results in low density lipoprotein modification in blood. Relevance to atherogenesis caused by hemodialysis. J Biol Chem. 1999;274:18916–18924. doi: 10.1074/jbc.274.27.18916. [DOI] [PubMed] [Google Scholar]
- 24.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 25.Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ, Shih DM, Van Lenten BJ, Frank JS, Demer LL, Edwards PA, Fogelman AM. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol. 1996;16:831–842. doi: 10.1161/01.atv.16.7.831. [DOI] [PubMed] [Google Scholar]
- 26.Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci U S A. 1987;84:2995–2998. doi: 10.1073/pnas.84.9.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proc Natl Acad Sci U S A. 1998;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran TN, Kim SH, Gallo C, Amaya M, Kyees J, Narayanaswami V. Biochemical and biophysical characterization of recombinant rat apolipoprotein E: similarities to human apolipoprotein E3. Arch Biochem Biophys. 2012;529:18–25. doi: 10.1016/j.abb.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 31.Sreerama N, Venyaminov SY, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal Biochem. 2000;287:243–251. doi: 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- 32.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 33.Patel AB, Khumsupan P, Narayanaswami V. Pyrene fluorescence analysis offers new insights into the conformation of the lipoprotein-binding domain of human apolipoprotein E. Biochemistry. 49:1766–1775. doi: 10.1021/bi901902e. [DOI] [PubMed] [Google Scholar]
- 34.Wetterau JR, Aggerbeck LP, Rall SC, Jr, Weisgraber KH. Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. J Biol Chem. 1988;263:6240–6248. [PubMed] [Google Scholar]
- 35.Pace CN, Vanderburg KE. Determining globular protein stability: guanidine hydrochloride denaturation of myoglobin. Biochemistry. 1979;18:288–292. doi: 10.1021/bi00569a008. [DOI] [PubMed] [Google Scholar]
- 36.Weers PM, Abdullahi WE, Cabrera JM, Hsu TC. Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: destabilization by threonine 31. Biochemistry. 2005;44:8810–8816. doi: 10.1021/bi050502v. [DOI] [PubMed] [Google Scholar]
- 37.Fisher CA, Narayanaswami V, Ryan RO. The lipid-associated conformation of the low density lipoprotein receptor binding domain of human apolipoprotein E. J Biol Chem. 2000;275:33601–33606. doi: 10.1074/jbc.M002643200. [DOI] [PubMed] [Google Scholar]
- 38.Narayanaswami V, Szeto SS, Ryan RO. Lipid association-induced N-and C-terminal domain reorganization in human apolipoprotein E3. J Biol Chem. 2001;276:37853–37860. doi: 10.1074/jbc.M102953200. [DOI] [PubMed] [Google Scholar]
- 39.Raussens V, Drury J, Forte TM, Choy N, Goormaghtigh E, Ruysschaert JM, Narayanaswami V. Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem J. 2005;387:747–754. doi: 10.1042/BJ20041536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher C, Abdul-Aziz D, Blacklow SC. A two-module region of the low-density lipoprotein receptor sufficient for formation of complexes with apolipoprotein E ligands. Biochemistry. 2004;43:1037–1044. doi: 10.1021/bi035529y. [DOI] [PubMed] [Google Scholar]
- 41.Kiss RS, Weers PM, Narayanaswami V, Cohen J, Kay CM, Ryan RO. Structure-guided protein engineering modulates helix bundle exchangeable apolipoprotein properties. J Biol Chem. 2003;278:21952–21959. doi: 10.1074/jbc.M302676200. [DOI] [PubMed] [Google Scholar]
- 42.Tamamizu-Kato S, Wong JY, Jairam V, Uchida K, Raussens V, Kato H, Ruysschaert JM, Narayanaswami V. Modification by acrolein, a component of tobacco smoke and age-related oxidative stress, mediates functional impairment of human apolipoprotein E. Biochemistry. 2007;46:8392–8400. doi: 10.1021/bi700289k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vedhachalam C, Narayanaswami V, Neto N, Forte TM, Phillips MC, Lund-Katz S, Bielicki JK. The C-terminal lipid-binding domain of apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry. 2007;46:2583–2593. doi: 10.1021/bi602407r. [DOI] [PubMed] [Google Scholar]
- 44.Bielicki JK, Zhang H, Cortez Y, Zheng Y, Narayanaswami V, Patel A, Johansson J, Azhar S. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J Lipid Res. 2010;51:1496–1503. doi: 10.1194/jlr.M003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y, Patel AB, Narayanaswami V, Hura GL, Hang B, Bielicki JK. HDL mimetic peptide ATI-5261 forms an oligomeric assembly in solution that dissociates to monomers upon dilution. Biochemistry. 2011;50:4068–4076. doi: 10.1021/bi2002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang G, Ueberheide BM, Waldemarson S, Myung S, Molloy K, Eriksson J, Chait BT, Neubert TA, Fenyo D. Protein quantitation using mass spectrometry. Methods Mol Biol. 2010;673:211–222. doi: 10.1007/978-1-60761-842-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao B, Fu X, McDonald TO, Green PS, Uchida K, O'Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280:36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- 48.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 49.Lambert C, Li J, Jonscher K, Yang TC, Reigan P, Quintana M, Harvey J, Freed BM. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J Biol Chem. 2007;282:19666–19675. doi: 10.1074/jbc.M611527200. [DOI] [PubMed] [Google Scholar]
- 50.Furuhata A, Ishii T, Kumazawa S, Yamada T, Nakayama T, Uchida K. N(epsilon)-(3-methylpyridinium)lysine, a major antigenic adduct generated in acrolein-modified protein. J Biol Chem. 2003;278:48658–48665. doi: 10.1074/jbc.M309401200. [DOI] [PubMed] [Google Scholar]
- 51.Jairam V, Uchida K, Narayanaswami V. Pathophysiology of Lipoprotein Oxidation [Google Scholar]
- 52.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 53.Grundy SM. Role of low-density lipoproteins in atherogenesis and development of coronary heart disease. Clin Chem. 1995;41:139–146. [PubMed] [Google Scholar]
- 54.Parthasarathy S, Rankin SM. Role of oxidized low density lipoprotein in atherogenesis. Prog Lipid Res. 1992;31:127–143. doi: 10.1016/0163-7827(92)90006-5. [DOI] [PubMed] [Google Scholar]
- 55.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 56.Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 57.Raffai RL, Dong LM, Farese RV, Jr, Weisgraber KH. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc Natl Acad Sci U S A. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 59.Ji ZS, Brecht WJ, Miranda RD, Hussain MM, Innerarity TL, Mahley RW. Role of heparan sulfate proteoglycans in the binding and uptake of apolipoprotein E-enriched remnant lipoproteins by cultured cells. J Biol Chem. 1993;268:10160–10167. [PubMed] [Google Scholar]
- 60.Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005;74:535–562. doi: 10.1146/annurev.biochem.74.082803.133354. [DOI] [PubMed] [Google Scholar]
- 61.Fisher C, Beglova N, Blacklow SC. Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol Cell. 2006;22:277–283. doi: 10.1016/j.molcel.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 62.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 63.Morrow JA, Arnold KS, Dong J, Balestra ME, Innerarity TL, Weisgraber KH. Effect of arginine 172 on the binding of apolipoprotein E to the low density lipoprotein receptor. J Biol Chem. 2000;275:2576–2580. doi: 10.1074/jbc.275.4.2576. [DOI] [PubMed] [Google Scholar]
- 64.Weisgraber KH, Innerarity TL, Mahley RW. Role of lysine residues of plasma lipoproteins in high affinity binding to cell surface receptors on human fibroblasts. J Biol Chem. 1978;253:9053–9062. [PubMed] [Google Scholar]
- 65.Blacklow SC. Versatility in ligand recognition by LDL receptor family proteins: advances and frontiers. Curr Opin Struct Biol. 2007;17:419–426. doi: 10.1016/j.sbi.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prevost M, Raussens V. Apolipoprotein E-low density lipoprotein receptor binding: study of protein-protein interaction in rationally selected docked complexes. Proteins. 2004;55:874–884. doi: 10.1002/prot.20080. [DOI] [PubMed] [Google Scholar]
- 67.Guttman M, Prieto JH, Croy JE, Komives EA. Decoding of lipoprotein-receptor interactions: properties of ligand binding modules governing interactions with apolipoprotein E. Biochemistry. 49:1207–1216. doi: 10.1021/bi9017208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mahley RW, Huang Y, Rall SC., Jr Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949. [PubMed] [Google Scholar]
- 69.Futamura M, Dhanasekaran P, Handa T, Phillips MC, Lund-Katz S, Saito H. Two-step mechanism of binding of apolipoprotein E to heparin: implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J Biol Chem. 2005;280:5414–5422. doi: 10.1074/jbc.M411719200. [DOI] [PubMed] [Google Scholar]
- 70.Libeu CP, Lund-Katz S, Phillips MC, Wehrli S, Hernaiz MJ, Capila I, Linhardt RJ, Raffai RL, Newhouse YM, Zhou F, Weisgraber KH. New insights into the heparan sulfate proteoglycan-binding activity of apolipoprotein E. J Biol Chem. 2001;276:39138–39144. doi: 10.1074/jbc.M104746200. [DOI] [PubMed] [Google Scholar]
- 71.Weisgraber KH, Rall SC, Jr, Mahley RW, Milne RW, Marcel YL, Sparrow JT. Human apolipoprotein E. Determination of the heparin binding sites of apolipoprotein E3. J Biol Chem. 1986;261:2068–2076. [PubMed] [Google Scholar]
- 72.Lund-Katz S, Zaiou M, Wehrli S, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC. Effects of lipid interaction on the lysine microenvironments in apolipoprotein E. J Biol Chem. 2000;275:34459–34464. doi: 10.1074/jbc.M005265200. [DOI] [PubMed] [Google Scholar]
- 73.Lund-Katz S, Wehrli S, Zaiou M, Newhouse Y, Weisgraber KH, Phillips MC. Effects of polymorphism on the microenvironment of the LDL receptor-binding region of human apoE. J Lipid Res. 2001;42:894–901. [PubMed] [Google Scholar]
- 74.Zheng Y, Kim SH, Patel AB, Narayanaswami V, Iavarone AT, Hura GL, Bielicki JK. The positional specificity of EXXK motifs within an amphipathic alpha-helix dictates preferential lysine modification by acrolein: implications for the design of high-density lipoprotein mimetic peptides. Biochemistry. 2012;51:6400–6412. doi: 10.1021/bi300626g. [DOI] [PubMed] [Google Scholar]
- 75.Nofer JR, Kehrel B, Fobker M, Levkau B, Assmann G, von Eckardstein A. HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis. 2002;161:1–16. doi: 10.1016/s0021-9150(01)00651-7. [DOI] [PubMed] [Google Scholar]
- 76.Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, Genest J., Jr Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res. 2004;45:839–848. doi: 10.1194/jlr.M300418-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer HB. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44:828–836. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 78.Fitzgerald ML, Morris AL, Rhee JS, Andersson LP, Mendez AJ, Freeman MW. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J Biol Chem. 2002;277:33178–33187. doi: 10.1074/jbc.M204996200. [DOI] [PubMed] [Google Scholar]
- 79.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 80.Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG, Eggerman TL, Patterson AP, Duverger NJ, Santamarina-Fojo S, Brewer HB., Jr Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280:818–823. doi: 10.1006/bbrc.2000.4219. [DOI] [PubMed] [Google Scholar]
- 81.Hara H, Yokoyama S. Interaction of free apolipoproteins with macrophages. Formation of high density lipoprotein-like lipoproteins and reduction of cellular cholesterol. J Biol Chem. 1991;266:3080–3086. [PubMed] [Google Scholar]
- 82.Oram JF, Yokoyama S. Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res. 1996;37:2473–2491. [PubMed] [Google Scholar]
- 83.Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS. Cyclic AMP induces apolipoprotein E binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. J Biol Chem. 1996;271:30647–30655. doi: 10.1074/jbc.271.48.30647. [DOI] [PubMed] [Google Scholar]
- 84.Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler Thromb Vasc Biol. 2007;27:1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 85.Singh R, Tucek M, Maxa K, Tenglerova J, Weyand EH. A rapid and simple method for the analysis of 1-hydroxypyrene glucuronide: a potential biomarker for polycyclic aromatic hydrocarbon exposure. Carcinogenesis. 1995;16:2909–2915. doi: 10.1093/carcin/16.12.2909. [DOI] [PubMed] [Google Scholar]
- 86.Cominacini L, Garbin U, Davoli A, Micciolo R, Bosello O, Gaviraghi G, Scuro LA, Pastorino AM. A simple test for predisposition to LDL oxidation based on the fluorescence development during copper-catalyzed oxidative modification. J Lipid Res. 1991;32:349–358. [PubMed] [Google Scholar]
- 87.Shao B, O'Brien K D, McDonald TO, Fu X, Oram JF, Uchida K, Heinecke JW. Acrolein modifies apolipoprotein A-I in the human artery wall. Ann N Y Acad Sci. 2005;1043:396–403. doi: 10.1196/annals.1333.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.