

Abstract
Staphylococcus aureus a natural inhabitant of nasopharyngeal tract mainly survives as biofilms and possess complete Krebs cycle which plays major role in its pathogenesis. This TCA cycle is regulated by Isocitrate dehydrogenase (IDH) we have earlier cloned, sequenced (HM067707), expressed and characterized this enzyme from S. aureus ATCC12600. We have observed only one type of IDH in all the strains of S. aureus which dictates the flow of carbon thereby controlling the virulence and biofilm formation, this phenomenon is variable among bacteria. Therefore in the present study comparative structural and functional analysis of IDH was undertaken. As the crystal structure of S. aureus IDH was not available therefore using the deduced amino sequence of complete gene the 3D structure of IDH was built in Modeller 9v8. The PROCHECK and ProSAweb analysis showed the built structure was close to the crystal structure of Bacillus subtilis. This structure when superimposed with other bacterial IDH structures exhibited extensive structural variations as evidenced from the RMSD values correlating with extensive sequential variations. Only 24% sequence identity was observed with both human NADP dependent IDHs (PDB: 1T09 and 1T0L) and the structural comparative studies indicated extensive structural variations with an RMSD values of 14.284Å and 10.073Å respectively. Docking of isocitrate to both human IDHs and S. aureus IDH structures showed docking scores of -11.6169 and -10.973 respectively clearly indicating higher binding affinity of isocitrate to human IDH.
Keywords: Isocitrate dehydrogenase, NADP, RMSD, TCA Cycle
Background
Staphylococcus aureus is a Gram positive human pathogen causes severe community and hospital acquired infections which range from minor skin infections to life threatening diseases like endocarditis, toxic shock syndrome and pneumonia [1]. One of the main virulence factors of S. aureus is its ability to form biofilms which also makes the organism to resist antibiotics, with the occurrence of multidrug resistant strains of S. aureus where high reductive conditions prevails the elevated biofilm formation [2, 3] this has warranted urgent need for the development of effective drugs which are highly narrow and pathogen specific as it would minimize the transfer of drug resistance among the bacteria [4]. In S. aureus TCA cycle is suppressed upon depletion of rapidly catabolizable carbon sources; this coincides with the transition to producing only formylated δ-toxin and results in an increased inflammatory response and subsequent biofilm formation suggesting that there is an important linkage between bacterial TCA cycle and pathogenesis [3, 5]. Carbon flow between the Krebs cycle and the glyoxylate cycle is controlled by isocitrate dehydrogenase (IDH) via its own activation and inactivation mechanism [6]. Oxidative decarboxylation of isocitrate to α-ketoglutarate is catalysed by IDH enzyme with the release of CO2 and NADPH. It is existed as homodimer with its monomeric form having 40- 70 kDa in various bacteria [6]. Though some bacteria contain the NAD-IDH (EC 1.1.1.41) but most of the bacteria have NADPdependent enzyme (EC 1.1.1.42) [7], [8]. IDH enzyme from bacteria lacks the βαβαβ motif [6, 9]. The IDH activation drives the flow of carbon through the TCA cycle inducing a decrease in the intracellular level of isocitrate and an increase in the level of α-ketoglutarate thereby regulating redox status in the bacteria which is a vital factor in the virulence of S. aureus [10]. These features are best explained through docking of substrate to its 3D structure of the enzyme and correlating with its kinetics [10, 11]. Therefore, the present study the structural and functional characterization of S. aureus IDH and also its comparative structural analysis with other bacterial and human IDH to predict the role of IDH in the pathogenesis.
Methodology
3D Structure building for S. aureus IDH:
IDH gene was amplified from chromosomal DNA of S. aureus by using the IDH1 5'-CATGCTGATGTAGAAACT-3' and IDH2 5'-TCAAGGAACTGAAGGAT-3' primers, which were designed from the IDH gene sequence of S. aureus Mu 50 strain. The cocktail reaction mixture consists of 10 mM Tris–HCl (pH 8.8), 1.5mMMgCl2, 100 µmol of dNTPS mix, 100 ρmoles of each primer, 1 U of Taq DNA polymerase (Mereck Biosciences Pvt Ltd) and 0.5 µg of chromosomal DNA. Amplification conditions included an initial denaturation step for 10 min at 94 °C; 35 cycles of each having denaturation at 94 °C for 60 s, annealing at 33.1 °C for 60 s, amplification at 72 °C for 100 s and final extension step at 72 °C for 5 min in a Mastercycler gradient Thermocycler (Eppendorf). The amplicons were purified by NP-PCR kit (Taurus Scientific, USA). After purification the products were sequenced and deposited at GenBank (www.ncbi.nlm.nih.gov/genbank/submt.html). The 3D model of the S. aureus IDH was built by using Modeller 9v8 tool [12, 13]. The S. aureus IDH complete protein sequence (YP_041160.1) was submitted to BLASTp against PDB [14] and the putative IDH crystal structure from Bacillus subtilis (PDB ID: 1HQS) which showed 80% identity was taken as template to build the 3D structure. In the same way, the IDH protein sequences of H. pylori and Mycobacterium tuberculosis were Submitted to BLASTp against PDB and the putative IDH crystal structure from E. coli (PDB ID: 1BL5) showed 70% identity with H. pylori and was taken as template while, for M. tuberculosis 61% identity with Azatobacter vinelandii (PDB ID:1ITWA) was observed and was chosen as template. In Mycobacterium tuberculosis there are two isoforms of NADP-dependent IDH [7, 15] since both the isoforms are NADP -dependent we took one isoform to built the structure for comparative analysis. Clustal X tool was used for generating alignment files in PIR format for Query and template sequences [16]. The Python script was written and 20 best models were generated. The best predicted model with least DOPE (discrete optimized protein energy) score was selected for further investigation.
Validation of S. aureus IDH Model:
The stereo chemical quality of the predicted model was validated by PROCHECK and ProSAweb servers. Both can read the atomic co-ordinates of the 3D model and judge the quality of the structure. PROCHECK validation server [17] was used to generate the Ramachandran plot, whereas overall quality of the predicted model and its identity nearest to crystal X-ray structures was determined from ProSA web server which is expressed as Z score.
Structural and comparative Analysis between S. aureus IDH with Human and other bacterial IDH:
The comparative structural analysis to ensure the identity and variability of IDH structure from S. aureus with other IDH structures were carried out using Pymol software. This program has distinctive features where it can define the structural resemblance score as the log-odds of two probabilities using a scheme similar to Dayhoff׳s amino acid replacement score. In the program we have crystallized structures by inputting the PDB code and by loading the PDB format files in the user׳s computer. The validated 3D model of S. aureus IDH was super imposed with bacterial IDH: Bacillus subtilis (PDB ID: 1HQS) and Escherichia coli (PDB ID: 91CD) were retrieved from PDB database while H. pylori IDH and Mycobacterium tuberculosis IDH structures were generated following the protocol mentioned earlier [12, 13]. Further, the study was extended to understand the structural differences with human NADPdependent IDH isoforms (PDB ID: 1TOL, 1TO9).
Molecular Docking analysis of Human IDH and S. aureus IDH:
Docking was performed for S. aureus IDH and human IDH with isocitrate by using Molecular Operating Environment (MOE 2011.10) software. Three dimensional molecule of isocitrate was retrived from PubChem [18] and its structure was optimized in MOE. Structures of S. aureus IDH and Human IDH were loaded individually into MOE tool for removing hetero atoms, H2O molecules and polar hydrogens were added. Protonation (at temperature of 300K, pH 7) and energy minimization were carried out in OPLS (optimized for potential for Liquid simulation) force field at a gradient of 0.05 to calculate the atomic coordinates of the protein that are local minima of molecular energy function and in order to determine low energy conformations for molecular dynamics simulations. Docking was performed for S. aureus IDH and Human IDH with isocitrate to find out the binding modes and affinity variations. After completion of the process the conformation with least docking score was taken from the total docked conformations in each docking process and the binding conformations of isocitrate was analyzed in the binding sites of S. aureus and human IDHs [11, 19, 20].
Results
Structural analysis of IDH:
The deduced amino acid sequence of S. aureus IDH gene was used to built the structure of IDH. Total 20 best models were generated for IDH using Modeller 9v8 tool using the structure of IDH from Bacillus subtilis (PDB ID: 1HQS) as template which showed 80% identity. The lowest DOPE score of -30582.42383 was found with the model 11 among 20 models and it was considered for further study. The stereochemistry of the final model was verified by submitting to PROCHECK validation server and Ramachandran plot showed 91.3% of the residues in most favorable region and no residues were found in disallowed region (Figure 1). The ProSaWeb evaluation of IDH model revealed a compatible Z-Score value of -8.24 that falls in the range of native conformation of X- ray crystal structure (Figure 2). Similarly, the IDH structures for H. pylori and M. tuberculosis were built using E. coli and Azatobacter vinelandii IDH crystal structures as templates respectively. These structures were validated by using PROCHECK and ProSaweb servers and the results indicated that the built structures were highly reliable Table 1 (see supplementary material). Superimposing of S. aureus IDH structure with other bacterial IDH structures revealed extensive structural differences as evidenced from the RMSD values (Figure 3).
Figure 1.
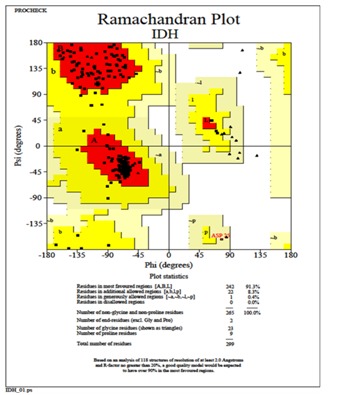
Ramachandran plot generated by PROCHECK validation server showing the stereochemical quality of the S. aureus IDH structure.
Figure 2.
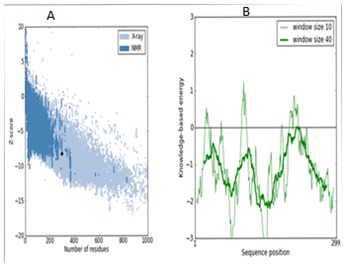
ProSA-web and Z-score of S. aureus IDH structure in PDB. The energy plots were presented with window size 10 (light green) and window size 40 (dark green). The Z-score plot of IDH model (left). Energy plot of IDH model (right).
Figure 3.
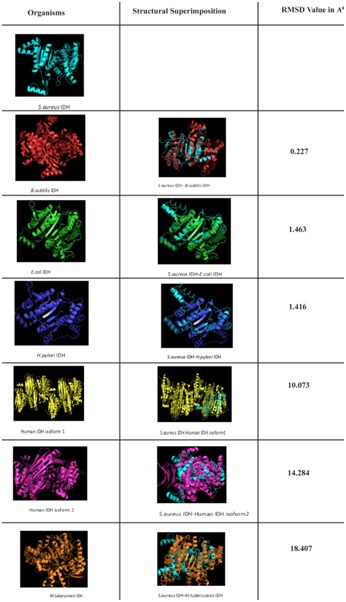
Structural comparison of NADP dependent S. aureus IDH with other bacterial NADP dependent IDH and Human NADP dependent IDH isoforms.
In order to assess the same we have also compared S. aureus IDH structure with human IDH structure which showed extensive variations both in the domain and non domain regions as indicated from RMSD values of 14.284Å and 10.073Å (Figure 3). The identical regions were random throughout the alignment especially in the substrate binding regions which showed much extent of variation and completely showing different conformations (Figure 4). The results of MOE docking studies showed that isocitrate was placed close to the residues D187, D 187, N207 and R20 by means of hydrogen bonds in S. aureus with a docking score of -10.963. Here D 187 exclusively formed two hydrogen bonds with isocitrate indicating strong involvement of D 187 in the substrate binding region. While human IDH was found to be interacting with V276, V276, V276 and N275 with a docking score of -11.6169 respectively Table 2 (see supplementary material) (Figure 5). In human IDH V276 was found to be playing a predominant role forming 3 Hydrogen bonds with isocitrate providing much extent of strength to the docking complex.
Figure 4.
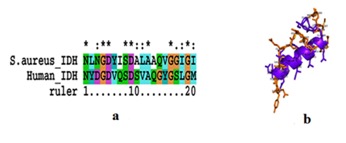
A) Alignment of active site residues of Human NADP dependent IDH and NADP dependent S. aureus IDH; B) Superimposed active site region structures of Human NADP dependent IDH and NADP dependent S. aureus IDH.
Figure 5.
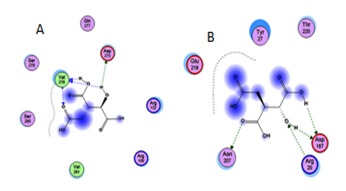
A) Docking of isocitrate with the Human IDH and S. aureus IDH. The dotted lines indicated the hydrogen bond interactions between IDH active site and Isocitrate 1. Human IDH interaction with isocitrate; B) S. aureus IDH interaction with isocitrate.
Discussion
In S. aureus the TCA cycle which plays pivotal role in the growth of the organism and this cycle is regulated by IDH whose expression makes carbon to enter into Krebs cycle in post-exponential growth phase [21]. In this phase bacteria tends to synthesize more exopolysaccharides and PIA (Polysacharide Intracellular Adhesin) molecules which are very critical in biofilm formation and these features are common in multidrug resistant strains of S. aureus (Figure 6) [2, 3, 22], therefore IDH expression plays critical role in the pathogenesis of S. aureus [1] and bacteria have placed Krebs cycle to cater to its environment needs therefore, comparative structural analysis of IDH which regulates TCA cycle in both pathogen and host will probably through light in understanding the host pathogen interactions in the establishment of infection. Keeping in view we have carried out comparative structural and functional studies on NADP dependent IDH in both S. aureus and human [7, 9]. Bacteria utilize TCA cycle differently and IDH must be playing crucial role in their pathogenesis and biofilm formation [7]. The S. aureus IDH structure was built using the homology modeling method was close to the crystal structure of Bacillus subtilis. The multiple sequence alignment results showed very high sequence homology exists between S. aureus IDH sequence and with other bacterial IDH sequences however, the comparative structural analysis showed extensive variations as indicated RMSD values and the regions showing identity were spread randomly in the structures (Figure 3) which correlated with the IDH kinetic results [21] suggesting that in S. aureus TCA cycle is uniquely placed which also correlated with the growth pattern and biofilm formation [10]. The structural superimposition of S. aureus IDH with both human IDH isoforms (PDB ID: 1T09, 1TOL) showed considerable variations in both domain and non domain regions and these differences were also observed in the sequence and active site structures (Figure 4) [12, 13]. The docking of isocitrate to both human and S. aureus IDH structures revealed higher affinity for human IDH compared to S. aureus [7, 8] correlating with enzyme kinetics [21, 23] Table 2 (Figure 5). This pathogen colonizes mostly in the nasopharyngeal tract as biofilms by adjusting its metabolic flux [1, 3, 22] and regulates TCA cycle to produce virulence factors thus spreads its infection.
Figure 6.
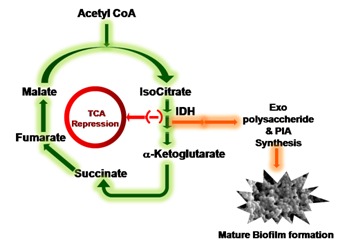
An explanation of TCA cycle repression through altered IDH metabolic status of S. aureus resulting in its massive redirection into Exopolysaccharide and PIA synthesis and finally leads to Biofilm formation.
Supplementary material
Acknowledgments
We sincerely acknowledge Sri Venkateswara Institute of Medical Sciences and University for providing funds and facilities under SBAVP scheme (SBAVP/Ph.D/02) to carry out this work and this paper forms a part of Ph.D work going to be submitted to SVIMS University.
Footnotes
Citation:Prasad et al, Bioinformation 10(2): 081-086 (2014)
References
- 1.Lowy FD, et al. N Engl J Med. 1998;33:520. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Nelson JL, et al. Antimicrob Agents Chemother. 2007;51:616. doi: 10.1128/AAC.01057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y, et al. Infect Immun. 2009;77:4256. doi: 10.1128/IAI.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee VJ, et al. Curr Opin Microbiol. 1999;2:475. doi: 10.1016/s1369-5274(99)00003-x. [DOI] [PubMed] [Google Scholar]
- 5.Somerville GA, et al. J Bacteriol. 2003;185:6686. doi: 10.1128/JB.185.22.6686-6694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurley JH, et al. Proc Natl Acad Sci U S A. 1989;86:8635. doi: 10.1073/pnas.86.22.8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steen IH, et al. J Biol Chem. 2001;276:43924. doi: 10.1074/jbc.M105999200. [DOI] [PubMed] [Google Scholar]
- 8.Dean AM, Koshland DE. Biochemistry. 1993;32:9302. doi: 10.1021/bi00087a007. [DOI] [PubMed] [Google Scholar]
- 9.Xu X, et al. J Biol Chem. 2004;279:33946. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 10.Yeswanth S, et al. Anaerobe. 2013;24:43. doi: 10.1016/j.anaerobe.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Yellapu NK, et al. Biotechnol Res Int. 2013;2013:264793. doi: 10.1155/2013/264793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eswar N, et al. Curr Protoc Bioinformatics. 2006;5:5. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bramucci E, et al. BMC Bioinformatics. 2012;13:S2. doi: 10.1186/1471-2105-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee S, et al. BMC Biochem. 2005;6:20. doi: 10.1186/1471-2091-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JD, et al. Nucleic Acids Res. 1997;25:4876. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laskowski RA, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 18.Wang Y, et al. Nucleic Acids Res. 2009;37:W623. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valasani KR, et al. J Chem Inf Model. 2013;53:2033. doi: 10.1021/ci400196z. [DOI] [PubMed] [Google Scholar]
- 20.Valasani KR, et al. Chem Biol Drug Des. 2013;81:238. doi: 10.1111/cbdd.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad UV, et al. Appl Biochem Biotechnol. 2013;169:862. doi: 10.1007/s12010-012-0027-8. [DOI] [PubMed] [Google Scholar]
- 22.Somerville GA, et al. Infect Immun. 2002;70:6373. doi: 10.1128/IAI.70.11.6373-6382.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi H, et al. J Mol Model. 2012;18:1037. doi: 10.1007/s00894-011-1140-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.