

Abstract
MicroRNAs (miRNAs) are the group of ∼22 nucleotides long noncoding small endogenous and evolutionary conserved post-transcriptional regulatory RNAs, which show an enormous role in various biological and metabolic processes in both animals and plants. To date not a single miRNA has been identified in coffee (Coffea arabica), which is an economically important plant of Rubiaceae family. In this study a well-developed, powerful and comparative computational approach, EST-based homology search is applied to find potential miRNA of coffee. We blasted publicly available EST sequences obtained from NCBI GenBank against previously known plant miRNAs. For the first time, one potential miRNA from a large miRNA family with appropriate fold back structures was identified through a series of filtration criteria. A total of six potential target genes in Arabidopsis were identified based on their sequence complementarities. The target genes mainly encode transport inhibitor like protein, transcription factor, DNA-binding protein, and GRR1-like protein, and these genes play an important role in various biological processes like response to chitin, cold, salt stress, water deprivation etc. Overall, findings from this study will accelerate the way for further researches of miRNAs and their functions in coffee.
Keywords: Expressed sequence tags, MicroRNA, Computational approach, Coffea arabica
1. Introduction
Coffee (Coffea arabica) is one of the major beverage crops and one of the most worthful agricultural commodities and treated as second ranked trade exchanges on international community. Coffee represents one of the key export and cash crops in tropical and subtropical developing countries at present date. The genus Coffea belongs to the Rubiaceae family, which also includes some other important plants. Many researchers suggest coffee consumption reduces the risk of Alzheimer’s disease, Parkinson’s disease, heart disease, diabetes mellitus type 2, gout, and cirrhosis of the liver (Klatsky et al., 2006). It is also a valuable source of some secondary metabolites like polyphenols, caffeic acid, chlorogenic acid and more impotently caffeine which is an antioxidant and which has some medicinal properties against type 2 diabetes (Cheng et al., 2011; Pereira et al., 2006).
MicroRNAs (miRNAs) are a class of endogenous small regulatory RNAs consisting of about 18–22 nucleotides derived from their precursor sequences, which negatively regulate gene expression at the posttranscriptional levels by binding to target mRNAs for initiating mRNA cleavage or inhibition of mRNA translation (Zhang et al., 2006a). Many investigations experimentally prove that these miRNAs play an important role in different biological and metabolic processes in plants and animals (Ambros and Chen, 2007; Carrington and Ambros, 2003; Zhang et al., 2007a). In plants, miRNAs function to control tissue (leaf, root, stem, and flower) differentiation and development, phase change from vegetative growth to reproductive growth, signal transduction, and the response to environmental conditions such as biotic (pathogens) and abiotic stress (e.g. water deprivation, salinity, and drought) (Chen, 2005; Zhang et al., 2006a). In the year of 2002 first miRNAs were discovered in plants (Park et al., 2002), but at the present date several hundred miRNAs have been identified in plants by computational and experimental approaches.
A comparative genomics study across hugely contradictory taxa has shown that many miRNAs are highly evolutionarily conserved from species to species in the plant and animal kingdom. This feature of extensive evolutionary conservation of these miRNAs among themselves renders a powerful approach to their identification using comparative genomics. On the basis of this strategy, researchers developed an expressed sequence tag (EST) and a genome survey sequence (GSS) approach to identify miRNAs (Zhang et al., 2005) from plants or even from various animals. EST analysis has some substantial advantages over the other approaches such as: (1) conserved miRNAs can be identified whose whole genome sequences have not been well known, (2) provides direct evidence for miRNA expression that cannot be inferred from genomic sequence surveys, and (3) miRNA identification can be conducted without highly specialized software. Many miRNAs are successfully identified in different plant species, such as soybean (Chen et al., 2009; Zhang et al., 2008), maize (Zhang et al., 2006b), tomato (Yin et al., 2008), tobacco (Frazier et al., 2010), potato (Xie et al., 2011; Zhang et al., 2009), wheat (Han et al., 2009), Brassica napus (Xie et al., 2007), citrus (Song et al., 2009), switch grass (Xie et al., 2010) apple (Gleave et al., 2008) and Asiatic cotton (Wang et al., 2012) by using this approach.
Directional cloning and sequencing are two early approaches for miRNA identification but recently high-throughput sequencing is very much effective. However, the identification of novel miRNAs from the large pool of small RNA sequences and analysis of such large volume of data is still a great challenge. Recently, in silico computation approach is used to reduce the setback and support the wet lab approaches.
According to miRBase (a public miRNA database) a total of 18,226 miRNAs have been deposited (Griffiths-Jones et al., 2006) of which 4014 plant miRNAs have been currently identified from 52 different species. Unfortunately, there has been no report on miRNAs in coffee despite its enormous economic significance throughout the world. In this study, we applied an EST based homology search to identify miRNAs in coffee using currently available 1,72,326 coffee expressed sequence tags (ESTs) from the NCBI Genbank database. Based on our findings, only one potential miRNA, belonging to a large family has been identified for the first time in coffee. This newly identified miRNA also plays important roles in different biological processes. We also report the major features of this newly identified coffee miRNA.
2. Materials and methods
2.1. Coffee ESTs, cDNAs and mRNAs sequence
Coffee mRNA, cDNA and EST sequences were obtained from the GenBank nucleotide databases at NCBI (<http://ftp.ncbi.nlm.nih.gov>; March 2012). A total of 1,72,326 coffee ESTs were deposited in the EST database (dbEST) and all of these ESTs were screened against the known plant miRNAs which were used as reference miRNA.
2.2. Reference set of miRNA
To search potential miRNAs in coffee a total of previously known 989 mature miRNA sequences from Arabidopsis thaliana and Oryza sativa were obtained from miRNA Registry Database, miRBase (Release 18.0, May, 2012) (Griffiths-Jones, 2004). These miRNAs were defined as the reference set of miRNA sequences by using the previous work on computational prediction of miRNAs by Zhang et al. (2007b). To avoid redundant or overlapping miRNAs, the repeated sequences of miRNAs within the above species were removed and only the unique ones were used as query.
2.3. Availability of softwares
Comparative software mpiBLAST-1.6.0 (Darling et al., 2003) is a freely available, open-source; parallel implementation of NCBI BLAST (Altschul et al., 1997) which use algorithm similar to BLAST-2.2.22 of NCBI GenBank. Zuker folding algorithm MFOLD 3.2 <http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form> was used online to analyze the secondary structure of RNAs (Zuker, 2003). The Plant Small RNA Analysis Server (psRNATarget) formerly known as miRU was used for miRNA target analysis (Dai and Zhao, 2011).
2.4. Identification of potential miRNAs
Fig. 1 summarizes the procedure for predicting potential miRNAs from coffee. The reference miRNA sequences from A. thaliana and O. sativa were used as a query for homology search against our local coffee EST sequence database. The mature sequences of all miRNAs from A. thaliana and O. sativa were subjected to BLASTn search (Altschul et al., 1997) in the coffee EST databases using mpiBLAST-1.6.0 algorithm (Darling et al., 2003). This mpiBLAST program was run in a LINUX based 32 core cluster system, which shows result similar to the NCBI BLAST program and it is relatively fast, takes about 12 min on average to analyze this data. Here we created three different datasets at three different e-value thresholds < 0.001, <0.01 and <0.1, respectively. The target sequences were forced to contain no more than three to four mismatches; the adjusted BLASTn parameter settings were as follows: low complexity was chosen as the sequence filter; the number of descriptions and alignments was kept as default; the default word-match size between query and database sequences was 7. After the BLASTn step we got 89 (at e-value < 0.001), 322 (at e-value < 0.01) and 497 (at e-value < 0.1) sequences respectively for our three datasets. From these three sets finally we obtained 89 EST sequences which were common in all three sets.
Figure 1.
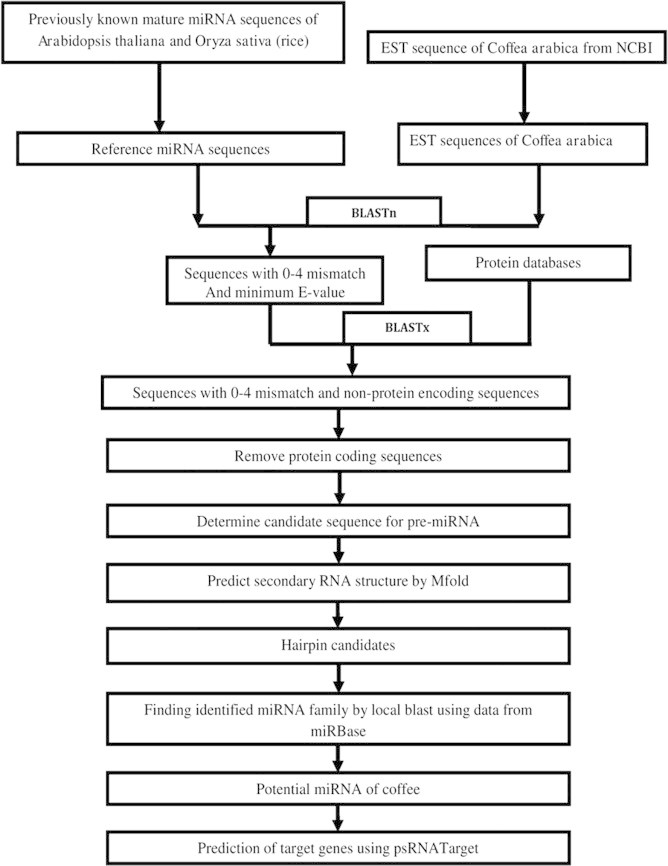
Schematic representation of the coffee miRNA search procedure used to identify homologs of known plant miRNA.
These sequences were considered as potential miRNA candidates only if they fulfill the following criteria with slight modification from a previous study by Zhang et al. (2007b): (1) at least 18–20 nt length was assumed between the predicted mature miRNAs, and (2) allowed to have 0–4 nt mismatches in sequence with all previously known plant mature miRNAs.
These whole EST sequences were used for BLASTx analysis using NCBI BLAST-2.2.26 + against NCBI nr (non-redundant protein) database on May, 2012 for removing the protein-coding sequences and retaining only the non-protein-coding sequences (Altschul et al., 1997). About 67 EST sequences were found that were non-protein-coding sequences.
2.5. Prediction of secondary structure
The secondary structure of candidate pre-miRNA sequences of these potential miRNA homologs was predicted using the Zuker folding algorithm with MFOLD-3.2 (Zuker, 2003). All parameters were set to default values. All outputs obtained from mfold were recorded into an excel file, which included the EST ID numbers, respective miRNA homologs, total length of the sequences, the number of each nucleotide (A, G, C and U), the number of arms per structure, location of the matching regions, percentage (%) of (A + U/T) and (G + C) content and minimal folding free energy (MFE, ΔG in kcal/mol). Then, the adjusted minimal folding free energy (AMFE) and the minimal folding free energy index (MFEI) were calculated according to the following equation from a previous report (Zhang et al., 2006c).
Previous studies of Zhang et al. (2006c), Ng Kwang and Mishra (2007) showed that miRNA precursor sequences have significantly higher negative minimal folding free energies (MFEs) and minimal folding free energy indexes (MFEIs) than other types of RNA e.g. tRNA, rRNA. To avoid designating other RNAs as miRNA candidates, these two characteristics and G + C content were considered when predicting secondary structures.
In brief an EST was considered as a potential miRNA candidate if it fulfills the following criteria described by Wang et al. (2011): (1) the predicted mature miRNAs had no more than three nucleotide substitutions compared with a known reference mature miRNAs; (2) the EST sequence must fold into an appropriate and proper stem-loop hairpin secondary structure; (3) the mature miRNA must be localized in one arm of the stem-loop structure; (4) there were no more than 6 mismatches between the predicted mature miRNA sequence and its opposite miRNA∗ sequence in the secondary structure; and (5) the predicted secondary structure must contain high negative MFE and high MFEI values.
2.6. Phylogenetic analysis
Small RNAs are conserved naturally; because of this reason ortholog discovery can be done through bioinformatics analysis. A homology search of candidate coffee miRNA was done against all plant miRNAs using a Bioedit sequence alignment editor tool (version 7.0.9.0) (Hall, 1999) allowing maximum of two mismatches and e-value < 0.001. The corresponding precursor sequences of homolog small RNAs were identified and collected, then these homolog sequences of various plant species were aligned with homolog coffee miRNA using Jalview 2.6.1 (Waterhouse et al., 2009). A query of coffee small RNAs against known miRNA families (miRBase, release 18, <http://www.mirbase.org>) allowed us to identify the family of our predicted miRNA and then, the maximum likelihood trees were constructed for the identified family based on the Tamura-Nei model (Tamura and Nei, 1993) with default bootstrap values using MEGA5.0 (Tamura et al., 2011) to illustrate the evolutionary relationships with other members of the same family.
2.7. Potential miRNA targets
Only few gene sequences are available for coffee, thus we used Arabidopsis as a reference organism for determining the targets of the newly identified miRNAs. The predicted coffee miRNAs were used as query against the A. thaliana DFCI gene index (AGI) release 15 and A. thaliana TAIR10, cDNA, removed miRNA gene (release date 14th December 2010) using psRNATarget <http://plantgrn.noble.org/psRNATarget/> with following parameters: (1) maximum expectation value 3; (2) multiplicity of target sites 2; (3) range of central mismatch for translational inhibition 9–11 nucleotide; (4) maximum mismatches at the complementary site ⩽4 without any gaps. psRNATarget provides reverse complementary matching between miRNAs and its target transcript and finds the target site accessibility by calculating unpaired energy (UPE) necessary for opening the secondary structure around the miRNA target site (Dai and Zhao, 2011).
3. Results and discussion
3.1. Identification of potential miRNAs in C. arabica
For predicting new miRNAs in coffee, we used homology searched based computational methods. Sequence and structural properties of known miRNAs are used to screen candidate miRNAs in the EST database of coffee against known 989 mature miRNAs of a dicotyledonous plant Arabidopsis and monocotyledonous rice. Fig. 1 shows the search and filtering procedure for identifying potential miRNAs in coffee. It is possible to carry out a computational exploration for new miRNAs because most of the known mature miRNAs are evolutionarily conserved within the different species within the plant kingdom (Zhang et al., 2006d).
After circumspectly considering the Blastn and Blastx results of about 1,72,326 EST from NCBI EST database (dbEST) we were able to identify 67 unique EST sequences that show homology with known reference plant mature miRNA and also were protein non-coding sequences. Then these EST sequences were subjected to secondary structure prediction by using MFOLD. The results obtained by MFOLD were inspected manually for determining the sequence of a miRNA precursor and appropriate stem-loop structure using the criteria described in Section 2. After analyzing all outputs we identified only one potential miRNA that fulfills all criteria described by Wang (Wang et al., 2011) (Figs. 2 and 3). The normal frequency of miRNA from EST is 0.01% (1 in 10,000 ESTs) (Zhang et al., 2006d) but in the case of coffee it was approximately 0.00058%. This is because EST sequences retrieved from database mostly are cloned from leaf, flower bud and first or second stages of fruit regions but there are very few sequences that were cloned from the apical region and the root of the plant. In some other plants it was shown that miRNAs are expressed more frequently in the root and apical regions than the other part of the plant (Breakfield et al., 2012). While performing a BLAST search we also set the e-value very low to find out a more significant matching region among the ESTs and reference mature plant miRNAs, thus we got only sequences with 100% similarity and few mismatches. The length of the identified precursor miRNA and mature sequences was about 172 nt and 22 nt respectively. Table 1 shows a summary of newly identified coffee miRNA.
Figure 2.

EST sequence containing the miRNA encoded within the cluster. Shadowed sequences represent pre-miRNA; underlined red colored sequences represent the mature miRNA.
Figure 3.
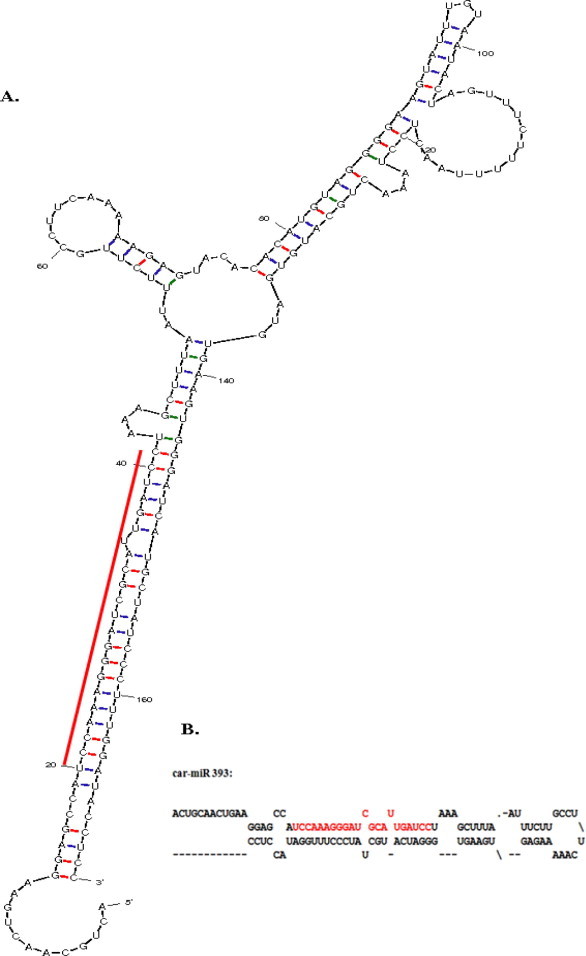
(a) Predicted stem-loop hairpin secondary structure of the coffee miRNA identified in this study. Mature miRNA sequence is in red color. The length of the accurate miRNA precursor may be slightly longer than what is presented here; (b) Predicted fold-back secondary structure of the coffee miRNA obtained from MFOLD output.
Table 1.
Properties of newly identified miRNA in coffee.
| Properties | Value |
|---|---|
| EST ID | Gi|315791179|gb| GW432396.1| GW432396 |
| Length of EST sequences | 554 nt |
| Precursor coordinates | 88–259 |
| Precursor length | 172 nt |
| Mature miRNA length | 22 nt |
| Mature miRNA sequence | TCCAAAGGGATCGCATTGATCC |
| MFE | −60.93 kcal/mol |
| AMFE | 35.3056 |
| MFEI | 0.88 |
| Nucleotide mismatch | 02 |
| Number of nucleotides in pre-miRNA sequences | A = 50; T/U = 59; G = 34; C = 35 |
| (A + T/U)% | 59.88 |
| (G + C)% | 40.12 |
| Family of miRNA | miR 393 |
Note: MFE, minimal folding free energy; AMFE, adjusted minimal folding free energy; MFEI, minimal folding free energy index.
Minimal folding free energy (MFE) is a prominent characteristic for determining the secondary structure of nucleic acids such as DNA and RNA. The lower the MFE the more thermodynamically stable is the secondary structure of the corresponding DNA or RNA sequence (Bonnet et al., 2004). The MFE index (MFEI) for each sequence was calculated as previously reported by Yin et al. (2008). In our present study, the MEFI value of predicted miRNA was 0.88. It supports the Ambros et al. (2003) finding, miRNA precursor sequences have significantly higher MFEI value than other non-coding or coding RNAs e.g. tRNA, rRNA. To evade false prediction of other RNAs as miRNA candidates, the MFEI is a result of structure prediction, but not considered during prediction (Zhang et al., 2007b).
Though there are a few computational approaches to identify miRNAs of plants, animals or even microorganisms, each and every approach has its own potential but using EST analysis has some advantages over other methods (Frazier et al., 2011; Zhang et al., 2008). It has been suggested that most of the miRNAs predicted by EST analysis can be obtained by a high-throughput deep sequencing process (Kwak et al., 2009).
Mature miRNA sequences are in the stem portion of the hairpin structures (Fig. 3). The predicted miRNA hairpin structure shows that there are at least 20 nt engaged in Watson–Crick or G/U pairings between the miRNA/miRNA∗ in the stem region and does not contain large internal loops or bulges. These also support Zhang et al. (2006d) findings. The newly identified coffee miRNA was subjected to BLAST search against all mature miRNA sequences deposited in the database to predict the family where it belongs and shows that the newly identified miRNA belong to an independent large highly conserved miRNA family denoted as “miR 393” (Fig. 4). The comparisons of the predicted mature miRNA sequences with other members in the same family showed that most members could be found to have a high degree of sequence similarity with others. The phylogenetic tree among the members of this family illustrated the evolutionary relationships of coffee miRNA (Fig. 5). Newly known microRNA from coffee was named as “car-miR 393” by following the procedure of nomenclature of microRNA proposed by miRBase (Griffiths-Jones et al., 2006).
Figure 4.

Conservation of coffee miRNA (car-miR) with diverse plant species. Conserved portion is highlighted.
Figure 5.

Phylogenetic tree for the newly identified miRNA in coffee (car-mir) that showing homology with several published miRNAs of miR393 family. The tree was constructed using the maximum likelihood method based on the Tamura-Nei model. Identified coffee miRNA is shown in red box.
3.2. Prediction of potential targets of putative miRNA in C. arabica
For understanding the role of miRNAs in various cellular functions and gene regulation networks the miRNA target gene identification is an important step. The conserved nature of miRNAs in different organisms suggests their conserved function (Zhang et al., 2006d). Their targets are mostly transcription factors which affect plant development and specific genes. To identify potential miRNA targets for known plant miRNAs, we used the plant small RNA analysis server and coffee mRNA/cDNA from NCBI along with the parameters as mentioned in Section 2. In this study, we identified a total of six targets for this one miRNA family which belongs to several gene families with different biological functions (Table 2).
Table 2.
List of the potential targets of newly identified miRNA in coffee.
| Target protein | Biological functions and molecular process | e-Value | Target Acc. | Predicted inhibition type |
|---|---|---|---|---|
| Auxin transporter protein 1 | Auxin signaling pathway | 2.0 | TC390317 | Cleavage |
| Ethylene signaling pathway | ||||
| Plant defense | ||||
| WRKY transcription factor 33 | Transcription | 3.0 | TC364349 | Cleavage |
| Transcription regulation | ||||
| Transcription factor bHLH7 (contains similarity to gene_id:MYM9.3) | Regulation of transcription | 3.0 | TC391347 | Cleavage |
| DNA-dependent transcription | ||||
| GRR1-like protein 1 | Auxin mediated signaling pathway | 3.0 | TC362644 | Cleavage |
| Negative regulation of transcription by glucose | ||||
| Stamen development | ||||
| Inositol hexakisphosphate binding | ||||
| Auxin signaling F-box 2 (AFB2) | Response to molecule of bacterial origin | 1.0 | AT3G26810.1 | Cleavage |
| Auxin signaling pathway | ||||
| Plant defense | ||||
| Auxin signaling F-box 3 (AFB3) | Auxin signaling pathway | 1.0 | AT1G12820.1 | Cleavage |
| Plant defense | ||||
| Ubl conjugation pathway | ||||
Our prediction of target genes for the coffee miRNA discovered that more than one gene was regulated by individual miRNA. This result shows similarity with some recent research in other plant species (Jones-Rhoades and Bartel, 2004; Zhang et al., 2006b) which also suggest that miRNA research should be focused on regulatory networks rather than individual connections between miRNA and their predicted target genes or regulatory factors. Some miRNAs directly target transcription factors which directly or indirectly affect growth and development, and also specific genes which control metabolism of plants (Zhang et al., 2006a).
Genes targeted by our identified miRNA contain transport inhibitor response like proteins such as auxin transporter protein 1 which plays an important role in regulatory pathways like auxin polar transport, ethylene signaling pathway and also involved in auxin influx transmembrane transporter activity (Yang et al., 2006). Among the targets two are transcriptional factors. Transcriptional factors are important components in the transcriptional process and play an important role in a variety of biological functions including plant development, hormone signaling and metabolism. First transcriptional factor WRKY is an important protein involved in some biological processes such as camalexin biosynthetic process; cellular heat acclimation; defense response to bacterium and fungus; positive regulation of autophagy; response to chitin, cold salt stress, water deprivation, DNA-dependent transcription etc. (Jiang and Deyholos, 2009; Lai et al., 2011; Libault et al., 2007; Qiu et al., 2008; Zheng et al., 2006). Second transcriptional factor bHLH7 is involved in DNA-binding. Another important target of coffee miRNA is GRR 1-like protein which has also some important biological and molecular functions such as auxin mediated signaling pathway, negative regulation of transcription by glucose, stamen development, inositol hexakisphosphate binding etc. (Cecchetti et al., 2008; Thelander et al., 2002). Here we also found two auxin signaling proteins namely auxin signaling F-box 2 and auxin signaling F-box 3. These two proteins are the component of SCF (ASK-cullin-F-box) E3 ubiquitin ligase complexes, which may mediate the ubiquitination and subsequent proteasomal degradation of target proteins. These are also involved in embryogenesis regulation by auxin and some plant defense mechanisms like antibacterial resistance (Cecchetti et al., 2008; Dharmasiri et al., 2005; Thelander et al., 2002). Some studies mentioned that miRNAs of this large family (miR 393) show response against some stress related genes and development process (Gao et al., 2011; Sunkar et al., 2012).
Overall, these findings gave us some clear knowledge that the coffee miRNA 393 may target both transcription factors as well as some specific genes. These findings of miRNA in coffee will cover the way for understanding the function and processing of coffee small RNAs in future. Moreover, it shows an easy approach for the prediction and analysis of miRNAs to those species whose genomes are not available through bioinformatics approaches.
4. Conclusion
Though this type of miRNA–related research is one of the most frequent research topics in biological sectors but miRNA research on coffee is still very far from the other plant species. In this study we are trying to identify potential miRNAs in this highly economical plant species and we are able to identify one potential miRNA along with six potential target genes. In our research work we employed a well-developed and proven potential EST based homology search method to identify miRNA in coffee. Findings of our study will be supportive to understand the gene regulation mechanism and the biogenesis of miRNA in the near future. It also fortifies the present bioinformatics approach for new miRNAs identification from plant species whose genome is not yet fully known.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgements
We acknowledge Professor Dr. Muhammed Zafar Iqbal for his cooperation and supportive hand throughout the research. We would like to thank Md. Mokkaram Hossain for his valuable support during blasting in cluster computer. We also thank the Department of Genetic Engineering and Biotechnology, and the Department of Computer Science and Engineering Shahjalal University of Science and Technology, Sylhet 3114, Bangladesh for supporting this research.
Footnotes
Peer review under responsibility of King Saud University.

References
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V., Chen X.M. The regulation of genes and genomes by small RNAs. Development. 2007;134:1635–1641. doi: 10.1242/dev.002006. [DOI] [PubMed] [Google Scholar]
- Ambros V., Bartel B., Bartel D.P. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet E., Wuyts J., Rouze P., Peer Y.V. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004;20:2911–2917. doi: 10.1093/bioinformatics/bth374. [DOI] [PubMed] [Google Scholar]
- Breakfield N.W., Corcoran D.L., Petricka J.J., Shen J., Sae-Seaw J., Rubio-Somoza I., Weigel D., Ohler U., Benfey P.N. High-resolution experimental and computational profiling of tissue-specific known and novel miRNAs in Arabidopsis. Genome Res. 2012;22:163–176. doi: 10.1101/gr.123547.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington J.C., Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336–338. doi: 10.1126/science.1085242. [DOI] [PubMed] [Google Scholar]
- Cecchetti V., Altamura M.M., Falasca G., Costantino P., Cardarelli M. Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell. 2008;20:1760–1774. doi: 10.1105/tpc.107.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.M. MicroRNA biogenesis and function in plants. FEBS Lett. 2005;579:5923–5931. doi: 10.1016/j.febslet.2005.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Hu Z., Zhang H. Identification of MicroRNAs in Wild Soybean (Glycine soja) J. Integr. Plant Biol. 2009;51:1071–1079. doi: 10.1111/j.1744-7909.2009.00887.x. [DOI] [PubMed] [Google Scholar]
- Cheng B., Liu X., Gong H., Huang L., Chen H., Zhang X., Li C., Yang M., Ma B., Jiao L., Zheng L., Huang K. Coffee components inhibit amyloid formation of human islet amyloid polypeptide in vitro: possible link between coffee consumption and diabetes mellitus. J. Agric. Food Chem. 2011;59(24):13147–13155. doi: 10.1021/jf201702h. [DOI] [PubMed] [Google Scholar]
- Dai X., Zhao P.X. PsRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling, A., Carey, L., Feng, W., 2003. The Design Implementation, and Evaluation of mpiBLAST. In: 4th International Conference on Linux Clusters: The HPC Revolution 2003 in Conjunction with the Cluster World Conference & Expo. San Jose, CA.
- Dharmasiri N., Dharmasiri S., Weijers D., Lechner E., Yamada M., Hobbie L., Ehrismann J.S., Jürgens G., Estelle M. Plant development is regulated by a family of auxin receptor F box proteins. Dev. Cell. 2005;9:109–119. doi: 10.1016/j.devcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Frazier T.P., Xie F., Freistaedter A., Burklew C.E., Zhang B. Identification and characterization of microRNAs and their target genes in tobacco (Nicotiana tabacum) Planta. 2010;232:1289–1308. doi: 10.1007/s00425-010-1255-1. [DOI] [PubMed] [Google Scholar]
- Frazier T.P., Sun G., Burklew C.E., Zhang B. Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol. Biotechnol. 2011;49(2):159–165. doi: 10.1007/s12033-011-9387-5. [DOI] [PubMed] [Google Scholar]
- Gao P., Bai X., Yang L., Lv D., Pan X., Li Y., Cai H., Ji W., Chen Q., Zhu Y. Osa-MIR393: a salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 2011;38(1):237–242. doi: 10.1007/s11033-010-0100-8. [DOI] [PubMed] [Google Scholar]
- Gleave A.P., Ampomah-Dwamena C., Berthold S., Dejnoprat S., Karunairetnam S., Nain B., Wang Y.Y., Crowhurst R.N., MacDiarmid R.M. Identification and characterisation of primary microRNAs from apple (Malus domestica cv. Royal Gala) expressed sequence tags. Tree Genet. Genomes. 2008;4:343–358. [Google Scholar]
- Griffiths-Jones S. The microRNA registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. MiRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T.A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Han Y., Luan F., Zhu H., Shao Y., Chen A., Lu C., Luo Y., Zhu B. Computational identification of microRNAs and their targets in wheat (Triticum aestivum L.) Sci. China C Life Sci. 2009;52:1091–1100. doi: 10.1007/s11427-009-0144-y. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Deyholos M.K. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 2009;69:91–105. doi: 10.1007/s11103-008-9408-3. [DOI] [PubMed] [Google Scholar]
- Jones-Rhoades M.W., Bartel D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Klatsky A.L., Morton C., Udaltsova N., Friedman G.D. Coffee, cirrhosis, and transaminase enzymes. Arch. Intern. Med. 2006;166(11):1190–1195. doi: 10.1001/archinte.166.11.1190. [DOI] [PubMed] [Google Scholar]
- Kwak P.B., Wang Q.Q., Chen X.S., Qiu C.X., Yang Z.M. Enrichment of a set of microRNAs during the cotton fiber development. BMC Genomics. 2009;10:457. doi: 10.1186/1471-2164-10-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z., Wang F., Zheng Z., Fan B., Chen Z. A critical role of autophagy in plant resistance to necrotrophic fungal pathogens. Plant J. 2011;66:953–968. doi: 10.1111/j.1365-313X.2011.04553.x. [DOI] [PubMed] [Google Scholar]
- Libault M., Wan J., Czechowski T., Udvardi M., Stacey G. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol. Plant Microbe. Interact. 2007;20:900–911. doi: 10.1094/MPMI-20-8-0900. [DOI] [PubMed] [Google Scholar]
- Ng Kwang L.S., Mishra S.K. Unique folding of precursor microRNAs: quantitative evidence and implications for de novo identification. RNA. 2007;13:170–187. doi: 10.1261/rna.223807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W., Li J., Song R., Messing J., Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002;12:1484–1495. doi: 10.1016/s0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira M.A., Parker E.D., Folsom A.R. Coffee consumption and risk of type 2 diabetes mellitus: an 11-year prospective study of 28, 812 postmenopausal women. Arch. Intern. Med. 2006;166(12):1311–1316. doi: 10.1001/archinte.166.12.1311. [DOI] [PubMed] [Google Scholar]
- Qiu J.L., Fiil B.K., Petersen K., Nielsen H.B., Botanga C.J., Thorgrimsen S., Palma K., Suarez-Rodriguez M.C., Sandbech-Clausen S., Lichota J., Brodersen P., Grasser K.D., Mattsson O., Glazebrook J., Mundy J., Petersen M. Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 2008;27:2214–2221. doi: 10.1038/emboj.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C., Fang J., Li X., Liu H., Thomas C.C. Identification and characterization of 27 conserved microRNAs in citrus. Planta. 2009;230:671–685. doi: 10.1007/s00425-009-0971-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R., Li Y.F., Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Tamura K., Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelander M., Fredriksson D., Schouten J., Hoge J.H., Ronne H. Cloning by pathway activation in yeast: identification of an Arabidopsis thaliana F-box protein that can turn on glucose repression. Plant Mol. Biol. 2002;49:69–79. doi: 10.1023/a:1014440531842. [DOI] [PubMed] [Google Scholar]
- Wang L., Liu H., Li D., Chen H. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genomics. 2011;12:154. doi: 10.1186/1471-2164-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Wang Q., Wang B. Identification and characterization of MicroRNAs in Asiatic cotton (Gossypium arboreum L.) PLoS ONE. 2012;7(4):e33696. doi: 10.1371/journal.pone.0033696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview Version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F., Huang S.Q., Guo K., Xiang A.L., Zhu Y.Y., Nie L., Yang Z.M. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007;581:1464–1474. doi: 10.1016/j.febslet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- Xie F., Frazier T.P., Zhang B. Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum) Planta. 2010;232:417–434. doi: 10.1007/s00425-010-1182-1. [DOI] [PubMed] [Google Scholar]
- Xie F., Frazier T.P., Zhang B. Identification, characterization and expression analysis of MicroRNAs and their targets in the potato (Solanum tuberosum) Gene. 2011;473:8–22. doi: 10.1016/j.gene.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Yang Y., Hammes U.Z., Taylor C.G., Schachtman D.P., Nielsen E. High-affinity auxin transport by the AUX1 influx carrier protein. Curr. Biol. 2006;16:1123–1127. doi: 10.1016/j.cub.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Yin Z., Li C., Han X., Shen F. Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum) Gene. 2008;414:60–66. doi: 10.1016/j.gene.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Pan X.P., Wang Q.L., Cobb G.P., Anderson T.A. Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005;15:336–360. doi: 10.1038/sj.cr.7290302. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Pan X.P., Cobb G.P., Anderson T.A. Plant microRNA: a small regulatory molecule with big impact. Dev. Biol. 2006;289:3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Pan X.P., Cobb G.P., Anderson T.A. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006;580:3753–3762. doi: 10.1016/j.febslet.2006.05.063. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Pan X.P., Cox S.B., Cobb G.P., Anderson T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006;63:246. doi: 10.1007/s00018-005-5467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.H., Pan X.P., Cox S.B., Cobb G.P., Anderson T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006;46:243–259. doi: 10.1111/j.1365-313X.2006.02697.x. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Wang Q., Pan X. MicroRNAs and their regulatory roles in animals and plants. J. Cell. Physiol. 2007;210:279–289. doi: 10.1002/jcp.20869. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Wang Q., Wang K., Pan X., Liu F., Guo T., Cobb G.P., Anderson T.A. Identification of cotton miRNA and their targets. Gene. 2007;397:26–37. doi: 10.1016/j.gene.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Zhang B.H., Pan X., Stellwag E.J. Identification of soybean microRNAs and their targets. Planta. 2008;229:161–182. doi: 10.1007/s00425-008-0818-x. [DOI] [PubMed] [Google Scholar]
- Zhang W., Luo Y., Gong X., Zeng W., Li S. Computational identification of 48 potato microRNAs and their targets. Comput. Biol. Chem. 2009;33:84–93. doi: 10.1016/j.compbiolchem.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Zheng Z., Qama r.S.A., Chen Z., Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48:592–605. doi: 10.1111/j.1365-313X.2006.02901.x. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]