

Abstract
Aims
Imbalance between pro- and antioxidant species (e.g. during aging) plays a crucial role for vascular function and is associated with oxidative gene regulation and modification. Vascular aging is associated with progressive deterioration of vascular homeostasis leading to reduced relaxation, hypertrophy, and a higher risk of thrombotic events. These effects can be explained by a reduction in free bioavailable nitric oxide that is inactivated by an age-dependent increase in superoxide formation. In the present study, mitochondria as a source of reactive oxygen species (ROS) and the contribution of manganese superoxide dismutase (MnSOD, SOD-2) and aldehyde dehydrogenase (ALDH-2) were investigated.
Methods and results
Age-dependent effects on vascular function were determined in aortas of C57/Bl6 wild-type (WT), ALDH-2−/−, MnSOD+/+, and MnSOD+/− mice by isometric tension measurements in organ chambers. Mitochondrial ROS formation was measured by luminol (L-012)-enhanced chemiluminescence and 2-hydroxyethidium formation with an HPLC-based assay in isolated heart mitochondria. ROS-mediated mitochondrial DNA (mtDNA) damage was detected by a novel and modified version of the fluorescent-detection alkaline DNA unwinding (FADU) assay. Endothelial dysfunction was observed in aged C57/Bl6 WT mice in parallel to increased mitochondrial ROS formation and oxidative mtDNA damage. In contrast, middle-aged ALDH-2−/− mice showed a marked vascular dysfunction that was similar in old ALDH-2−/− mice suggesting that ALDH-2 exerts age-dependent vasoprotective effects. Aged MnSOD+/− mice showed the most pronounced phenotype such as severely impaired vasorelaxation, highest levels of mitochondrial ROS formation and mtDNA damage.
Conclusion
The correlation between mtROS formation and acetylcholine-dependent relaxation revealed that mitochondrial radical formation significantly contributes to age-dependent endothelial dysfunction.
Keywords: Vascular dysfunction, Mitochondrial oxidative stress, Manganese superoxide dismutase, Mitochondrial aldehyde dehydrogenase, 8-oxodG
1. Introduction
Cardiovascular diseases, such as myocardial infarction, stroke, arteriosclerosis, or peripheral occlusive diseases increase in prevalence with age. The aging vasculature displays typical morphological, and molecular alterations leading to the increased vascular stiffness, reduced compliance and most importantly, endothelial dysfunction.1 In the aged endothelium, the multimodal regulation of vascular tone, smooth muscle cell proliferation, and cell adhesion by the endothelium-derived autacoids such as prostacyclin (PGI2) and nitric oxide (•NO) becomes unbalanced.2,3 These mediators are adversely affected by enhanced free radical formation. Superoxide (•O2−) has been shown to react in a nearly diffusion-controlled reaction with •NO to form the highly reactive peroxynitrite (ONOO−).4 Thus, •O2− not only reduces the bioavailability of •NO, but also leads to the inactivation of enzymes such as prostacyclin synthase or manganese superoxide dismutase via tyrosine nitration. Various aging models revealed that prostacyclin synthase and MnSOD were impaired in parallel to an increase in •O2− and protein tyrosine nitration that results from enhanced ONOO− formation.5 Vascular •O2− sources include the NADPH-oxidases, uncoupled endothelial NO-synthase, and the mitochondrial electron transport chain.
MnSOD−/− mice best demonstrate the detrimental effects of superoxide and resulting ROS and RNS formation in mitochondria. Newborn MnSOD−/− mice suffer from severe oxidative stress and die within a few days due to dilated cardiomyopathy or neurodegenerative processes. It is currently unknown whether the long-term treatment with antioxidants can prevent ROS-mediated mitochondrial DNA (mtDNA) mutations.6 Heterozygous MnSOD+/− animals survive, but show age-dependent endothelial dysfunction and enhanced arteriosclerosis.7–9 Major alterations were found in mitochondria such as reduced antioxidant capacity, increased mtDNA damage and reduced activity of enzymes of the respiratory chain, and citric acid cycle. These findings suggest that chronic unopposed ROS/RNS formation results in a higher mtDNA mutation rate.
Several studies have also demonstrated that ALDH-2 deficiency contributes to the oxidative stress-related diseases10–12 or parallels a decreased antioxidant capacity.13–15 Recently, we could demonstrate that ALDH-2 deficiency contributes to cardiovascular oxidative stress and dysfunction in the setting of nitrate tolerance, acetaldehyde overload, or doxorubicin-induced cardio-toxicity.16 ALDH-2 is redox-sensitive and inactivated under conditions of mitochondrial oxidative stress,17,18 after treatment with nitroglycerol or in aging. The dual role in the cardiovascular system as the bioactivating enzyme for nitroglycerol and the removal of reactive aldehydes has great impact on the development of endothelial dysfunction. It has been shown that lipid peroxidation and one of the major reactive aldehydes formed, hydroxynonenal (HNE), increase with age. Endogenous aldehydes react with all biological macromolecules and may elevate ROS formation by causing mitochondrial dysfunction.
Based on these findings, we postulate that aging leads to an increase in mtDNA damage as a result of augmented oxidant formation and that reactive oxygen species (ROS) generated from mitochondria lead to impaired nitrate induced-relaxation. We demonstrate that in two models of increased mitochondrial oxidative stress, trough deletion of mitochondrial aldehyde dehydrogenase (ALDH-2−/−) or heterozygous deficiency in manganese superoxide dismutase (MnSOD+/−) leads to the age-related vascular dysfunction.
2. Methods
2.1. Materials
For isometric tension studies with glycerol trinitrate (GTN), a nitrolingual infusion solution (1 mg/mL) was obtained from G. Pohl-Boskamp (Hohenlockstedt, Germany). L-012 (8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H)dione sodium salt) was purchased from Wako Pure Chemical Industries (Osaka, Japan). All other chemicals were of analytical grade and obtained from Sigma–Aldrich, Fluka or Merck. The ALDH-2−/−19 and MnSOD+/−9 mice were generated as published previously.
2.2. Animals
All animal treatments were in accordance with the Guide for the Care and the Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The Ethics Committee of the University Hospital Mainz approved the animal protocol. Male mice in a C57/Bl6 background [B6 wild-type (WT) or ALDH-2−/−] were divided into three age groups, 2 (young), 6 (middle-aged), and 12 months (old), of which each group consisted of 11–20 mice. Additional studies were performed with male mice in a C57/Bl6x129/Ola mixed background (MnSOD+/+ and MnSOD+/−)9 at 7 or 16 months (7–8 mice in each group). The different strains were investigated at different time points (except those for 2-hydroxyethidium determination), but animals from the same strain at different age were analyzed on the same day.
2.3. Isometric tension studies
Isometric tension recording to assess the vasodilator responses to ACh, GTN, and diethylamine NONOate (DEA/NO) were performed in isolated mouse aortic rings preconstricted with prostaglandin F2α (to reach 60–70% of the maximal tone induced by KCl bolus) as described previously.20,21 The stocks of DEA/NO were prepared directly before use in a 1 mM NaOH solution.
2.4. Isolation of mitochondria
Crude mitochondrial preparations were obtained from mouse hearts by gentle homogenization in a glass homogenizer followed by several differential centrifugation steps according to published methods.21–23 For detection of 8-oxodG in mtDNA, mitochondria were further purified by isopycnic gradient centrifugation.24 Purified mitochondria were resuspended in suspension buffer (0.25 M meso-inositol; 10 mM sodium phosphate, pH 7.4; 1 mM magnesium chloride) and used for fluorescent-detection alkaline DNA unwinding (FADU)-analysis.
2.5. Detection of mitochondrial oxidative stress
Mitochondrial suspensions were diluted to a final protein concentration (measured by Lowry) of 0.1 mg/mL in 0.5 mL of PBS buffer containing L-012 (100 µM). ROS production was determined after stimulation with succinate (5 mM final concentration, complex II substrate). L-012-enhanced (100 µM) chemiluminescence was measured at 30 s intervals over 5 min with a chemiluminometer (Berthold Techn., Bad Wildbad, Germany) and the signal was expressed as photon counts per minute. The assay was previously validated.21,25 L-012 ECL is a measure of oxidative stress (the sum of all ROS/RNS produced, with a superior sensitivity for peroxynitrite).
As a specific measure of mitochondrial superoxide formation, 2-hydroxyethidium levels were determined as previously described.26 Briefly, cardiac mitochondria (0.5 mg/mL) were incubated with 50 µM DHE for 20 min at 37°C in PBS buffer. Mitochondria were snap-frozen and stored at −80°C. For analysis, samples were diluted with 50% acetonitrile, centrifuged and 50 µL of the supernatant were subjected to HPLC analysis. The system consisted of a control unit, two pumps, mixer, detectors, column oven, degasser and an autosampler (AS-2057 plus) from Jasco (Groß-Umstadt, Germany), and a C18-Nucleosil 100-3 (125 × 4) column from Macherey–Nagel (Düren, Germany). A high-pressure gradient was employed with acetonitrile and 25 mM citrate buffer, pH 2.2, as mobile phases with the following percentages of the organic solvent: 0 min, 36%; 7 min, 40%; 8–12 min, 95%; 13 min, 36%. The flow was 1 mL/min and DHE was detected by its absorption at 355 nm, whereas 2-hydroxyethidium and ethidium were detected by fluorescence (Ex. 480 nm/Em. 580 nm).
The protocols for the detection of 3-nitrotyrosine and reduced peroxiredoxin-3 (Prx-3) are provided in Supplementary material online.
2.6. Detection of 8-oxodG in mitochondrial DNA with the fluorescence-detection alkaline DNA unwinding-assay
The basic assay format underlying the detection of DNA strand breaks by the automated FADU-assay has been described elsewhere (Moreno-Villanueva et al., 2008 submitted for publication). Here, a modified version of this assay was used by employing the Fpg enzyme to convert oxidative DNA base lesions into strand breaks. Resuspended mitochondria were lysed for 15 min in suspension buffer containing 0.5% NP40. Lysates were split into three reactions: T0 as background signal omitting alkaline unwinding, P0 representing existing strand breaks, and P1+Fpg measuring oxidative DNA lesions. Two microlitre of 8-oxoguanine DNA glycosylase (Fpg) (New England BioLabs, Frankfurt, Germany) was added to the P1 reaction only. All reactions of mitochondrial lysates were supplemented with 10× Fpg reaction buffer and incubated at 30°C for 45 min. Samples were processed with an automated FADU-assay on a Genesis RSP 100 robot system (Tecan, Hombrechtikon, Switzerland). Briefly, 70 µL of lysis buffer (9 M urea, 10 mM NaOH, 2.5 mM cyclohexyl-diamin-tetraacatate, and 0.1% sodium dodecyl sulphate) was pipetted to 70 µL of mitochondrial sample at 0°C. After 30 s of lysis, the alkaline solution (0.425 parts lysis buffer in 0.2 M NaOH) at 0°C was injected and after 30 s of unwinding samples were neutralized with 140 µL of neutralization buffer (14 mM β-mercaptoethanol; 1 M glucose). T-samples were neutralized with 140 µL of neutralization buffer prior to the addition of the alkaline solution, representing total amount of double-stranded DNA (100% control). Double-stranded DNA content was measured by SybrGreen (10 000×, MoBiTec, Göttingen, Germany; 156 µL of a 3:25 000 in H2O) and fluorescence was detected in a 96-well-plate fluorescence reader (Tecan Spectraflour, Crailsheim, Germany) at an excitation of 492 nm and an emission of 520 nm.
2.7. Statistical analysis
Results are expressed as means ± SEM. One-way ANOVA (with Bonferroni's or Dunn's correction for the comparison of multiple means) was used for the comparisons of vasodilator potency and efficacy; L-012-derived chemiluminescence, HPLC-based detection of 2-hydroxyethidium and DNA strand breaks. The EC50 value for each experiment was obtained by log-transformation. P < 0.05 were considered significant.
3. Results
3.1. Mitochondrial oxidative stress
In parallel to the progressive age-dependent impairment of vascular functions in B6WT mice, a significant increase in mitochondrial ROS formation by 33% or by 80% was observed in the middle-aged or aged group, respectively (Figure 1A). In contrast, ALDH-2 deficiency already significantly increased ROS formation by 1.5-fold in young ALDH-2−/− mice over young B6WT mice and reached a maximum in middle-aged animals (P < 0.05 vs. young ALDH-2−/− mice), which was not further enhanced with age (Figure 1B). Aged MnSOD+/+ (B6xOla) mice exerted ROS levels comparable to the B6WT mice, but MnSOD+/− mice (Figure 1C) showed the highest level of free radicals formed that was further increased by 20% compared with middle-aged ALDH-2−/− mice (P < 0.05). The middle-aged MnSOD+/+ mice showed ROS levels comparable to those in middle-aged B6WT mice, which surprisingly were not significantly increased in middle-aged MnSOD+/−. The data obtained with L-012 ECL (measure of oxidative stress) detection were confirmed by a more specific superoxide measurement with HPLC-based detection of 2-hydroxyethidium (Figure 2). This further substantiates the fact that MnSOD is an indispensable mitochondrial antioxidant protein important to compensate for the age-dependent increase in mitochondrial ROS formation and to preserve mitochondrial function over the lifespan.
Figure 1.

Effects of aging in aldehyde dehydrogenase (ALDH-2)- or manganese superoxide dismutase (MnSOD) deficient mice on mitochondrial oxidative stress. Reactive oxygen species (ROS) formation in cardiac mitochondria (0.1 mg/mL final protein) was measured by L-012 (100 µM) enhanced chemiluminescence in the presence of succinate (5 mM). Mitochondria from young, middle-aged, and old B6WT mice (A), ALDH-2−/− mice (B6WT background) (B) and middle-aged vs. old MnSOD+/+/MnSOD+/− mice (B6xOla background) (C) were compared with respect to ROS formation. Data are means ± SEM of indicated number of independent experiments performed with mitochondria from two to three pooled hearts from animals from the same group on the same day. Different strains were measured at different time points. *P < 0.05 vs. young group or MnSOD+/+ mice and #P < 0.05 vs. middle-aged group.
Figure 2.

Effects of aging in aldehyde dehydrogenase (ALDH-2)- or manganese superoxide dismutase (MnSOD)-deficient mice on mitochondrial superoxide formation. Superoxide formation in cardiac mitochondria (0.1 mg/mL final protein) containing 50 µM dihydroethidium was measured by HPLC-based detection of 2-hydroxyethidium (2-HE) in the presence of succinate (5 mM). (A) Mitochondria from young and old B6WT mice, old ALDH-2−/− mice (B6WT background) and middle-aged vs. old MnSOD+/+/MnSOD+/− mice (B6xOla background) were compared with respect to 2-HE formation. (B) Representative chromatograms for 2-HE formation in mitochondria from 7-month MnSOD+/− mice in the presence and absence of succinate (5 mM) or with antimycin A (10 µg/mL) (inhibition of complex III stimulates mitochondrial •O2− formation). (C) Chromatograms of the standards 2-HE and ethidium (each 500 nM). Data are means ± SEM of six measurements performed with mitochondria from three pooled hearts from the same group of animals on the same day. *P < 0.05 vs. young B6WT group, #P < 0.05 vs. old B6WT group and §P < 0.05 vs. 7-month MnSOD+/+ mice. 16-month MnSOD+/− was significant different vs. 16-month +/+ and 7-month +/− groups.
Overall cardiac oxidative stress was also assessed by 3-nitrotyrosine formation, which was increased in old C57/Bl6 and ALDH-2−/− mice (see Supplementary material online, Figure S1) but also by MnSOD deficiency as previously published.9 Aortic mitochondrial oxidative stress was detected by the level of Prx-3 in aortic tissue. Prx-3 is a specific marker for mitochondrial oxidative stress (it exists in a monomeric, reduced thiol form and a dimeric, oxidized disulfide form) and the level of its reduced form decreased in old C57/Bl6 and ALDH-2−/− mice (see Supplementary material online, Figure S2).
The age-dependent increase in mitochondrial ROS formation in B6WT mice parallels mtDNA strand breaks and 8-oxodG content (P1+Fpg) (Figure 3A). However, in young ALDH-2−/− animals (Figure 3B), the frequency of mtDNA strand breaks and the 8-oxodG content were already doubled compared with B6WT. MtDNA strand breaks had a statistically insignificant propensity to increase with age, but only 8-oxodG was significantly elevated in these animals (Figure 3B). Surprisingly, MnSOD-deficiency had no effect on mtDNA damage in the middle aged group nor did the frequency of strand breaks increase in the 16-month-old MnSOD+/+ animals (Figure 3C). This may be related to the different background mouse strain of MnSOD+/+ and MnSOD+/− mice as compared with B6WT or ALDH-2−/− mice (see also Section 4.4). Nonetheless, aged MnSOD deficient mice exerted pronounced mtDNA damage compared with their MnSOD+/+ littermates (Figure 3C).
Figure 3.

Effects of aging in aldehyde dehydrogenase (ALDH-2)- or manganese superoxide dismutase (MnSOD)-deficient mice on mitochondrial DNA lesions. DNA damage in mitochondria from heart tissue from B6WT (A), ALDH-2−/− (B) or MnSOD+/+ vs. MnSOD+/− (C) was determined by the FADU-assay. P0 represents the basic strand break content and P1 samples were pre-treated with Fpg to induce single strand breaks at 8-oxodG sites. Data are means ± SEM of six independent experiments with heart samples each pooled from six mice (B6WT and ALDH-2−/− mice) or three to six independent experiments with heart samples each pooled from three to six mice (MnSOD+/+/MnSOD+/− mice). *P < 0.05 vs. young group or MnSOD+/+ mice and #P < 0.05 vs. middle-aged group.
3.2. Endothelial and smooth muscle function
Endothelium-dependent ACh-induced relaxation of aortas from B6WT mice (Figure 4A, left panel) was significantly deteriorated with age (see Supplementary material online, Table S1). Middle-aged B6xOla control blood vessels (MnSOD+/+) responded similarly to ACh induced relaxation as aortas of B6WT of young and middle-aged animals (compare left and right panel of Figure 4A). However, aged B6xOla mice manifested pronounced endothelial dysfunction and reduced efficacy of ACh than B6WT animals. Interestingly, relaxation of middle-aged MnSOD deficient mice (MnSOD+/−) remained unaltered compared with middle-aged B6xOla control mice (Figure 4A, right panel and Supplementary material online, Table S1), but endothelial dysfunction was most progressed in old MnSOD+/−animals. Middle-aged ALDH-2−/− mice had already developed endothelial-dysfunction (Figure 4A, middle panel and Supplementary material online, Table S1), which was not further impaired in vessels of old ALDH-2−/− mice. The differences between B6WT and ALDH-2−/− mice were not statistically significant, but endothelial dysfunction in MnSOD-deficient mice was significantly different in comparison to all other groups.
Figure 4.
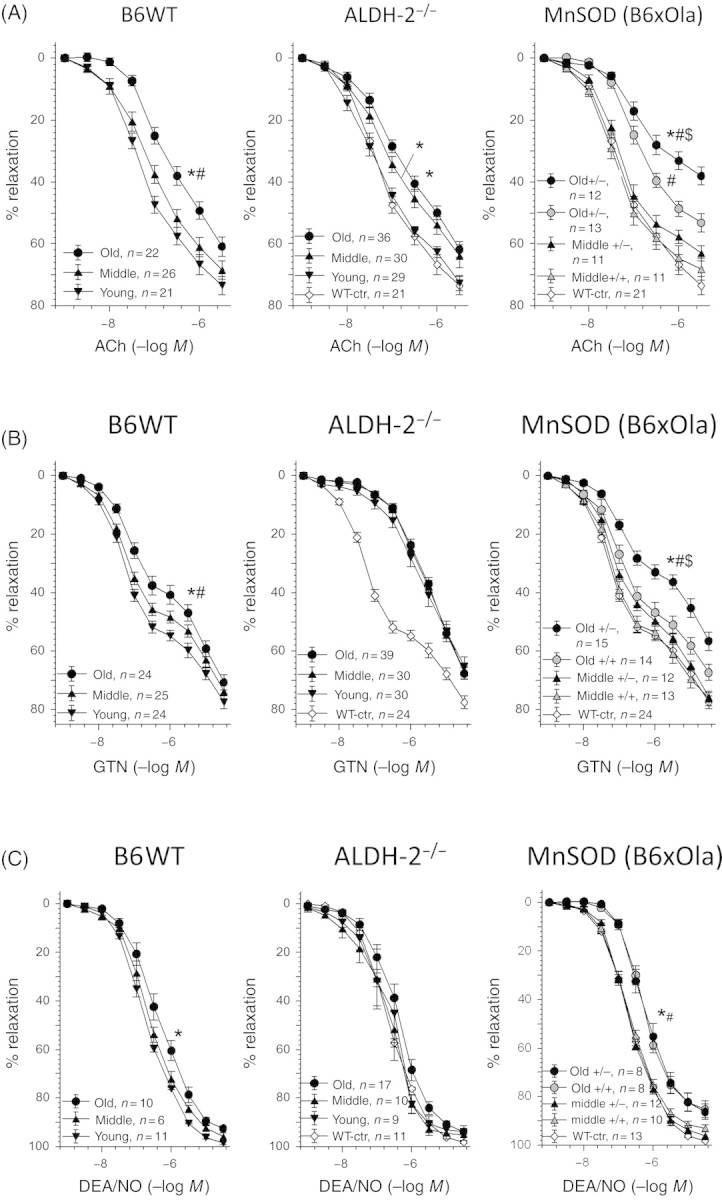
Effects of aging in aldehyde dehydrogenase (ALDH-2)- or manganese superoxide dismutase (MnSOD)-deficient mice on vascular function. Concentration–relaxation curves for acetylcholine (ACh, A), nitroglycerin (GTN, B) and diethylamine NONOate (DEA/NO, C) were obtained by isometric tension recordings of aortic ring segments from young, middle-aged, and aged B6WT mice (left panel), ALDH-2−/− mice (middle panel) or middle-aged vs. old MnSOD+/+/MnSOD+/− mice (right panel). Data are means ± SEM of 40 (B6WT), 50 (ALDH-2−/−) and 7–11 (MnSOD+/+ and MnSOD+/−) animals or more precisely the indicated numbers of aortic rings (see figure inset). *P < 0.05 vs. young group or MnSOD+/+ mice, #P < 0.05 vs. middle-aged group, and $P < 0.05 vs. old B6WT and old ALDH-2−/−.
Endothelium-independent vasodilation was assessed by nitroglycerin (GTN). Impairment of GTN-dependent relaxation of aortas from B6WT mice showed a clear correlation with age (Figure 4B, left panel and Supplementary material online, Table S1). Blood vessels from aged MnSOD+/+ (B6xOla) mice exerted comparable effects to the aged B6WT group (although not significantly different to the middle-aged MnSOD+/+ group), but MnSOD deficiency further (Figure 4B, right panel and Supplementary material online, Table S1) impaired the GTN-response (P < 0.05 vs. old B6WT group). Again, MnSOD-deficiency had only a marginal impact on vascular function in middle-aged animals. However, GTN-dependent relaxation was generally impaired in ALDH-2−/− mice and no significant alterations in the aging vasculature were observed. As previously shown,27,28 GTN-dependent relaxation depends on the bioactivation of the organic nitrate by mitochondrial ALDH-2 but, importantly, aging had no further effect on the GTN response in the vascular smooth muscle (Figure 4B, middle panel and Supplementary material online, Table S1). We therefore propose GTN-dependent relaxation or, more precisely, ALDH-2-mediated GTN-bioactivation as a useful marker for mtROS formation in aging induced vascular dysfunction.
To address endothelium-independent relaxation without relying on ALDH-2-dependent bioactivation, the •NO donor diethylamine NONOate (DEA/NO) was used. •NO-triggered vasodilation revealed a slight, but significant, impairment in old B6WT mice (Figure 4C, left panel). Old B6xOla mice showed a clear impairment of the •NO-dependent vasodilation compared with middle-aged mice, which was not affected by MnSOD-deficiency (Figure 4C, right panel). Nevertheless, DEA/NO-induced relaxation remained unaffected by aging in ALDH-2−/− mice (Figure 4C, middle panel).
3.3. Correlations
In order to explore the relationship between mitochondrial oxidative stress, mtDNA damage, and vascular dysfunction in the knockout mouse models of ALDH−/− and MnSOD+/−, each of these parameters were correlated in the diagrams shown in Figure 5A–C. The best correlation was observed between mitochondrial oxidative stress and vascular function (assessed by acetylcholine-dependent relaxation). Statistical analysis of these parameters revealed a linear relationship with a correlation coefficient of 0.881. This implies that mitochondrial ROS formation is directly related to vascular function. The plot of mtROS vs. mtDNA damage or mtDNA damage vs. vascular dysfunction was less striking, but still resulted in correlation coefficients of 0.684 and 0.590, respectively.
Figure 5.

Correlations between mitochondrial oxidative stress (mtROS), mitochondrial DNA (mtDNA) damage, and vascular (endothelial) function (ACh-induced maximal relaxation). (A) mtROS formation was plotted for all age-groups and mouse strains vs. the corresponding maximal efficacy in response to acetylcholine (ACh). (B) mtROS was plotted for all age-groups and mouse strains vs. the corresponding mtDNA damage. (C) mtDNA damage was plotted for all age-groups and mouse strains vs. the corresponding maximal efficacy in response to acetylcholine. r, correlation coefficient. The groups are 1 = B6WT, 2 months; 2 = B6 WT, 6 months; 3 = ALDH-2−/−, 2 months; 4 = MnSOD+/+, 7 months; 5 = MnSOD+/−, 7 months; 6 = WTB6, 12 months; 7 = ALDH-2−/−, 12 months; 8 = MnSOD+/+, 16 months; 9 = ALDH-2−/−, 6 months; 10 = MnSOD+/−, 16 months. P-values for linear regressions are 0.001 (A), 0.03 (B) and 0.07 (C).
4. Discussion
4.1. Free radical and mitochondrial hypothesis of aging and endothelial dysfunction
In 1954, Harman expressed for the first time the free radical theory of aging: ‘the reaction of active free radicals, normally produced in the organism, with cellular constituents’.29 The participation of ROS in cardiovascular disorders and aging is well-documented. Acutely, ROS overproduction can interfere in important signalling cascades such as inactivation of bioavailable •NO,2 alterations in prostaglandin metabolism or dysregulation of calcium and phosphorylation cascades. Chronically, ROS lead to irreversible oxidations and accumulation of oxidized biological macromolecules, e.g. increase in DNA mutations.30
This hypothesis was extended on mitochondria as the most abundant cellular source of ROS and the resulting increase in the mutation rate of the mitochondrial genome. In general, the biogenesis of respiratory chain components requires concerted contributions from two physically separated genomes, the nuclear DNA and the maternally inherited mtDNA. Mutations of the mitochondrial genome are assumed to impair mitochondrial physiology and ATP-synthesis, which is accompanied by enhanced ROS formation and increased apoptosis.31
Age-dependent interference in vascular redox regulation is best demonstrated by •NO-bioavailability in various rodent models5 and in humans.32,33 With age, •NO is gradually reduced and therefore serves as a useful biomarker for age-dependent endothelial dysfunction. The prevailing hypothesis is that •O2− reacts with •NO and consequently reduces endothelium-derived free •NO, thus impairing vaso-relaxation. We can confirm previously published data that •NO-mediated relaxation, triggered by acetylcholine, declines with age and is severely aggravated in MnSOD+/− or ALDH-2−/− knockout animals. These findings parallel mitochondrial ROS levels that were elevated in an age-dependent manner, and highest in MnSOD+/− and ALDH-2−/− mice. These results suggest that •O2−, as the primary radical generated, inactivates free endothelium-derived •NO. Furthermore, from the interaction of •O2− with •NO, the highly reactive ONOO− is formed. This reactive nitrogen species targets and inactivates enzymes that are important for vascular relaxation such as prostacyclin synthase, soluble guanylyl cyclase, sarcoplasmic endoplasmic reticulum calcium ATPase34, or vital antioxidant defence enzymes such as ALDH-2 or MnSOD.5
4.2. The importance of manganese superoxide dismutase for the cardiovascular system
The importance of mitochondrial •O2− formation is best demonstrated by MnSOD−/− mice, which die postnataly due to dilated cardiomyopathy or neurodegenerative processes.35,36 Major alterations were found in mitochondria such as reduced antioxidant capacity, increased mtDNA damage, and reduced activities of enzymes of the respiratory chain and citric acid cycle. We can confirm that the heterozygous mice developed a severe endothelial-dysfunction, a blunted vasodilatory response to GTN, probably due to the reduced ALDH-2 activity and ROS formation. The close proximity of mtDNA to the formation of deleterious ROS suggests that mtDNA is particularly sensitive to mutations and oxidative lesions. MtDNA lesions were age-dependently increased and severely augmented in both knockout animal models, MnSOD+/− and ALDH−/−mice. These findings suggest that chronic ROS formation, exceeding the mitochondrial antioxidant and repair capacities, result in a higher mutation rate of mtDNA. MtDNA deterioration and oxidative protein modifications may initiate a vicious cycle in which mitochondrial dysfunction further increases oxidative stress resulting in endothelial dysfunction, cell senescence and apoptosis.
4.3. Importance of aldehyde dehydrogenase in the aging vasculature
In the cardiovasculature, mitochondrial ALDH-2 plays a dual role in detoxification of reactive aliphatic aldehydes such as 4-hydroxy-2-nonenal (4-HNE) or xenobiotics37 and bioactivation of organic nitrates.38 4-HNE as an endogenously formed reactive aldehyde is a lipid peroxidation-derived product that reacts with free sulfhydryls and amino groups of proteins or enhances DNA mutagenesis. Lipid peroxidation and the derived toxic aldehydes are particularly detrimental to the mitochondrial inner membrane promoting mtDNA damage, apoptosis,37 (reactive aldehydes can induce mitochondrial permeability transition) and impairment of the electron transport chain. Therefore, as part of the antioxidant defence system, ALDH-2 plays an important role in the preservation of mitochondrial and endothelial physiology as well as the response to organic nitrates. We have recently demonstrated that ALDH-2 deficiency contributes to cardiovascular oxidative stress and dysfunction since chronic doxorubicin, acetaldehyde and GTN-triggered mitochondrial ROS formation and vascular dysfunction were significantly increased in ALDH-2−/− mice.16 Partially, these effects were compensated for by the induction of antioxidant proteins, which demonstrate that compensatory backup systems are highly effective.39 Excessive formation of aldehydes may lower glutathione levels particularly in mitochondria, because aldehydes can be alternatively detoxified by glutathione transferases. The altered GSH/GSSG ratio may induce counter-regulatory mechanisms or impair enzyme function via S-glutathiolation.
Moreover, ALDH-2 is the most important enzyme in the cardiovasculature for the bioactivation and the beneficial effects of organic nitrates. The presence of reactive active site sulfhydryls, which are important for catalysis, renders the enzyme sensitive to ROS. Aging and oxidative stress related diseases such as diabetes are associated with impairment of ALDH-activity, reduced ethanol detoxification and enhanced tolerance to organic nitrates. Importantly, the ALDH-2−/− mice show a blunted response to GTN, which remains unaffected by the aging process. This strongly suggests that the age-dependent attenuation of the GTN-mediated vaso-relaxation is mainly caused by the ROS mediated inactivation of mitochondrial ALDH-2.
4.4. Limitations
The major limitation of the study is the use of different mouse strains (C57/Bl6 and B6xOla), because the background strain may play an important role for MnSOD-associated vascular dysfunction as reported by Brown et al.7 In addition, old MnSOD+/+/MnSOD+/− (16 months) mice differed in age from old B6WT and ALDH-2−/− mice (12 months). However, age-matched controls for each set of experiments suggested a correlation between mtROS and vascular dysfunction, independent of mouse strain or age-group (Figure 5A). The study is further limited by the use of cardiac, instead of aortic mitochondria, which may behave quite distinctly in cardiovascular diseases.40 Nevertheless, we have previously shown that rat cardiac and aortic oxidative stress (mitochondria and NADPH oxidase activity) exert a very similar oxidative stress response in experimental diabetes,41 hypertension42, and nitrate tolerance.38
To address endothelium-independent relaxation without relying on ALDH-2-dependent GTN bioactivation, we also tested the •NO donor diethylamine NONOate (DEA/NO). •NO-triggered vasodilation revealed a slight, but significant, impairment in old B6WT mice (Figure 4C, left panel). Old B6xOla mice showed a clear impairment of the •NO-dependent vasodilation compared with middle-aged mice, which was not affected by MnSOD-deficiency (Figure 4C, right panel). However, DEA/NO-induced relaxation remained unaffected by aging in ALDH-2−/− mice (Figure 4C, middle panel).
4.5. Conclusion
In the present study, we provide evidence that mitochondrial oxidative stress increases with age and contributes to age-related vascular dysfunction. Importantly, two knockout mouse models with increased mitochondrial oxidative stress (ALDH-2−/− and MnSOD+/− mice) demonstrated that mitochondrial ROS generation and oxidative mtDNA lesions are an important determinant for age-associated vascular dysfunction. Moreover, mitochondrial ALDH-2 exerts crucial vasoprotective effects and reduces age-dependent dysfunction, since middle-aged ALDH-2−/− mice have already developed age-dependent endothelial dysfunction.
We have recently reported on the cross-talk between mtROS and cytosolic ROS/RNS in a model of increased mitochondrial oxidative stress (nitroglycerin-induced tolerance). In this system, endothelial dysfunction (sensitive to NADPH oxidases) and vascular dysfunction (sensitive to mitochondria) were dependent on the activation of distinct oxidant sources.26 This cross-talk was blocked by in vivo and ex vivo administration of the mitochondrial permeability transition pore inhibitor cyclosporine A, which selectively improved endothelial dysfunction, whereas nitrite tolerance was not affected. In contrast, the respiratory complex I inhibitor rotenone improved endothelial dysfunction and tolerance. Conversely, in vivo or ex vivo treatment with the KATP opener diazoxide caused a nitrate tolerance-like phenomenon in control animals, whereas the KATP inhibitor glibenclamide improved tolerance in nitroglycerin-treated animals. gp91phox−/− and p47phox−/− mice developed tolerance but no endothelial dysfunction in response to nitroglycerin treatment. We propose that a similar cross-talk exists in the aging vasculature and that aging-induced mtROS can activate cytosolic ROS/RNS sources leading to age-related vascular dysfunction (Figure 6).
Figure 6.

Hypothetic scheme of aging-induced vascular dysfunction and the role of mitochondria in this process. Aging-induced mitochondrial dysfunction triggers mitochondrial reactive oxygen species (mtROS) formation from respiratory complexes I, II, and III (Q = ubiquinone). Break-down of mtROS is catalyzed by glutathione peroxidase (GPx, for H2O2) or manganese superoxide dismutase (MnSOD), the latter is in turn inhibited by mitochondrial peroxynitrite (ONOO−) formation. mtROS increase the levels of toxic aldehydes and inhibit the mitochondrial aldehyde dehydrogenase (ALDH-2), the detoxifying enzyme of those aldehydes. Increase in mtROS and toxic aldehydes also leads to mtDNA strand breaks which leads to augmented dysfunction in respiratory chain complexes and further increase in mtROS since mtDNA encodes mainly for those respiratory complexes. mtROS also activates mitochondrial permeability transition pore (mPTP), which upon opening releases mtROS to the cytosol leading to protein kinase C (PKC)-dependent NADPH oxidase activation, eNOS uncoupling and finally to endothelial dysfunction.26 Cytosolic reactive oxygen and nitrogen species (ROS/RNS) in turn were demonstrated to activate KATP channels, which causes alterations in mitochondrial membrane potential (Ψ) and further augments mtROS levels.43 Effects of rotenone (Rot), cyclosporine A (CsA), diazoxide (Diaz) and glibenclamide (Glib) have been recently demonstrated.26,43
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
The present work was supported by continuous funding by the German Research Foundation (DFG) (SFB 553—C17 to A.D.; BU 698/6-1 to A.B.) and by the Robert-Müller-Foundation, the ‘Forschungsfonds 2006’ of the University Mainz and MAIFOR grants of the University Hospital Mainz (to A.D.). K.S.-K is supported by the German Research Foundation within the Clinical Research Group KFO142. P.W. is supported by the German Heart Foundation/German Foundation of Heart Research.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank Volker Ullrich for helpful discussions and Jörg Schreiner, Merle Götz and Nicole Schramm for expert technical assistance. We acknowledge the help of Rebecca Zee and Dave Pimentel in preparing the manuscript.
References
- 1.Burke GL, Evans GW, Riley WA, Sharrett AR, Howard G, Barnes RW, et al. Arterial wall thickness is associated with prevalent cardiovascular disease in middle-aged adults. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke. 1995;26:386–391. doi: 10.1161/01.str.26.3.386. [DOI] [PubMed] [Google Scholar]
- 2.Vanhoutte PM. Ageing and endothelial dysfunction. Eur Heart J Suppl. 2002;49:A8–A17. [Google Scholar]
- 3.Nakajima M, Hashimoto M, Wang F, Yamanaga K, Nakamura N, Uchida T, et al. Aging decreases the production of PGI2 in rat aortic endothelial cells. Exp Gerontol. 1997;32:685–693. doi: 10.1016/s0531-5565(97)00089-2. [DOI] [PubMed] [Google Scholar]
- 4.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 5.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 7.Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007;27:1941–1946. doi: 10.1161/ATVBAHA.107.146852. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2331–2336. doi: 10.1161/01.ATV.0000238347.77590.c9. [DOI] [PubMed] [Google Scholar]
- 9.Strassburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A, et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. 2005;38:1458–1470. doi: 10.1016/j.freeradbiomed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Ohsawa I, Kamino K, Nagasaka K, Ando F, Niino N, Shimokata H, et al. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J Hum Genet. 2003;48:404–409. doi: 10.1007/s10038-003-0046-y. [DOI] [PubMed] [Google Scholar]
- 11.Ohta S. Roles of mitochondrial dysfunctions in Alzheimer's disease–contribution of deficiency of ALDH 2. Rinsho Shinkeigaku. 2000;40:1231–1233. [PubMed] [Google Scholar]
- 12.Ren J. Acetaldehyde and alcoholic cardiomyopathy: lessons from the ADH and ALDH2 transgenic models. Novartis Found Symp. 2007;285:69–76. doi: 10.1002/9780470511848.ch5. Discussions 76–69, 198–199. [DOI] [PubMed] [Google Scholar]
- 13.Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann N Y Acad Sci. 2004;1011:36–44. doi: 10.1007/978-3-662-41088-2_4. [DOI] [PubMed] [Google Scholar]
- 14.Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J Neurochem. 2003;84:1110–1117. doi: 10.1046/j.1471-4159.2003.01619.x. [DOI] [PubMed] [Google Scholar]
- 15.Szocs K, Lassegue B, Wenzel P, Wendt M, Daiber A, Oelze M, et al. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J Mol Cell Cardiol. 2007;42:1111–1118. doi: 10.1016/j.yjmcc.2007.03.904. [DOI] [PubMed] [Google Scholar]
- 16.Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress—evidence for indirect antioxidative properties. Biochem Biophys Res Commun. 2008;367:137–143. doi: 10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- 17.Wenzel P, Hink U, Oelze M, Schuppan S, Schaeuble K, Schildknecht S, et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (aldh-2) activity: Implications for mitochondrial oxidative stress and nitrate tolerance. J Biol Chem. 2007;282:792–799. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- 18.Daiber A, Oelze M, Coldewey M, Kaiser K, Huth C, Schildknecht S, et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem Biophys Res Commun. 2005;338:1865–1874. doi: 10.1016/j.bbrc.2005.10.106. [DOI] [PubMed] [Google Scholar]
- 19.Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, et al. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–311. doi: 10.1016/s0014-5793(00)01710-5. [DOI] [PubMed] [Google Scholar]
- 20.Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daiber A, Oelze M, Coldewey M, Bachschmid M, Wenzel P, Sydow K, et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: a comparison of pentaerythritol tetranitrate with other organic nitrates. Mol Pharmacol. 2004;66:1372–1382. doi: 10.1124/mol.104.002600. [DOI] [PubMed] [Google Scholar]
- 22.Raha S, McEachern GE, Myint AT, Robinson BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med. 2000;29:170–180. doi: 10.1016/s0891-5849(00)00338-5. [DOI] [PubMed] [Google Scholar]
- 23.Daiber A, Oelze M, Sulyok S, Coldewey M, Schulz E, Treiber N, et al. Heterozygous Deficiency of Manganese Superoxide Dismutase in Mice (Mn-SOD+/−): A Novel Approach to Assess the Role of Oxidative Stress for the Development of Nitrate Tolerance. Mol Pharmacol. 2005;68:579–588. doi: 10.1124/mol.105.011585. [DOI] [PubMed] [Google Scholar]
- 24.Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daiber A, Oelze M, August M, Wendt M, Sydow K, Wieboldt H, et al. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radic Res. 2004;38:259–269. doi: 10.1080/10715760410001659773. [DOI] [PubMed] [Google Scholar]
- 26.Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Muller J, et al. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal. 2008;10:1435–1447. doi: 10.1089/ars.2007.1969. [DOI] [PubMed] [Google Scholar]
- 27.Wenzel P, Hink U, Oelze M, Seeling A, Isse T, Bruns K, et al. Number of nitrate groups determines reactivity and potency of organic nitrates: a proof of concept study in ALDH-2-/- mice. Brit J Pharmacol. 2007;150:526–533. doi: 10.1038/sj.bjp.0707116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 30.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 31.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 32.Chauhan A, More RS, Mullins PA, Taylor G, Petch C, Schofield PM. Aging-associated endothelial dysfunction in humans is reversed by L-arginine. J Am Coll Cardiol. 1996;28:1796–1804. doi: 10.1016/s0735-1097(96)00394-4. [DOI] [PubMed] [Google Scholar]
- 33.Gerhard M, Roddy MA, Creager SJ, Creager MA. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension. 1996;27:849–853. doi: 10.1161/01.hyp.27.4.849. [DOI] [PubMed] [Google Scholar]
- 34.Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, et al. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol. 2006;290:H2220–H2227. doi: 10.1152/ajpheart.01293.2005. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 36.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen JJ, Yu BP. Detoxification of reactive aldehydes in mitochondria: effects of age and dietary restriction. Aging (Milano) 1996;8:334–340. doi: 10.1007/BF03339590. [DOI] [PubMed] [Google Scholar]
- 38.Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J. 2005;19:1143–1145. doi: 10.1096/fj.04-3084fje. [DOI] [PubMed] [Google Scholar]
- 40.Rosen P, Wiernsperger NF. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto-Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes Metab Res Rev. 2006;22:323–330. doi: 10.1002/dmrr.623. [DOI] [PubMed] [Google Scholar]
- 41.Wendt MC, Daiber A, Kleschyov AL, Mulsch A, Sydow K, Schulz E, et al. Differential effects of diabetes on the expression of the gp91(phox) homologues nox1 and nox4. Free Radic Biol Med. 2005;39:381–391. doi: 10.1016/j.freeradbiomed.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Oelze M, Warnholtz A, Faulhaber J, Wenzel P, Kleschyov AL, Coldewey M, et al. NADPH oxidase accounts for enhanced superoxide production and impaired endothelium-dependent smooth muscle relaxation in BKbeta1−/− mice. Arterioscler Thromb Vasc Biol. 2006;26:1753–1759. doi: 10.1161/01.ATV.0000231511.26860.50. [DOI] [PubMed] [Google Scholar]
- 43.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.