

Abstract
In the genomic and proteomic era, efficient and automated analyses of sequence properties of protein have become an important task in bioinformatics. There are general public licensed (GPL) software tools to perform a part of the job. However, computations of mean properties of large number of orthologous sequences are not possible from the above mentioned GPL sets. Further, there is no GPL software or server which can calculate window dependent sequence properties for a large number of sequences in a single run. With a view to overcome above limitations, we have developed a standalone procedure i.e. PHYSICO, which performs various stages of computation in a single run based on the type of input provided either in RAW-FASTA or BLOCK-FASTA format and makes excel output for: a) Composition, Class composition, Mean molecular weight, Isoelectic point, Aliphatic index and GRAVY, b) column based compositions, variability and difference matrix, c) 25 kinds of window dependent sequence properties. The program is fast, efficient, error free and user friendly. Calculation of mean and standard deviation of homologous sequences sets, for comparison purpose when relevant, is another attribute of the program; a property seldom seen in existing GPL softwares.
Availability
PHYSICO is freely available for non-commercial/academic user in formal request to the corresponding author akbanerjee@biotech.buruniv.ac.in
Keywords: UNIX, physicochemical properties, FASTA, software, protein sequence
Background
In post genomic era, bioinformatics taking the lion share of research in the field of computational biology. Physicochemical properties of amino acids (building block of protein) can be used to study protein sequence profiles, structures and functions [1]. Various types of chromatographic procedures need apriori information about physicochemical properties such as molecular weight, net charge and binding affinity for the isolation of proteins. In general, Isoelectric point, Molecular weight, Aliphatic index and GRAVY are most popular among all physicochemical properties. However, these values do not reveal anything about the code of proteins i.e. the sequence property. Sequence properties such as Polarity, hydrophobicity, hydrophilicity, relative mutability, bulkiness, refractivity and others, which are computed using experimentally determined amino acids index values, are most important. Determination of physicochemical properties of unknown proteins and finding its correlation with known ones is very important as it can provide insight into their structure and hence function. Three dimensional structures of proteins, which are more conserve, are determined by more diverse amino acids sequences [2]. Several online GPL softwares are present which analyze physicochemical properties of proteins. Most authentic among them is from ExPASy [3]. However, major bottle neck in this is that one has to input one sequence at a time for computation of above properties, which is time consuming and limited by the speed of internet. Further, managing outputs may create confusion. Propas [4] a standalone software, which can perform calculation on a number of sequences but only of three kinds of physicochemical properties. Thus, available software tools suffer from one or the other limitations. We, therefore, have developed PHYSICO which not only computes 25 kinds of sequence properties and physicochemical properties of large set of protein sequences but also computes mean properties as well as standard deviation of all input sequences for comparison. Our program thus would be helpful for researchers in the field of bioinformatics and computational biology.
Program input
PHYSICO takes protein FASTA file (raw or block) as input. User needs to be on the same directory where the FASTA file is located. In the command prompt, the program is to be run with the name of FASTA file as argument. Upon thorough check of the input file, PHYSICO starts computation and output redirection.
Preparation of RAW-FASTA file:
In this, full length sequence is to be kept in one line, following the title line (FASTA FORMAT). Any number of sequences of any length can be kept in the file. Sequences are to be retrieved from an authentic database and fed in a local text file along with header line.
Preparation of BLOCK-FASTA file:
In block FASTA file all sequences are of same length. Analysis of this file is useful for comparison among different orthologous sets adapted in different environment (such as mesophilic, thermophilic etc). Firstly a raw FASTA file is made using all available orthologous sequences as above and then it is subjected for multiple sequence alignment. Indels are to be removed and FASTA format is to be made. The block FASTA file thus obtained can be subdivided based on environment specific proteins (thermophilic or mesophilic etc) or can be separated in the same FASTA file by putting a specific marker (such as ##1, ##2 etc) just after > in the header line. Program can automatically identify the marker if it exists and process accordingly.
Program output
PHYSICO writes itemized excel outputs for block or raw FASTA files as input. In block FASTA file homologous position based computations are relevant and thus the program redirects extra relevant outputs for block FASTA files apart from common outputs. Figure 1 show details of the program.
Caveats and future development
PHYSICO is written in GAWK (interactive) language which can preferably run in any C shell UNIX prompt and also be made run on B shell Linux and window command prompts. We are developing a web based interface for PHYSICO and other tools developed in our lab.
Figure 1.
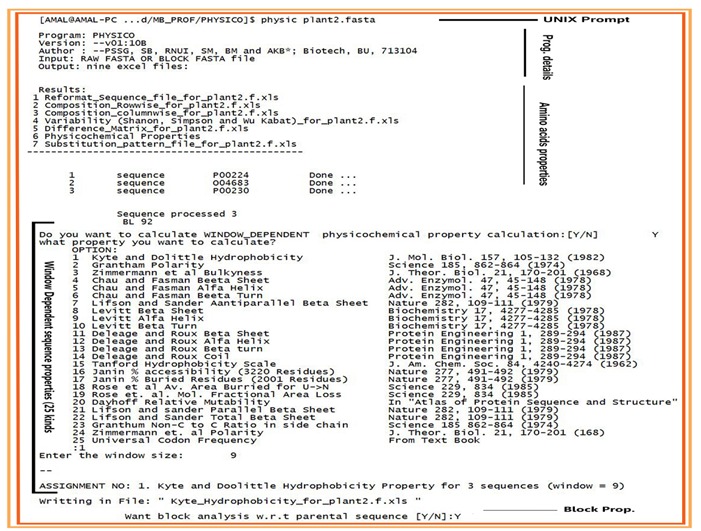
Details and execution of the program in UNIX environment.
Acknowledgments
We thankfully acknowledge the computational facility Laboratory of the Department of Biotechnology, Govt. of India, The University of Burdwan.
Footnotes
Citation:Gupta et al, Bioinformation 10(2): 105-107 (2014)
References
- 1.Cai CZ, et al. Nucleic Acids Res. 2003;31:3692. doi: 10.1093/nar/gkg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cozzone Alain J, et al. Nature. 2002 www.els.net. [Google Scholar]
- 3.Wilkins MR, et al. Methods Mol Biol. 1999;112:531. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 4.Songfeng Wu, et al. Bioinformation. 2012;8:167. doi: 10.6026/97320630008167. [DOI] [PMC free article] [PubMed] [Google Scholar]