

Abstract
Acute air pollutant inhalation is linked to adverse cardiac events and death, and hospitalizations for heart failure. Diesel engine exhaust (DE) is a major air pollutant suspected to exacerbate preexisting cardiac conditions, in part, through autonomic and electrophysiologic disturbance of normal cardiac function. To explore this putative mechanism, we examined cardiophysiologic responses to DE inhalation in a model of aged heart failure–prone rats without signs or symptoms of overt heart failure. We hypothesized that acute DE exposure would alter heart rhythm, cardiac electrophysiology, and ventricular performance and dimensions consistent with autonomic imbalance while increasing biochemical markers of toxicity. Spontaneously hypertensive heart failure rats (16 months) were exposed once to whole DE (4h, target PM2.5 concentration: 500 µg/m3) or filtered air. DE increased multiple heart rate variability (HRV) parameters during exposure. In the 4h after exposure, DE increased cardiac output, left ventricular volume (end diastolic and systolic), stroke volume, HRV, and atrioventricular block arrhythmias while increasing electrocardiographic measures of ventricular repolarization (i.e., ST and T amplitudes, ST area, T-peak to T-end duration). DE did not affect heart rate relative to air. Changes in HRV positively correlated with postexposure changes in bradyarrhythmia frequency, repolarization, and echocardiographic parameters. At 24h postexposure, DE-exposed rats had increased serum C-reactive protein and pulmonary eosinophils. This study demonstrates that cardiac effects of DE inhalation are likely to occur through changes in autonomic balance associated with modulation of cardiac electrophysiology and mechanical function and may offer insights into the adverse health effects of traffic-related air pollutants.
Key Words: echocardiography, air pollution, cardiac function, autonomic, cardiovascular, electrocardiography, heart rate variability, arrhythmia, rats, heart failure
Exposure to combustion-derived air pollutants has been linked to adverse cardiovascular health outcomes, especially in individuals with preexisting cardiac disease. Epidemiological studies suggest the involvement of multiple air pollutants, including fine and ultrafine particulate matter (PM2.5 and UFP, with diameters < 2.5 μm and < 0.1 μm, respectively), nitrogen dioxide (NO2), carbon monoxide (CO), and sulfur dioxide (SO2) (Brook et al., 2004). Diesel engine exhaust (DE) is a major urban source of these pollutants, as well as volatile and semivolatile organics and carbonyls. Moreover, DE is an important contributor to vehicular emissions attendant to adverse clinical outcomes near roadways. For instance, ischemic heart disease hospitalizations in eight European cities have been attributed to DE exposure (Le Tertre et al., 2002), and increases in mortality have been shown to parallel increased traffic particle levels (Maynard et al., 2007). Clinical studies indicate that DE may impart toxicity by adversely altering cardiovascular function. Recent research has demonstrated that acute DE inhalation increases systolic blood pressure, impairs vasodilation, and/or enhances vasoconstriction in humans (Cosselman et al., 2012; Mills et al., 2011). In addition, Mills et al. (2007) found that DE exposure increased exercise-induced electrocardiographic ST depression (indicative of myocardial ischemia or altered cardiac repolarization) in volunteers with known coronary heart disease and prior myocardial infarction.
Investigations into the adverse health effects of short-term air pollutant exposure indicate that individuals with heart failure are a particularly susceptible subgroup (Dominici et al., 2006; Goldberg et al., 2003; Pope et al., 2008). For example, elevations in daily particulate matter (PM) concentrations have been associated with increased heart failure-related hospitalizations (Dominici et al., 2006; Pope et al., 2008) and mortality (Goldberg et al., 2000), and rising PM10 levels increase the rate of new heart failure diagnoses and deaths in survivors of myocardial infarction (Zanobetti and Schwartz, 2007). The multiple biochemical and physiological responses demonstrated within susceptible subgroups such as heart failure patients highlight several candidate mechanisms of toxicity, including changes in autonomic balance, electrophysiological properties, vascular function, hemostasis and thrombosis, and systemic inflammation (Anselme et al., 2007; Brook, 2008; Campen et al., 2005; Lucking et al., 2011; Mills et al., 2007; Peretz et al., 2008). Yet, the mechanisms accounting for increased vulnerability of the failing heart to the effects of air pollution remain unclear.
Evidence demonstrating that DE inhalation can alter cardiac dimensions and performance (key indices of normal mechanical function) is limited. Yan et al. (2008) and Huang et al. (2010) found suggestions of such effects in rodents, but these studies were restricted to DE particles and used intratracheal instillation rather than inhalation. Others demonstrated in rats with postinfarct heart failure that DE-induced spontaneous arrhythmia accompanied and followed decreased heart rate variability (HRV) (Anselme et al., 2007), indicating possible autonomic mediation of effects. In contrast, DE exposure of younger, healthier rats with minimal cardiac hypertrophy caused only modest changes in HRV and electrocardiography (Carll et al., 2012), demonstrating the importance of modeling susceptibility in animal studies. To further elucidate the impact of short-term DE exposure on cardiac function in heart failure and its relation to autonomic nervous regulation, we examined the effects of a single inhalation exposure to DE on cardiac performance, ventricular chamber dimension, arrhythmia, repolarization, and HRV measures of autonomic modulation, as well as pulmonary and systemic injury and inflammation in an aged rat model prone to heart failure.
Materials and Methods
Animals and radiotelemetry implantation
Lean male spontaneously hypertensive heart failure (SHHF) rats (MccCrl-Lepr cp; n = 20) were shipped at 6 weeks of age from Charles River Laboratories, (Kingston, NY) to the U.S. Environmental Protection Agency (EPA)’s AAALAC-approved animal facility, housed in pairs in 42- × 21- × 20-cm Plexiglas cages with pine-shave bedding in a room (22±1°C, 50±5% relative humidity, 12-h light:dark cycle 0600:1800h), and provided standard Purina rat chow (5001; Brentwood, MO) and water ad libitum. At 15 months of age, 16 rats were implanted with radiotelemeters (model TA11CTA-F40; Data Sciences International, St Paul, MN) for the purpose of recording ECG, heart rate (HR), core body temperature, and activity wirelessly as previously described (Lamb et al., 2012). Lean male SHHF rats acquire cardiac hypertrophy by 3 months of age and transition into dilated cardiomyopathy and heart failure at 18 months of age (Carll et al., 2011b). All studies conformed to the guidelines of the U.S. EPA Institutional Animal Care and Use Committee. Three weeks after surgery, rats were weighed, monitored by ECG, and omitted from telemetric monitoring if they differed from the mean body weight by more than 1 SD (n = 2) or if their cardiac electrograms were unsuitable for HRV analysis due to frequent arrhythmias (n = 1) or a displaced lead (n = 1). Rats were transferred to a satellite facility and maintained under the same conditions as previously stated but in 33- × 18- × 19-cm Plexiglas cages. Rats were then habituated to 3min of manual restraint on two consecutive days in preparation for echocardiographic assessments. Animals were assigned semiblindly to one of two exposure groups (clean filtered air, “Air”; or whole diesel exhaust, “DE”) while maintaining equivalent mean body weights per group.
Diesel exhaust exposure and generation.
Rats were acclimated twice to exposure conditions within the clean filtered air chamber for 1h on two separate days before exposure. On the exposure day, rats were allowed to acclimate to the chambers for 20min, and then baseline data were recorded for the next 30min. Rats were then exposed to DE (target PM2.5 of 500 µg/m3) or Air for 4h in whole-body exposure chambers (Table 1). Thereafter, DE exposures were stopped for a 30-min recovery period in which clean filtered air was circulated through exposure chambers. Rats were returned to home cages immediately thereafter. DE exposures were at UFP and NO2 concentrations comparable with those found in traffic tunnels and roadways within the United States and Europe (Anselme et al., 2007; Svartengren et al., 2000; Zhu et al., 2007). DE was generated using a 4.8-kW (6.4-hp) direct injection single-cylinder 0.320-l displacement Yanmar L70V diesel generator operated at a constant 3600rpm on low sulfur diesel fuel (32 ppm) at a constant load of 3 kW as previously described (Carll et al., 2012). From the engine, the exhaust was mixed with clean air previously passed through high-efficiency particulate air (HEPA) filters. Air dilution of DE was adjusted periodically to maintain target PM2.5 mass concentration. The diluted DE was delivered to a Hazelton 1000 (984 l) exposure chamber. Control animals were placed in a second chamber supplied with the same HEPA-filtered room air as that used to dilute DE. The chambers were operated at the same temperature, pressure, and flow rate (424 l/min; approximating 25 air exchanges per hour). Chamber concentrations of PM, O2, CO, NO, NO2, and SO2 were measured as previously described (Carll et al., 2012). Chamber temperatures, relative humidity, and noise were also monitored and maintained within acceptable ranges (< 80 dB, 30–70%, and 73±5°F).
Table 1.
Inhalation Exposure Characterization
| Air | DE | |
|---|---|---|
| PM2.5 (µg/m3) | 21 | 515 |
| PM2.5 number (n/cm3) | 9.7×103 | 2.3×106 |
| Number median diameter of PM (nm) | 96 | 58 |
| Volume median diameter of PM (nm) | 184 | 89 |
| O2 (%) | 20.7±0.0 | 20.2±0.0 |
| CO (ppm) | 0.0±0.1 | 16.6±1.4 |
| NO (ppm) | 0.06±0.00 | 15.9±1.2 |
| NO2 (ppm) | 0.07±0.00 | 0.66±0.09 |
| SO2 (ppm) | BDL | BDL |
Notes. Data represent mean ± SD generated from measurements made continuously (concentrations of O2, CO, NO, and NO2), once (PM2.5 mass concentration), or six times (DE PM2.5 number) per exposure. Number median diameter was based on exposure day particle size distributions ± SD. Volume diameter was calculated from number-based mobility diameters and assumed spherical particles. Air indicates filtered air; DE, diesel exhaust; PM2.5, fine particulate matter; BDL, below detectable limit.
Left ventricular echocardiography.
One hour after the second exposure acclimation (1 day before DE or Air exposure), rats were held supine at 45° for no more than 3min while being measured for baseline left ventricular (LV) function and dimensions by transthoracic echocardiography (Nemio 30; Toshiba, Duluth, GA) using a 14-MHz linear array transducer (PLM 1204AT). Measures were repeated on the day of inhalation exposure, 1.5–2h after cessation of DE exposure (1–1.5h after removal from exposure chambers). Handling, measurements, and data analyses were performed in random order with laboratory personnel blinded to treatment groups. Two-dimensional long-axis images of the LV were obtained in parasternal long- and short-axis views with M-mode recordings at the midventricular level in both views. At least three consecutive LV contraction cycles were used to determine HR, fractional shortening (FS), ejection fraction (EF), cardiac output, stroke volume (SV), internal LV diameter at the ends of diastole (EDD) and systole (ESD), and thickness of the posterior wall and the interventricular septum (IVS). End diastolic and end systolic volumes (EDV and ESV) were determined using the area-length method as validated previously (Joho et al., 2007). LV dimensions were used to calculate FS ([EDD−ESD]/EDD), SV (EDV−ESV), and EF (SV/EDV).
Radiotelemetry data acquisition and analysis.
Radiotelemetry was used to track changes in cardiovascular and thermoregulatory function by continuously monitoring ECG, core body temperature, and activity in awake, unrestrained rats beginning at 3 days before inhalation exposure and continuing through exposure until euthanasia 24h after exposure. Data were obtained as described previously (Carll et al., 2012), and ECG waveforms were sampled at a rate of 1000 Hz in 2-min streams every 10min within home cages and 1-min streams every 5min within exposure chambers (DataART 3.01; Data Sciences International). ECG waveforms were analyzed with computer software (ECGauto 2.5.1.35; EMKA Technologies, Falls Church, VA) that enabled user identification and exclusion of arrhythmias and artifacts from automated analysis of HRV and ECG morphology parameters as previously detailed (Carll et al., 2012). ECG morphology parameters were based on P, Q, R, S, and T waves and included the following: intervals of PR, QRS, ST, QT, QTe (onset of Q to end of T), QTc (HR-corrected QT, using Fridericia correction), T-peak to T-end (Tp-Te), and amplitudes of Q, R, T-peak, and ST (mean amplitude between S and T-peak), relative to the isoelectric line (15milliseconds [ms] before Q). ST area was calculated as total negative area underneath the isoelectric line from S-nadir to T-peak.
HRV analysis generated HR and time-domain measures, including mean time between adjacent QRS-complex peaks (RR interval), standard deviation of the RR interval (SDNN), square root of the mean of squared differences of adjacent RR intervals (RMSSD), triangular index, and percent of adjacent normal RR intervals differing by ≥ 15ms (pNN15). pNN15 is a measure of parasympathetic tone. SDNN and triangular index represent overall HRV, whereas RMSSD represents parasympathetic influence over HR (Rowan et al., 2007). HRV analysis also provided frequency-domain parameters, including low frequency (LF: 0.200–0.750 Hz) and high frequency (HF: 0.75–2.00 Hz), and the ratio of these two frequency domains (LF/HF). For frequency-domain analysis, the signal was analyzed with a Hanning window for segment lengths of 512 samples with 50% overlapping. LF is generally believed to represent a combination of sympathetic and parasympathetic tone, whereas HF indicates cardiac vagal (parasympathetic) tone, and LF/HF serves as an index of sympathovagal balance (Rowan et al., 2007).Arrhythmias were identified while blinded to treatment group according to previously described criteria (Carll et al., 2012). To facilitate statistical analysis of each arrhythmia type and allow the data to converge under the Poisson distribution, zero values for each arrhythmia type within a sample interval were converted to 0.1. Arrhythmia frequencies were calculated over specific periods in home cages (pre-exposure and postexposure, 7h each) and in exposure chamber (baseline, midexposure, recovery), normalized to adjust for time differences between periods and gaps in data, and presented as number of events per hour of theoretically continuous ECG waveforms. Each premature beat was counted individually as a single event (e.g., 1 bigeminy = 2 ventricular premature beat [VPB] events), whereas atrioventricular (AV) or sinoatrial block arrhythmias were counted as one event regardless of duration or neighboring events.
HRV and ECG morphologic analyses were conducted on ECG waveforms collected while rats resided in home cages at 3 days pre-exposure and immediately postexposure (on both days, 1:00 p.m–8:00 p.m.), which were time matched to control for physiologic effects of circadian rhythm. The postexposure period of 1:30–2:30 p.m. was excluded from HRV and arrhythmia analyses to allow animals to recover from handling during echocardiographic measurements. To adjust for this gap in sampling, data collected at 1:00–1:30 p.m. and 2:30–3:00 p.m. were used to represent 1 and 2h postexposure, respectively. ECG data collected within the exposure chamber (5h total) were also analyzed according to the following periods: baseline (7:50–8:20 a.m.), exposure (8:20 a.m–12:20 p.m.), and recovery (12:20–12:50 p.m.). All ECG streams with less than 10 s of identifiable conduction cycles were excluded from ECG parameter calculation, and streams with less than 30 s of identifiable RR intervals were excluded from HRV analysis. Thorough visual inspection was conducted to identify and exclude arrhythmias and artifacts.
Tissue collection and analysis.
At approximately 24h after onset of the 4-h inhalation exposure, rats were deeply anesthetized with an ip injection of a sodium pentobarbital/phenytoin solution. Blood, lung lavage fluid, and tissue samples (heart and lungs) were collected, processed, and analyzed as previously described (Carll et al., 2012). Heart weight was normalized by right tibia length. To examine for indications of cardiopulmonary inflammation, injury, oxidative stress, and risk, multiple biochemical markers were assayed. Lavage supernatants were analyzed for albumin, gamma-glutamyl transferase, lactate dehydrogenase, N-acetyl-b-d-glucosaminidase (NAG), total antioxidant status, and total protein (Carll et al., 2010). Serum supernatants were analyzed for creatine kinase, C-reactive protein (CRP), total protein, and glutathione peroxidase, glutathione reductase, and glutathione-S-transferase; supernatants from plasma were assayed for angiotensin converting enzyme (ACE), albumin, blood urea nitrogen, creatinine, and total protein (Carll et al., 2011a). Serum was also analyzed for α-hydroxybutyrate dehydrogenase, D-dimer, ferritin, glucose, insulin, lipoprotein (a), total cholesterol, triglycerides, alanine aminotransferase, aspartate aminotransferase, lactate dehydrogenase-1, lipase, high- and low-density lipoprotein cholesterol, myoglobin, sorbitol dehydrogenase, superoxide dismutase (SOD), manganese SOD, and copper-zinc SOD according to previous procedures (Carll et al., 2010 , 2012).
Statistics.
The statistical analyses for all data in this study were performed using Prism version 4.03 (GraphPad Software Inc., San Diego, CA). One-way ANOVA with Tukey post hoc test was used to detect significant differences between groups in biochemical endpoints and tissue weight. Repeated measures two-way ANOVA with Bonferroni’s post hoc test was performed on (1) arrhythmia frequency data, which were collected at pre-, mid-, and postexposure periods (spanning approximately 4h each) and normalized by sampling duration; (2) HRV and ECG morphology parameters during the exposure period, including exposure at 1–4h, baseline (30min), and recovery periods (30min); and (3) HRV and ECG morphology to analyze for intragroup differences between time-matched periods (separated by exactly 24h) collected on the exposure day and on a previous day. Two-way ANOVA with Bonferroni’s post hoc test was also used to analyze for intergroup differences in change in HRV and ECG parameters at postexposure relative to the day before exposure. A value of p < 0.05 was considered statistically significant. Linear regressions were performed to test for correlations between various physiologic endpoints.
Results
Physiological Responses to Exposure by Inhalation
HR, HRV, and ECG morphology.
At pre-exposure and baseline, no significant differences emerged between the groups for HR, HRV parameters, or ECG morphology (Table 2). All rats in each group were active at the beginning of the exposure and became inactive during exposure to Air or DE. As expected with decreased activity, HR decreased for both the Air and DE groups over the exposure period (Fig. 1). Also, HR increased for both groups during baseline measurements after transfer of animals to the exposure chamber (Table 2). Only DE exposure altered HRV parameters significantly during exposure relative to baseline. SDNN, RMSSD, triangular index, LF, HF, and pNN15 increased at 3 and 4h and recovery, consistent with parasympathetic activation (p < 0.05). The groups did not differ from each other in HR at any time during exposure; yet, DE exposure increased pNN15 (at 4h) and triangular index (at recovery) relative to the Air group (all p < 0.05). Exposure to DE did not affect any measures of ECG morphology relative to baseline or the Air group during the exposure period.
Table 2.
HRV and ECG Morphology Parameters Prior to Exposure to DE
| Preexposure (home cages) | Baseline (exposure chamber) | |||
|---|---|---|---|---|
| Air | DE | Air | DE | |
| mean (SE) | mean (SE) | mean (SE) | mean (SE) | |
| HRV | ||||
| HR (beats/min) | 277 (7.5) | 282 (5) | 355 (13) | 334 (6) |
| SDNN (ms) | 10.5 (0.7) | 9.9 (0.9) | 8.1 (0.5) | 8.0 (0.6) |
| RMSSD (ms) | 5.7 (0.9) | 5.7 (1.4) | 3.8 (0.8) | 4.1 (1.1) |
| Triangular index | 1.25 (0.04) | 1.18 (0.04) | 1.20 (0.08) | 1.20 (0.05) |
| LF/HF | 0.92 (0.15) | 1.15 (0.32) | 1.42 (0.33) | 1.17 (0.11) |
| LF (ms2) | 1.64 (0.58) | 2.35 (0.74) | 0.66 (0.16) | 0.68 (0.21) |
| HF (ms2) | 2.30 (0.75) | 2.8 (1.3) | 0.78 (0.34) | 0.93 (0.47) |
| pNN15 (%) | 5.7 (0.9) | 5.7 (1.4) | 1.6 (1.5) | 2.3 (2.2) |
| ECG morphology | ||||
| PR (ms) | 61.0 (1.8) | 59.7 (2.3) | 57.7 (1.9) | 56.3 (1.7) |
| T amplitude (mV) | 0.058 (0.011) | 0.063 (0.089) | 0.074 (0.021) | 0.100 (0.013) |
| QTc (ms) | 80.1 (3.3) | 78.3 (0.8) | 83.3 (2.1) | 80.9 (2.0) |
| ST area (mV·ms) | −0.92 (0.4) | −0.93 (0.19) | −0.55 (0.45) | −0.72 (0.18) |
| Tp-Te (ms) | 28.9 (3.1) | 27.7 (1.7) | 30.3 (4.1) | 30.3 (2.0) |
Fig. 1.
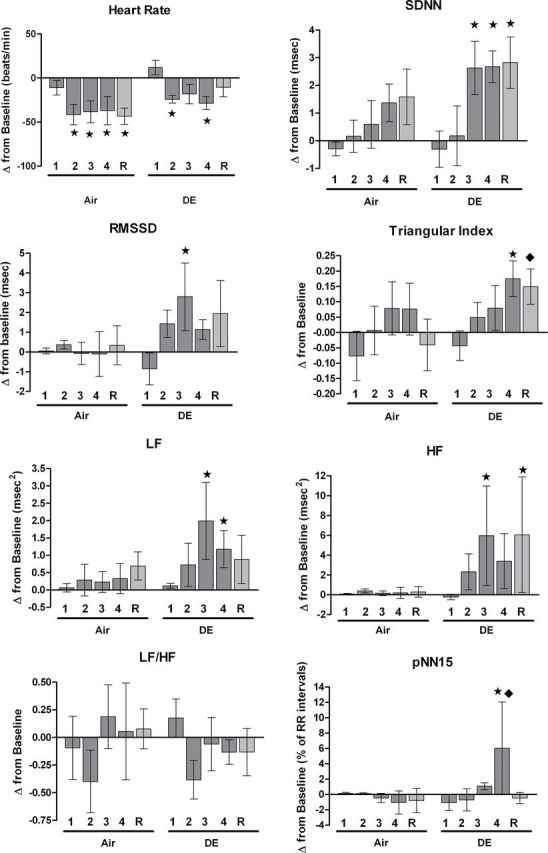
Change from baseline in HR and HRV endpoints (mean ± SE) during whole-body exposure. 1, 2, 3, 4, and R represent hours 1 through 4 of exposure and recovery (postexposure) within the chamber. All measurements were taken from unrestrained conscious rats temporarily housed within exposure chambers. Stars and diamonds mark significant differences from baseline (in chambers) and the air group (at the same hour), respectively (p < 0.05). See Table 2 for baseline values. N = 6 per group.
Cardiac arrhythmia.
During exposure, the DE and Air groups did not differ from each other in their rates of second-degree AV block events or premature beats, including VPBs (P = NS). The rate of VPBs increased during midexposure to Air when compared with pre-exposure, baseline, and recovery (Supplementary fig. 1, p < 0.05).
Physiological Responses After Exposure by Inhalation
HR and HRV.
There were notable differences in HRV over the first 4h after animals were returned to home cages after inhalation exposure. The DE group exceeded the Air group in change in triangular index and LF from pre-exposure (Fig. 2; all p < 0.05). Change in SDNN was also higher in the DE group relative to Air during 1–4h of postexposure (Fig. 2; p = 0.06).
Fig. 2.
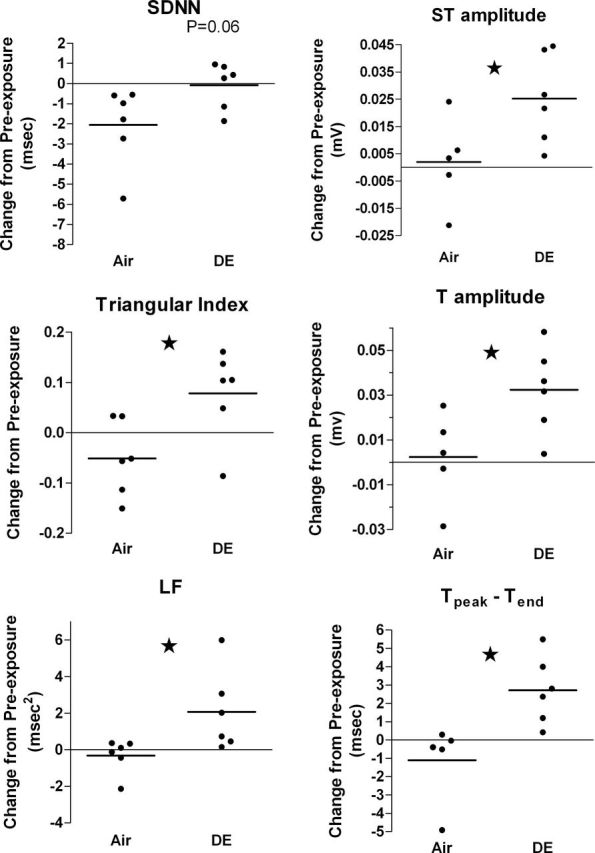
Change in HRV (left column) and ECG (right column) over the first 4h after removal from exposure chambers. Horizontal bars and circles represent group means and individual rats, respectively. Data were collected within home cages at 1–4h of postexposure, excluding the first 30min after echocardiography and at corresponding times on a preceding day. Stars indicate p < 0.05. N = 5–6 per group.
ECG morphology.
Relative to air exposure, DE caused several differences in ECG morphology over the first 4h of postexposure (in home cages). The DE group exceeded the Air group in change from pre-exposure ST area, T amplitude, and Tp-Te (Fig. 2; all p < 0.05). DE also exceeded Air in ST amplitude change (mean ± SE: 24.2±7 vs. 2.0±7 µV; p = 0.04). These alterations in T-wave and S-wave area, amplitude, and duration (Fig. 3) indicate that DE altered repolarization. No other aspects of ECG morphology were affected by DE in the hours following exposure. All four measures of ventricular repolarization at postexposure (ST amplitude, ST area, T amplitude, and Tp-Te) positively correlated with midexposure HRV (Table 3; all p < 0.05), indicating a possible relationship between changes in repolarization and preceding autonomic imbalance. Postexposure triangular index also correlated with postexposure Tp-Te (p = 0.03) and had a near-significant correlation with ST amplitude (Supplementary fig. 2; p = 0.07).
Fig. 3.

ECG waveforms before (gray) and after (black) DE exposure in a single rat. Each waveform represents the average cardiac electrogram from a 2-min ECG sampled at 10-min intervals. Data were collected from an individual rat within its home cage at 1–4h of postexposure, excluding the first 30min after echocardiography and at corresponding times on a preceding day. Horizontal time mark indicates 20-ms interval.
Table 3.
Correlations Between Midexposure Change in HRV and Postexposure Change in ECG Measures of Ventricular Repolarization (ECG Values Limited to 1–4h of Postexposure)
| HRV parameter | Exposure hour | ECG parameter | Postexposurea hour | r | p value |
|---|---|---|---|---|---|
| 3 | Tp-Te | 1 | 0.7 | 0.03 | |
| SDNN | Recovery | Tp-Te | 2 | 0.66 | 0.04 |
| ST area | 2 | 0.62 | 0.06 | ||
| 4 | Tp-Te | 1–4 | 0.67 | 0.03 | |
| Triangular index | ST amplitude | 1–4 | 0.6 | 0.06 | |
| Tp-Te | 4 | 0.74 | 0.01 | ||
| T amplitude | 4 | 0.69 | 0.03 | ||
| ST amplitude | 4 | 0.66 | 0.04 | ||
| Recovery | T amplitude | 1–4 | 0.63 | 0.05 | |
| Tp-Te | 1–4 | 0.61 | 0.06 | ||
| T amplitude | 2 | 0.67 | 0.03 | ||
| ST area | 4 | 0.66 | 0.04 | ||
| Tp-Te | 4 | 0.62 | 0.05 |
Notes. Change in HRV during individual hours of exposure (relative to baseline) significantly correlated with a subsequent change in ECG during 1–4h of postexposure. r indicates Pearson correlation coefficient. N = 5–6 per group.
aWhen animals were monitored in home cages.
Cardiac arrhythmia.
DE exposure increased the rate of Mobitz type II second-degree AV block events over the first 4h after removal of animals from exposure chambers. The DE group’s rate of Mobitz II AV block events was increased relative to itself at all prior periods and relative to the Air group at postexposure (Fig. 4; p < 0.05). Among the eight individual Mobitz II AV block events observed at postexposure in the DE-exposed group, four happened among four rats within 1h of return to home cages (1.5h postexposure), and the remaining half occurred in a single rat 4h into the postexposure period within home cages. Because most DE-exposed rats (four of six) had an AV block arrhythmia at 1h of postexposure, we looked at this time point for associations with changes in HRV and ECG morphology. At 1h of postexposure, AV block events among all rats positively correlated with change in RMSSD and SDNN, whereas AV block negatively correlated with HR (Supplementary table 1; p < 0.05).
Fig. 4.
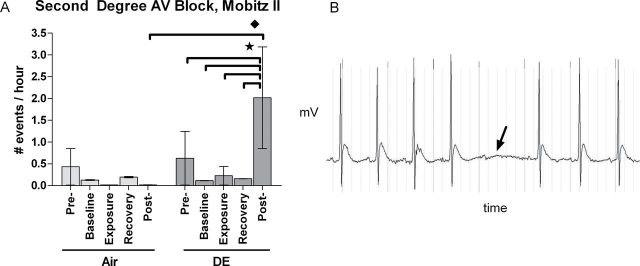
DE increased hourly rate of Mobitz II AV block per rat (mean ± SE) at postexposure (A). ECG waveform with representative second-degree AV block Mobitz II arrhythmia (arrow) following DE exposure (B). Baseline, exposure, and recovery were all measured within exposure chambers. Stars and diamonds indicate significant differences between periods and groups, respectively (p < 0.05). Vertical grey lines behind ECG waveform indicate time in 50-ms intervals. N = 6 per group.
Echocardiography.
At pre-exposure, LV systolic function in both groups was normal as indicated by mean EF (90%) and FS (54%). DE exposure altered several measures of LV diameter, volume, and wall thickness concomitant with an increase in cardiac output (Fig. 5). At postexposure, end diastolic and end systolic volumes (EDV and ESV) increased in the DE group relative to pre-exposure (178 and 34 µl, respectively), leading to an increase in SV (144 µl) and cardiac output (39.4ml/min) (Fig. 5, p < 0.05). These increases in LV volumes and output corresponded with LV wall thinning, including decreased posterior wall thickness at diastole (−13%) and systole (−10%) and decreased interventricular septal thickness at diastole (−11%) and systole (−10%) relative to pre-exposure (all p < 0.05). At postexposure, DE also increased SV, cardiac output (body weight normalized and raw), and EDV while decreasing posterior wall thickness relative to Air (all p < 0.05). DE did not affect HR during echocardiographic measures relative to Air (339±7 vs. 332±14 beats/min; P = NS), but both groups decreased in HR relative to their own pre-exposure values (Fig. 5, p < 0.05). All of the cardiac parameters that were affected by DE exposure also correlated with changes in HRV during the exposure period (Table 4, p < 0.05).
Fig. 5.
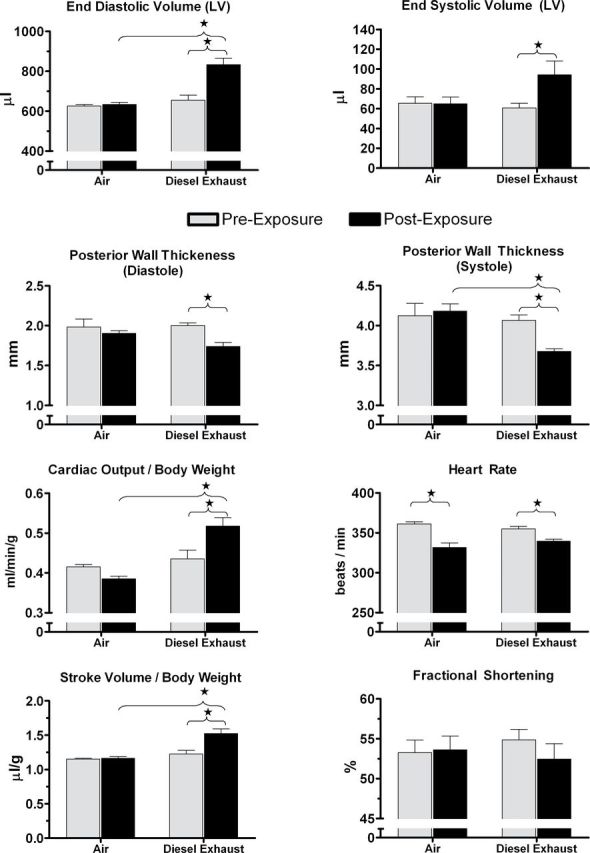
A single 4-h exposure to diesel exhaust (500μg/m3) increased left ventricular (LV) chamber volume, SV, and cardiac output and decreased LV wall thickness in aged SHHF rats. The groups did not differ from each other in HR before or after exposure. Echocardiographic measures of LV volume, thickness, and function were performed on conscious rats before and after exposure to filtered air or diesel exhaust. Stars and brackets indicate significant differences (p < 0.05). N = 6–8 per group.
Table 4.
Correlations Between Midexposure Change in HRV and Postexposure Change in Left Ventricular Function and Dimensions
| Echo parameter | HRV parameter | Exposure hour | r | p value |
|---|---|---|---|---|
| LVPW (D) | RMSSD | 3 | −0.60 | 0.05 |
| 4 | −0.82 | < 0.01 | ||
| R | −0.64 | 0.04 | ||
| SDNN | 3 | −0.76 | < 0.01 | |
| 4 | −0.74 | < 0.01 | ||
| pNN15 | 4 | −0.65 | 0.04 | |
| LVPW (S) | SDNN | 3 | −0.71 | 0.02 |
| 4 | −0.76 | < 0.01 | ||
| RMSSD | 4 | −0.82 | < 0.01 | |
| LVEDV | RMSSD | 3 | 0.74 | < 0.01 |
| HF | 3 | 0.63 | 0.04 | |
| pNN15 | 4 | 0.84 | < 0.01 | |
| LVESV | pNN15 | 4 | 0.73 | 0.02 |
| SV | RMSSD | 3 | 0.77 | < 0.01 |
| pNN15 | 4 | 0.71 | 0.02 | |
| HF | 3 | 0.70 | 0.02 | |
| HF | R | 0.66 | < 0.01 | |
| Cardiac output | RMSSD | 3 | 0.65 | 0.03 |
| pNN15 | 4 | 0.65 | 0.04 |
Notes. Changes in echocardiographic measures after the exposure period correlated with changes in HRV during exposure. Change was calculated as difference from baseline. N = 6 per group. 3, 4, and R represent the third and fourth hour of exposure and the recovery hour immediately following inside exposure chambers. PW (D) and PW (S), posterior wall thickness at end diastole and end systole, respectively.
Pulmonary and Systemic Markers of Inflammation and Injury
The air-exposed aged SHHF rats in this study had cardiac hypertrophy relative to 10-week-old SHHF, 19-week-old Wistar Kyoto (WKY), and 15-month-old WKY rats (Fig. 6; p < 0.05). Exposure to DE did not alter cardiac or lung weights (Supplementary table 2). DE exposure increased pulmonary eosinophils in aged SHHF rats (Supplementary table 2, p < 0.05). Relative to the Air group, the DE group increased serum CRP (+ 6.5%; p = 0.01) and plasma total protein (+ 7%; p = 0.05). Decreases in NAG and lipase of uncertain significance were evident in the DE group relative to Air control (p < 0.05). There were no other significant changes in circulating or pulmonary biochemical or cellular endpoints.
Fig. 6.
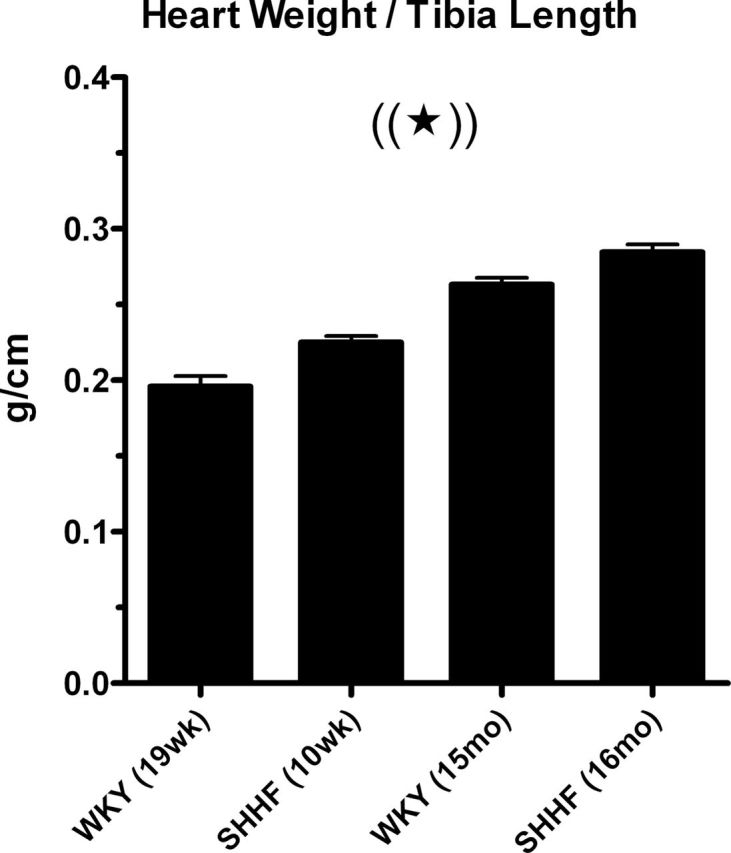
Age- and strain-dependent cardiac hypertrophy in SHHF rats. Mean (+ SE) heart weight to tibia length ratio of 16-month-old, air-exposed SHHF rats in this study compared with that of air-exposed animals in similar studies conducted in our laboratory (Carll et al., 2012; Lamb et al., 2012). Star and parentheses indicate that all groups are significantly different from each other (p < 0.05).
Discussion
In this study, a single 4-h exposure to DE by inhalation altered multiple cardiac endpoints in aged heart failure–prone rats with cardiac hypertrophy but without overt signs or symptoms of heart failure. Principal among these was DE-induced LV dilation, which may correspond with myocardial stretch and attendant electrophysiologic effects (Franz et al., 1992) and changes in cardiac repolarization. This effect may bear particular implications for the mechanisms underlying air pollutant–induced hospitalizations for heart failure. In the 16-month-old SHHF rats of our current study, the absence of congestive symptoms at baseline, maintenance of normal LV EF (90%) and FS (relative to Tamura et al., 1999), and elevated cardiac weight indicate a state of compensated LV hypertrophy prior to exposure. FS at baseline (53%) and dimensional changes after DE exposure compared closely with conscious measurements in terminally senescent mice exposed to carbon black particles (Tankersley et al., 2008). In these mice, a 4-day inhalation exposure (PM2.5 = 400 µg/m3) caused LV dilation and evidence of myocardial stretch (increased gene expression of natriuretic peptides), whereas it decreased FS and activated proteases responsible for myocardial remodeling. Similarly, others have demonstrated decreased contractility accompanied by increased LV volume and pressure after a single, high, 1–2mg/kg intratracheal dose of DE particles (Huang et al., 2010; Yan et al., 2008). Importantly, LV dilation can increase filling pressures and wall stress (Tkacova et al., 1997), which promotes parasympathetic reflexes (Wang et al., 1995), cardiac arrhythmia (Huang et al., 2009), and signaling pathways for LV remodeling (Force et al., 2002). Thus, it seems increasingly plausible that repeat exposures could cause LV remodeling and systolic degradation, in part, through ventricular dilation. Incidentally, Wold and colleagues recently found that long-term (9 months) exposures to lower concentrations of PM2.5 (85 µg/m3) significantly decreased contractility in mice. Collectively, these findings merit further investigation into the influence of ventricular dilation on air pollutant–induced cardiac remodeling and dysfunction.
The mechanisms that underlie DE-induced increases in cardiac output remain uncertain but may involve alterations in venous tone. The increase in SV and LV volume concurrent with normal HR, EF, and FS indicates that DE increased ventricular preload. Accordingly, Knuckles et al. (2008) previously found that DE enhances endothelin-1-induced constriction of veins (but not arterioles) through uncoupling of nitric oxide synthase (NOS). Tankersley et al. (2008) found in restrained, conscious mice that PM-induced LV dilation was mediated by NOS uncoupling. Importantly, physical restraint causes acute stress, which stimulates endothelin-1 release (Treiber et al., 2000), and could trigger DE-enhanced venoconstriction. Although we found no effects of DE on ACE, the observations of others (Ghelfi et al., 2010) implicate the renin-angiotensin system as a mediator of DE-induced increases in cardiac output. Our findings merit further investigation to determine their mechanistic origins.
DE exposure in aged hypertrophic SHHF rats within the current study led to robust increases in HRV both during and after exposure. These results are in contrast to our recent findings in similarly exposed young adult SHHF, normotensive WKY, and hypertensive (SH) rats, in which HRV effects were limited and included a small decrease in RMSSD (−0.8ms) at recovery in the SHHF rats (Carll et al., 2012; Lamb et al, 2012). Although this is our strongest evidence to date that DE exposure can cause a relative parasympathetic dominance over cardiac function, we have seen similar HRV effects with ozone inhalation (Farraj et al., 2012). Likewise, Tankersley et al. (2004) saw marked increases in HRV with repeated (4 days) inhalation exposure to carbon particles in terminally senescent mice. Previous studies have shown that exposure to DE or DE particles causes pulmonary irritant receptor activation (Deering-Rice et al., 2011; Hazari et al., 2011; Wong et al., 2003), which is known to cause parasympathetic reflexes (Widdicombe and Lee, 2001). The multiple effects of whole DE on arrhythmia and HRV in the aged SHHF rat versus the minimal effects of this same exposure on young SHHF rats (Carll et al., 2012) support epidemiologic evidence that age and progression of cardiac disease heighten susceptibility to air pollutant exposure (Brook et al., 2004). Moreover, these effects indicate that parasympathetic reflexes may factor into this susceptibility. Increased parasympathetic neural input to the heart can provoke second-degree AV block (Drici et al., 2000; Massie et al., 1978). We found correlations between postexposure second-degree AV block events and changes in HRV (Supplementary table 1) that accord with previous demonstrations of parasympathetic-mediated AV block (Castellanos et al., 1974; Drici et al., 2000; Massie et al., 1978). Beyond this effect, the clinical health implications of enhanced parasympathetic tone in response to DE exposure remain uncertain and unexplored.
The effects of DE that we observed on autonomic balance, bradyarrhythmia due to AV block, ventricular repolarization, and LV dilation may interrelate (Fig. 7). Changes in HRV and LV volume correlated, consistent with the known link between parasympathetic excitation and the activation of myocardial stretch receptors (Crawford, 2003). Parasympathetic activation provokes a K+ channel (I KACh) that augments repolarizing currents, decreases spontaneous depolarization, increases HRV, and promotes AV block (Drici et al., 2000; Moreno-Galindo et al., 2011). Likewise, the changes in spatiotemporal heterogeneity of repolarization that we observed may have resulted from several factors, including myocardial stretch (Tan et al., 2004; Xian Tao et al., 2006), autonomic mechanisms (Drici et al., 2000; Kanda et al., 2011; Moreno-Galindo et al., 2011), inflammation (Zhang et al., 2011), or changes in HR, electrolyte balance, and metabolism (Channer and Morris, 2002). Ultimately, our findings of altered repolarization, increased HRV, LV dilation, and AV block collectively suggest an important role for parasympathetic mechanisms in the adverse cardiophysiologic effects of DE.
Fig. 7.
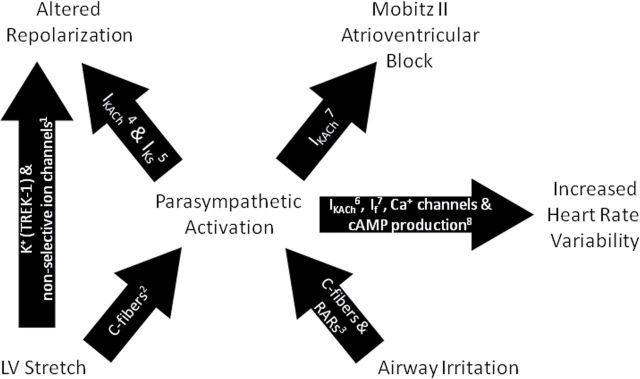
Proposed pathways accounting for electrophysiological effects of DE. cAMP, cyclic adenosine monophosphate; I f, hyperpolarization-activated current; I KACh, acetylcholine-sensitive K+ channel; I Ks, delayed rectifier K+ channel; RARs, rapidly activated receptors; TREK-1, two-pore-domain potassium channel. 1 Tan et al., 2004; Xian Tao et al., 2006; 2 Crawford, 2003; Wang et al., 1995; 3 Widdicombe and Lee, 2001; 4 Moreno-Galindo et al., 2011; 5 Kanda et al., 2011; 6 Wickman et al., 1998; 7 Drici et al., 2000; 8 Harvey and Belevych, 2003.
Our findings of DE-induced ECG changes correspond with prior observations and bear notable implications for the health effects of DE exposure. Rich et al. (2012) recently found in patients with prior coronary events that Tp-Te (a measure of the heterogeneity of transmural depolarization [Castro Hevia et al., 2006]) increased with exposure to fine mode particles. We similarly observed an increase in Tp-Te in the first 4h after DE exposure, suggesting that DE may desynchronize repolarization between the different regions or cell types of the ventricular myocardium. Tp-Te prolongation has been demonstrated to correlate with postinfarct LV remodeling (Szydlo et al., 2010) and ventricular tachycardia and sudden cardiac death in patients with hypertrophic cardiomyopathy (Shimizu et al., 2002). Similarly, the effects of DE on ST area and ST- and T-wave amplitudes were not unprecedented. Both elevation and depression of the ST interval and T wave may indicate myocardial ischemia (Channer and Morris, 2002). Several researchers have reported decreased T wave or ST amplitudes with air pollutant exposure. Such findings occurred with exposure to ambient particles in ischemic heart disease patients (Henneberger et al., 2005), inhalation of DE in exercised coronary artery disease patients (Mills et al., 2007) and sedentary atherosclerotic mice (Campen et al., 2005), and inhalation of particle-free DE gases in young adult SH and SHHF rats (Lamb et al., 2012; Carll et al., 2012). Although these changes in repolarization may result from ischemia, they may also stem from alterations in transmembane K+ balance by myocardial stretch and parasympathetic activation as previously mentioned.
Unlike our previous findings in young adult WKY, SH, and SHHF rats (Carll et al., 2012; Lamb et al., 2012), DE increased markers of cardiopulmonary inflammation in aged SHHF rats, suggesting that age and advancement toward heart failure mediate enhanced proclivity to these effects. The DE-induced increase in serum CRP is consistent with the findings of Rich et al. (2012), which along with their aforementioned observations of Tp-Te prolongation included positive correlations between CRP and particle concentrations within the preceding 2–4 days. The increase in pulmonary eosinophils was unexpected; however, similar effects of DE have been observed in healthy humans (Sehlstedt et al., 2010). The relationship between these findings and the changes in cardiac physiology require further study.
In summary, a single inhalation exposure to DE in aged heart failure–prone rats without evidence of heart failure caused LV dilation and changes in cardiac repolarization. In light of our prior study (Carll et al., 2012), our findings demonstrate that age in a heart failure–prone rat strain confers overt susceptibility to the effects of air pollutant exposure on repolarization, HRV, and the occurrence of bradyarrhythmia. Most of the observed physiologic changes correlated with increased HRV markers of parasympathetic influence, suggesting autonomic modulation played an important role in the observations. The mechanism by which increased parasympathetic influence may relate to such effects requires further study. Taken together, these findings may provide new insight on the health effects of traffic-related air pollutants.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
Funding
EPA-NHEERL/UNC-DESE Cooperative Training in Environmental Sciences & Research (CR83323601); EPA/UNC Toxicology Training Agreement (CR-83515201-0).
Supplementary Material
Acknowledgments
The authors thank Drs Kevin L. Dreher, Urmila P. Kodavanti, and Jane Ellen Simmons of the U.S. Environmental Protection Agency for reviewing this article, Judy H. Richards of the U.S. EPA for her extensive biochemical analyses, Dr Eric Bair of the University of North Carolina at Chapel Hill for his statistical guidance, and Dr M. Ian Gilmour of the U.S. EPA for his critical role in developing the inhalation exposure facility used in this study.
References
- Anselme F., Loriot S., Henry J. P., Dionnet F., Napoleoni J. G., Thuillez C., Morin J. P. 2007. Inhalation of diluted diesel engine emission impacts heart rate variability and arrhythmia occurrence in a rat model of chronic ischemic heart failure. Arch. Toxicol. 81, 299–307 [DOI] [PubMed] [Google Scholar]
- Brook R. D. 2008. Cardiovascular effects of air pollution. Clin. Sci. 115, 175–187 [DOI] [PubMed] [Google Scholar]
- Brook R. D., Franklin B., Cascio W., Hong Y., Howard G., Lipsett M., Luepker R., Mittleman M., Samet J., Smith S. C., Jr, et al. 2004. Air pollution and cardiovascular disease: A statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 109, 2655–2671 [DOI] [PubMed] [Google Scholar]
- Campen M. J., Babu N. S., Helms G. A., Pett S., Wernly J., Mehran R., McDonald J. D. 2005. Nonparticulate components of diesel exhaust promote constriction in coronary arteries from ApoE-/- mice. Toxicol. Sci. 88, 95–102 [DOI] [PubMed] [Google Scholar]
- Carll A. P., Haykal-Coates N., Winsett D. W., Rowan W. H., 3rd, Hazari M. S., Ledbetter A. D., Nyska A., Cascio W. E., Watkinson W. P., Costa D. L., et al. 2010. Particulate matter inhalation exacerbates cardiopulmonary injury in a rat model of isoproterenol–induced cardiomyopathy. Inhal. Toxicol. 22, 355–368 [DOI] [PubMed] [Google Scholar]
- Carll A. P., Haykal-Coates N., Winsett D. W., Hazari M. S., Nyska A., Richards J. H., Willis M. S., Costa D. L., Farraj A. K. (2011). Dietary salt exacerbates isoproterenol-induced cardiomyopathy in rats. Toxicol. Pathol. 39, 925–937 [DOI] [PubMed] [Google Scholar]
- Carll A. P., Hazari M. S., Perez C. M., Krantz Q. T., King C. J., Winsett D. W., Costa D. L., Farraj A. K. 2012. Whole and particle-free diesel exhausts differentially affect cardiac electrophysiology, blood pressure, and autonomic balance in heart failure–prone rats. Toxicol. Sci. 128, 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carll A. P., Willis M. S., Lust R. M., Costa D. L., Farraj A. K. (2011). Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc. Toxicol. 11, 91–112 [DOI] [PubMed] [Google Scholar]
- Castellanos A., Sung R. J., Cunha D., Myerburg R. J. 1974. His bundle recordings in paroxysmal atrioventricular block produced by carotid sinus massage. Br. Heart J. 36, 487–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro Hevia J., Antzelevitch C., Tornés Bárzaga F., Dorantes Sánchez M., Dorticós Balea F., Zayas Molina R., Quiñones Pérez M. A., Fayad Rodríguez Y. 2006. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J. Am. Coll. Cardiol. 47, 1828–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channer K., Morris F. 2002. ABC of clinical electrocardiography: Myocardial ischaemia. BMJ. 324, 1023–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosselman K. E., Krishnan R. M., Oron A. P., Jansen K., Peretz A., Sullivan J. H., Larson T. V., Kaufman J. D. 2012. Blood pressure response to controlled diesel exhaust exposure in human subjects. Hypertension. 59, 943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford M. H. 2003. Current Diagnosis and Treatment in Cardiology. 2nd ed McGraw-Hill, New York, NY. [Google Scholar]
- Deering-Rice C. E., Romero E. G., Shapiro D., Hughen R. W., Light A. R., Yost G. S., Veranth J. M., Reilly C. A. 2011. Electrophilic components of diesel exhaust particles (DEP) activate transient receptor potential ankyrin-1 (TRPA1): A probable mechanism of acute pulmonary toxicity for DEP. Chem. Res. Toxicol. 24, 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici F., Peng R. D., Bell M. L., Pham L., McDermott A., Zeger S. L., Samet J. M. 2006. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA. 295, 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drici M. D., Diochot S., Terrenoire C., Romey G., Lazdunski M. 2000. The bee venom peptide tertiapin underlines the role of I(KACh) in acetylcholine-induced atrioventricular blocks. Br. J. Pharmacol. 131, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farraj A. K., Hazari M. S., Winsett D. W., Kulukulualani A., Carll A. P., Haykal-Coates N., Lamb C. M., Lappi E., Terrell D., Cascio W. E., et al. 2012. Overt and latent cardiac effects of ozone inhalation in rats: Evidence for autonomic modulation and increased myocardial vulnerability. Environ. Health Perspect. 120, 348–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force T., Michael A., Kilter H., Haq S. 2002. Stretch-activated pathways and left ventricular remodeling. J. Card. Fail. 8,(Suppl. 6)S351–S358 [DOI] [PubMed] [Google Scholar]
- Franz M. R., Cima R., Wang D., Profitt D., Kurz R. 1992. Electrophysiological effects of myocardial stretch and mechanical determinants of stretch-activated arrhythmias. Circulation. 86, 968–978 [DOI] [PubMed] [Google Scholar]
- Ghelfi E., Wellenius G. A., Lawrence J., Millet E., Gonzalez-Flecha B. 2010. Cardiac oxidative stress and dysfunction by fine concentrated ambient particles (CAPs) are mediated by angiotensin-II. Inhal. Toxicol. 22, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M. S., Bailar J. C., 3rd, Burnett R. T., Brook J. R., Tamblyn R., Bonvalot Y., Ernst P., Flegel K. M., Singh R. K., Valois M. F. 2000. Identifying subgroups of the general population that may be susceptible to short-term increases in particulate air pollution: A time-series study in Montreal, Quebec. Res. Rep. Health. Eff. Inst. 97, 7–113; discussion 115 [PubMed] [Google Scholar]
- Goldberg M. S., Burnett R. T., Valois M. F., Flegel K., Bailar J. C., 3rd, Brook J., Vincent R., Radon K. 2003. Associations between ambient air pollution and daily mortality among persons with congestive heart failure. Environ. Res. 91, 8–20 [DOI] [PubMed] [Google Scholar]
- Harvey R. D., Belevych A. E. 2003. Muscarinic regulation of cardiac ion channels.. Br. J. Pharmacol.. 139,, 1074–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazari M. S., Haykal-Coates N., Winsett D. W., Krantz Q. T., King C., Costa D. L., Farraj A. K. 2011. TRPA1 and sympathetic activation contribute to increased risk of triggered cardiac arrhythmias in hypertensive rats exposed to diesel exhaust. Environ. Health Perspect. 119, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger A., Zareba W., Ibald-Mulli A., Rückerl R., Cyrys J., Couderc J. P., Mykins B., Woelke G., Wichmann H. E., Peters A. 2005. Repolarization changes induced by air pollution in ischemic heart disease patients. Environ. Health Perspect. 113, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. H., Lin L. Y., Tsai M. S., Hsu C. Y., Chen H. W., Wang T. D., Chang W. T., Cheng T. J., Chen W. J. 2010. Acute cardiac dysfunction after short-term diesel exhaust particles exposure. Toxicol. Lett. 192, 349–355 [DOI] [PubMed] [Google Scholar]
- Huang H., Wei H., Liu P., Wang W., Sachs F., Niu W. 2009. A simple automated stimulator of mechanically induced arrhythmias in the isolated rat heart. Exp. Physiol. 94, 1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho S., Ishizaka S., Sievers R., Foster E., Simpson P. C., Grossman W. 2007. Left ventricular pressure-volume relationship in conscious mice. Am. J. Physiol. Heart Circ. Physiol. 292, H369–H377 [DOI] [PubMed] [Google Scholar]
- Kanda V. A., Purtell K., Abbott G. W. 2011. Protein kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm. 8, 1641–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckles T. L., Lund A. K., Lucas S. N., Campen M. J. 2008. Diesel exhaust exposure enhances venoconstriction via uncoupling of eNOS. Toxicol. Appl. Pharmacol. 230, 346–351 [DOI] [PubMed] [Google Scholar]
- Lamb C. M., Hazari M. S., Haykal-Coates N., Carll A. P., Krantz Q. T., King C., Winsett D. W., Cascio W. E., Costa D. L., Farraj A. K. 2012. Divergent electrocardiographic responses to whole and particle-free diesel exhaust inhalation in spontaneously hypertensive rats. Toxicol. Sci. 125, 558–568 [DOI] [PubMed] [Google Scholar]
- Le Tertre A., Medina S., Samoli E., Forsberg B., Michelozzi P., Boumghar A., Vonk J. M., Bellini A., Atkinson R., Ayres J. G., et al. 2002. Short-term effects of particulate air pollution on cardiovascular diseases in eight European cities. J. Epidemiol. Community Health. 56, 773–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking A. J., Lundbäck M., Barath S. L., Mills N. L., Sidhu M. K., Langrish J. P., Boon N. A., Pourazar J., Badimon J. J., Gerlofs-Nijland M. E., et al. 2011. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation. 123, 1721–1728 [DOI] [PubMed] [Google Scholar]
- Massie B., Scheinman M. M., Peters R., Desai J., Hirschfeld D., O’Young J. 1978. Clinical and electrophysiologic findings in patients with paroxysmal slowing of the sinus rate and apparent Mobitz type II atrioventricular block. Circulation. 58, 305–314 [DOI] [PubMed] [Google Scholar]
- Maynard D., Coull B. A., Gryparis A., Schwartz J. 2007. Mortality risk associated with short-term exposure to traffic particles and sulfates. Environ. Health Perspect. 115, 751–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills N. L., Miller M. R., Lucking A. J., Beveridge J., Flint L., Boere A. J., Fokkens P. H., Boon N. A., Sandstrom T., Blomberg A., et al. 2011. Combustion-derived nanoparticulate induces the adverse vascular effects of diesel exhaust inhalation. Eur. Heart J. 32, 2660–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills N. L., Törnqvist H., Gonzalez M. C., Vink E., Robinson S. D., Söderberg S., Boon N. A., Donaldson K., Sandström T., Blomberg A., et al. 2007. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N. Engl. J. Med. 357, 1075–1082 [DOI] [PubMed] [Google Scholar]
- Moreno-Galindo E. G., Sánchez-Chapula J. A., Sachse F. B., Rodríguez-Paredes J. A., Tristani-Firouzi M., Navarro-Polanco R. A. 2011. Relaxation gating of the acetylcholine-activated inward rectifier K+ current is mediated by intrinsic voltage sensitivity of the muscarinic receptor. J. Physiol. (Lond.). 589,(Pt 7)1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A., Kaufman J. D., Trenga C. A., Allen J., Carlsten C., Aulet M. R., Adar S. D., Sullivan J. H. 2008. Effects of diesel exhaust inhalation on heart rate variability in human volunteers. Environ. Res. 107, 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C. A., 3rd, Renlund D. G., Kfoury A. G., May H. T., Horne B. D. 2008. Relation of heart failure hospitalization to exposure to fine particulate air pollution. Am. J. Cardiol. 102, 1230–1234 [DOI] [PubMed] [Google Scholar]
- Rich D. Q., Zareba W., Beckett W., Hopke P. K., Oakes D., Frampton M. W., Bisognano J., Chalupa D., Bausch J., O’Shea K., et al. 2012. Are ambient ultrafine, accumulation mode, and fine particles associated with adverse cardiac responses in patients undergoing cardiac rehabilitation?. Environ. Health Perspect. 120, 1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan W. H., 3rd, Campen M. J., Wichers L. B., Watkinson W. P. 2007. Heart rate variability in rodents: Uses and caveats in toxicological studies. Cardiovasc. Toxicol. 7, 28–51 [DOI] [PubMed] [Google Scholar]
- Sehlstedt M., Behndig A. F., Boman C., Blomberg A., Sandström T., Pourazar J. 2010. Airway inflammatory response to diesel exhaust generated at urban cycle running conditions. Inhal. Toxicol. 22, 1144–1150 [DOI] [PubMed] [Google Scholar]
- Shimizu M., Ino H., Okeie K., Yamaguchi M., Nagata M., Hayashi K., Itoh H., Iwaki T., Oe K., Konno T., et al. 2002. T-peak to T-end interval may be a better predictor of high-risk patients with hypertrophic cardiomyopathy associated with a cardiac troponin I mutation than QT dispersion. Clin. Cardiol. 25, 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svartengren M., Strand V., Bylin G., Järup L., Pershagen G. 2000. Short-term exposure to air pollution in a road tunnel enhances the asthmatic response to allergen. Eur. Respir. J. 15, 716–724 [DOI] [PubMed] [Google Scholar]
- Szydło K., Wita K., Trusz-Gluza M., Tabor Z. 2010. Late phase of repolarization (TpeakTend) as a prognostic marker of left ventricle remodeling in patients with anterior myocardial infarction treated with primary coronary intervention. Cardiol. J. 17, 244–248 [PubMed] [Google Scholar]
- Tamura T., Said S., Gerdes A. M. 1999. Gender-related differences in myocyte remodeling in progression to heart failure. Hypertension. 33, 676–680 [DOI] [PubMed] [Google Scholar]
- Tan J. H., Liu W., Saint D. A. 2004. Differential expression of the mechanosensitive potassium channel TREK-1 in epicardial and endocardial myocytes in rat ventricle. Exp. Physiol. 89, 237–242 [DOI] [PubMed] [Google Scholar]
- Tankersley C. G., Campen M., Bierman A., Flanders S. E., Broman K. W., Rabold R. 2004. Particle effects on heart-rate regulation in senescent mice. Inhal. Toxicol. 16, 381–390 [DOI] [PubMed] [Google Scholar]
- Tankersley C. G., Champion H. C., Takimoto E., Gabrielson K., Bedja D., Misra V., El-Haddad H., Rabold R., Mitzner W. 2008. Exposure to inhaled particulate matter impairs cardiac function in senescent mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R252–R263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkacova R., Hall M. J., Liu P. P., Fitzgerald F. S., Bradley T. D. 1997. Left ventricular volume in patients with heart failure and Cheyne-Stokes respiration during sleep. Am. J. Respir. Crit. Care Med. 156, 1549–1555 [DOI] [PubMed] [Google Scholar]
- Treiber F. A., Jackson R. W., Davis H., Pollock J. S., Kapuku G., Mensah G. A., Pollock D. M. 2000. Racial differences in endothelin-1 at rest and in response to acute stress in adolescent males. Hypertension. 35, 722–725 [DOI] [PubMed] [Google Scholar]
- Wang S. Y., Sheldon R. S., Bergman D. W., Tyberg J. V. 1995. Effects of pericardial constraint on left ventricular mechanoreceptor activity in cats. Circulation. 92, 3331–3336 [DOI] [PubMed] [Google Scholar]
- Wickman K., Nemec J., Gendler S. J., Clapham D. E. 1998. Abnormal heart rate regulation in GIRK4 knockout mice.. Neuron.. 20,, 103–114 [DOI] [PubMed] [Google Scholar]
- Widdicombe J., Lee L. Y. 2001. Airway reflexes, autonomic function, and cardiovascular responses. Environ. Health Perspect. 109,(Suppl. 4)579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. S., Sun N. N., Keith I., Kweon C. B., Foster D. E., Schauer J. J., Witten M. L. 2003. Tachykinin substance P signaling involved in diesel exhaust-induced bronchopulmonary neurogenic inflammation in rats. Arch. Toxicol. 77, 638–650 [DOI] [PubMed] [Google Scholar]
- Xian Tao L., Dyachenko V., Zuzarte M., Putzke C., Preisig-Muller R., Isenberg G., Daut J. 2006. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res. 69, 86–97 [DOI] [PubMed] [Google Scholar]
- Yan Y. H., Huang C. H., Chen W. J., Wu M. F., Cheng T. J. 2008. Effects of diesel exhaust particles on left ventricular function in isoproterenol-induced myocardial injury and healthy rats. Inhal. Toxicol. 20, 199–203 [DOI] [PubMed] [Google Scholar]
- Zanobetti A., Schwartz J. 2007. Particulate air pollution, progression, and survival after myocardial infarction. Environ. Health Perspect. 115, 769–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sun X., Zhang Y., Wang J., Lu Y., Yang B., Wang Z. 2011. Potential therapeutic value of antioxidants for abnormal prolongation of QT interval and the associated arrhythmias in a rabbit model of diabetes. Cell. Physiol. Biochem. 28, 97–102 [DOI] [PubMed] [Google Scholar]
- Zhu Y., Eiguren-Fernandez A., Hinds W. C., Miguel A. H. 2007. In-cabin commuter exposure to ultrafine particles on Los Angeles freeways. Environ. Sci. Technol. 41, 2138–2145 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.