

Introduction
The truth of a theory lies in the deductive methods used to establish it and the experimental demonstration of its fundamental premises and consequences—Jacques Monod
I had the good fortune, early on, to be gripped by a scientific problem, gene regulation, that had ramifications beyond what I imagined. Its unfoldings have kept me enthralled ever since. We began with bacteria, and especially with bacteriophage λ, and then moved to work with yeast and mammalian cells. We always sought coherent descriptions, ideas that would apply to apparently disparate cases, regulation of prokaryotic and eukaryotic genes, for example, despite the fact that the latter, but not the former, are sequestered in a nucleus and wrapped in nucleosomes.
My goal here is to put the various stages of our understanding, from the beginning of my involvement, in an overarching context. I might not say anything that has not been said by others or me, but my hope is that otherwise obscure connections and simplifications will be made clear. I hope I avoid distortions, but I cannot cite all the important contributions of others. More detailed explanations for various experiments and arguments, along with more extensive references, can be found in my two books (1, 2) and in the references here. Where I refer to “we,” I of course mean to include the crucial role of this or that student or postdoctoral fellow. They are identified in the reference list but not in the text.
What Is the “Repressor?”
I spent the summer of 1961 working in the laboratory of Aaron Novick and Frank Stahl at the University of Oregon, down the road from where I was finishing my undergraduate degree at Reed College in Portland, Oregon. I was sent to that lab by Ed Novitski, the master Drosophila geneticist, after a blissful summer of fly work at the Rocky Mountain Biological Laboratory in Crested Butte, Colorado. Major personalities appeared in Crested Butte: Ed himself, of course, H. J. Muller, Bruce Baker, Charles Remington, and so on, all in serious good humor. Ed's motives in sending me to Aaron and Frank are suspect: he despised the rumblings of molecular biology, the end of intelligent science. Perhaps he thought I would mix up the tubes and bring the whole enterprise to a halt. I did in fact mix up the tubes, but the effect was more of a hiccup than a demolition.
Frank, who I later learned was in a transient tiff with his pal Matt Meselson, took the clue from Ed and told me there was only one person under whom I should do my Ph.D.: Matt. Thus, the next year, I followed Matt's move, orchestrated by Jim Watson, from Caltech to Harvard (the college, not the medical school), where I entered graduate school. Only at Harvard did I realize what a serious place Reed had been. I should have noticed: it seemed that half the students entering Reed, and often the most interesting ones, dropped out. There was a limit, apparently, to which serious self-indulgence (on the one hand) and serious work (on the other) successfully mixed. At Reed, you actually had to be able to discuss the two-hundred or so pages of reading that you had been assigned. At Harvard, the key question usually worked: will that be on the exam?
In Oregon, I had encountered dazzling ideas emanating from the Institut Pasteur in Paris. François Jacob, Jacques Monod, and colleagues, working with bacteria, had proposed the existence of regulatory molecules called “repressors” that would turn off expression of specific genes unless inactivated by specific extracellular signals. The word “specific” requires attention, as it will come up often in this essay. Here, it signifies that one repressor, the λ phage repressor, was proposed to maintain most of the forty or so genes of the virus in a dormant state (i.e. “off”) in a lysogenic bacterium. And a different repressor, the Lac repressor, was proposed to control expression of the lac genes, genes required for metabolism of the sugar lactose. The signal that inactivated each repressor was specific, too: UV light in the λ case and lactose (or a metabolic derivative thereof) in the Lac case. Inactivation of the λ repressor, it was proposed, switched on expression of dormant phage genes and thereby initiated a lytic cycle of phage growth; in the Lac case, the signal (lactose) elicited expression of the lac genes only when needed (i.e. in the presence of lactose). I recently have touched upon our indebtedness to the French scientists in presenting this picture to us in my reflections on Jacob (3).
We were inspired by the dream, a bit vague at the time, that the repressor (what it is, what it does, and how) would illuminate development of a complex organism from a fertilized egg. Even then, we surmised that formation of different body parts requires differential expression of common genes and that different organisms can develop using essentially the same set of genes. λ was especially interesting because it presented us with the “memory” problem: once lysogeny was established in a bacterium, that state of gene expression was perpetuated for very many generations in the absence of an inducing signal. Neither “remembering” nor switching required any mutation. The switch is thus “epigenetic”: I had not thought the matter through when I incorrectly called it a “genetic” switch (1). Analogous switches had to underlie intermediate and final stages of development, we thought.
However, work on the repressor had to wait. As Frank put it, I needed a license (i.e. a Ph.D. degree) to do experimental science on my own. So I went to work, learning the ropes, on a tangential problem involving the growth of phage λ. Amazingly, by the time I had completed work for my Ph.D. degree in Matt's lab in 1965 and despite rumors of serious efforts, no one had managed to get their hands on one of these putative repressors. I say “amazingly” because four years is a long time in this field. We were thereby given a gift in the form of a difficult but, as it turned out, not unsolvable task.
Isolation of the Repressor
As a junior fellow of the Society of Fellows at Harvard (from 1965 to 1968), not only did I get a free dinner every week, but I also was given my own lab, right next door to E. O. Wilson, the great ant man himself. (I think Wilson took a dim view of our operation; he had not planned to study the behavior of ants labeled at random with 32P.) So we began the assault. At that point, the repressor was defined as the product of a phage gene (called c1) based on the effects of mutations in that gene. The repressor had no known enzymatic activity and was inferred to be present in low amounts, perhaps as few as 10–100 molecules/cell.
Nowadays, of course, one would overexpress the protein in cells using recombinant DNA techniques, and isolating the gene product would not be a serious challenge. We used the opposite approach to raise the relative amount of repressor synthesis in cells: we in effect destroyed (or nearly so) all bacterial and phage genes except c1 and fed the damaged cells radioactive amino acids. We detected a radiolabeled protein that was absent or had changed properties if the c1 gene was deleted or damaged (4). Bingo!
“We” here is meant to include Nancy Hopkins, whose name, consistent with the blinkered custom of the times, does not appear on the first few λ repressor papers. She was, at the time and before becoming a graduate student, a technician who made crucial contributions to the work.
Was the product of the c1 gene actually a repressor, or might it have been, for example, an enzyme that converted some other molecule into the real repressor (an unlikely but possible scenario)? Could the c1 gene product on its own regulate a gene, or was it part of some more complicated apparatus? Did the repressor work directly on the DNA, the favored view, or at some other stage of gene expression? Specificity was the key.
The Repressor Binds DNA
We mixed radiolabeled repressor with λ DNA, sedimented the mixture in a velocity gradient, and saw (with great excitement) that some of the repressor was bound to the fast-sedimenting DNA. (Nowadays gel electrophoresis or, so-called “chromatin immuno-precipitation,” and not centrifugation, would be used for such an experiment. It saves a lot of space and money.) This binding was specific in the following sense. We knew, thanks to the French scientists, that a phage closely related to λ, called 434, made its own repressor that had no effect on λ gene expression, and, correspondingly, the λ repressor had no effect on 434. We found, first, that the λ repressor did not bind to 434 DNA under conditions in which it bound λ DNA (5). Then, even better, we showed that the 434 repressor bound to 434 DNA but not to λ DNA (6).
The demonstration that the two repressors, λ and 434, bound with opposite specificities to the two different DNA molecules, consistent with their behaviors in vivo, left little room for doubt: these proteins could bind to specific DNA sites (called “operators”), and a surmise later confirmed (7), they prevent transcription of target genes. These experiments illustrate the power of working with pairs and of combining genetics and biochemistry, lessons I learned, among many others, from Matt.
This description omits the drama: in an enormously stimulating competition, Wally Gilbert and his postdoctoral fellow Benno Müller-Hill raced us to be the first to “solve the repressor,” as Jim Watson would say. Let us call it a tie: they were marginally faster in detecting their repressor, the Lac repressor (8, 9), and we were, by a respectable margin, the first to show specific DNA binding of a repressor.
The experiments demonstrating specific DNA binding did not reveal how this binding was achieved. Might unusual DNA sequences be involved? Might the DNA have to unwind to expose base pairs or form some unusual structure such as Z-DNA, a popular item back then? Had the repressor turned out to be an RNA molecule or to be attached to one, the problem would have seemed simpler. However, the repressor was a pure protein, and a series of indirect experiments soon indicated that it recognizes its affined specific DNA sequence in the standard helical form (10). This left us with a mystery.
By now I had moved, spiritually if not physically, from junior fellow to lecturer at Harvard. There was some good bureaucratic reason for the odd title, but I cannot recall it now. Along with the rare postdoctoral fellow, a few students joined up, some of whom would not leave until they had done something interesting. Fortunately, no one had yet invented the “four (or even five) years and you are out” rule. We were not very specialized back then. With only a modest-sized lab, we performed genetic, physiological, and biophysical experiments with everyone thinking about everyone else's experiments. Our department included Matt Meselson, Jim Wang, Jim Watson, Steve Harrison, Don Wiley, Paul Doty, Wally Gilbert (newly arrived from physics), Guido Guidotti, Nancy Kleckner, Konrad Bloch, Jack Strominger, and, later (after finishing his postdoctoral research with me), Tom Maniatis. Group meetings were open, and the criticisms and brainstorming could be fierce, a kind of scientific paradise.
How the Repressor Binds DNA: The “Recognition Helix” and the Helix-Turn-Helix (HTH) Motif
X-ray crystallography and model building (first of a related protein, discussed below, called Cro) (11–13) suggested that an α-helix (we called it the “recognition helix”) could insert into the groove of ordinary B-form DNA. Amino acid functional groups extending from the helix would make specific contacts with edges of base pairs, we thought. A slew of structural and genetic experiments, including the crystal structure of the 434 repressor with its operator, confirmed this idea. For example, in a helix-swap experiment, we replaced the putative recognition helix on one repressor with that found on another and thereby changed the specificity of binding as predicted (14). Our experiments on and thinking about structural problems were very much influenced by conversations and collaborations with Steve Harrison and Jim Wang at Harvard.
An imaginative bit of combined sequence/structural analysis, performed by three former lab members in collaboration with R. F. Doolittle at the University of California, San Diego, pointed to a few critical residues that were present at a characteristic spacing in several known and putative DNA-binding proteins. This pattern, they correctly surmised, signaled the presence of a common HTH motif that presented the recognition helix for DNA binding (15). This soon led to the identification of HTH motifs in homeodomain proteins, factors crucial for development of higher eukaryotes (16).
However, these structural insights only partially solved our problem. Specificity is a matter of degree. Put roughly (pretty much the best we can do even now), this means that, at the concentration of repressor found in a lysogen, the repressor would be found bound much more frequently at the operator than at other places in the genome. The information read by a single recognition α-helix is obviously not sufficient to achieve this degree of selectivity; too many sequences identical to the five or so base pairs recognized by that α-helix would be strewn about by chance. In addition, even if the site were unique, the repressor would spend so much time on the sea of related sites that it would never find the right one (see Appendix 1 in Ref. 1). Two protein-protein interactions, in addition to the protein-DNA interactions, turned out to be critical as follows.
Repressor Dimers Bind 2-fold Rotationally Symmetric Sites
In the first of these protein-protein interactions, repressor dimers form in concentration-dependent equilibrium with monomers; the higher the monomer concentration, the higher the fraction (approximately quadratically) is present as dimers. This aspect of repressor behavior was initially revealed by velocity gradient centrifugation of the repressor, which by then we could purify (17).
This made sense: because protein dimers are typically 2-fold rotationally symmetric, the preferred binding site would itself be 2-fold symmetric, thereby allowing DNA contacts with both recognition helices (Fig. 1). Only where two copies of the recognized sequence lie adjacent to each other and arranged with 2-fold symmetry would the protein bind significantly. Indeed, it emerged that each repressor dimer recognizes a 17-bp sequence that is nearly, if not perfectly, 2-fold symmetric. Experiments that I will not recount indicated that the major path of DNA binding was, first, formation of a dimer (or conceivably of a higher order oligomer) and, second, binding to DNA (18). We soon learned, however, that even the relative uniqueness of a 17-bp 2-fold symmetric site did not suffice.
FIGURE 1.
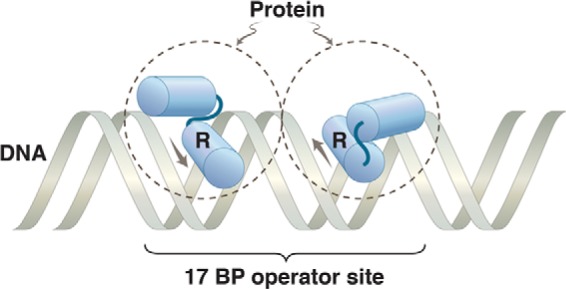
Protein dimer bound to DNA. The dashed circles represent two identical subunits of a protein bound to a 2-fold rotationally symmetric operator. The HTH motif on each monomer is indicated, with the recognition helix labeled R. The arrows show the direction N → C of the recognition helix in each monomer. We are dealing here with a simple binding reaction, so the protein rapidly comes off and rebinds. The image represents, in effect, a snapshot of the protein and DNA at an instant of binding.
By 1971, I was a bona fide Harvard professor. I had survived a hilarious initiation interview with the bow-tied Dean John Dunlop. Gazing at the ceiling, he informed me he was reluctantly granting this promotion despite the “grave reservations” of certain prominent types who “doubted my loyalty to this institution.” You see, I had, in a quixotic fervor, put forth a motion to the Harvard faculty that it express opposition to the Vietnam War. In the ensuing chaos and dismay, I think the motion actually passed, but with curiously little effect on either Harvard or the war.
Reiterated Sites and Cooperativity
Anyway, we set about isolating a λ operator by digesting 32P-labeled λ DNA with DNase in the presence of pure repressor. We expected that the repressor would cover and thus protect from nuclease digestion a fragment of ∼20 bp. The surprising result was that the repressor protected, in addition to the expected fragment, other longer ones as well (10). I soon met Fred Sanger on a ski lift somewhere (a proper scientific meeting, as I recall) and quickly arranged to travel to Cambridge (England) with Tom Maniatis (then a postdoctoral fellow in my lab) to determine the sequences of some of these DNA fragments.
Fred's early sequencing methods were cumbersome compared with the slick ones that followed, but they worked. We found, to our surprise, that the operator (called OR, for operator right; there is also an OL) bears not one but, apparently, three repressor-binding sites. The sites are similar but not identical 17-bp sequences, and each is ∼2-fold rotationally symmetric. We labeled these sites OR1, OR2, and OR3 (19–21). This was the only period after matriculation into graduate school that I “worked” (i.e. watched Tom work) in a laboratory other than my own. Note that, as typically represented, the sites read left to right OR3, OR2, OR1. These sites, especially OR1 and OR2, were mutated, it turned out, in various λ “virulent” mutants (i.e. mutants that ignore the repressor and grow lytically in its presence) (22).
Why three repressor dimer-binding sites in the operator? Why not just one? Part of the answer lies in the phenomenon called cooperative repressor binding or, simply, cooperativity. We discovered the effect when, back at Harvard, we performed a modified version of the DNase protection experiment called footprinting. That experiment enabled us to visualize repressor “filling” (protecting) the individual sites in OR as a function of increasing repressor concentration. Two adjacent operator sites (OR1 and OR2) were filled first, and the third site (OR3) was filled only at higher repressor concentrations. However, similar experiments with DNA bearing single sites, i.e. DNA fragments bearing each of the three sites separated from its neighbors, showed that sites OR2 and OR3 had identical intrinsic affinities for the repressor, an affinity some 10-fold lower than that of site OR1. Somehow, the presence of OR1 increases the apparent affinity of OR2, but not that of OR3, for the repressor (23).
In other words, on WT DNA, the repressor binds cooperatively to sites 1 and 2 (Fig. 2). We surmised that the concentration of repressor in a lysogen is such that OR1 and OR2 are usually occupied by repressor. The third site, OR3, is occupied less frequently, a matter I will return to below. The shorthand summary of the affinity of sites in OR for the repressor is OR1 = OR2 > OR3 when the sites are adjacent and OR1 > OR2 = OR3 when the sites are on separate DNA fragments.
FIGURE 2.
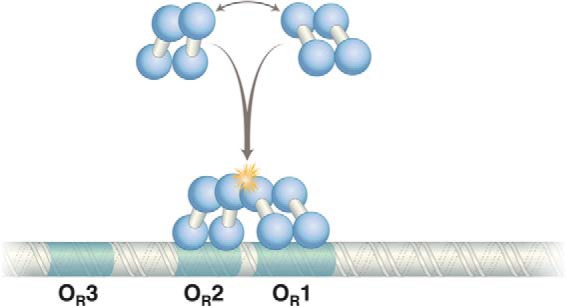
Cooperative binding of two λ repressor dimers to DNA. The N-terminal domains bind DNA, and contacts between C-terminal domains mediate dimerization and cooperative binding of dimers (starred) to DNA. The contact mediating the λ repressor N-terminal domain is separated from the C-terminal domain by a linker of 40 amino acids.
Cooperative binding of the repressor to DNA, we realized, is formally analogous to the classic case of cooperative binding of oxygen to hemoglobin. However, in the latter case, the helping effect of one oxygen molecule on the binding of another is affected by a conformational change in the protein. The four O2-binding sites are equivalent, and cooperativity is inferred from the shape of the curve describing affinity for oxygen as a function of oxygen concentration. In our case, we could literally see one repressor molecule (a dimer) binding to the preferred site, OR1, helping another repressor dimer bind to the second site, OR2.
Mechanism of Cooperativity
How does a repressor dimer binding to site OR1 help another bind to OR2? Recall, as discussed above, that the repressor has little or no effect on DNA structure, so the class of models that would invoke structural changes transmitted along the DNA (analogous to conformation changes in Hb upon O2 binding) seemed unlikely. We suspected, rather, that cooperativity was mediated by contacts (touching) between DNA-binding repressors. This view was confirmed by two quite different kinds of experiments, one biochemical and the other genetic.
In collaboration with Julian Sturtevant at Yale University and using his scanning calorimeter, we found that the repressor underwent two well separated “melts” (denaturations) as the temperature was raised, suggesting that the protein consists of two domains that can denature independently. This was confirmed by the finding that two fragments were produced by papain cleavage of the repressor (one that includes the N terminus (residues 1–92) and the other the C terminus (residues 132–236)) and by the subsequent finding that the fragments denatured at two different temperatures as predicted (24).
The most important further result of this series of experiments, in the current context, was that the separated N-terminal domain (which contains the HTH motif) dimerized and bound to the three sites in OR but did so non-cooperatively, i.e. the affinity order of binding of the N-terminal domain to the three adjacent sites in OR mimicked their intrinsic affinities as described above: OR1 > OR2 = OR3 (23, 25). These results indicated that the cooperativity function must be provided by the C-terminal domains. This surmise was later confirmed by the isolation of point mutants of the repressor that had lost the cooperativity function. Such mutants change residues on a surface of the C-terminal domain. The interaction between C-terminal domains of repressor dimers is weak in chemical terms (1 or 2 kcal; one or two amino acid contacts) but is sufficient to increase the stability of repressor-DNA complexes some 10–50-fold (26–28).
DNA Looping
We also learned, early on, that repressor-binding sites need not lie immediately adjacent to each other for cooperative binding. When the sites are separated, a DNA loop, which apposes the interacting proteins, must accommodate the reaction. For example, a repressor binding to a single site could, we found, help another bind to a site positioned some 100 bp away. The reaction produced DNA loops that were visualized under the electron microscope. As the sites were positioned ever more closely to each other, only those separated by integral numbers of turns of the DNA helix (i.e. modulo 10) were bound cooperatively (29, 30). This was as expected if the repressor binds DNA in essentially its B-form and if DNA has limited flexibility such that repressors bound on opposite faces of the helix cannot touch each other. We called this a “side of the helix” experiment.
At the time, all of this was rather surprising, as it was believed, or assumed, that the persistence length of DNA was such that looping would be impossible (given the weak interactions between DNA-bound repressors) over short distances and very unlikely over large distances thanks to entropic considerations. However, the evidence was undeniable. Later, thanks to others, we encountered a loop between repressor sites separated by 3000 bp on the λ genome (see “Lysogeny: Maintenance” below).
The protein-protein interactions mediating cooperativity do not have to be strong to be biologically important. The magnitude of the dimer-dimer interaction at λ OR is particularly small: an increase in repressor dimer concentration of, say, 10-fold (and therefore an increase in monomer concentration of just some 3-fold) overrides the effect. Thus, at even modestly increased concentrations, the repressor will tend to bind sites without the cooperative effect that ordinarily helps that binding. Looked at from the opposite point of view, we see that cooperativity ensures specificity of binding at lower concentrations of repressor than otherwise would be required.
Sites that are bound cooperatively do not have to bind the same protein at both sites, of course. Where non-identical proteins bind cooperatively, we have a simple way to integrate signals: only when both (or all of the) proteins are present and able to bind DNA (each responding to a separate signal), would any bind significantly. Binding sites can be (and often are, especially in higher eukaryotes) widely separated; the interactions between binding proteins in those cases must be strong enough to overcome the entropic costs of DNA looping. These arguments are worth keeping in mind later as we discuss the mechanism of transcription activation.
We next faced these questions. What precisely does the DNA-bound repressor do, and how does it do it? The newcomer might get derailed momentarily here. The λ repressor was (like its Lac counterpart) called a repressor because genetic experiments first had revealed its role in preventing expression of lytic phage genes. However, we now know that the protein works equally as an activator and as a repressor, as discussed below.
The λ Repressor: An Activator as Well as a Repressor
Genetic experiments of others indicated that the repressor regulates expression of its own gene (31–34). We created a regulatory circuit that measured the effects of the repressor, as a function of its concentration, on the activity of PRM (promoter for repressor maintenance), that which drives transcription of the repressor gene in lysogens. The results were dramatic: as the concentration of repressor was increased, transcription emanating from PRM at first increased and then, at higher concentrations, decreased. By selectively mutating one or more sites in the operator, we determined which sites were used for each effect. These and other experiments (34–36) revealed the mechanisms of activation and repression as follows.
Mechanism of Activation: pc Mutants and Recruitment
Activation is a form of cooperative binding of proteins to DNA: DNA-bound repressor touches, and thereby helps, RNA polymerase bind and work at the adjacent promoter. We say that the repressor recruits polymerase to PRM. Two crucial findings that revealed the mechanism were as follows. First, the repressor must bind DNA to work as an activator. Thus, repressor mutants that have lost the DNA-binding function by mutation cannot activate nor can the repressor activate a promoter in the absence of the proper DNA-binding sequence. Second, the repressor must bear a so-called “activating region” on its surface. This region was defined by repressor mutants (called pc, for positive control) that bind DNA normally but cannot activate (35, 36). Further experiments revealed that the operator site required for the repressor to work as an activator of its own gene is OR2. Model building showed that that repressor bound there would touch, with its activating region, RNA polymerase bound at PRM (Figs. 3 and 4) (37, 40).
FIGURE 3.
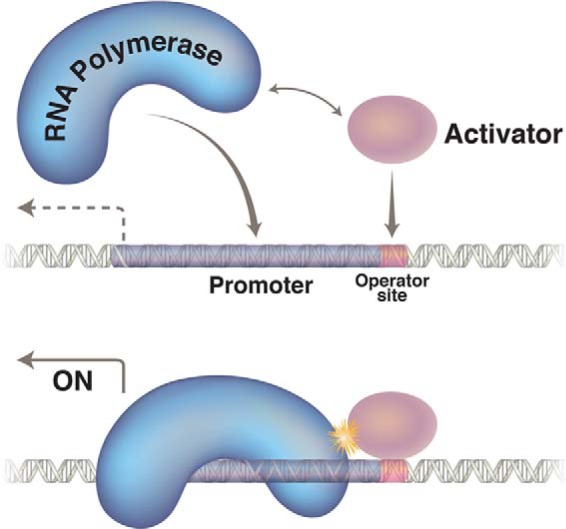
Transcription activation is a form of cooperative binding of proteins to DNA. The polymerase has a weak affinity for the promoter, and the activator (shown here binding to a single site) recruits it to a nearby promoter by virtue of a protein-protein contact.
FIGURE 4.
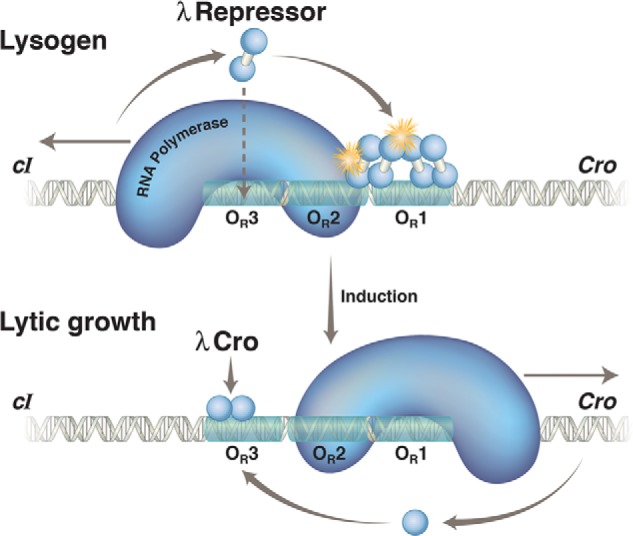
λ repressor and Cro action at OR in a lysogen and following induction. Upper, in a lysogen, the λ repressor, encoded by the cI gene, preferentially occupies two adjacent sites, OR1 and OR2, in the operator OR. In this state, the repressor activates transcription of its own gene (which proceeds leftward in the figure) as it represses transcription of the cro gene (which would otherwise proceed rightward). With lower efficiency, the repressor also binds the weak site 3 and thereby turns off transcription of its own gene. Binding of the third site is facilitated by interaction with other repressor dimers bound to a site (called OL) some 3000 bp away, in an example of cooperative binding accommodated by DNA looping (not shown). The stars indicate protein-protein contacts of about equal strengths, one mediating cooperative binding of repressor dimers and the other recruitment of RNA polymerase and activation of transcription of the repressor gene. Lower, the repressor is destroyed (cleaved) upon exposure to UV irradiation. This relieves repression of cro and other genes required for lytic growth. Cro binds first to OR3 and abolishes transcription of the repressor gene. Later in the lytic cycle, Cro binds to sites 1 and 2 and represses transcription of its own gene (not shown). Not explicitly shown are the two relevant promoters, each of which covers ∼50 bp. PRM, which directs transcription of the cI gene in a lysogen, lies immediately adjacent to the operator site OR2, so the repressor and polymerase contact one another as they bind to their respective sites. In contrast, site OR3 overlaps this promoter, and hence, repressor bound there excludes polymerase. PR, which directs transcription of cro and other lytic genes, overlaps OR1 and OR2, so repressor bound to either site represses PR.
Just as the “helping” effect of one repressor dimer on the binding of another is evident only at repressor concentrations below a certain level, so too is “activation” observed only when polymerase is held below a certain level. Thus, we found that purified λ repressor activated transcription of its own gene when mixed with pure RNA polymerase and λ DNA but did so only if the polymerase concentration was held below a certain value. At higher polymerase levels, the gene was transcribed at a high rate without added repressor, and the stimulatory effect of the repressor was no longer observed (37). Confirming another prediction of the recruitment idea, we found that a different protein, ordinarily a repressor, suitably repositioned on DNA, can be made to work as an activator if a few residues making up an activating region are added to its surface (38).
I know that “recruitment” sounds simple, and it seems to take the frisson out of the word “activate,” but there it is. Two further implications of the mechanism follow. First, keeping a gene “on” requires maintenance in the form of the continuing action of the activator or of some other activator that might take its place. Activation, per se, does not entail memory. Second, although removing an activator decreases the rate of transcription of a gene, it does not suffice to turn the gene off entirely. The activator simply increases the rate of a reaction (polymerase binding to a promoter) that occurs spontaneously in the absence of the activator. Thus, in the absence of an activator and at the concentration of RNA polymerase in a bacterium, the gene will be transcribed at a lower basal rate unless a repressor turns the gene off entirely.
Repression
Repression is the effect of a repressor excluding binding of RNA polymerase to a promoter. This can occur where a repressor-binding site overlaps a promoter. The degree of repression will then depend on the concentrations of repressor and polymerase and their affinities for their sites (operators and promoters) on DNA.
The conceptually important points here are that repression and activation are reflections of simple binding interactions between proteins and DNA and between DNA-bound proteins. There is no activation in a traditional sense, just a binding reaction (between a DNA-bound repressor activating region and RNA polymerase) that increases the efficiency of a reaction that otherwise proceeds spontaneously at a lower level. Whether a protein activates (or represses) transcription of its own or of other genes is a function of circuitry, i.e. the position on DNA of its operator sites, and on whether or not the protein bears an activating region on its surface. The magnitudes of all such effects are, of course, determined by the relative concentrations and affinities of the relevant proteins. How do these mechanisms work together to make a switch?
The Biphasic λ Switch
Recall the two opposing states of λ gene expression. In a lysogenic bacterium, all of the phage genes are silent except the repressor gene itself; and, upon induction (elicited by, for example, UV irradiation), virtually every cell switches so that the repressor gene is off and the lytic genes are on. Soon thereafter, the cells burst and release a new crop of phage. Maintenance of lysogeny is a kind of memory: once lysogeny is established, the progeny cells remember that state unless instructed otherwise by an extracellular signal. The phages that emerge upon induction are identical copies of those that established lysogeny in the first place, and so no mutations are involved. These properties, taken together, as I have noted, define the switch as epigenetic (39, 40): the new state of gene expression (as in lysogeny) is remembered (self-perpetuated), in this case, over very many generations, unless and until a signal (in this case, UV light) instructs otherwise.
Understanding how the switch works requires no mechanistic principles beyond those already discussed, but we do need to consider one more regulatory protein encoded by λ: Cro. The cro gene lies just adjacent to the repressor gene, with the operator OR lying in between the two genes. cro, transcribed from PR (promoter rightward), is off in a lysogen and is the first gene expressed upon induction. The action of cro was demonstrated dramatically by creating lysogens deleted for all of the phage genes except the fragment containing cI and cro. These bacteria variegated: at any given time or condition, they expressed one or the other, but not both genes (41). Just how cro manages to work contra repressor (and vice versa) soon became clear as the complete workings of the switch emerged. We need first describe the crucial distinction between maintenance of lysogeny and its establishment.
Lysogeny
Maintenance
The repressor does two things to maintain lysogeny: it activates expression of its own gene as it represses transcription of lytic genes, including that of cro. These repressor functions require repressor binding (cooperatively, please recall) at sites OR1 and OR2. The disposition of these two operator sites is such that repressor bound to either site excludes polymerase from PR (the lytic gene promoter), and, as noted, repressor bound to OR2 recruits polymerase to PRM. It is only because phage genomes are highly compact (presumably, the effect of selection for rapid growth) that the same linked operator sites, OR1 and OR2, are involved in both functions of the repressor. A separate set of sites, for example, could have been used to control PR and would have been had PR been positioned some distance from PRM. The repressor is passively transmitted in the cytoplasm as the cells divide; thus lysogeny is perpetuated. This state is extremely stable, rarely flipping to the alternative state (lytic growth), in the absence of an extracellular signal (42).
The repressor has an additional function in a lysogen: it maintains its own level below a specified value by binding to the weaker site (OR3). This negative feedback loop maintains the repressor level 2–3-fold lower than it otherwise would be, an effect important for efficient induction. We now know (thanks to others) that repressor binding to OR3 (and hence, negative autoregulation) is helped by interactions between repressors bound at OR and at a second λ operator, OL (also comprising three 17-bp repressor-binding sites), located some 3000 bp (!) away. A large DNA loop accommodates the interaction (43, 44).
Establishment
The work of others added a crucial missing piece: how does transcription of the repressor gene get started in the first place? Upon phage infection, a transcriptional activator called CII is expressed. CII works just as does the repressor in its guise as an activator, but it does so at a site that prompts transcription from the distal promoter PRE (promoter for repressor establishment.) The dollop of repressor thereby produced triggers the positive feedback loop that maintains lysogeny. cII is shut off along with cro and other genes by repressor bound at OR1 and OR2 (Fig. 5) (45).
FIGURE 5.
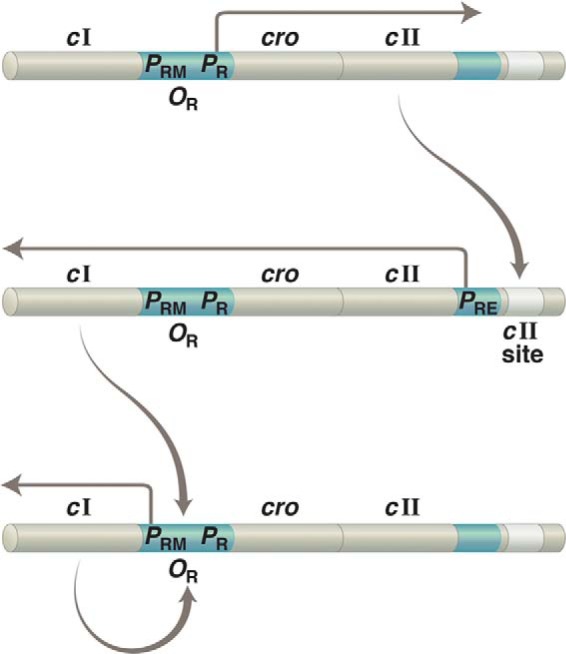
Establishment of lysogeny. The same gene, cI, is transcribed from two different promoters: from PRE to establish lysogeny and from PRM to maintain that state. Upper, upon infection of a bacterium with phage λ, transcription rightward from PR results in production of the transcriptional activator CII. Middle, CII binds to the “cII site” on DNA and activates leftward transcription from PRE. Lower, newly made repressor binds OR1 and OR2 and turns off transcription of cII (as well as of other genes required for lytic growth) as it simultaneously activates leftward transcription from PRE. As implied by this figure, PR controls not only lytic genes (as indicated in the text) but also cII, which is required to establish lysogeny. In addition, PR is an unusual promoter in that it requires no activator for full activity.
Induction
UV light causes reversal of the states, with lytic growth now replacing lysogenic growth, by (indirectly) causing cleavage of the repressor. The cleavage separates the N-terminal domain from the C-terminal domain, thereby eliminating the cooperativity function between repressor dimers. As the effective concentration of repressor drops, so too does the rate of new repressor synthesis. As the repressor is destroyed and the operator is vacated, repression of PR is lifted, and cro (and other genes) are expressed. Understanding the mechanism of action of cro now becomes crucial.
We found, to our surprise, that Cro binds to the same three sites in OR as does the repressor, but it does so with an opposite order of affinity. Thus, as shown by experiments performed both in vivo and in vitro, many similar to those used to dissect repressor action, Cro binds most tightly to OR3 and less tightly to OR1 and OR2. Therefore, its first and strongest effect is to repress transcription of the repressor gene. There is nothing particularly mysterious about the binding order differences of the repressor and Cro; the residues on the Cro recognition helix, some different from those on the repressor recognition helix, prefer, as it were, the sequence at OR3 to those at OR1 and OR2 (23, 46).
We might have anticipated the need for cro for induction based on our picture of the mechanism of activation. We surmise that destruction of the λ repressor (and hence, loss of activation of the repressor gene) would not suffice to eliminate production of more repressor, albeit at a low level. Turning off that basal transcription requires a specific repressor (Cro) that excludes polymerase from the promoter. Absent Cro, induction is inefficient. Most bacterial activators work like the λ repressor (i.e. by recruitment) and, like the λ repressor, are paired with specific repressors. Understanding the mechanism of activation explains the near-ubiquitous requirement for repressors associated with activators in bacteria (47–49). We will encounter an interesting twist to this idea in dissecting transcription regulation in eukaryotes.
Cooperative binding of the repressor to DNA stabilizes both the lysogenic and lytic states. A not insignificant fraction of repressor must be destroyed (say 50%) for induction to be triggered irreversibly; and, in the lytic state, a significant level of repressor must be made to trigger binding to the operators (and hence, lysogeny). Of course, the transition between the states, the switch-like effect, as repressor concentration drops or increases, is more dramatic than it would be in the absence of cooperativity (50).
An Integrated System
The switch was not deduced from general observations or theoretical or mathematical models. Rather, its parts were assembled as we went along, its glorious integrated working revealed only near the end. At every stage, we could test this or that aspect, challenging with genetics and biochemistry, trying to ensure that each bit was well in hand before going on. The reader will recognize that this approach is nowadays rather out of fashion. Instead, we have the “big picture,” many genes, obscure words, and mathematical formulations. λ remains the best understood integrated system we have, and perhaps one should ponder how we got it.
Most of the time, we biologists (if I may call myself that) work on isolated pieces of some extensive and complex set of reactions. I guess that the dream of systems biologists is to grasp them all at once and see how perturbations in one part might affect the workings of other parts. Our picture of the λ switch became so complete and so precisely defined that we would pounce on any detail that seemed out of place. Here is an example: recall that I have said that the repressor must be bound to OR2 to activate transcription from the adjacent promoter PRM and that the repressor ordinarily binds to OR1 (a strong site) cooperatively with the repressor at OR2 (a weak site). Only at higher repressor concentrations would the repressor bind site OR3 and repress transcription of its own gene. However, in an experiment using an operator damaged only in site OR1, not only was the promoter not activated, it was repressed (below its basal level), even at low repressor concentrations. Those in vivo results were explained by the observation that, in vitro, in the absence of OR1, the repressor binds cooperatively to sites OR2 and OR3 and thus covers and thereby represses PRM. We called this phenomenon “alternate pairwise cooperativity,” and a bizarre result now made sense (23).
Irreducible Complexity?
Were one to encounter the λ switch presented whole, it might seem irreducibly complex. One might think that, because every feature we discussed contributes to the workings of the switch, it is hard to imagine how it might have evolved. To the contrary! Experiments in which parts of the switch have been removed by mutation (autogenous negative control, cooperativity, and so on) reveal that these mutations, while damaging the switch more or less, do not destroy it. Thus, parts of the switch can be regarded as evolutionary add-ons, features that make a basic system that works work better (51).
The λ World: An Overview
Regulation of transcription is effected by proteins that bind specific sites on DNA. Cooperativity is an essential feature of this specific binding: one protein can help another bind DNA by simply touching it. This helping effect is also the disarmingly simple mechanism underlying activation of transcription: a transcriptional activator is a specific DNA-binding protein that has the appropriate residues on its surface to contact RNA polymerase and thereby help it bind and work at a promoter. These cooperativity and activating surfaces require only a few amino acids, and the interactions they engage in are weak (1 or 2 kcal; factors of 10–50 in binding constants). Where a protein is working as a repressor, its binding site overlaps that of RNA polymerase at a promoter, and the repressor excludes binding of the enzyme. It is not hard to see, therefore, how a protein can activate expression of certain genes as it represses transcription of others; the λ repressor is such a regulator.
The very simplicity of the mechanism of activation means that any gene can be brought under control of any activator simply by properly apposing the activator and polymerase-binding sites. Thus, we have the solution to the memory problem: where an activator works on its own gene, that state of gene expression, once established, will tend to be self-perpetuating; as cells divide, the activator is distributed to daughter cells, and the state is maintained. Memory is not a property of any single element per se, but rather it is a property of the system of the basic elements suitably arranged. By combining these elements, nature can produce sophisticated switches, allowing genes to be expressed in alternative states, with sensitive and dramatic transitions between them in response to signals.
The λ epigenetic switch includes these salient aspects. 1) The repressor and Cro comprise a double negative loop in which one or the other, but not both, is expressed (the former in a lysogen and the latter during lytic growth). 2) The repressor activates transcription of its own gene in a positive feedback loop in a lysogen. Positive feedback loops also tend to be found in one or another alternative state, either on or off. Even a transient interruption of a positive feedback loop will switch it off. 3) Cooperative repressor binding to DNA helps ensure stabilities of the alternative states and the dramatic “all or none” transition upon induction. 4) Negative autoregulation by the repressor in a lysogen helps ensure the relevance of this cooperativity by maintaining the repressor concentration below a specified level. 5) A separate gene regulatory circuit establishes repressor synthesis in the first place. Once set in the lysogenic mode, the switch is extremely stable and, once turned to the lytic mode, is irreversible.
I hasten to mention another aspect of the λ world. I was lucky to be surrounded by imaginative and generous scientists (younger every year) who brought their critical intelligences to bear on our shared λ problems. It might surprise newcomers to hear that every year, at this or that gathering, we were as interested to hear that a troublesome finding (reported the previous year) could now be discounted, as we were to hear about something entirely new, progress in either case. Every year, it seemed, produced a new vocabulary that described new mutants and/or that conceptualized the new state of affairs. Skip a year, and you were lost! Many of the people who contributed in important ways, only some of whom are mentioned here, wrote articles for the two classic λ books (52, 53). I have described elsewhere the bracing effect of surviving the editorial comments of Al Hershey (the editor of the first volume) (54, 55).
So now we faced the question of how our insights might apply or not in eukaryotes. Is a different mechanism for DNA binding required? Is the mechanism of activation different, perhaps more complicated? And so on. So we turned to yeast, a bona fide eukaryote, nucleus, nucleosomes, and all, but an organism that can be manipulated genetically almost as easily as bacteria: mutants selected, plasmids carrying extra genes added to cells, and so on. They double every 90 min or so, slower than bacteria, but we could live with that.
In reading the rest of this essay, please keep in mind what was always in our mind: the ideas we had developed in studying λ. Someone once said that “the most important results of basic science are ideas,” and the following might be read as an illustration of this notion. A certain degree of abstraction is, of course, required in formulating mechanisms that apply in organisms as diverse as bacteria and yeast, but that is the fun of it. I should mention that it was not all fun. A special kind of grief, one familiar to some readers I suspect, was soon in store: the National Institutes of Health refused to renew my longstanding grant because I had “switched fields.” Of course, I had not switched fields; in fact, I was working in exactly the same field. Try explaining that!
Eukaryotes
We stuck to the rule: our subject was (and is) regulation of transcription, not transcription per se. Thus, the issue is how this gene versus that gene is specifically instructed to begin transcription and not the machinery that is required to carry out these instructions. The strategy is particularly important when shifting one's attention from a bacterial to a eukaryotic gene because, unlike in bacteria, transcribing a eukaryotic gene requires, in addition to RNA polymerase itself, a multisubunit protein transcription complex, the various activities of which are still not clear. Of course, eukaryotic DNA is wrapped in nucleosomes.
Yeast GAL Genes
The GAL gene products (Gal1 and Gal10, in particular) metabolize the sugar galactose. The addition of galactose to cells in culture induces their transcription of those genes by >1000-fold. Two key control genes determine this regulation: GAL4 and GAL80. Deletion of the former eliminates induction, and (separately) deletion of the latter renders GAL gene expression constitutive (i.e. the genes are fully on even in the absence of galactose). Perhaps Gal4 is an activator and Gal80 a repressor (making them analogous to the λ repressor and Cro)? Not quite.
Gal4: An Activator-“like” λ Repressor
Gal4, we soon showed, is indeed a specific DNA-binding protein (Fig. 6). It recognizes four sites in the genetically defined operator (here, a UASg (upstream activating sequence galactose)) (56). Placing the UASg near other genes brought those genes under the control of galactose, identically to the control of the GAL genes (57). So, Gal4 apparently (obviously) is an activator. Unlike in the λ case, however, rather than lying immediately adjacent to the relevant promoter, the activator-binding sites lie some 300 bp upstream (hence, the name UASg). How does Gal4 work?
FIGURE 6.
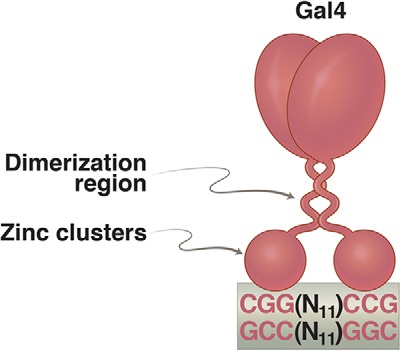
Gal4 dimer bound to a 17-bp 2-fold rotationally symmetric DNA site. Each of the “zinc clusters,” as they are called, recognizes a base pair triplet. These triplets are separated by 11 bp. Activators related to Gal4 bear identical zinc clusters, but the spacing between the triplets is unique in each case. The formal name for these zinc clusters is the Zn(II)Cys6 binuclear cluster. Such DNA-binding domains are not found in bacteria.
Recall our findings that the activating and DNA-binding functions of the λ repressor are genetically separable: mutations in the repressor can abolish activation without damaging DNA binding. Could that be true for Gal4? Yes, it turned out, and distinguishing these two functions was actually easier with Gal4; whereas the DNA-binding function and activating regions lie on the same domain of the λ repressor (the N-terminal domain), they lie on different domains of Gal4 and are readily physically separable, as follows.
We expressed in yeast a gene encoding just the N-terminal 145 residues (from a total of 881) of Gal4 and found that this little protein mimicked a λ repressor-positive control mutant: it dimerizes and binds DNA normally (to sites in the UASg, in this case) but cannot activate transcription. In contrast, the C-terminal domain had no detectable function on its own (58). Perhaps the C-terminal domain contains an activating region analogous to the λ repressor activating region? If so, then tethering it to DNA in some way (any way) might enable it to work. Our first domain-swap experiment, a dramatic event at the time, confirmed this idea as follows (Fig. 7).
FIGURE 7.
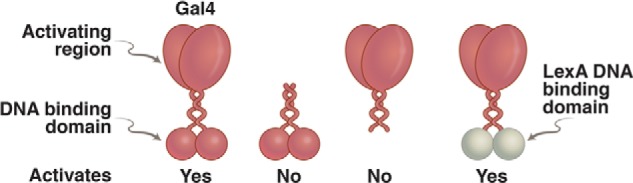
Domain-swap experiment. On the left is an intact Gal4, which binds DNA and activates transcription. To its right is the N-terminal domain alone, which dimerizes and binds DNA but cannot activate transcription. Second from the right is the separated C-terminal domain, which cannot bind DNA and cannot activate. On the far right is a hybrid protein in which the DNA-binding domain of the bacterial repressor LexA has been swapped for that of Gal4. This protein activates transcription in a wide array of eukaryotes, provided each target gene has been modified so as to bear a LexA-binding site nearby.
A Domain Swap
We fused a bacterial repressor called LexA to the C-terminal portion of Gal4 and found that the hybrid protein (ectopically expressed) activated a gene in yeast if, and only if, LexA-binding sites had been inserted near the beginning of the gene. Thus, we had replaced one DNA-binding domain with another (one from a bacterium!) and had thereby maintained the activator's function but had changed its specificity (i.e. which gene it would work on) (59). Other DNA-binding domains were swapped in as well, and each of these was found to determine a unique specificity. The LexA-Gal4 hybrid, like Gal4 itself, could work in a wide array of higher eukaryotes, including mammalian and plant cells, provided a suitable operator sequence was inserted near the target gene. Indeed, it turned out that the Gal4 activating region, tethered to DNA, works universally in the eukaryotic world (60–62).
Gal80 and the Two-hybrid Assay
In contrast to Gal4, Gal80 is not a DNA-binding protein, and it is not a repressor; rather, it is an inhibitor. Gal80 attaches to and covers the activating region on the C-terminal domain of Gal4. Thus, in cells grown in the absence of galactose, Gal4 can be produced and bind to DNA, but it remains inactive thanks to the inhibitory effect of Gal80. Galactose then frees the Gal4 activating region from this inhibitor. A prediction of these ideas was that attaching an activating region to Gal80 would turn it into an activator, so long as it could be tethered to DNA by interaction with Gal4. This turned out to be true (63). This result, which reinforced the idea that activating regions must be tethered to DNA to work, triggered development by others of the “two-hybrid” system (64).
We still did not know what eukaryotic activating regions actually do. Could it be that they (a) work as does the λ repressor, i.e. by touching and thereby recruiting to a promoter, RNA polymerase, or some other component of the eukaryotic machinery, or (b) by interacting with the machinery in some more specialized way, causing, for example, a change in structure of some protein that then triggers transcription? Analysis of an odd yeast mutant argued against b and for a.
Mechanism of Activation: A Crucial Mutant
I noted above that the Gal4 N-terminal domain, on its own, dimerizes and binds DNA but does not activate transcription. We isolated a rare mutant yeast strain in which the Gal4 N-terminal domain alone does activate transcription. The mutation changes a residue in Gal11, one of the components (it turned out) of the protein complex called the Mediator. The Mediator, in turn, can associate with RNA polymerase. Like other Mediator components, Gal11 is required for full expression of many genes. It is a historical accident that Gal11 had been identified in a screen for mutants that affected GAL gene expression (hence, its name).
We soon learned that our mutation, which we called Gal11P (potentiator), created a site of contact with the dimerization region of Gal4. In a further set of experiments, various Gal11P-like alleles (constructed by changing the identity of the amino acid changed in the original Gal11P) were found to bind the Gal4 dimerization region, and the strength of the interaction measured in vitro was correlated with the degree of activation observed in vivo (65, 66).
Thus, a simple protein-protein contact between a DNA-tethered peptide and a component of the transcriptional machinery suffices for activation of transcription. The requirement for an activating region can be obviated, as predicted by the recruitment model, as show in the following.
Activator-Mediator Fusions
We fused the Gal4 DNA-binding domain directly to Gal11 and found that the fusion protein worked as a strong activator in yeast of genes bearing Gal4-binding sites upstream. We later found that certain other Mediator components can also work as activators when tethered to DNA as part of a fusion protein bearing a DNA-binding domain. In all of these cases, the fused Mediator subunit evidently is inserted into that large protein complex, and the whole thing, along with whatever else might be required for transcription, is brought to the DNA by the exposed DNA-binding domain (Fig. 8) (66–68). We then discovered, counterintuitively at first, that activators have a general negative effect when expressed at high levels, an effect explained by recruitment, the mechanism of activation as follows.
FIGURE 8.
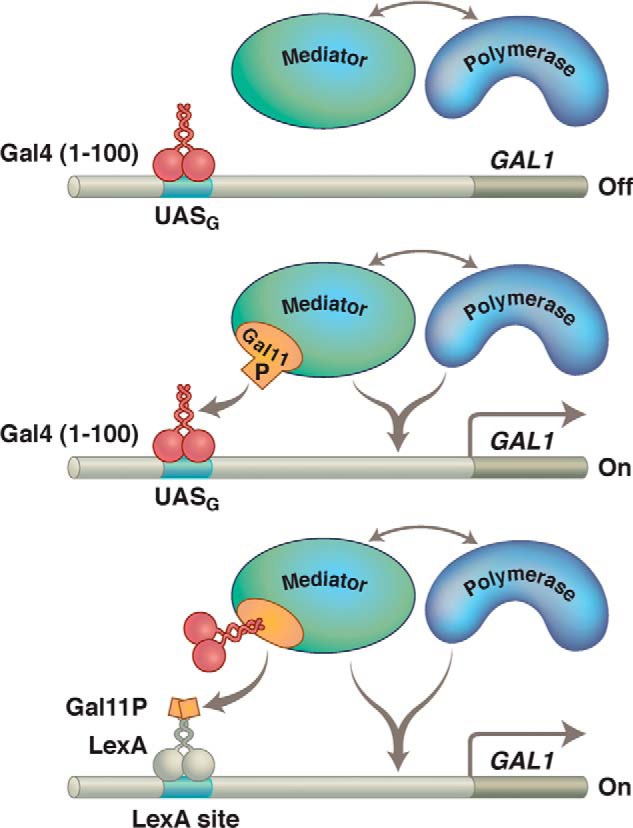
Gal11P effect. Gal11 is a component of the protein complex called the Mediator, shown here interacting with RNA polymerase. Upper, Gal4(1–100) (its N-terminal domain) does not contain an activating region and makes no contact with the polymerase-associated (i.e. Mediator) complex. This Gal11 fragment therefore does not activate transcription in a WT cell. Middle, the P (potentiator) mutation in the protein Gal11 creates a simple binding interaction with the Gal4 fragment, and that interaction (provided the Gal4 fragment is bound to DNA) results in activation of transcription. Lower, a bit of Gal11 bearing the P mutation has been fused to LexA, and that fragment activates only if the N-terminal domain of Gal4 has been fused to Gal11. There are many ways to effect recruitment.
Squelching
As the concentration of Gal4 is increased, transcription of its target genes at first goes up, and, at artificially high concentrations, it goes down. Unlike the positive effect, this negative effect extends to genes other than those bearing Gal4-binding sites nearby. We call the general negative effect “squelching.” The effect is as expected if the target of activating regions (e.g. a component of the transcriptional machinery) is not prebound to DNA but must be recruited to DNA. Thus, as the concentration of activating region(s) in cells increases, these targets are titrated and effectively sequestered, so they no longer can be brought to the DNA. As predicted, squelching does not require that an activating region be fused to a DNA-binding domain (69). Squelching can be observed in vitro, and relief of squelching by high concentrations of a presumed target of an activating region (i.e. the Mediator) was used as an early assay to isolate the Mediator (70).
Two factors determine the degree of squelching: the concentration of the activating region and its “strength,” i.e. the affinity with which it engages its target. The herpes viral protein VP16 bears an unusually strong activating region, for example, and Gal4-VP16 is an unusually strong activator. VP16 is also a strong “squelcher” in mammalian cells. Duplications and higher reiterations of even a small segment of this activating region (attached to a DNA-binding domain) produced ever more powerful activators/squelchers, consistent with the idea that activating regions are unstructured and work (as shown explicitly in other cases) approximately proportional to their lengths (71, 72). These and related results prompted the realization that nature must limit strengths/concentrations of activating regions in nuclei to limit squelching. It is not surprising, therefore, that unusually strong activating regions (e.g. that on VP16) are introduced only transiently by viruses.
Please note that squelching is a phenomenon distinct from autogenous negative regulation by the repressor discussed above. That latter effect requires specific binding of the repressor to an operator site (OR3) as discussed. Squelching, a general inhibition of transcription by titration of activating regions targets by overexpressed activating regions, has not been observed in bacteria, probably because the bacterial activating region-target interactions are, for any given case, too weak to manifest the effect. For more on the interaction of classic activating regions with components of the transcriptional machinery, see Refs. 73–76.
Activating Regions: As Many as You Like
Many different peptides (unstructured, evidently) work as activators in eukaryotes when attached to DNA-binding domains or to proteins that bind DNA-bound proteins (77, 78). Activating regions apparently can touch any of various surfaces on the transcriptional machinery to effect recruitment and trigger transcription. The apparent promiscuity of these interactions may explain why activating regions that work in yeast work in higher eukaryotes as well. As might be expected from these considerations, the reaction is not highly stereospecific; for example, Gal4 will work when bound to any of a large variety of positions around the DNA helix (77, 79, 80). In contrast, in bacteria, individual activators bear unique activating surfaces that touch this or that part of RNA polymerase, depending on the geometry of the activator-polymerase complex.
In sum, the archetypical eukaryotic transcriptional activator (like the λ repressor in its guise as an activator) comprises two essential functions: a DNA-binding domain and an activating region. The former determines specificity and can be swapped for any of a wide array of other DNA-binding domains (with consequent changes in specificity), and the latter can be replaced by any of a wide array of peptides or even by components of the transcriptional machinery itself. A purely “artificial” eukaryotic activator can be constructed by, for example, fusing a novel peptide to a synthesized compound that binds DNA (81). Could not be simpler! Beware: the very simplicity of the mechanism (recruitment) means that, unless strengths and concentrations of activating regions are controlled, negative (not positive) effects can ensue.
It was 1997, time to give in to Paul Mark's offer and move to the Sloan-Kettering Institute in New York! This is a change I never regretted: to quote Henri Matisse in a letter to his wife mailed from New York as he was on his way to Tahiti, “In New York, you see humanity at work.” It was time to get serious.
Nucleosomes and the Logic of Gene Regulation
Although we did not make particular note of it when we began our yeast studies, regulation of the GAL genes differs in an interesting way from regulation of bacterial genes. In bacteria, as I have noted, removing an activator does not suffice to fully turn off transcription of a gene. Instead, a specific repressor is required for the silencing (elimination of even basal levels of transcription) of a gene. However, in yeast, the inhibitory effect of Gal80 on Gal4 is sufficient to turn the target genes fully off, with no specific repressor required. Perhaps, as suggested by others (82), nucleosome formation (a distinguishing feature of eukaryotes) somehow substitutes for specific repressors.
Nucleosomes comprise, typically, 150-bp segments of DNA wrapped around protein (histone) cores. They form apparently ubiquitously on eukaryotic genomes and would be expected to sequester DNA safe from the transcriptional machinery. Nucleosomes covering promoters (i.e. regions where the transcriptional machinery assembles to transcribe a gene) would be expected to prevent spontaneous transcription and so eliminate the basal level problem. If so, we have new problems. How, for example, might nucleosomes have that effect while not blocking binding of activators to DNA? Might activator-binding sites be inherently nucleosome-free? If so, what would determine that scenario? How does recruitment (the mechanism of activation) help solve this problem?
Having obsessed for so many years about binding reactions, we decided to treat nucleosomes as, in effect, DNA-binding proteins. Taking hints from a long history of experiments in this area, we set about measuring the tendency of any specified DNA segment to form a nucleosome in vivo. We called our assay a “nucleosome occupancy” assay (83). The assay measures the fractional occupancy (in the population) by a nucleosome at any given position on the genome at any given instant. Moreover, because we can change DNA sequences, we can determine the relationship between nucleosome occupancy and DNA sequence (84). Using this assay, we have provided scenarios, consistent with many experiments of others, for how nucleosome formation can obviate the need for specific repressors.
There are two overarching principles. First, there are various strategies by which activators overcome the nucleosome obstacle that might prevent access to DNA. Probably the most common of these is cooperativity. As I have stressed previously, activator-binding sites in eukaryotes, seemingly invariably, come in multiple (sometimes identical and sometimes different) sites. Should these sites be covered by a nucleosome, activators will work cooperatively, even if they do not interact with each other, to displace that nucleosome(s) to access their DNA sites. The cooperative effect presumably would be even stronger were the proteins to interact. For another strategy facilitating activator binding, see discussion of the GAL genes below.
Second, although there is a significant range of affinities for nucleosomes along DNA (higher GC content imposing higher affinities), even the lower affinity regions form nucleosomes sufficiently tightly that removing them quickly, upon command, requires an ATP-utilizing enzyme. These promoter nucleosomes otherwise block assembly of the transcriptional machinery (a large multiprotein complex that apparently makes few contiguous contacts with DNA); thus, they eliminate basal transcription. These principles are illustrated in two different cases: one in yeast (85) and the other in mammalian cells (86, 87).
Yeast: The GAL Genes
The UASg, which, as noted, contains four Gal4-binding sites, also bears a site recognized by a protein complex called RSC. RSC, which has so-called “nucleosome-remodeling” activity, holds in place, at the UASg, a partially unwound nucleosome that exposes the Gal4 sites for ready access. This structure is found constitutively at the UASg in yeast, so Gal4 can quickly bind its sites. DNA-bound Gal4 will remain inactive in the absence of galactose thanks to the inhibitory effect of Gal80. Upon the addition of galactose and exposure of its activating region, Gal4 recruits the enzyme SWI/SNF, which strips off nucleosomes bound to the adjacent promoters, and Gal4 then recruits the transcriptional machinery that transcribes the genes. Absent SWI/SNF, these genes are activated but more slowly than in its presence. The nucleosomes covering the promoter evidently suffice to keep basal transcription very low (Fig. 9).
FIGURE 9.
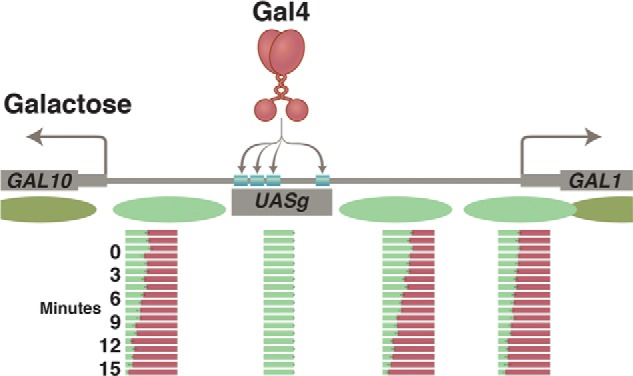
Nucleosomes at the GAL1/GAL10 genes before and after activation by Gal4. Prior to the addition of galactose, nucleosomes (green ovals) form in the promoters of the GAL1 and GAL10 genes. Upon addition of galactose, the exposed activating region of Gal4 recruits the enzyme SWI/SNF, which removes the promoter nucleosomes. The increasingly red bars show, at time points listed in minutes, the course of nucleosome removal. This step is followed by recruitment of the transcriptional machinery and transcription of both genes. Gal4 has acquired access to its four sites in the UASg, as explained in the text.
Gal4 also can access its sites in the absence of the RSC structure ordinarily formed at the UASg, a revealing scenario that can be created by deleting the RSC-binding site from the UASg. In this case, Gal4 must compete with occluding nucleosomes to find its sites, a feat it accomplishes, but more slowly than in the presence of the facilitating structure. If conclusions from our ongoing experiments are correct, successful competition with occluding nucleosomes in this mutant case requires that more than one Gal4 site be present, presumably to foster cooperative binding.
Higher eukaryotes, we believe, do not express an RSC that forms the facilitating structure at the UASg; so where Gal4 and UASg are used to control genes in those organisms, Gal4 must compete with occluding nucleosomes for DNA access. It seems to work fine.
Mammalian Cells: The kit Gene
Expression of the kit gene in murine mast cells requires the effect of proteins bound to an enhancer positioned some 150 kb (!) upstream of the promoter. These proteins (including GATA1 and GATA2) cover some 500 bp and, with the aid of a giant loop, replace a nucleosome at the promoter with the transcriptional machinery. As in other cases, loop formation is evidently fostered by the cohesin protein complex. The enhancer (as evidenced by its appearance in other cells in which the gene is silent) does not bear a structure (such as that seen at the UASg in yeast) that facilitates binding of the activators. Rather, the activators compete with and displace enhancer nucleosomes, a process likely helped by the fact that the GC content of the enhancer is such that nucleosomes form there with only modest “affinities.” In contrast, the nucleosomes at the promoter (a CpG island, as it is called) form with very high avidities and thereby, we suggest, prevent even low levels of basal transcription (Fig. 10).
FIGURE 10.

The murine kit locus. Upper, the promoter, which lies in the CpG island, is covered by avidly forming nucleosomes. Formation of these promoter nucleosomes, we believe, ensures an extremely low basal level of transcription. The enhancer, positioned some 150 kb upstream, has a lower GC content, so the nucleosomes form there less avidly. Lower, specific DNA-binding proteins bind the enhancer, displacing enhancer nucleosomes. Then, accommodated by a giant DNA loop (not drawn), those DNA-bound activators replace one promoter nucleosome with the transcriptional machinery and thereby activate transcription. The remaining nucleosomes at the promoter now are phased passively thanks, we believe, to the barrier erected by the transcriptional machinery. (Only the presence of the polymerase is indicated at the promoter.) Pol II, polymerase II.
Logic of Gene Regulation
The picture that emerges is that the primary form of gene regulation in eukaryotes is positive (activation). By this, I mean that eukaryotic genes are typically off, with very low basal levels, in the absence of activators and specific repressors, thanks to ubiquitous nucleosome formation. This conclusion, if correct, means that maintaining differentiated states in eukaryotes could be simpler than predicted on the λ model. Thus, an activator that worked on specified genes as well as on its own gene (in a positive feedback loop) could suffice to maintain the differentiated state; no negative effect on other genes would be required. From the opposite point of view, even transient dips in transcription generally, such as has been observed as an early step in transdetermination experiments (88),1 could switch off positive feedback loops that maintain, at least in part, differentiated states.
These ideas are supported by cases described by others in which the differentiated state of a tissue in a higher organism requires constant maintenance by an activator working in a positive feedback loop (89). Perhaps stem cells are similarly maintained by activators. Consistent with this idea, in collaboration with a former student now in Denmark, we performed an experiment indicating that Oct4 and, we since have learned, Sox2, DNA-binding proteins that help in the formation and maintenance of stem cells, work as activators, not as repressors (90).
This analysis, if correct, presents a new problem: if repressors are not needed for full inactivation of a gene, what are the roles of the eukaryotic repressors described in the literature? Perhaps some of these actually are inhibitors that, like Gal80, prevent a specific activator from working. In other cases, bona fide repressors would turn off (or down) a gene or genes while leaving the relevant activators free to work on other genes. For example, at the “silent mating-type loci” in yeast, specific DNA-binding repressors recruit proteins (Sir proteins) that keep the nearby genes off despite the presence, presumably, of the activators that otherwise would work on those genes (91). A curiosity of yeast silencing is that it is rather easily overcome by the action of a strong activator, as is observed readily if activator-binding sites are inserted near the silenced region. Polycomb, a repressor of genes in higher eukaryotes, is similarly easily overcome by a strong activator (92). Do such repressors work only to counteract weak activators? Might they be used only transiently during development to allow sequential binding of activators to enhancers, countering intermediate effects? This remains to be seen.
Yet a Bigger Picture
Sometime around 2002, Alex Gann (a former postdoctoral fellow of mine; by then at the Cold Spring Harbor Laboratory and on his way to becoming dean of the graduate school there) and I realized that the principles of gene regulation apply to a broad range of biological regulatory mechanisms (93). One way to state the general problem goes like this. Nature uses a common set of enzymes (polymerases, kinases, ubiquitylators, etc.) to different ends: to make hands and feet in a single individual, for example, and more generally to make different organisms, humans and mice, for example. An important mechanism by which these and other enzymes are used to different ends is, as for transcription control, recruitment (Fig. 11).
FIGURE 11.
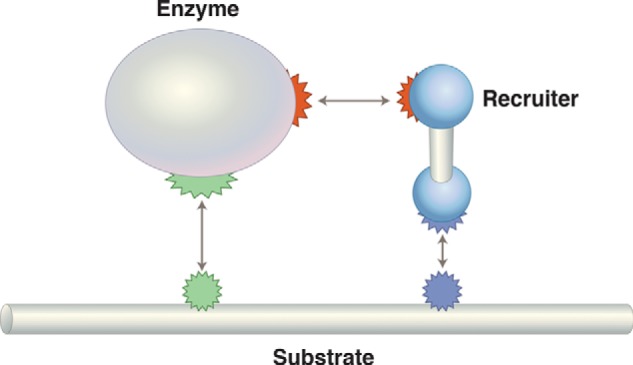
General regulatory scheme for regulation by recruitment. If the enzyme is RNA polymerase, then the recruiter is a transcriptional activator, and the substrate is DNA. To take another case, consider specific ubiquitylation: the enzyme would be an E2 ligase, the recruiter an E3 ligase, and the substrate a specific protein.
This becomes obvious, I think, if we characterize the role of a typical transcriptional activator as a specificity factor: activators impart specificity to RNA polymerase not by changing its inherent enzymatic activity but rather by determining which gene (or genes) will be transcribed under its direction. Thus, rather than evolving multiple RNA polymerases, nature uses essentially just one, and directs its activity to different genes by recruitment. This, of course, is also how specificity of ubiquitylation (and hence, usually of protein degradation) is determined. The recruiting reactions of so-called E3 ligases appose the ubiquitylating enzyme with specific substrates.
In many cases, binding reactions, working cooperatively with one another, play essential roles at every step from signal (e.g. growth hormone) recognition to transcription. Sometimes, the recruitment requires a separate recruiter (the λ repressor, and E3 ligases are examples). In other cases, enzymatic active sites are parts of proteins that bear separate surfaces that direct the enzymatic activity to a particular substrate constitutively or upon command (e.g. upon phosphorylation). There is a good reason for this common regulatory strategy: it is rather easy to evolve (select) new specificities. All you need to do, for example, is change surface residues on recruiters to give them new specificities or fuse an enzymatic activity to a new recruiting domain. As I mentioned above, it is not hard to see that the binding reactions comprising the λ switch could have been constantly improved by small evolutionary add-ons (2, 94–96).
Coherency
I mentioned at the outset of this article that, as we focused on mechanistic details, we sought coherent explanations and abstractions that would apply to apparently disparate cases. The extent to which one searches for this kind of coherency is, I guess, a matter of taste, and, for any given endeavor, it remains to be seen how appropriate such a bent might be. We had no way of knowing, at the start, that studying the λ repressor and its action would yield a coherent picture of a regulatory switch and even less indication that the principles of protein-DNA interaction and gene regulation, gleaned from the λ studies, would apply even in eukaryotes.
It turns out that the very simplicity of the design and mechanism of action of transcriptional regulators makes it easy to see how natural variation can throw up many regulatory circuit options for natural selection to consider. It is not surprising, now, to see diagrams of gene regulatory circuits in higher eukaryotes that look like “a lot of λs” (97). We see regulatory proteins everywhere, usually working cooperatively, turning on sets of genes. Some of these genes encode inhibitors that block the effects of those or other activators, and so on. Once established, positive feedback loops maintain states of gene expression, including in differentiated cells, unless/until perturbed (e.g. by a signal), allowing the system to move on to the next phase of gene expression. Perhaps evolution is the key: selection must occur in small steps, and the simplest mechanism that works will be used over and over again, sometimes in so many guises that the underlying similarities are at first hard to see.
Specificity and Memory
Two intertwined words summarize a great many aspects of gene regulation: specificity and memory. The former can be imposed by specific DNA-binding proteins, and the latter is a systems property. “Systems” sounds fancy, but it is conceptually simple. As I have described, where the activated gene encodes the activator itself, we have memory: a self-perpetuating state of gene expression transmitted by regulatory proteins distributed to daughter cells as cells divide.
These now obvious ideas seem to be hard to accept for some. Ignoring the specificity problem and in the search for some alternative solution to the memory problem, they have created an incoherent and counterfactual world, one in which chromatin structure determines the activity of transcription factors (recruiters) rather than the other way around. Chromatin structure is usually meant to imply histone modifications, which somehow have acquired the name epigenetic modifications. The literature is replete with studies of histone modifications presented as studies of “epigenetics.”
As I and others have pointed out elsewhere (39, 40, 98–100) and without subsequent contradiction to my knowledge, tests repeatedly have shown that such modifications are not self-perpetuating. Moreover, the idea that such modifications regulate gene expression is incoherent. The enzymes that impose such modifications lack the requisite specificity: every nucleosome (and hence, every gene) looks the same. As you would expect from the principles discussed in this article, both the establishment and maintenance of such modifications for any given case (for every case studied, to my knowledge) require the action of specificity determinants in the form of recruiters, usually proteins and in some cases RNA molecules. Take away the specificity determinants, and the modifications go away.
The modifications, where they might be relevant, like the many other events occurring associated with gene transcription, are an effect, not a cause, of the action of recruiters. I add the words “where they might be relevant” because it remains to be seen for many of these modifications whether histones are even the physiologically important substrates of the modifying enzymes. For example, it has been recently shown that two histone methylases have, in fact, a broad array of substrates in vivo, and which is relevant remains to be seen (101).
I find that discussing this matter is not entirely straightforward because sometimes it is hard to be sure what authors mean to say. Consider this recent example from a “Perspectives” article (published in the journal Science) that provides an overview of findings described in a research article in the same issue: “DNA variants influence a layer of gene regulation called epigenetics through the sequence-specific activity of transcription factors” (102). What, one wonders, do these authors mean by “a layer of gene regulation called epigenetics?” I think they mean “histone modifications,” but why use the word epigenetics? What do they mean by “influence,” and what would happen absent this influence?
In a bizarre twist, one of the papers being previewed by this “Perspectives” article comes to the cogent conclusion that (even in human cells!) “transcription factors mediate sequence-specific gene regulation, with histone modifications reflecting their activities” (103). Note that the authors of the research article never used the word epigenetic but rather explain in direct terms what they mean.
Another example is a recent review article in the journal Nature Review Genetics (104), which focuses on “mechanisms of cellular reprogramming mediated by transcription factors.” A promising start, as we know of numerous examples of cellular reprogramming (105) effected by forced expression of transcription factors (i.e. specific DNA-binding proteins). What, then, are we to make of the very first highlighted definition: “Epigenome: Heritable changes in chromatin such as histone modifications …that affect gene expression?” Do the authors mean to say that such modifications are self-perpetuating, as this definition seems to imply? Do they mean to say that such modifications determine gene expression? Maybe not, but then what is their image of how genes are regulated? And so on. Muddy waters can be deep, but often are not.
What Is Fundamental?
I hope that one point illustrated by this essay is that one can make ever broader generalizations by solving basic problems, sometimes in near-fanatical detail, and then seeing where those solutions can lead. That is as opposed to looking at problems in general, but even that description seems to miss an important aspect of this kind of science. No part of the world can simply be read; it always must be interpreted, and those interpretations are subject to constant re-evaluation. For example, only in retrospect did we realize the importance of the fact that DNA-binding regulators do not, need not, change DNA structure to work. It is true that there are many kinds of DNA-binding domains and that, if examined closely enough, some may have detectable effects on DNA structure, but the function selected in evolution was positioning, location on DNA. Exactly how that is accomplished does not matter. Nature uses (selects) whatever is around the kitchen, so long as it does the job. However, this was not immediately obvious. What is fundamental often emerges only in retrospect. I love Karl Popper's remark: “Basic models tell us more than we can at first know.” Animations illustrating some of the points of this paper can be found at http://www.mskcc.org/research/lab/mark-ptashne.
Acknowledgments
I thank Alex Gann, Wally Gilbert, Max Gottesman, Ann Hochschild, Sandy Johnson, Tom Maniatis, and members of my lab for comments.
Footnotes
S. Narayan and M. Ptashne, unpublished data.
REFERENCES
- 1. Ptashne M. (2004) A Genetic Switch: Phage Lambda Revisited, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Ptashne M., Gann A. (2002) Genes & Signals, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3. Ptashne M. (2013) François Jacob (1920–2013). Cell 153, 1180–1182 [DOI] [PubMed] [Google Scholar]
- 4. Ptashne M. (1967) Isolation of the λ phage repressor. Proc. Natl. Acad. Sci. U.S.A. 57, 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ptashne M. (1967) Specific binding of the lambda phage repressor to lambda DNA. Nature 214, 232–234 [DOI] [PubMed] [Google Scholar]
- 6. Pirrotta V., Ptashne M. (1969) Isolation of the 434 phage repressor. Nature 222, 541–544 [DOI] [PubMed] [Google Scholar]
- 7. Steinberg R. A., Ptashne M. (1971) In vitro repression of RNA synthesis by purified lambda phage repressor. Nat. New Biol. 230, 76–80 [DOI] [PubMed] [Google Scholar]
- 8. Gilbert W., Müller-Hill B. (1966) Isolation of the Lac repressor. Proc. Natl. Acad. Sci. U.S.A. 56, 1891–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilbert W., Müller-Hill B. (1967) The lac operator is DNA. Proc. Natl. Acad. Sci. U.S.A. 58, 2415–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maniatis T., Ptashne M. (1973) Multiple repressor binding at the operators in bacteriophage λ. Proc. Natl. Acad. Sci. U.S.A. 70, 1531–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson W. F., Ohlendorf D. H., Takeda Y., Matthews B. W. (1981) Structure of the cro repressor from bacteriophage λ and its interaction with DNA. Nature 290, 754–758 [DOI] [PubMed] [Google Scholar]
- 12. Pabo C. O., Lewis M. (1982) The operator-binding domain of λ repressor: structure and DNA recognition. Nature 298, 443–447 [DOI] [PubMed] [Google Scholar]
- 13. Aggarwal A. K., Rodgers D. W., Drottar M., Ptashne M., Harrison S. C. (1988) Recognition of a DNA operator by the repressor of phage 434: a view at high resolution. Science 242, 899–907 [DOI] [PubMed] [Google Scholar]
- 14. Wharton R. P., Ptashne M. (1985) Changing the binding specificity of a repressor by redesigning an α-helix. Nature 316, 601–605 [DOI] [PubMed] [Google Scholar]
- 15. Sauer R. T., Yocum R. R., Doolittle R. F., Lewis M., Pabo C. O. (1982) Homology among DNA-binding proteins suggests use of a conserved super-secondary structure. Nature 298, 447–451 [DOI] [PubMed] [Google Scholar]
- 16. Laughon A., Scott M. P. (1984) Sequence of a Drosophila segmentation gene: protein structure homology with DNA-binding proteins. Nature 310, 25–31 [DOI] [PubMed] [Google Scholar]
- 17. Chadwick P., Pirrotta V., Steinberg R., Hopkins N., Ptashne M. (1970) The λ and 434 phage repressors. Cold Spring Harb. Symp. Quant. Biol. XXXV, 283–294 [Google Scholar]
- 18. Pirrotta V., Chadwick P., Ptashne M. (1970) Active form of two coliphage repressors. Nature 227, 41–44 [DOI] [PubMed] [Google Scholar]
- 19. Maniatis T., Ptashne M. (1973) Structure of the λ operators. Nature 246, 133–136 [DOI] [PubMed] [Google Scholar]
- 20. Maniatis T., Ptashne M., Backman K., Kield D., Flashman S., Jeffrey A., Maurer R. (1975) Recognition sequences of repressor and polymerase in the operators of bacteriophage λ. Cell 5, 109–113 [DOI] [PubMed] [Google Scholar]
- 21. Maniatis T., Ptashne M., Barrell B. G., Donelson J. (1974) Sequence of a repressor-binding site in the DNA of bacteriophage λ. Nature 250, 394–397 [DOI] [PubMed] [Google Scholar]
- 22. Hopkins N., Ptashne M. (1971) Genetics of virulence. in The Bacteriophage Lambda (Hershey A. D., ed) Vol. 2, pp. 571–574, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 23. Johnson A. D., Meyer B. J., Ptashne M. (1979) Interactions between DNA-bound repressors govern regulation by the λ phage repressor. Proc. Natl. Acad. Sci. U.S.A. 76, 5061–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pabo C. O., Sauer R. T., Sturtevant J. M., Ptashne M. (1979) The lambda repressor contains two domains. Proc. Natl. Acad. Sci. U.S.A. 76, 1608–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sauer R. T., Pabo C. O., Meyer B. J., Ptashne M., Backman K. C. (1979) Regulatory functions of the λ repressor reside in the amino-terminal domain. Nature 279, 396–400 [DOI] [PubMed] [Google Scholar]
- 26. Whipple F. W., Kuldell N. H., Cheatham L. A., Hochschild A. (1994) Specificity determinants for the interaction of λ repressor and P22 repressor dimers. Genes Dev. 8, 1212–1223 [DOI] [PubMed] [Google Scholar]
- 27. Whipple F. W., Hou E. F., Hochschild A. (1998) Amino acid-amino acid contacts at the cooperativity interface of the bacteriophage λ and P22 repressors. Genes Dev. 12, 2791–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burz D. S., Ackers G. K. (1994) Single-site mutations in the C-terminal domain of bacteriophage λ cI repressor alter cooperative interactions between dimers adjacently bound to OR. Biochemistry 33, 8406–8416 [DOI] [PubMed] [Google Scholar]
- 29. Hochschild A., Ptashne M. (1986) Cooperative binding of λ repressors to sites separated by integral turns of the DNA helix. Cell 44, 681–687 [DOI] [PubMed] [Google Scholar]
- 30. Griffith J., Hochschild A., Ptashne M. (1986) DNA loops induced by cooperative binding of λ repressor. Nature 322, 750–752 [DOI] [PubMed] [Google Scholar]
- 31. Eisen H., Brachet P., Pereira da Silva L., Jacob F. (1970) Regulation of repressor expression in λ. Proc. Natl. Acad. Sci. U.S.A. 66, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heinemann S. F., Spiegelman W. G. (1970) Control of transcription of the repressor gene in bacteriophage λ. Proc. Natl. Acad. Sci. U.S.A. 67, 1122–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reichardt L., Kaiser A. D. (1971) Control of λ repressor synthesis. Proc. Natl. Acad. Sci. U.S.A. 68, 2185–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reichardt L. F. (1975) Control of bacteriophage lambda repressor synthesis: regulation of the maintenance pathway of the cro and cI products. J. Mol. Biol. 93, 289–309 [DOI] [PubMed] [Google Scholar]
- 35. Guarente L., Nye J. S., Hochschild A., Ptashne M. (1982) Mutant λ phage repressor with a specific defect in its positive control function. Proc. Natl. Acad. Sci. U.S.A. 79, 2236–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hochschild A., Irwin N., Ptashne M. (1983) Repressor structure and the mechanism of positive control. Cell 32, 319–325 [DOI] [PubMed] [Google Scholar]
- 37. Meyer B. J., Ptashne M. (1980) Gene regulation at the right operator (OR) of bacteriophage λ: III. λ repressor directly activates gene transcription. J. Mol. Biol. 139, 195–205 [DOI] [PubMed] [Google Scholar]
- 38. Bushman F. D., Ptashne M. (1988) Turning λ Cro into a transcriptional activator. Cell 54, 191–197 [DOI] [PubMed] [Google Scholar]
- 39. Ptashne M. (2007) On the use of the word ‘epigenetic’. Curr. Biol. 17, R233–R236 [DOI] [PubMed] [Google Scholar]
- 40. Ptashne M. (2013) Epigenetics: core misconcept. Proc. Natl. Acad. Sci. U.S.A. 110, 7101–7103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neubauer Z., Calef E. (1970) Immunity phase-shift in defective lysogens: non-mutational hereditary change of early regulation of λ prophage. J. Mol. Biol. 51, 1–13 [DOI] [PubMed] [Google Scholar]
- 42. Lin L. L., Little J. W. (1989) Autodigestion and RecA-dependent cleavage of Ind− mutant LexA proteins. J. Mol. Biol. 210, 439–452 [DOI] [PubMed] [Google Scholar]
- 43. Dodd I. B., Perkins A. J., Tsemitsidis D., Egan J. B. (2001) Octamerization of λ CI repressor is needed for effective repression of PRM and efficient switching from lysogeny. Genes Dev. 15, 3013–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dodd I. B., Shearwin K. E., Perkins A. J., Burr T., Hochschild A., Egan J. B. (2004) Cooperativity in long-range gene regulation by the λ CI repressor. Genes Dev. 18, 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schmeissner U., Court D., Shimatake H., Rosenberg M. (1980) Promoter for the establishment of repressor synthesis in bacteriophage λ. Proc. Natl. Acad. Sci. U.S.A. 77, 3191–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hochschild A., Douhan J., 3rd, Ptashne M. (1986) How λ repressor and λ Cro distinguish between OR1 and OR3. Cell 47, 807–816 [DOI] [PubMed] [Google Scholar]
- 47. Meyer B. J., Maurer R., Ptashne M. (1980) Gene regulation at the right operator (OR) of bacteriophage λ: II. OR1, OR2, and OR3: their roles in mediating the effects of repressor and cro. J. Mol. Biol. 139, 163–194 [DOI] [PubMed] [Google Scholar]
- 48. Johnson A., Meyer B. J., Ptashne M. (1978) Mechanism of action of the cro protein of bacteriophage lambda. Proc. Natl. Acad. Sci. U.S.A. 75, 1783–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Svenningsen S. L., Costantino N., Court D. L., Adhya S. (2005) On the role of Cro in λ prophage induction. Proc. Natl. Acad. Sci. U.S.A. 102, 4465–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johnson A. D., Poteete A. R., Lauer G., Sauer R. T., Ackers G. K., Ptashne M. (1981) λ Repressor and cro–components of an efficient molecular switch. Nature 294, 217–223 [DOI] [PubMed] [Google Scholar]
- 51. Little J. W. (2010) Evolution of complex gene regulatory circuits by addition of refinements. Curr. Biol. 20, R724–R734 [DOI] [PubMed] [Google Scholar]
- 52. Hershey A. D. (ed) (1971) in The Bacteriophage Lambda, Vol. 2, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 53. Hendrix R. W., Roberts J. W., Stahl F. W., Weisberg R. A. (eds) (1983) Lambda II, Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 54. Ptashne M. (2007) On learning to write. Curr. Biol. 17, R394–R395 [DOI] [PubMed] [Google Scholar]
- 55. Ptashne M. (2007) On speaking, writing and inspiration. Curr. Biol. 17, R348–R349 [DOI] [PubMed] [Google Scholar]
- 56. Giniger E., Varnum S. M., Ptashne M. (1985) Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 40, 767–774 [DOI] [PubMed] [Google Scholar]
- 57. Guarente L., Yocum R. R., Gifford P. (1982) A GAL10-CYC1 hybrid yeast promoter identifies the GAL4 regulatory region as an upstream site. Proc. Natl. Acad. Sci. U.S.A. 79, 7410–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keegan L., Gill G., Ptashne M. (1986) Separation of DNA binding from the transcription-activating function of a eukaryotic regulatory protein. Science 231, 699–704 [DOI] [PubMed] [Google Scholar]
- 59. Brent R., Ptashne M. (1985) A eukaryotic transcriptional activator bearing the DNA specificity of a prokaryotic repressor. Cell 43, 729–736 [DOI] [PubMed] [Google Scholar]
- 60. Fischer J. A., Giniger E., Maniatis T., Ptashne M. (1988) GAL4 activates transcription in Drosophila. Nature 332, 853–856 [DOI] [PubMed] [Google Scholar]
- 61. Kakidani H., Ptashne M. (1988) GAL4 activates gene expression in mammalian cells. Cell 52, 161–167 [DOI] [PubMed] [Google Scholar]
- 62. Ma J., Przibilla E., Hu J., Bogorad L., Ptashne M. (1988) Yeast activators stimulate plant gene expression. Nature 334, 631–633 [DOI] [PubMed] [Google Scholar]
- 63. Ma J., Ptashne M. (1988) Converting a eukaryotic transcriptional inhibitor into an activator. Cell 55, 443–446 [DOI] [PubMed] [Google Scholar]
- 64. Fields S., Song O. (1989) A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 [DOI] [PubMed] [Google Scholar]
- 65. Himmelfarb H. J., Pearlberg J., Last D. H., Ptashne M. (1990) GAL11P: a yeast mutation that potentiates the effect of weak GAL4-derived activators. Cell 63, 1299–1309 [DOI] [PubMed] [Google Scholar]
- 66. Barberis A., Pearlberg J., Simkovich N., Farrell S., Reinagel P., Bamdad C., Sigal G., Ptashne M. (1995) Contact with a component of the polymerase II holoenzyme suffices for gene activation. Cell 81, 359–368 [DOI] [PubMed] [Google Scholar]
- 67. Wang X., Muratani M., Tansey W. P., Ptashne M. (2010) Proteolytic instability and the action of nonclassical transcriptional activators. Curr. Biol. 20, 868–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Farrell S., Simkovich N., Wu Y., Barberis A., Ptashne M. (1996) Gene activation by recruitment of the RNA polymerase II holoenzyme. Genes Dev. 10, 2359–2367 [DOI] [PubMed] [Google Scholar]
- 69. Gill G., Ptashne M. (1988) Negative effect of the transcriptional activator GAL4. Nature 334, 721–724 [DOI] [PubMed] [Google Scholar]
- 70. Kelleher R. J., 3rd, Flanagan P. M., Kornberg R. D. (1990) A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 61, 1209–1215 [DOI] [PubMed] [Google Scholar]
- 71. Triezenberg S. J., LaMarco K. L., McKnight S. L. (1988) Evidence of DNA: protein interactions that mediate HSV-1 immediate early gene activation by VP16. Genes Dev. 2, 730–742 [DOI] [PubMed] [Google Scholar]
- 72. Ma J., Ptashne M. (1987) A new class of yeast transcriptional activators. Cell 51, 113–119 [DOI] [PubMed] [Google Scholar]
- 73. Brzovic P. S., Heikaus C. C., Kisselev L., Vernon R., Herbig E., Pacheco D., Warfield L., Littlefield P., Baker D., Klevit R. E., Hahn S. (2011) The acidic transcription activator Gcn4 binds the Mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol. Cell 44, 942–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Herbig E., Warfield L., Fish L., Fishburn J., Knutson B. A., Moorefield B., Pacheco D., Hahn S. (2010) Mechanism of Mediator recruitment by tandem Gcn4 activation domains and three Gal11 activator-binding domains. Mol. Cell. Biol. 30, 2376–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reeves W. M., Hahn S. (2005) Targets of the Gal4 transcription activator in functional transcription complexes. Mol. Cell. Biol. 25, 9092–9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wu Y., Reece R. J., Ptashne M. (1996) Quantitation of putative activator-target affinities predicts transcriptional activating potentials. EMBO J. 15, 3951–3963 [PMC free article] [PubMed] [Google Scholar]
- 77. Giniger E., Ptashne M. (1987) Transcription in yeast activated by a putative amphipathic α helix linked to a DNA binding unit. Nature 330, 670–672 [DOI] [PubMed] [Google Scholar]
- 78. Ruden D. M., Ma J., Li Y., Wood K., Ptashne M. (1991) Generating yeast transcriptional activators containing no yeast protein sequences. Nature 350, 250–252 [DOI] [PubMed] [Google Scholar]
- 79. Giniger E., Ptashne M. (1988) Cooperative DNA binding of the yeast transcriptional activator GAL4. Proc. Natl. Acad. Sci. U.S.A. 85, 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ruden D. M., Ma J., Ptashne M. (1988) No strict alignment is required between a transcriptional activator binding site and the “TATA box” of a yeast gene. Proc. Natl. Acad. Sci. U.S.A. 85, 4262–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mapp A. K., Ansari A. Z., Ptashne M., Dervan P. B. (2000) Activation of gene expression by small molecule transcription factors. Proc. Natl. Acad. Sci. U.S.A. 97, 3930–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Struhl K. (1999) Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell 98, 1–4 [DOI] [PubMed] [Google Scholar]
- 83. Bryant G. O., Prabhu V., Floer M., Wang X., Spagna D., Schreiber D., Ptashne M. (2008) Activator control of nucleosome occupancy in activation and repression of transcription. PLoS Biol. 6, 2928–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang X., Bryant G. O., Floer M., Spagna D., Ptashne M. (2011) An effect of DNA sequence on nucleosome occupancy and removal. Nat. Struct. Mol. Biol. 18, 507–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Floer M., Wang X., Prabhu V., Berrozpe G., Narayan S., Spagna D., Alvarez D., Kendall J., Krasnitz A., Stepansky A., Hicks J., Bryant G. O., Ptashne M. (2010) A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141, 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Berrozpe G., Bryant G. O., Warpinski K., Ptashne M. (2013) Regulation of a mammalian gene bearing a CpG island promoter and a distal enhancer. Cell Rep. 4, 445–453 [DOI] [PubMed] [Google Scholar]
- 87. Wang X., Bai L., Bryant G. O., Ptashne M. (2011) Nucleosomes and the accessibility problem. Trends Genet. 27, 487–492 [DOI] [PubMed] [Google Scholar]
- 88. Polo J. M., Anderssen E., Walsh R. M., Schwarz B. A., Nefzger C. M., Lim S. M., Borkent M., Apostolou E., Alaei S., Cloutier J., Bar-Nur O., Cheloufi S., Stadtfeld M., Figueroa M. E., Robinton D., Natesan S., Melnick A., Zhu J., Ramaswamy S., Hochedlinger K. (2012) A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wenick A. S., Hobert O. (2004) Genomic cis-regulatory architecture and trans-acting regulators of a single interneuron-specific gene battery in C. elegans. Dev. Cell 6, 757–770 [DOI] [PubMed] [Google Scholar]
- 90. Hammachi F., Morrison G. M., Sharov A. A., Livigni A., Narayan S., Papapetrou E. P., O'Malley J., Kaji K., Ko M. S., Ptashne M., Brickman J. M. (2012) Transcriptional activation by Oct4 is sufficient for the maintenance and induction of pluripotency. Cell Rep. 1, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Madhani H. (2007) From a to α: Yeast as a Model for Cellular Differentiation, Cold Spring Harbor Laboratory Press, Cold Spring Harbor Laboratory Press, NY [Google Scholar]
- 92. Taberlay P. C., Kelly T. K., Liu C. C., You J. S., De Carvalho D. D., Miranda T. B., Zhou X. J., Liang G., Jones P. A. (2011) Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 147, 1283–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ptashne M., Gann A. (1998) Imposing specificity by localization: mechanism and evolvability. Curr. Biol. 8, R897. [DOI] [PubMed] [Google Scholar]
- 94. Ptashne M., Gann A. (2003) Signal transduction. Imposing specificity on kinases. Science 299, 1025–1027 [DOI] [PubMed] [Google Scholar]
- 95. Ptashne M., Gann A. (2001) Transcription initiation: imposing specificity by localization. Essays Biochem. 37, 1–15 [DOI] [PubMed] [Google Scholar]
- 96. Ptashne M., Gann A. (1998) Imposing specificity by localization: mechanism and evolvability. Curr. Biol. 8, R812–R822 [DOI] [PubMed] [Google Scholar]
- 97. Davidson E. H. (2006) The Regulatory Genome: Gene Regulatory Networks In Development And Evolution, 1st Ed., Academic Press, San Diego, CA [Google Scholar]
- 98. Ptashne M. (2013) Faddish stuff: epigenetics and the inheritance of acquired characteristics. FASEB J. 27, 1–2 [DOI] [PubMed] [Google Scholar]
- 99. Ptashne M. (2009) Binding reactions: epigenetic switches, signal transduction and cancer. Curr. Biol. 19, R234–R241 [DOI] [PubMed] [Google Scholar]
- 100. Ptashne M., Hobert O., Davidson E. (2010) Questions over the scientific basis of epigenome project. Nature 464, 487. [DOI] [PubMed] [Google Scholar]
- 101. Islam K., Chen Y., Wu H., Bothwell I. R., Blum G. J., Zeng H., Dong A., Zheng W., Min J., Deng H., Luo M. (2013) Defining efficient enzyme-cofactor pairs for bioorthogonal profiling of protein methylation. Proc. Natl. Acad. Sci. U.S.A. 110, 16778–16783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Furey T. S., Sethupathy P. (2013) Genetics. Genetics driving epigenetics. Science 342, 705–706 [DOI] [PubMed] [Google Scholar]
- 103. Kilpinen H., Waszak S. M., Gschwind A. R., Raghav S. K., Witwicki R. M., Orioli A., Migliavacca E., Wiederkehr M., Gutierrez-Arcelus M., Panousis N. I., Yurovsky A., Lappalainen T., Romano-Palumbo L., Planchon A., Bielser D., Bryois J., Padioleau I., Udin G., Thurnheer S., Hacker D., Core L. J., Lis J. T., Hernandez N., Reymond A., Deplancke B., Dermitzakis E. T. (2013) Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342, 744–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Buganim Y., Faddah D. A., Jaenisch R. (2013) Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 14, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Graf T., Enver T. (2009) Forcing cells to change lineages. Nature 462, 587–594 [DOI] [PubMed] [Google Scholar]