

Abstract
Objective
Tuberous Sclerosis Complex (TSC) represents one of the most common genetic causes of epilepsy. TSC gene inactivation leads to hyperactivation of the mammalian target of rapamycin (mTOR) signaling pathway, raising the intriguing possibility that mTOR inhibitors might be effective in preventing or treating epilepsy in patients with TSC. Mice with conditional inactivation of the Tsc1 gene primarily in glia (Tsc1GFAPCKO mice) develop glial proliferation, progressive epilepsy, and premature death. Here, we tested whether rapamycin could prevent or reverse epilepsy as well as other cellular and molecular brain abnormalities in Tsc1GFAPCKO mice.
Methods
Tsc1GFAPCKO mice and littermate controls were treated with rapamycin or vehicle starting at postnatal day 14 (early treatment) or six weeks of age (late treatment), corresponding to times before and after onset of neurological abnormalities in Tsc1GFAPCKO mice. Mice were monitored for seizures by serial video-EEG and for long-term survival. Brains were examined histologically for astrogliosis and neuronal organization. Expression of phospho-S6 and other molecular markers correlating with epileptogenesis was measured by Western blotting.
Results
Early treatment with rapamycin prevented the development of epilepsy and premature death observed in vehicle-treated Tsc1GFAPCKO mice. Late treatment with rapamycin suppressed seizures and prolonged survival in Tsc1GFAPCKO mice that had already developed epilepsy. Correspondingly, rapamycin inhibited the abnormal activation of the mTOR pathway, astrogliosis, neuronal disorganization, and increased brain size in Tsc1GFAPCKO mice.
Interpretation
Rapamycin has strong efficacy for preventing seizures and prolonging survival in Tsc1GFAPCKO mice.
Introduction
Tuberous sclerosis complex (TSC) is caused by mutation of the TSC1 or TSC2 gene and characterized by tumor or hamartoma formation in multiple organs.1–3 Neurological involvement accounts for the most disabling clinical problems, including seizures, autism, and mental retardation. Epilepsy in TSC is particularly severe, affecting between 60–90% of patients and is often refractory to all available medical and surgical therapies. Thus, more effective treatments with true “anti-epileptogenic” actions are clearly needed for TSC patients with epilepsy.
Recent excitement has been generated by the finding that the TSC genes regulate the mammalian target of rapamycin (mTOR) signaling pathway, which controls cell growth and differentiation.7–10 Mutation of either TSC gene results in hyperactivation of the mTOR pathway, leading to abnormal cell growth, proliferation and tumorigenesis. Animal and preliminary clinical studies already suggest that mTOR inhibitors, such as rapamycin, decrease growth of TSC-related tumors, such as renal angiomyolipomas and subependymal giant cell astrocytomas.11–14 However, the utility of mTOR inhibitors in treating epilepsy in TSC has not been investigated in either clinical trials or animal models. We recently described a mouse model of TSC with conditional inactivation of the Tsc1 gene in GFAP-positive cells (Tsc1GFAPCKO mice), which develops progressive epilepsy, encephalopathy, and premature death,15,16 as well as cellular and molecular brain abnormalities likely contributing to epileptogenesis.17–19 In the present study, we test whether treatment with rapamycin can prevent or reverse epilepsy and the associated brain abnormalities in Tsc1GFAPCKO mice.
Materials and Methods
Animals and Drug Protocols
All experiments were conducted according to an approved Washington University animal protocol. Tsc1flox/flox-GFAP-Cre knockout (Tsc1GFAPCKO) mice with conditional inactivation of the Tsc1 gene in glia were generated as described previously.15 Tsc1flox/+-GFAP-Cre and Tsc1flox/flox littermates have previously been found to have no abnormal phenotype and were used as controls in these experiments. Rapamycin (LC Labs, Woburn, MA) was initially dissolved in 100% ethanol, stored at −20°C, and diluted in a vehicle solution containing 5% Tween 80, 5% PEG 400 (Sigma, St. Louis, MO) and 4% ethanol immediately before injection. In “early treatment” studies, rapamycin or vehicle treatment was initiated at postnatal day 14, which precedes the onset of seizures and other neurological abnormalities in Tsc1GFAPCKO mice. In “late treatment” studies, drug treatment was initiated at six weeks of age, which is typically after onset of seizures in these mice.16 Tsc1GFAPCKO mice and control mice were administrated i.p. rapamycin or vehicle 5 days a week, which continued until death or the pre-defined endpoint of the experiment. In one set of studies, mice were monitored daily for survival and weekly for body weight without further interventions. Other studies involved video-EEG monitoring, Western blotting, or histological analysis at defined time points, as described below.
Video-EEG Monitoring
Vehicle- and rapamycin-treated Tsc1GFAPCKO mice underwent weekly video-EEG monitoring starting at 4 weeks of age in early treatment studies and 6 weeks of age in late treatment studies. Bilateral epidural electrode placement and continuous digital EEG and video recordings were performed with established methods, as described previously.16 Forty-eight hour epochs of continuous video-EEG data were obtained once a week from each mouse, until the animal died or the electrodes malfunctioned, and were analyzed for interictal abnormalities and seizures, as reported previously.16 Briefly, interictal spikes were defined as fast (<200 ms) epileptiform waveforms at least twice the amplitude of the background activity. Electrographic seizures were identified by their characteristic pattern of discrete periods of rhythmic spike discharges that evolved in frequency and amplitude lasting at least 10 seconds, typically ending with repetitive burst discharges and voltage suppression. Seizure frequency (# seizures/48 hr period, based on analysis of the entire EEG record) and interictal spike frequency (# spikes/min) were calculated from each 48 hr epoch. Average interictal grade (scale of 1–4) based on multiple samples from each epoch was scored based on a previously-described scale16,20,21: 1 - normal background activity (+/− 6–8 Hz sinusoidal theta rhythm), no epileptiform spikes; 2 - mostly normal background activity, few epileptiform spikes; 3 - mostly abnormal background activity, many spikes; 4 - burst-suppression pattern.
Western Blot Analysis
After 5 weeks (1–3 weeks in preliminary studies) of rapamycin or vehicle treatment, Western blotting was performed to assay expression of phospho-S6 and Glt1 by standard methods. Briefly, neocortex and hippocampus were dissected, sonicated, and centrifuged. Equal amounts of total protein extract were separated by SDS-PAGE and transferred to nitrocellulose membrane. After incubating with primary antibody to phospho-S6 (1:1000, Cell Signaling, Beverly, MA), S6 (1:1000, Cell Signaling), actin (1:5000, Sigma) and Glt-1 (1:1000, Alpha Diagnostics, San Antonio, TX), the membranes were reacted with peroxidase-conjugated secondary antibody. Signals were detected by using ECL reagent (Pierce, Rockford IL) and quantitatively analyzed with ImageJ software.
Histology/Immunohistochemistry
After 3 weeks (late treatment) or 5 weeks (early treatment) of treatment, histological analysis was performed to assess glial proliferation and neuronal organization by standard methods. Briefly, brains were perfusion-fixed with 4% paraformaldehyde and cut into 50 μm sections with a vibratome. Some sections were stained with 0.5% cresyl violet. Other sections were labeled with GFAP antibody (anti-rabbit, 1:500, Sigma) and then rhodamine-conjugated anti-rabbit IgG (1:500, Sigma). Images were acquired with a Zeiss LSM PASCAL confocal microscope. GFAP-immunoreactive cells in neocortex and hippocampus were counted by an investigator blinded to the treatment of the mice. In images from coronal sections at ~ 2 mm posterior to bregma and ~ 1 mm from midline, regions of interest were marked in neocortex by a 200 μm wide box spanning from the neocortical surface to the bottom of layer VI and in hippocampus by a 200x200 μm2 box within the striatum radiatum of CA1 and dentate gyrus. GFAP-immunoreactive cells were quantified in the regions of interest from 2 sections per mouse from a total of 4–5 mice per group. Due to the robust differences in GFAP-positive cells between treatment groups, a more formal stereological approach was not employed. In addition, some sections were counter-stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma), a non-specific nuclear stain for all cells, and quantified for total cell number using similar methods as described above.
Statistics
SigmaStat (Systat Software, San Jose, CA) was used for statistical analysis. For quantitative comparisons (expressed as mean ± SEM), student’s t-test, one-way analysis of variance (ANOVA) with Tukey multiple comparisons post-tests, non-parametric (Kruskal-Walis) ANOVA, or 2-way repeated measured ANOVA was used, depending on the number of group comparisons and conformity to a normal distribution. Kaplan Meier LogRank test was used for survival analysis. Statistical significance was defined as P<0.05.
Results
Rapamycin blocks activation of the mTOR pathway in a dose-dependent manner
Pilot studies in control mice first documented that rapamycin treatment i.p. for 1 week caused a dose-dependent decrease in S6 phosphorylation, a marker of mTOR pathway activation, with 3 mg/kg almost completely inhibiting S6 phosphorylation (Fig. 1A,B). We next tested the effect of 3 mg/kg rapamycin on mTOR activation in Tsc1GFAPCKO mice, with “early treatment” starting at P14 before the onset of seizures in the mice. Vehicle-treated Tsc1GFAPCKO mice have elevated phospho-S6 expression compared to controls, and 3 mg/kg rapamycin for 5 weeks blocked S6 phosphorylation in neocortex and hippocampus of control and Tsc1GFAPCKO mice (Fig. 1C, D). Similar results with phospho-S6 expression were also obtained in Tsc1GFAPCKO mice after 1 and 3 weeks of rapamycin treatment (86% and 89% decrease in the phospho-S6/total S6 ratio in neocortex after 1 and 3 week treatment, respectively).
Figure 1.

Rapamycin treatment antagonizes activation of the mTOR pathway in a dose-dependent fashion. (A) Western blotting shows total S6 and phospho-S6 (P-S6) expression in neocortex and hippocampus of control mice administered different daily doses of rapamycin for one week. (B) Quantitative summary demonstrates that rapamycin blocked the activation (phosphorylation) of S6 in a dose-dependent fashion. The ratio of P-S6/total S6 was normalized to the vehicle-treated control group. (C) Western blotting shows total S6 and P-S6 expression in Tsc1GFAPCKO mice administered 3 mg/kg rapamycin or vehicle for five weeks starting at P14. (D) Quantitative summary demonstrates that vehicle-treated Tsc1GFAPCKO mice have significantly elevated P-S6 levels compared to controls and 3 mg/kg rapamycin inhibits the activation of P-S6 in both Tsc1GFAPCKO and control mice. The ratio of P-S6/total S6 was normalized to the vehicle-treated control group. * p<0.05, ** p<0.01, *** p<0.001 by ANOVA (n = 6 mice per group).
Rapamycin prevents progressive astrogliosis, increased brain size and abnormal neuronal organization in Tsc1GFAPCKO mice
Tsc1GFAPCKO mice develop progressive increases in astrocyte number (astrogliosis) beginning at about 3 weeks of age diffusely throughout the brain, but most notably in neocortex and hippocampus.15 To determine whether early rapamycin treatment could prevent astrogliosis, we treated Tsc1GFAPCKO and control mice with rapamycin or vehicle for 5 weeks starting at P14. In control mice, rapamycin had no effect on the number of GFAP-immunoreactive cells (Fig. 2A–D). In contrast, while vehicle-treated Tsc1GFAPCKO mice showed the expected increase in GFAP-immunoreactive cells compared to control mice, rapamycin treatment prevented astrogliosis in Tsc1GFAPCKO mice in both neocortex and hippocampus (Fig. 2A–D). Paralleling the differences in GFAP-positive cells, similar differences were also seen in the number of DAPI-positive cells between different groups (Fig. 2D), indicating that rapamycin inhibited the total cell (astrocyte) number, not simply the level of GFAP-expression by existing astrocytes.
Figure 2.

Rapamycin treatment prevents histological abnormalities of astrogliosis, increased brain size, and neuronal disorganization in Tsc1GFAPCKO mice. (A–D) Consistent with previous studies,15 vehicle-treated Tsc1GFAPCKO mice displayed striking increases in GFAP-positive cells in neocortex (A,C) and CA1 and dentate gyrus (DG) of hippocampus (B,D) compared to control mice. Rapamycin treatment for 5 weeks starting at P14 prevented this increase in GFAP-positive cells in Tsc1GFAPCKO mice. Similarly, rapamycin prevented an increase in DAPI-positive cells, indicating that the differences in GFAP-positive cells reflect actual differences in overall cell (astrocyte) number, not merely changes in GFAP expression by existing astrocytes. * p<0.001 by ANOVA, between vehicle-treated Tsc1GFAPCKO mice and all other groups (n = 5 mice per group). (E) Correlated with glial proliferation, vehicle-treated Tsc1GFAPCKO mice developed dramatic, diffuse megencephaly compared to control mice, which was prevented by rapamycin treatment. Total brain weight and ratio of brain/body weight were significantly increased in vehicle-treated Tsc1GFAPCKO mice compared to control mice. Rapamycin treatment prevented this increase in brain weight in Tsc1GFAPCKO mice, but had no effect on control mice. * p<0.001 by ANOVA, between vehicle-treated Tsc1GFAPCKO mice and all other groups (n = 10 mice per group). (F) Consistent with previous studies,15 cresyl violet staining demonstrated an obvious dispersion of the pyramidal cell layer (marked by arrows) in hippocampus of vehicle-treated Tsc1GFAPCKO mice compared to controls, which was partially prevented by rapamycin. Scale bars = 200 μm.
The diffuse increase in astrocyte number in Tsc1GFAPCKO mice is also reflected in a generalized increase in brain size. At 7 weeks of age, vehicle-treated Tsc1GFAPCKO mice had grossly larger brains compared to control mice (Fig. 2E, top). Rapamycin treatment prevented this enlargement of Tsc1GFAPCKO brains, while rapamycin had no obvious effect on brain size of control mice. Quantitative analysis of both brain weight and ratio of brain/body weight (Fig. 2E, bottom) confirmed that rapamycin prevented the abnormal brain enlargement observed in vehicle-treated Tsc1GFAPCKO mice.
In addition to the striking abnormalities in astrocyte proliferation, Tsc1GFAPCKO mice have previously been shown to have disorganization and dispersion of the pyramidal cell layer of hippocampus.15 Consistent with previous studies, vehicle-treated Tsc1GFAPCKO mice displayed widely-dispersed pyramidal cell layers in all regions of hippocampus (CA1-CA4) compared to control mice (Fig. 2F). Rapamycin partially preserved a more compact organization to the hippocampal pyramidal neurons (Fig. 2F).
Rapamycin reverses the reduction of Glt-1 expression in Tsc1GFAPCKO mice
Tsc1GFAPCKO mice have previously been shown to have reduced expression of astrocyte-specific glutamate transporters, Glt-1 and GLAST, which represents a potential glial mechanism of epileptogenesis.17 Western blotting of forebrain lysates showed that rapamycin increased the expression of Glt-1 in Tsc1GFAPCKO mice back toward control levels (Fig. 3). Interestingly, rapamycin also increased Glt-1 expression in control mice.
Figure 3.
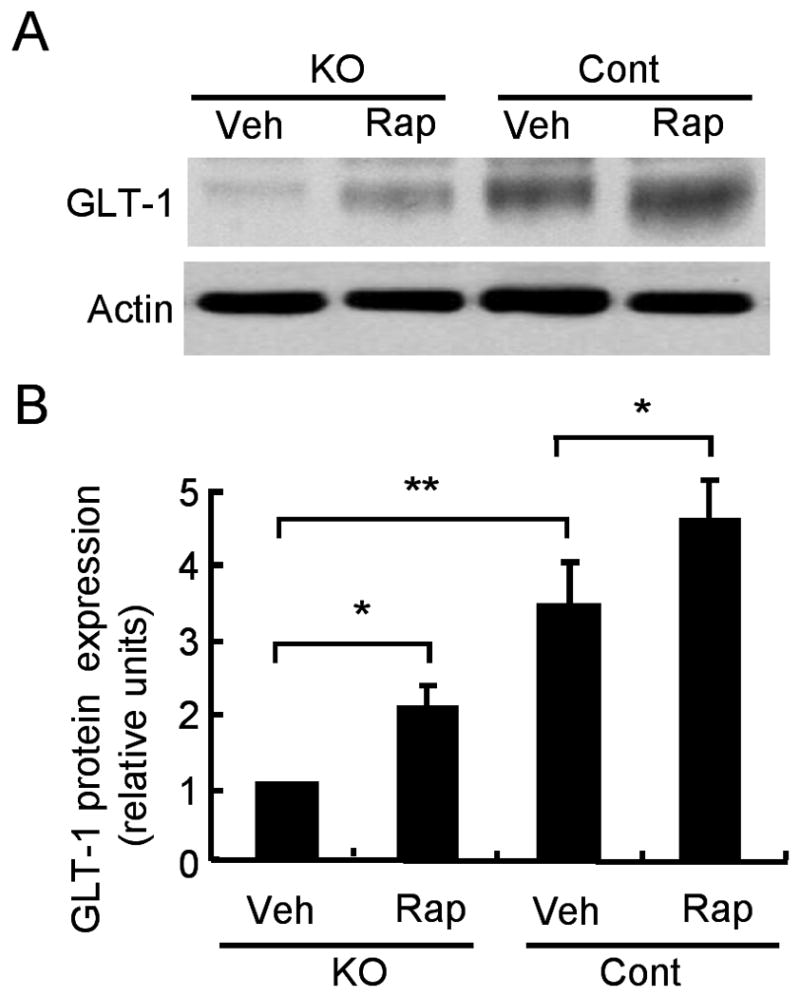
Rapamycin treatment reverses the reduction of Glt-1 expression in Tsc1GFAPCKO mice. (A) Western blotting of cortical homogenates shows Glt-1 expression in vehicle and rapamycin-treated Tsc1GFAPCKO and control mice. (B) Consistent with previous studies,17 quantitative summary demonstrates that vehicle-treated Tsc1GFAPCKO mice have significantly lower Glt-1 expression compared to control mice. Rapamycin treatment for 5 weeks starting at P14 increased the expression of Glt-1 in Tsc1GFAPCKO mice, as well as control mice. Glt-1 was normalized to actin. * p<0.05, ** p<0.01 by ANOVA (n = 6 mice per group).
Early rapamycin treatment prevents the development of epilepsy and premature death in pre-symptomatic Tsc1GFAPCKO mice
Next, we asked if early rapamycin treatment, beginning at P14, could prevent the development of seizures, which typically start between 1–2 months of age in Tsc1GFAPCKO mice. Vehicle-treated Tsc1GFAPCKO mice developed progressive seizures as described previously.15,16 In contrast, no seizures were detected in rapamycin-treated mice by weekly video EEG monitoring starting at 4 weeks of age until 17 weeks of age, when the study was terminated (Fig. 4A).
Figure 4.

Early rapamycin treatment prevents the development of epilepsy and premature death in pre-symptomatic Tsc1GFAPCKO mice. (A) Seizures started to develop in vehicle-treated Tsc1GFAPCKO mice between 1–2 months of age and became progressively more frequent with age (Note that premature death precluded analysis of seizure frequency beyond 10 weeks in vehicle-treated Tsc1GFAPCKO mice). Early rapamycin treatment starting at P14 completely prevented the development of seizures in Tsc1GFAPCKO mice (n = 8 mice per group). ** p<0.01, *** p<0.001 by ANOVA. (B) Interictal spike frequency progressively increased with age in vehicle-treated Tsc1GFAPCKO mice, but was stabilized by rapamycin. * p<0.05, ** p<0.01 by ANOVA, comparing vehicle and rapamycin groups (n = 8 mice per group). (C,D) Interictal EEG background activity, graded by a documented rating scale,16,20,21 became progressively more abnormal with age in vehicle-treated Tsc1GFAPCKO mice, but did not worsen in rapamycin-treated Tsc1GFAPCKO mice. * p<0.05, *** p<0.001 by ANOVA, comparing vehicle and rapamycin groups (n = 8 mice per group). (E) Reflective of a progressive encephalopathic process involving decreased feeding behavior, vehicle-treated Tsc1GFAPCKO mice start to exhibit weight loss after 7 weeks of age. Rapamycin-treated Tsc1GFAPCKO mice did not demonstrate such weight loss, but gained weight at a rate comparable to rapamycin-treated control mice. Rapamycin-treated control mice exhibited slightly slower weight gain compared to vehicle-treated control mice. * p<0.001 by ANOVA, involving all group comparisons except KO-Rap versus Cont-Rap (n = 10–14 mice per group). (F) Survival analysis showed that 50% of vehicle-treated Tsc1GFAPCKO mice died by 9 weeks of age, with all dead by 4 months (n = 10). In contrast, almost all rapamycin-treated Tsc1GFAPCKO mice (n = 10 of 11) survived during the entire treatment period, starting at P14 and extending until 6 months of age (p<0.001 by Kaplan-Meier LogRank test).
In addition to seizures, untreated Tsc1GFAPCKO mice exhibit a progressive encephalopathy and abnormalities in interictal EEG background, also starting between 1–2 months of age.15,16 Rapamycin treatment prevented the progressive abnormalities in interictal EEG background compared to vehicle-treated Tsc1GFAPCKO mice (Fig. 4B–D). Behavioral correlates to this progressive encephalopathic process are decreasing feeding behavior with age, associated with poor weight gain. Rapamycin-treated Tsc1GFAPCKO mice gained weight at a similar rate as rapamycin-treated control mice, in contrast to vehicle-treated Tsc1GFAPCKO mice, which gradually lost weight after 7 weeks of age (Fig. 4E). Of note, rapamycin-treated control mice had slightly slower weight gain compared to vehicle-treated control mice, indicating that rapamycin treatment has systemic side effects.
The effect of early rapamycin treatment on long-term survival was also tested. No mortality was observed in littermate controls treated with rapamycin or vehicle. Vehicle-treated Tsc1GFAPCKO mice exhibited premature death as described previously,15 with all mice dying by 4 months. In contrast, 10 of 11 rapamycin-treated mice survived to at least 6 months (Fig. 4F). To determine whether this survival effect might persist after discontinuing rapamycin, at 6 months half of the rapamycin-treated Tsc1GFAPCKO mice were switched to vehicle treatment (n = 5), whereas the other half continued to receive rapamycin (n = 5). While all the rapamycin-treated mice continued to survive, all mice switched to vehicle at 6 months died after an average of 62.6 ± 7.8 days. All of these mice were also witnessed to have obvious clinical seizures only after being switched to vehicle. In a separate set of mice, Tsc1GFAPCKO mice converted from rapamycin treatment at 6 months to vehicle for 5 weeks had significantly larger brains (brain weight = 0.63 ± 0.01 g in mice converted to vehicle versus 0.43 ± 0.01 in mice on continued rapamycin treatment, p<0.001 by t-test), increased numbers of GFAP-positive cells, increased phospho-S6 expression, and pyramidal neuron dispersion in the hippocampus, compared to age-matched Tsc1GFAPCKO mice with continual rapamycin treatment (Supplemental Figure 1). Thus, the beneficial effects of rapamycin were dependent on continued, long-term treatment and reversed after stopping the drug.
Late rapamycin treatment decreases seizures and improves survival in already-symptomatic Tsc1GFAPCKO mice
To determine whether late treatment with rapamycin could reverse the phenotypic abnormalities in already symptomatic Tsc1GFAPCKO mice, we treated Tsc1GFAPCKO mice with rapamycin or vehicle starting at 6 weeks of age in mice that were already experiencing seizures. No differences in seizure frequency, duration or interictal EEG grade were present between the two groups at 6 weeks immediately before initiation of treatment. However, seizure frequency in the rapamycin group decreased dramatically after the first week of treatment, compared to vehicle-treated mice which had progressively more frequent seizures (Fig. 5A). After 3 weeks of treatment, half (4 of 8) of the rapamycin-treated Tsc1GFAPCKO mice became seizure-free for the duration of the study and the remaining mice only had 0.4 ± 0.2 seizures/day, indicating that late rapamycin treatment could suppress or reverse the seizure phenotype in already symptomatic mice. Similarly, late rapamycin treatment stabilized the progressive interictal EEG abnormalities observed in vehicle-treated Tsc1GFAPCKO mice (Fig. 5B,C). Furthermore, late-rapamycin treatment dramatically improved survival of Tsc1GFAPCKO mice, with no deaths observed during the duration of rapamycin treatment, compared with vehicle-treated Tsc1GFAPCKO mice, which all died by 4 months of age (Fig. 5D). Corresponding histological analysis showed that Tsc1GFAPCKO mice receiving late rapamycin treatment for three weeks had a lower number of GFAP-positive cells (Fig. 5E) and DAPI-positive cells (data not shown), decreased brain size/weight (Fig. 5F), and more compact hippocampal pyramidal cell organization (Fig. 5G), compared to vehicle-treated Tsc1GFAPCKO mice. However, the brain size and number of GFAP-positive cells from Tsc1GFAPCKO mice with late rapamycin treatment was still significantly greater than age-matched vehicle-treated control mice, suggesting that late rapamycin treatment did not completely reverse the histological abnormalities of Tsc1GFAPCKO mice.
Figure 5.

Late rapamycin treatment decreases seizures and improves survival in already-symptomatic Tsc1GFAPCKO mice. (A) Late rapamycin treatment starting at 6 weeks of age, when many mice have already developed seizures, reduced seizure frequency in Tsc1GFAPCKO mice. The arrow indicates that rapamycin or vehicle treatment was started immediately following the initial video-EEG session at 6 weeks. ** p<0.01, *** p<0.001 by ANOVA comparing vehicle and rapamycin groups (n = 8 mice per group). (B,C) Late treatment with rapamycin prevented the progressive worsening of interictal spike frequency and background EEG grade. * p<0.05, ** p<0.01, *** p<0.001 (n = 8 mice per group). (D) Late rapamycin treatment increased survival of Tsc1GFAPCKO mice (p<0.001 by Kaplan-Meier LogRank test, n = 10 mice per group). (E,F) Late treatment with rapamycin for 3 weeks decreased GFAP-positive cells in neocortex, brain weight, and brain/body weight ratio compared to vehicle-treated Tsc1GFAPCKO mice, but not back to the levels of vehicle-treated control mice. * p<0.05, ** p<0.01, *** p<0.001 (n = 4 mice per group). (G) Tsc1GFAPCKO mice receiving rapamycin exhibited decreased dispersion of the pyramidal neuron layer (marked by arrows) in hippocampus compared to age-matched vehicle-treated Tsc1GFAPCKO mice.
Discussion
Novel therapies for epilepsy in TSC are clearly needed, given the severity and intractability of seizures in many TSC patients. Currently available medical treatments for epilepsy suppress seizures by directly reducing neuronal excitability, but do not necessarily target the underlying process of epileptogenesis. In TSC, the immediate link between TSC gene inactivation and de-regulated mTOR signaling identifies a rational therapeutic strategy that may effectively reverse underlying pathophysiological mechanisms causing the clinical manifestations of TSC. Inhibition of mTOR pathway signaling is already being explored as a possible therapeutic approach for reducing tumor growth in TSC patients.11–14 As less is known about mechanisms of epileptogenesis in TSC, the potential role of mTOR inhibitors in treating epilepsy has not been thoroughly investigated. In the present study, we provide novel evidence using a genetically-engineered Tsc mouse model that rapamycin may be a very effective treatment for epilepsy in TSC. Early rapamycin treatment before the onset of neurological symptoms prevented the development of epilepsy and associated cellular and molecular brain abnormalities in Tsc1GFAPCKO mice, as well as dramatically prolonging survival. Late rapamycin treatment also decreased seizures and prolonged survival in already symptomatic mice, although it only partially improved the underlying histological abnormalities.
The mechanisms of action of rapamycin in treating neurological abnormalities in TSC are incompletely understood, but likely involve effects on cell growth and proliferation. The TSC1 and TSC2 gene products, hamartin and tuberin, constitutively inhibit the protein kinase, mTOR, which regulates cell growth and proliferation via downstream signaling pathways involved in protein translation.7–10 Inactivation of either TSC gene results in hyperactivation of mTOR and associated downstream effectors (e.g. S6, S6K), accounting for abnormalities in cell size and proliferation observed in multiple organs in TSC.22–26 Rapamycin directly inhibits mTOR and thus opposes the hyperactivation of the mTOR pathway and resulting abnormalities in cell growth and proliferation observed in TSC. Somewhat analogous to our present results with Tsc1GFAPCKO mice, a rapamycin analog counters abnormal cell growth and brain enlargement, as well as seizures and premature death, in Pten-deficient mice.27 Rapamycin may also have similar effects in Tsc1SynapsinCKO mice related to neuronal growth and differentiation, but these results have not yet been published in complete form and effects on seizures may be more difficult to monitor due to very early mortality in these mice.28
While the prevailing concept of brain pathogenesis in TSC primarily invokes an early developmental defect in glioneuronal differentiation and proliferation,29 some pathophysiological processes in TSC may remain or become active at later stages. This may be particularly true with astrocyte proliferation in focal TSC brain lesions, such as subependymal giant cell astrocytomas (SEGAs) and, to a lesser degree, tubers. In support of this view, rapamycin has been shown to cause regression of SEGAs in children and adults with TSC, a dramatic effect that could be due to cell loss (e.g. necrosis/apoptosis) or reduced cell size.14 In the Tsc1GFAPCKO mice, while late treatment did not necessarily reverse the increased brain size and astrogliosis back to control levels, early and late treatment with rapamycin was clearly effective in preventing further progression of astrocyte proliferation and brain growth starting from the time rapamycin was initiated. Similarly, rapamycin at least partially prevented progressive neuronal disorganization in the hippocampus of Tsc1GFAPCKO mice. While the specific mechanisms mediating this neuronal effect are unclear, rapamycin could counter pyramidal cell dispersion by inhibiting intercalating astrocyte proliferation, aberrant glial-mediated neuronal migration, or effects of seizures on neurons.
Although the effects of rapamycin in suppressing cell growth and proliferation are clear both in the Tsc1GFAPCKO mice and in TSC-related tumors,11–14 less is known about the specific cellular and molecular mechanisms underlying the anti-epileptic action of rapamycin observed in this study. Mechanisms of epileptogenesis in TSC are poorly understood, although numerous abnormalities have been described in human tissue and animal models of TSC that may influence neuronal excitability on the molecular, cellular, and network level. While the most attention is focused on neuronal defects that may cause seizures, studies in Tsc1GFAPCKO mice suggest that glia may also contribute to epileptogenesis.15–19 On a circuit level, glial proliferation observed in these mice could promote seizures by disrupting normally-balanced excitatory and inhibitory neuronal circuits. In addition, on a molecular level, defects in astrocyte glutamate transport may lead to neuronal excitability due to inadequate buffering of extracellular excitatory glutamate. In the present study, rapamycin prevented or reversed cellular and molecular abnormalities in glial proliferation and astrocyte glutamate transporters in parallel with the inhibition of seizures, suggesting that these changes might account for the beneficial effects of rapamycin on seizures. The rapamycin-induced increase in Glt-1 protein expression in both control and Tsc1GFAPCKO mice supports previous work linking the Akt/mTOR pathway to selective transcriptional regulation of Glt-1.30 Alternatively, rapamycin could have anti-epileptic actions that are independent on mTOR inhibition, although to our knowledge rapamycin has not been reported to have anti-epileptic effects in non-mTOR-related epilepsy models. Finally, rapamycin could have other, more direct effects on neuronal physiology, independent of glial-mediated mechanisms.31,32 Additional studies are required to more definitively identify the most critical mechanisms underlying epileptogenesis in Tsc1GFAPCKO mice and the anti-epileptogenic action of rapamycin.
In addition to the impressive effects of rapamycin in inhibiting seizures in the Tsc1GFAPCKO mice, rapamycin had equally dramatic effects on survival of these mice. While all untreated Tsc1GFAPCKO mice die by 3–4 months of age, all but one mouse from both the early and late rapamycin treatment groups survived for as long as they continued to receive rapamycin. The mechanisms of death in untreated Tsc1GFAPCKO mice likely involve multiple factors, including direct seizure-induced death in some cases (documented during video-EEG monitoring) and a more systemic “wasting” syndrome as reflected in decreased activity and feeding behavior, an encephalopathic EEG, and progressive weight loss. Rapamycin likely improves survival in the Tsc1GFAPCKO mice by a combination of direct effects of inhibiting seizures and indirect effects on the general behavior/health of the mice.
While one must be careful in extrapolating animal model data to human disease, the documented anti-epileptogenic efficacy of rapamycin in Tsc1GFAPCKO mice with both “early” and “late” treatment paradigms has obvious clinical applications. As some patients are diagnosed with TSC prior to the onset of neurological symptoms due to non-neurological findings or a positive family history, early treatment with rapamycin may be able to prevent the development of epilepsy, especially in patients at high risk (e.g. with an abnormal brain MRI). As other patients are only diagnosed with TSC after the onset of seizures, later rapamycin treatment may be able to improve the symptoms and prognosis of already symptomatic patients. Of important note, however, our data indicate that long-term continued treatment with rapamycin is required in order to maintain a therapeutic effect, as stopping rapamycin treatment resulted in a delayed emergence of the neurological phenotype in Tsc1GFAPCKO mice. Furthermore, the present study did not systematically examine the safety of rapamycin in detail. While both rapamycin-treated Tsc1GFAPCKO and control mice did well in terms of survival and gross behavior, rapamycin decreased weight gain slightly in control mice, indicating that rapamycin is not completely without side effects. Rapamycin might also impair synaptic plasticity,31,32 which may have detrimental effects on learning and other critical developmental processes of the brain. Thus, additional animal and clinical studies are required to determine the relative risk/benefit ratio of rapamycin treatment for neurological symptoms of TSC. Overall, however, the present study provides exciting pre-clinical data that support the initiation of clinical trials using rapamycin analogs for treating epilepsy in TSC.
Supplementary Material
The beneficial effects of rapamycin on the phenotype of Tsc1GFAPCKO mice are dependent on continued treatment and reverses after termination of drug treatment. Tsc1GFAPCKO mice were treated with rapamycin from P14 to 6 months of age, then half were switched to vehicle (KO-Veh) and half received continued rapamycin (KO-Rap) for an additional 5 weeks. Age-matched control mice received only vehicle (Cont-Veh). (A) Total S6 and phospho-S6 (P-S6) expression was measured by Western blotting in neocortex and hippocampus. Tsc1GFAPCKO mice that were switched from rapamycin to vehicle had significantly elevated P-S6 levels compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. The ratio of P-S6/total S6 was normalized to the vehicle-treated control group. *** p<0.001 by ANOVA (B) Tsc1GFAPCKO mice that were switched from rapamycin to vehicle also had significantly increased GFAP positive cells in neocortex and hippocampus compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. * p<0.05, ** p<0.01, *** p<0.001 by ANOVA. (C) Tsc1GFAPCKO mice that were switched from rapamycin to vehicle exhibited increased dispersion of the pyramidal neuron layers of hippocampus, compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. n = 3 mice/group for all experiments.
Acknowledgments
This work was supported by the National Institutes of Health (K02NS045583 and R01NS056872, MW) and the Tuberous Sclerosis Alliance (MW).
Footnotes
Statement of Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 2.Holmes GL, Stafstrom CE the Tuberous Sclerosis Study Group. Tuberous Sclerosis Complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48:617–630. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- 3.Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 4.Webb DW, Fryer AE, Osborne JP. On the incidence of fits and mental retardation in tuberous sclerosis. J Med Genet. 1991;28:395–397. doi: 10.1136/jmg.28.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sparagana SP, Delgado MR, Batchelor LL, Roach ES. Seizure remission and antiepileptic drug discontinuation in children with tuberous sclerosis complex. Arch Neurol. 2003;60:1286–1989. doi: 10.1001/archneur.60.9.1286. [DOI] [PubMed] [Google Scholar]
- 6.Devlin LA, Shepherd CH, Crawford H, Morrison PJ. Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495–499. doi: 10.1017/S0012162206001058. [DOI] [PubMed] [Google Scholar]
- 7.Gao X, Zhang Y, Arrazola P, et al. Tsc tumour suppressor proteins antagonize amino acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- 8.Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 9.Tee AR, Fingar DC, Manning BD, et al. Tuberous sclerosis complex-1 and –2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 11.Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res. 2005;57:67–75. doi: 10.1203/01.PDR.0000147727.78571.07. [DOI] [PubMed] [Google Scholar]
- 12.Lee L, Sudentas P, Donohue B, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 13.Wienecke R, Fackler I, Linsenmaier U, et al. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis. 2006;48:e27–29. doi: 10.1053/j.ajkd.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 15.Uhlmann EJ, Wong M, Baldwin RL, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 16.Erbayat-Altay E, Zeng LH, Xu L, et al. The natural history and treatment of epilepsy in a murine model of tuberous sclerosis. Epilepsia. 2007;48:1470–1476. doi: 10.1111/j.1528-1167.2007.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong M, Ess KE, Uhlmann EJ, et al. Impaired astrocyte glutamate transport in a mouse epilepsy model of tuberous sclerosis complex. Ann Neurol. 2003;54:251–256. doi: 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- 18.Jansen LA, Uhlmann EJ, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in Tuberous Sclerosis Complex-1 deficient astrocytes. Epilepsia. 2005;46:1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 19.Zeng LH, Ouyang Y, Gazit V, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007;28:184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffey MA, Wozniak D, Wong M, et al. CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther. 2006;13:538–547. doi: 10.1016/j.ymthe.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Kielar C, Maddox L, Bible E, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2007;25:150–162. doi: 10.1016/j.nbd.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito N, Rubin GM. Gigas, a Drosophila homolog of the tuberous sclerosis gene product-2, regulates the cell cycle. Cell. 1999;96:529–539. doi: 10.1016/s0092-8674(00)80657-1. [DOI] [PubMed] [Google Scholar]
- 23.Potter CJ, Huang H, Xu T. Drosophila TSC1 functions with TSC2 to antagonize insulin signaling in regulating cell growth, cell proliferation and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes & Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tapon N, Ito N, Dickson BJ, et al. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 26.Uhlmann EJ, Li W, Scheidenhelm DK, et al. Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia. 2004;47:180–188. doi: 10.1002/glia.20036. [DOI] [PubMed] [Google Scholar]
- 27.Kwon CH, Zhu X, Zhang J, Baker SJ. mTOR is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meikle LM, Talos DM, Onda H, et al. Characterization of a murine neuronal specific knockout of Tsc1. Epilepsia. 2005;46(Suppl 8):273. [Google Scholar]
- 29.Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia. 2007 doi: 10.1111/j.1528-1167.2007.01270.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li LB, Toan SV, Zelenaia O, Watson DJ, Wolfe JH, Rothstein JD, Robinson MB. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem. 2006;97:759–771. doi: 10.1111/j.1471-4159.2006.03743.x. [DOI] [PubMed] [Google Scholar]
- 31.Tang SJ, Reis G, Kang H, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cammalleri M, Lutjens R, Berton F, et al. Time-restricted role for dendritic activation of the mTOR-p70S6K pathways in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci USA. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The beneficial effects of rapamycin on the phenotype of Tsc1GFAPCKO mice are dependent on continued treatment and reverses after termination of drug treatment. Tsc1GFAPCKO mice were treated with rapamycin from P14 to 6 months of age, then half were switched to vehicle (KO-Veh) and half received continued rapamycin (KO-Rap) for an additional 5 weeks. Age-matched control mice received only vehicle (Cont-Veh). (A) Total S6 and phospho-S6 (P-S6) expression was measured by Western blotting in neocortex and hippocampus. Tsc1GFAPCKO mice that were switched from rapamycin to vehicle had significantly elevated P-S6 levels compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. The ratio of P-S6/total S6 was normalized to the vehicle-treated control group. *** p<0.001 by ANOVA (B) Tsc1GFAPCKO mice that were switched from rapamycin to vehicle also had significantly increased GFAP positive cells in neocortex and hippocampus compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. * p<0.05, ** p<0.01, *** p<0.001 by ANOVA. (C) Tsc1GFAPCKO mice that were switched from rapamycin to vehicle exhibited increased dispersion of the pyramidal neuron layers of hippocampus, compared to Tsc1GFAPCKO mice that received continuous rapamycin, and control mice. n = 3 mice/group for all experiments.