

Background: Lipid asymmetry is an important property of eukaryotic plasma membranes not present in commonly used model membrane vesicles.
Results: PFO insertion in asymmetric model membrane vesicles was distinct from that in symmetric vesicles. A novel transmembrane insertion “intermediate” was identified.
Conclusion: Lipid asymmetry can strongly influence protein behavior.
Significance: This report illustrates a new approach to study proteins in membranes with natural lipid asymmetry.
Keywords: Bacterial Toxins, Cholesterol, Cholesterol-binding Protein, Membrane Bilayer, Membrane Biophysics, Membrane Proteins
Abstract
Eukaryotic membrane proteins generally reside in membrane bilayers that have lipid asymmetry. However, in vitro studies of the impact of lipids upon membrane proteins are generally carried out in model membrane vesicles that lack lipid asymmetry. Our recently developed method to prepare lipid vesicles with asymmetry similar to that in plasma membranes and with controlled amounts of cholesterol was used to investigate the influence of lipid composition and lipid asymmetry upon the conformational behavior of the pore-forming, cholesterol-dependent cytolysin perfringolysin O (PFO). PFO conformational behavior in asymmetric vesicles was found to be distinct both from that in symmetric vesicles with the same lipid composition as the asymmetric vesicles and from that in vesicles containing either only the inner leaflet lipids from the asymmetric vesicles or only the outer leaflet lipids from the asymmetric vesicles. The presence of phosphatidylcholine in the outer leaflet increased the cholesterol concentration required to induce PFO binding, whereas phosphatidylethanolamine and phosphatidylserine in the inner leaflet of asymmetric vesicles stabilized the formation of a novel deeply inserted conformation that does not form pores, even though it contains transmembrane segments. This conformation may represent an important intermediate stage in PFO pore formation. These studies show that lipid asymmetry can strongly influence the behavior of membrane-inserted proteins.
Introduction
In eukaryotic plasma membrane, lipid species are not evenly distributed in the two leaflets of the lipid bilayer. Aminophospholipids (phosphatidylserine and phosphatidylethanolamine) are usually found in the inner (cytosolic) leaflet, whereas phosphatidylcholine and sphingolipids are predominantly located in the outer (exofacial) leaflet facing the external environment around the cell (1, 2). This transverse lipid asymmetry has prominent effects on numerous cellular functions and TM protein topology. For instance, lipid asymmetry determines the difference of lipid charge across the bilayer, with a higher anionic charge at the cytofacial surface of membranes. This may influence membrane protein topography. For example, transmembrane (TM)2 proteins often have a relatively higher positive charge on the cytofacial end of their TM helices, which is known as the positive-inside rule (3). Thus asymmetry may partly explain how TM protein orientation is established during biosynthesis, as well as influence the conformation of TM and cytofacial juxtamembrane sequences (4). Lipid asymmetry may also influence proteins via an effect on lipid packing. The sphingolipid-rich outer leaflet is likely to exist, at least partly, in the tightly packed liquid ordered state, whereas the unsaturated phospholipids of the inner leaflet may predominantly exist in the more loosely packed liquid disordered state. Very recently, a comprehensive comparison of TM domains suggested that TM segments in the plasma membrane have an amino acid asymmetry such that residues embedded in the exofacial leaflet of the bilayer have smaller side chains than in the cytofacial leaflet (5). It was proposed that this allows better packing of TM proteins with the more tightly packed exofacial leaflet of the plasma membrane.
To study the effects of lipid asymmetry on membrane protein structure, the conformational changes of perfringolysin O (PFO) were examined in asymmetric lipid vesicles. PFO is a member of the cholesterol-dependent cytolysin family. After cholesterol-dependent binding to membranes, PFO undergoes oligomerization (involving 30–50 monomers) and membrane insertion to form a TM β-barrel containing a very large, stable pore (6, 7). PFO is an ideal subject for study because its membrane interaction and conformation are highly cholesterol-dependent, and because several methods are available to follow the changes in PFO conformation at each stage of the multistep process by which it inserts into membranes and forms pores. Furthermore, lessons from PFO behavior may be applicable to other cholesterol-dependent cytolysins, which are often important virulence factors in bacterial infections (8–10), and applicable to pore formation by complement proteins and perforins, important proteins in the immune system that have spontaneously membrane-inserting domains structurally very similar to those of the cholesterol-dependent cytolysins (11).
In this report, PFO interaction with membranes was compared in symmetric and asymmetric vesicles. It was found that the lipid composition in each leaflet of the membrane influences the cholesterol threshold for protein binding, insertion, oligomerization, and pore formation, suggesting an important role for lipid asymmetry in lipid-protein interaction. A novel conformation of PFO that is deeply membrane-inserted and has TM segments was identified in asymmetric vesicles. This conformation may be a novel intermediate in the pore formation process.
EXPERIMENTAL PROCEDURES
Materials
1-Palmitoyl-2-oleoyl-phosphatidylcholine(POPC), 1-palmitoyl-2-oleoyl-phosphatidylethanolamine (POPE), 1-palmitoyl-2-oleoyl-phosphatidyl-l-serine (POPS), cholesterol, and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). Lipids were dissolved in chloroform and stored at −20 °C. Concentrations of lipids were measured by dry weight. (2-Hydroxypropyl)-α-cyclodextrin (HPαCD) and methyl-β-cyclodextrin (MβCD) were purchased from Sigma-Aldrich. The labeling reagents 6-acryloyl-2-dimethylaminonaphthalene (acrylodan), N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl iodoacetamide (BODIPY-FL-IA), N-(biotinoyl)-N′-(iodoacetyl)ethylenediamine (biotin-IA), and streptavidin BODIPY-FL conjugate (BOD-SA) were purchased from Invitrogen.
Ordinary (Symmetric) Vesicle Preparation
To prepare multilamellar vesicles, lipid mixtures were mixed and dried under nitrogen followed by high vacuum for at least 1 h. The dried lipid mixtures were then dispersed in PBS (1.8 mm KH2PO4, 10 mm Na2HPO4, 137 mm NaCl, and 2.7 mm KCl at pH 7.4) or PBS adjusted to pH 5.1 with acetic acid at 70 °C to give the desired final concentration and agitated at 55 °C for 15 min using a VWR multitube vortexer (Westchester, PA) placed within a convection oven (GCA Corp., Precision Scientific, Chicago, IL). Samples were cooled to room temperature before use.
Large unilamellar vesicles (LUV) were prepared from multilamellar vesicles by subjecting the multilamellar vesicles to seven cycles of freezing in a mixture of dry ice and acetone and thawing at room temperature and then passed through 100-nm polycarbonate filters (Avanti Polar Lipids) 11 times to obtain LUV of uniform vesicle size. In the case of “acceptor” LUV used to prepare asymmetric vesicles (see below), lipids were dispersed in a solution containing 25% (w/v) sucrose dissolved in water instead of PBS. To wash away the untrapped sucrose, the resulting LUV solutions were mixed with 3.5 ml of PBS and subjected to ultracentrifugation at 190,000 × g for 30 min using Beckman L8–55 M ultracentrifuge and a SW 60 rotor. After the supernatant was discarded, the LUV pellet was resuspended with PBS to the desired concentration (unless otherwise noted, 8 mm, assuming no loss during centrifugation) for further experiments. The size of LUV was measured by dynamic light scattering on a Protein Solutions DynaPro 99 (Wyatt Technology, Santa Barbara, CA), and data were analyzed using the Dynamics V5.25.44 software supplied by Wyatt Technology.
Asymmetric LUV Preparation
Asymmetric large unilamellar vesicles were formed using a protocol adapted from that described by Cheng and London (12). First, 8 μmol of POPC (“donor”) in chloroform were dried under nitrogen and high vacuum for at least 1 h. Then the dried lipids were hydrated with 150 μl of 420 mm HPαCD at 70 °C and diluted with 450 μl of PBS. This donor mixture was vortexed in the multitube vortexer at 55 °C overnight. The next day, the mixture was sonicated in a bath sonicator (Special Ultrasonic Cleaner model G1112SP1; Laboratory Supplies Co., Hicksville, NY) at room temperature for 15 min before adding 500 μl of 8 mm (total lipid) acceptor POPE/POPS/cholesterol LUVs (with entrapped 25% (w/v) sucrose; see above). The POPE:POPS ratio was 1:1 mol:mol. The amount of cholesterol used in the acceptor vesicles was varied between 0 and 50 mol % depending on the specific experiment. The mixture of donor POPC/HPαCD and acceptor POPE/POPS/cholesterol LUVs was vortexed at 55 °C for 30 min, overlaid onto 3 ml of a 10% (w/v) sucrose solution, and then subjected to ultracentrifugation at 190,000 × g for 30 min at a setting of 25 °C. After the supernatant was removed, the resulting pellet was resuspended with 1 ml of 10% (w/v) sucrose solution, overlaid onto 3 ml of a 10% (w/v) sucrose solution, and the ultracentrifugation step was repeated. The final pellet was resuspended in PBS and immediately used for further experiments.
Based on high performance TLC analysis, the POPC content as a percentage of total phospholipid in the exchange vesicles was ∼50% for the entire range (0–50 mol %) of cholesterol contents. In addition, two independent assays were carried out to assay lipid asymmetry in the exchange vesicles. The binding of the cationic peptide pL4A18 (acetyl-K2LA9LWLA9LK2-amide) was used to detect the extent of residual anionic lipid (POPS in this case) on the outer leaflet of exchange vesicles (13), and chemical labeling by trinitrobenzensulfonate was used to detect the residual POPE on the outer leaflet of exchange vesicles (14–16). Both assays revealed that the POPCo/POPE:POPSi/cholesterol exchange vesicles (exchange vesicle names have the format Xo/Yi/cholesterol, where Xo designates the original donor lipid(s), which should be in the outer leaflet, and Yi designates the original acceptor vesicle lipid(s), which should be in the inner leaflet) are highly asymmetric, with ∼80–90% of phospholipids in the outer leaflet being exchanged with POPC (16). Because HPαCD does not bind to cholesterol, the cholesterol content of the exchange vesicles was equivalent to that of the acceptor vesicles prior to exchange (16). By measurement of the intensity of lipid bands on high performance TLC relative to lipid standards, the lipid yield from these preparations was found to be generally ∼1 μmol. In cases in which lipid concentration was not explicitly measured, 1 μmol was assumed as the lipid yield per preparation unless otherwise noted.
Vesicle Scrambling
To prepare symmetric vesicles with the same lipid composition as asymmetric vesicles, a lipid scrambling process was carried out similarly as previously described (12). For most experiments ∼ 0.4 μmol of concentrated asymmetric LUVs (typically 80 μl from the asymmetric preparation pellet suspended in 200 μl of PBS) were dried by a nitrogen stream. Next 200 μl of ethanol were added to dissolve the lipids. After the ethanol was dried by nitrogen stream and high vacuum for 1 h, 200 μl of PBS was added to hydrate the lipids at 70 °C. The samples were then subjected to seven cycles of freeze-thawing in dry ice/acetone. The final samples were divided into four aliquots each with ∼ 0.1 μmol of lipid.
Fluorescence Intensity Measurements
Fluorescence emission intensity was measured (unless otherwise noted) at room temperature on a SPEX Fluorolog 3 spectrofluorimeter. For fixed wavelength measurements, the excitation/emission wavelength sets used were 280 nm/340 nm for tryptophan, 488 nm/515 nm for BOD-SA, and 490 nm/510 nm for BODIPY-labeled PFO. For acrylodan-labeled PFO emission spectra measurements, samples were excited at 350 nm, and emission spectra were acquired from 420–560 nm. Unless otherwise noted, fluorescence intensity in single background samples lacking fluorophore was subtracted from the reported data.
Purification and Fluorescent Labeling of PFO
A functional Cys-less derivative of wild type PFO (PFO C459A) and a Cys-less prepore mutant of PFO (PFO C459A Y181A) were kind gifts of A. Heuck (University of Massachusetts at Amherst). PFO mutants (PFO C459A/A215C WT, PFO C459A/Y181A/A215C “prepore,” PFO C459A/V214C, and PFO C459A/E204C) with Cys substitutions were generated by QuikChange site-directed mutagenesis (Stratagene) (17). PFO was expressed in Escherichia coli BL21(DE3)pLysS and purified similarly as described previously (18). Protein concentration was determined from absorbance at 280 nm using an extinction coefficient of 74,260 m−1cm−1. Labeling of PFO at Cys-214 or Cys-215 with acrylodan or BODIPY-FL was also carried out similarly as described previously (17). Biotinylation of PFO at Cys-204 was carried out by mixing protein and biotin-IA at 1:6 mole ratio. After 2 h of incubation at room temperature, the reaction mixture was applied to a Sephadex G-50 column to separate biotinylated protein and free biotin-IA similar as described previously (17).
Measurement of PFO Vesicle Binding by Tryptophan Fluorescence
The ability of PFO to associate with lipid vesicles and interact with cholesterol was assessed by measuring the increase in intrinsic Trp emission intensity, which occurs when Trp residues located at the tip of domain 4 come to contact with cholesterol-containing membranes (19). A small aliquot of PFO was added to 1 ml of symmetric, asymmetric, or scrambled LUV suspension containing ∼100 μm total lipids in PBS pH 5.1. The final PFO concentration was 55 nm. After 1 h incubation at room temperature, Trp emission intensity was measured as described above.
Measurement of Vesicle Insertion by Acrylodan Fluorescence
PFO insertion into membranes was monitored by the changes of acrylodan fluorescence intensity of PFO labeled with acrylodan on Cys-215 as previously described (17). Residue 215 becomes buried in the bilayer when PFO inserts into the bilayer and form TM β-hairpins (20). To do this, a small aliquot of acrylodan-labeled PFO was added to 1 ml of symmetric, asymmetric, or scrambled LUV suspension containing ∼100 μm total lipids in PBS, pH 5.1. The final acrylodan-labeled PFO concentration was 25 nm. After 1-h incubation at room temperature, acrylodan emission spectra were measured as described above.
Measurement of PFO Oligomerization by FRET
To measure PFO oligomerization in membranes, FRET was used. The donor was PFO acrylodan-labeled on residue 215, and the acceptor was PFO BODIPY-labeled on residue 215. An equimolar mixture of donor- and acceptor-labeled PFO (30 nm each; “F” sample) was incubated with 1 ml of symmetric, asymmetric, or scrambled LUV suspension containing ∼100 μm total lipids in PBS, pH 5.1. In “Fo” samples, equal amount of unlabeled PFO replaced the acceptor-labeled PFO. “Blank/background” samples contained 60 nm unlabeled PFO in place of labeled PFO. After 1-h incubation of samples at room temperature, acrylodan emission intensity was measured with an excitation wavelength at 350 nm and an emission wavelength at 450 nm. FRET exaggerates oligomerization because it mainly detects oligomerization of the bound molecules. The reason for this is that acrylodan is much more fluorescent when membrane-inserted than when in solution.
Assay for Pore Formation
PFO-induced pore formation was measured as previously described by assaying the reaction of vesicle-trapped biocytin with externally added BOD-SA via the increase in the BODIPY fluorescence emission intensity upon binding of biocytin that escapes from vesicles to BOD-SA located in the external solution (18). In these experiments, BOD-SA (10 μl from the 1 μm stock solution in terms of streptavidin tetramers dissolved in PBS) was added externally to vesicles with 537 μm internal concentration entrapped biocytin that had been diluted to 1 ml with PBS, pH 5.1 (∼100 μm total lipid). BODIPY emission intensity was then measured at room temperature. PFO was added to a concentration of 185 nm. Samples were briefly mixed, and then BODIPY intensity was monitored as a function of time for up to 30 min at room temperature.
Oligomerization Assay by SDS-Agarose Gel Electrophoresis
First, 20 μl of symmetric, asymmetric or scrambled LUV suspension containing ∼100 μm total lipids was incubated with 10 μg of PFO for 1 h at room temperature. Then samples were solubilized with 5 μl of SDS loading buffer (40% glycerol (v/v), 25% SDS (w/v), and 0.1% bromphenol blue (w/v)) and analyzed using denaturing SDS-agarose gel electrophoresis as described previously (18).
Interaction of Biotinylated PFO E204C with Externally Added and Vesicle-trapped BOD-SA
Symmetric vesicles composed of POPE/POPS/cholesterol containing trapped BOD-SA were prepared as described previously (21, 22). A 1-ml mixture containing 10 mm lipid composed of 1:1 POPE/POPS with various amounts of cholesterol, 0.002 mol % Rho-DOPE, plus 70 μg/ml BOD-SA, and 20 mg/ml n-octyl-β-glucoside was dissolved in PBS, pH 7.4. After removing n-octyl-β-glucoside by dialysis overnight against 4 liters of PBS at 4 °C, mixtures containing trapped protein were applied to a Sepharose CL-4B column to separate free from LUV-entrapped BOD-SA. Fractions containing LUV-entrapped BOD-SA were collected, and the final concentration of BOD-SA and lipids were determined by measurement of the BODIPY and Rho-DOPE fluorescence, respectively. Asymmetric vesicles containing trapped BOD-SA were prepared by dispersing the acceptor lipids (POPE/POPS/cholesterol) in a solution containing 25% sucrose and 70 μg/ml BOD-SA. Lipid exchange and ultracentrifugation were the same as described above.
To measure biotinylated PFO E204C interaction with vesicle-trapped BOD-SA, LUVs containing trapped BOD-SA were diluted with PBS, pH 5.1, to give a concentration of 0.1 μg/ml BOD-SA (which gave a lipid concentration roughly in the range 100–200 μm). BODIPY fluorescence was measured, and then a small aliquot of concentrated biotinylated PFO E204C was added to give a final protein concentration of 50 nm. After incubation for 30 min, BODIPY fluorescence was remeasured.
To measure biotinylated PFO E204C interaction with externally added BOD-SA, BODIPY fluorescence was first measured in a 950-μl sample containing 0.1 μg/ml BOD-SA in PBS, pH 5.1. Then 50 μl of the appropriate “biotinylated PFO E204C-LUV” mixture was added, and fluorescence was measured after an incubation of 30 min at room temperature. The “biotinylated PFO E204C-LUV” mixture contained biotinylated PFO E204C preincubated with LUVs from a symmetric vesicle preparation or from a concentrated asymmetric LUV preparation so as to give the same final PFO and lipid compositions and concentrations as the samples with trapped BOD-SA.
To evaluate the fraction of the exposure of the biotinylated E204C facing the lumen of the vesicles, the percentage of internal reactivity was calculated. The percentage internal reactivity = 100% × (F/Fo − 1)in/[(F/Fo − 1)ex + (F/Fo − 1)in], where Fo is the BOD-SA fluorescence intensity prior to the addition of biotinylated PFO E204C to the internally (in) or externally (ex) located BOD-SA containing solution, and F is the BOD-SA fluorescence intensity 30 min after incubation of biotinylated PFO E204C with BOD-SA. The percentage of internal reactivity should be equivalent to the percentage of biotin residues exposed to the internal solution (aqueous lumen) of the vesicles if biotin is equally reactive with BOD-SA in the internally exposed and externally exposed conformations and BOD-SA is in excess. Although we used a biotinylated PFO concentration higher than that of BOD-SA, the increase in BODIPY fluorescence was never over 50%, consistent with excess BOD-SA (22). This probably reflects incomplete labeling of PFO by biotin. In all cases, fluorescence intensities used were the values after background intensities from samples lacking BOD-SA were subtracted.
Measurement of Vesicle Association by Centrifugation
The ability of WT and prepore PFO to associate with vesicles was assessed by ultracentrifugation. Asymmetric vesicles (∼0.1 μmol of lipid, containing 0.01 mol % Rho-DOPE) were prepared in 1 ml of PBS, pH 5.1, and incubated with a small aliquot of BODIPY-labeled PFO A215C for 1 h at room temperature. The final BODIPY-labeled PFO concentration was 25 nm. To extract cholesterol from vesicles, 20 μl of a 0.5 m MβCD stock was added to the above solutions to give a final 10 mm MβCD concentration, and samples were incubated for 30 min at room temperature. Samples with or without MβCD were then spun for 30 min in a Beckman L8–85 ultracentrifuge at 84,000 × g at 4 °C. After spinning, supernatant containing the unbound PFO was removed, and pellet containing the vesicles and bound PFO was resuspended in 1 ml of PBS, pH 5.1. Then BODIPY and rhodamine fluorescence was measured for both supernatant and pellet. Vesicle pelleting was efficient as judged by the rhodamine fluorescence in the pellet. It should be noted that the values shown for the ratio of the percentage membrane-bound PFO in the presence of MβCD to the percentage membrane bound PFO in the absence of MβCD have not been corrected for the small change in BODIPY fluorescence upon PFO binding to vesicles containing cholesterol. There is an increase of 30% in BODIPY fluorescence for prepore (Y181A/A215C) PFO and a decrease of 20% for WT PFO upon binding to vesicles. However, corrected values are almost identical to uncorrected values (average difference between corrected and uncorrected was 0.02).
Quenching Experiments to Measure Membrane Penetration by Acrylodan-labeled Cysteine Residues
To measure the penetration of PFO TM segments into the bilayer, quenching of acrylodan fluorescence by 10-doxylnonadecane (10-DN) was measured. Samples were prepared with 25 nm acrylodan-labeled PFO incorporated into 1 ml of symmetric vesicles (100 μm lipid) or asymmetric vesicles (∼100 μm lipid) in PBS, pH 5.1. Vesicles in F samples had an additional 5 mol % 10-DN mixed with the lipids prior to vesicle formation. Vesicles in Fo samples contained no 10-DN. The ratio of acrylodan fluorescence in samples containing 10-DN to that in samples lacking 10-DN was then calculated. Acrylodan fluorescence intensity was measured with an excitation wavelength at 350 nm and an emission wavelength at 450 nm. Fluorescence intensities were corrected for fluorescence intensities in background samples lacking protein. F/Fo values were corrected for nonspecific quenching using the quenching of acrylodan-labeled glutathione in the presence of POPE/POPS LUV in PBS, pH 5.1.
RESULTS
Behavior of PFO Is Different in Asymmetric and Symmetric Vesicles
The biological question we wished to answer is whether lipid asymmetry of the type seen in mammalian membranes affects the conformation of membrane proteins, taking advantage of the ability to prepare asymmetric vesicles with controlled cholesterol concentration. To do this, we compared the conformational properties of PFO in symmetric and asymmetric vesicles. In sequential steps, PFO binds to cholesterol, oligomerizes, inserts deeply into lipid bilayers, and finally converts into a pore-forming state containing a TM β-barrel (6). PFO is a good subject for studies of the effect of lipid asymmetry and cholesterol because both its binding to membranes and the series of membrane-induced conformational changes that it undergoes require cholesterol and are strongly influenced by the overall lipid composition of the membrane (18, 23–25).
Furthermore, there are good assays for many aspects of PFO-membrane interaction, which makes it possible to define the effect of asymmetry upon several aspects of the insertion process. Interaction of PFO with liposomal vesicles was examined using the increase of PFO Trp emission intensity observed upon binding to cholesterol (19). PFO insertion into the lipid bilayer was detected by the membrane penetration of residue 215 on TM hairpin 1 (TMH1). Upon insertion, the fluorescence properties of labels attached to residue 215 are greatly altered (26), and this has been extensively used to detect the insertion of the PFO β-barrel (17, 23, 27–29). We followed the insertion of acrylodan-labeled A215C PFO by the large increase in acrylodan fluorescence and large blue shift in the λmax of acrylodan emission, upon insertion, and by fluorescence quenching methods (see below). PFO oligomerization was assayed by FRET from PFO containing acrylodan-labeled residue 215 to PFO containing BODIPY-labeled residue 215. PFO-induced pore formation was measured by assaying the leakage of vesicle-trapped biocytin, detected by its binding to externally added BOD-SA, which induces an increase in BODIPY fluorescence (18, 30, 31). Finally, like many other β-barrel proteins, the oligomeric pore-forming β-barrel of PFO is resistant to unfolding and disassembly in SDS (32). SDS resistance was monitored using SDS-agarose gel electrophoresis, comparing the intensity of the monomer and oligomer bands.
Using these assays, PFO in the presence of asymmetric POPCo/POPE:POPSi vesicles (with various mol % cholesterol) was found to exhibit a cholesterol threshold for interaction with membranes with a midpoint for binding, oligomerization and membrane penetration at ∼22 mol % cholesterol (Fig. 1A and Table 1). In contrast, pore formation and the formation of SDS-resistant oligomers required much more cholesterol, ∼40 mol %. Controls showed that this difference in midpoints was not affected by the somewhat higher concentration of protein generally used to detect pores (data not shown). These observations indicate that at ∼30 mol % cholesterol the predominant conformation of PFO is membrane-bound, oligomerized, and membrane-inserted but does not form a pore. We call this state, which has properties intermediate relative to PFO in solution and the pore-forming state, the “inserted intermediate state.”
FIGURE 1.

Change in PFO (PFO A215C) behavior as a function of cholesterol concentration in asymmetric vesicles and in symmetric vesicles. A, POPCo/POPE:POPSi/cholesterol asymmetric vesicles. B, scrambled vesicles. C, 1:1 POPE/POPS symmetric vesicles. D, 90:5:5 POPC/POPE/POPS symmetric vesicles. Various conformational parameters were assayed. See “Experimental Procedures” for other experimental conditions. Normalized values are shown with value at 0 mol % cholesterol assigned as 0 and maximal change at 50 mol % cholesterol assigned as 1. The change from 0 to 50 mol % cholesterol generally involves a 2.5–4-fold increase in Trp fluorescence, a 10–15-fold increase in acrylodan fluorescence, an increase in FRET (1 − F/Fo) from acrylodan to BODIPY-labeled residue 215 from 40% (which is observed without cholesterol and also in the absence of lipid and caused by dimerization in solution (45) to 80%, an increase in BOD-SA caused by pore formation of 1.5–4-fold, and an increase from 0 to 70–100% oligomer formation on SDS-agarose gels. The averaged sample points were fitted to Boltzmann Sigmoid curves (GraphPad Prism software, La Jolla, CA). Average (mean) values and S.D. from four samples are shown. Unless otherwise noted, all experiments in this figure and subsequent figures were performed at room temperature (21–23 °C).
TABLE 1.
Cholesterol midpoint for PFO interaction with asymmetric or scrambled or symmetric vesicles
The midpoint is defined as the midpoint of sigmoidal curves for each parameter shown versus cholesterol concentration (e.g., Fig. 1). The cholesterol concentration at which the increase of each parameter is half-maximal was identified by fitting the averaged sample points to Boltzmann sigmoid curves (Prism software; GraphPad, La Jolla, CA). FRET values are not corrected for the difference between the acrylodan F when in solution and inserted into membranes. This artifact shifts the apparent FRET midpoint to slightly lower cholesterol concentrations. Asym., POPCo/POPE:POPSi/cholesterol asymmetric vesicles; Scram., asymmetric vesicles after scrambling lipids to destroy asymmetry; Sym. PE/PS, POPE/POPS symmetric vesicles with cholesterol; Sym. 90% PC, 90:5:5 POPC/POPE/POPS symmetric vesicles with cholesterol; Sym. 80% PC, 8:1:1 POPC/POPE/POPS symmetric vesicles with cholesterol; ND, not determined; NA, not applicable. Prepore PFO does not insert into membranes or form pores.
| WT PFO (PFO A215C) |
Prepore PFO (PFO Y181A A215C) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Asym. | Scram. | Sym. PE/PS | Sym. 90% PC | Sym. 80% PC | Asym. | Scram. | Sym. PE/PS | Sym. 90% PC | |
| % | % | ||||||||
| Binding (Trp) | 22 | 20 | 10 | 29 | 27 | 28 | 19 | 10 | 29 |
| Oligomerization (FRET) | 17 | 18 | 6 | 26 | 23 | ND | ND | ND | ND |
| Insertion (acrylodan) | 23 | 19 | 9 | 31 | 29 | NA | NA | NA | NA |
| Pore formation | 40 | 28 | 20 | 33 | 31 | NA | NA | NA | NA |
| SDS-resistant oligomers | 38 | 28 | 22 | 34 | 31 | NA | NA | NA | NA |
A large difference in cholesterol dependence for different steps of membrane interaction was not observed in symmetric POPC/POPE/POPS/cholesterol vesicles produced by scrambling the lipids in asymmetric vesicles. Because scrambled vesicles were prepared from the asymmetric vesicles, they have the same lipid composition (13). For the scrambled vesicles, all the changes in PFO membrane interaction occurred over an overlapping range of cholesterol concentrations, between 15 and 30 mol % cholesterol. Thus, there was no concentration of cholesterol at which the inserted intermediate state predominated. However, although the midpoint for binding, oligomerization, and membrane penetration was ∼20 mol %, that for pore formation and SDS resistance showed a midpoint that was slightly higher, ∼28 mol % (Fig. 1B and Table 1). This suggests that in scrambled vesicles some amount of the inserted intermediate may be present in the range 20–30 mol % cholesterol.
It has been proposed that, at least in some situations, PFO acts within acidic endocytic vesicles (33, 34), and most experiments using PFO were carried out at low pH. This has the advantage that low pH enhances PFO interaction with cholesterol without otherwise changing PFO behavior (18). To determine whether pH and temperature make a difference, experiments were repeated at neutral pH and/or at 37 °C. Behavior at 37 °C was very similar to that at room temperature. With a combination of neutral pH and 37 °C, higher cholesterol concentrations were needed to induce the various conformational changes observed at low pH, with a smaller cholesterol range over which the inserted intermediate form predominated, but otherwise PFO behavior was similar to that observed at low pH (data not shown).
PFO Interaction with Asymmetric Vesicles Is Different from That with Vesicles Composed of Only Inner Leaflet or Outer Leaflet Lipids
These results show that lipid asymmetry strongly influences PFO membrane interaction. To determine whether these differences arose from either the inner leaflet or outer leaflet lipids, the behavior of the asymmetric vesicles was compared with that of symmetric vesicles composed of only the inner leaflet or outer leaflet lipids. As shown in Fig. 1D, in symmetric vesicles composed of the lipids found in the outer leaflet of the asymmetric vesicles (i.e., POPC with a little residual POPE and POPS), the midpoint for all aspects of PFO membrane interaction was ∼30 mol % cholesterol, with little evidence for the inserted intermediate. In contrast, for the inner leaflet lipids, 1:1 POPE/POPS, as in the asymmetric vesicles, there was a difference in the concentration of cholesterol required for insertion and pore formation. However, there was enhanced binding relative to the asymmetric vesicles such that the midpoint for cholesterol binding, oligomer formation, and membrane insertion was at ∼10 mol % cholesterol, whereas that for pore formation and SDS resistance was at ∼20 mol % cholesterol (Fig. 1C). Thus, the behavior of PFO in asymmetric vesicles matches neither that in vesicles with symmetric outer leaflet lipids nor that in vesicles with symmetric inner leaflet lipids. Instead, in the asymmetric vesicles, the stable formation of the membrane-inserted intermediate seems to depend upon the inner leaflet lipids, whereas the high midpoint for initial interaction with cholesterol and for insertion is mainly determined by the outer leaflet lipids. It should be noted that as judged by dynamic light scattering, vesicle size was similar (radius, 65–75 nm) in all of the lipid compositions used (data not shown).
The observation that asymmetric vesicles have a slightly lower midpoint for binding, oligomerization, and insertion than that for symmetric vesicles with the same outer leaflet composition suggests that phosphatidylethanolamine/phosphatidylserine in the inner leaflet influences these events, presumably because the PFO in this state contacts the inner leaflet lipids. The contrasting behavior of a noninserted prepore PFO mutant (the Y181A mutant, which cannot form the TM β-barrel and induce pore formation (29)), which does not contact inner leaflet lipids, was consistent with this hypothesis. The cholesterol midpoint for binding of the prepore PFO mutant in asymmetric vesicles was similar to that in symmetric vesicles with the same outer leaflet composition as the asymmetric vesicles (Table 1).
The Inserted Intermediate Conformation Is Similar to the Pore-forming Conformation
The fluorescence changes in residue 215 and the indications that the inserted intermediate contacts inner leaflet lipids suggest that this conformation has deeply membrane-inserted segments. To further investigate this, we carried out several additional experiments. To rule out the alterative hypothesis that the change in fluorescence of residue 215 reflected a movement of residue 215 into a hydrophobic site within the interior of the protein rather than membrane insertion, quenching of acrylodan fluorescence by the membrane-inserted nitroxide-bearing quencher 10-DN was measured (21). 10-DN is a short range quencher (quenching range, ∼10–15 Å), which quenches the fluorescence of groups buried within the core of the lipid bilayer (35). As shown in Fig. 2, in both POPCo/POPE:POPSi/cholesterol and POPE/POPS/cholesterol vesicles, the fluorescence of acrylodan-labeled residue 215 was strongly quenched by 10-DN at and above the cholesterol concentrations at which the inserted intermediate state forms. The extent of quenching at concentrations of cholesterol at which the pore-forming conformation predominates was very similar to that at which the inserted intermediate predominates, indicating that the acrylodan is inserted facing the core of the lipid bilayer at a similar depth in the membrane in both of these two states. Because the inserted β-strands in the pore-forming state are in a TM configuration, this suggests the same is true in the inserted intermediate state (see below).
FIGURE 2.
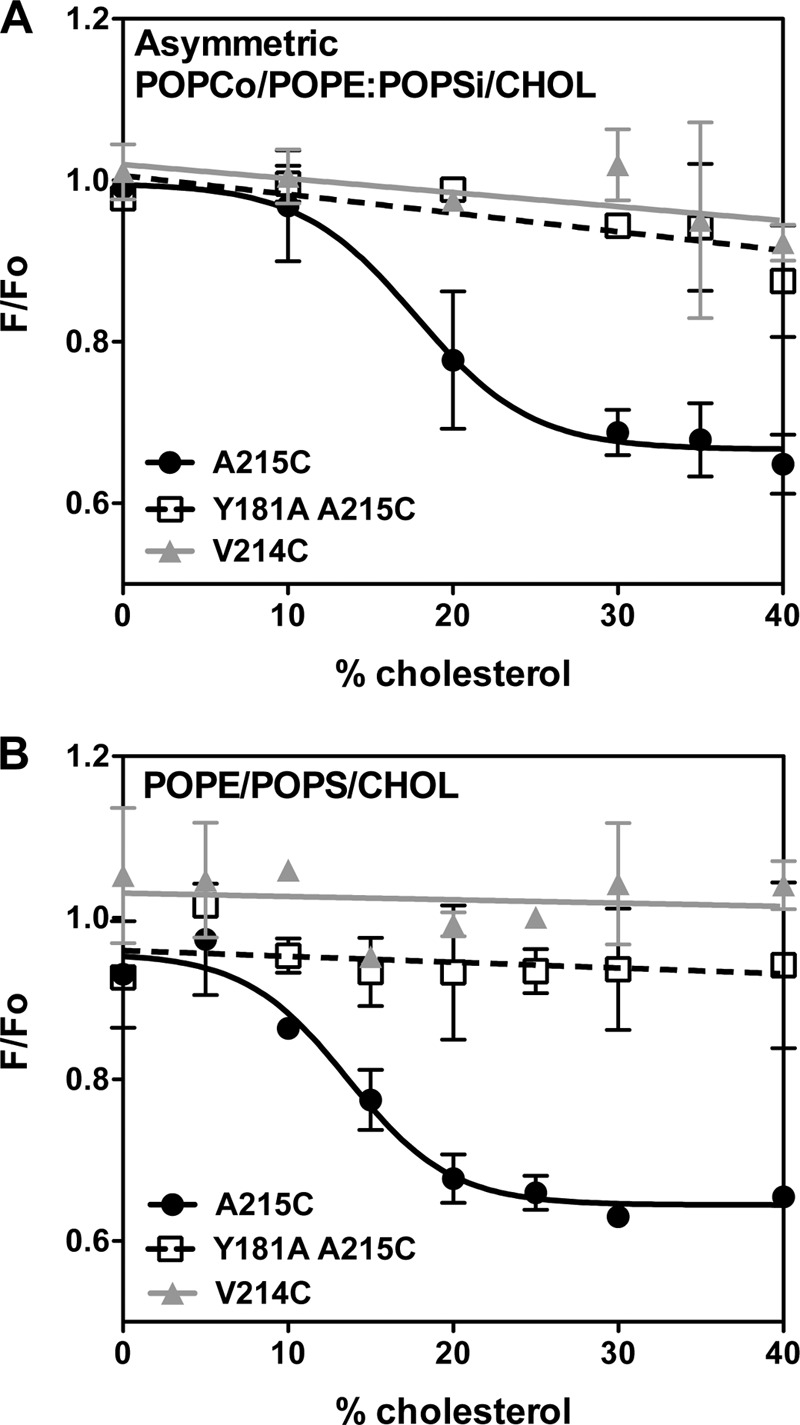
Quenching of acrylodan fluorescence by 10-DN. A, asymmetric POPCo/POPE:POPSi/cholesterol LUV. B, symmetric POPE/POPS/cholesterol LUV. Samples contained 25 nm acrylodan-labeled PFO A215C (●), PFO Y181A/A215C (□), or PFO V214C (▴) with LUVs (∼100 μm total lipid) in PBS pH 5.1. F/Fo is the ratio of acrylodan fluorescence intensity with an excitation wavelength at 350 nm and an emission wavelength at 450 nm in the presence of vesicles containing 5 mol % 10-DN (F) to that in the presence of vesicles lacking 10-DN (Fo). F/Fo values were corrected for nonspecific quenching using the quenching of acrylodan-labeled glutathione in the presence of POPE/POPS vesicles with or without 10-DN in PBS pH 5.1. Average (mean) values and S.D. from triplicates are shown.
Because TM β-strands have only every other residue exposed to the lipid bilayer, we also studied the behavior of residue 214 using an acrylodan-labeled PFO V214C mutant. In the pore-forming state, this residue has been shown to face the interior of the pore and is inaccessible to quenching by nitroxide-labeled phospholipids (23). Consistent with this, we found no quenching of acrylodan fluorescence by 10-DN in the pore-forming state at high cholesterol concentration. There was also no quenching of acrylodan fluorescence at the intermediate cholesterol concentrations at which the intermediate inserted conformation is present. This was true in both types of vesicles in which the inserted intermediate was observed: asymmetric vesicles and symmetric POPE/POPS/cholesterol vesicles. This suggests that in the inserted intermediate, residue 214 is exposed to a polar environment. For both the pore-forming state and inserted intermediate, the λmax of acrylodan emission for residue 214 was highly red-shifted relative to that of residue 215 (Fig. 3), also consistent with residue 214 being exposed to a polar/aqueous environment. However, the emission of residue 214 in the inserted intermediate was not as red-shifted as in the pore-forming state, suggesting a possible lower degree of exposure to water, which would be consistent with a TM state in lacking a pore, perhaps with the polar faces of TM strands in contact with each other (see “Discussion”). It is less consistent with β-strands sitting along the membrane surface with the polar face in contact with water.
FIGURE 3.

Acrylodan-labeled PFO TM segment residues interaction with asymmetric POPCo/POPE:POPSi/cholesterol vesicles (A and B) and with symmetric POPE/POPS/cholesterol vesicles (C and D). A and C, λmax of acrylodan fluorescence of labeled PFO A215C (black bars) and PFO V214C (striped bars) interacting with vesicles in PBS pH 5.1. B and D, acrylodan fluorescence intensity with an excitation wavelength at 350 nm and an emission wavelength at 450 nm for labeled PFO A215C (black bars) and PFO V214C (striped bars) interacting with vesicles in PBS pH 5.1. Samples contained 1 ml of LUVs (∼100 μm total lipid) and 25 nm acrylodan-PFO. Amount of cholesterol in different samples is given on the x axis. Average (mean) values and S.D. from triplicates are shown.
The Inserted Intermediate Conformation Has a Transmembrane Structure
Although the observations above are all consistent with the presence of TM β-strands in the inserted intermediate, they do not directly probe for TM orientation. To directly test whether the inserted intermediate has TM segments, the location of residue 204, which is in the loop between the two β-strands of TMH1, was examined. In the pore-forming TM state, this residue moves across the bilayer and would be exposed to the cytoplasm of the cell (27). To detect movement of residue 204 across the lipid bilayer, we monitored the reaction of a PFO E204C mutant biotinylated on Cys-204 with externally added or vesicle-entrapped BOD-SA (Fig. 4). The interaction of biotin with BOD-SA can be detected by an increase in BODIPY fluorescence (30). If the biotin is only exposed to the external solution, it should only react with externally added BOD-SA, and if only exposed to the internal solution, it should only react with vesicle-entrapped BOD-SA (22).
FIGURE 4.
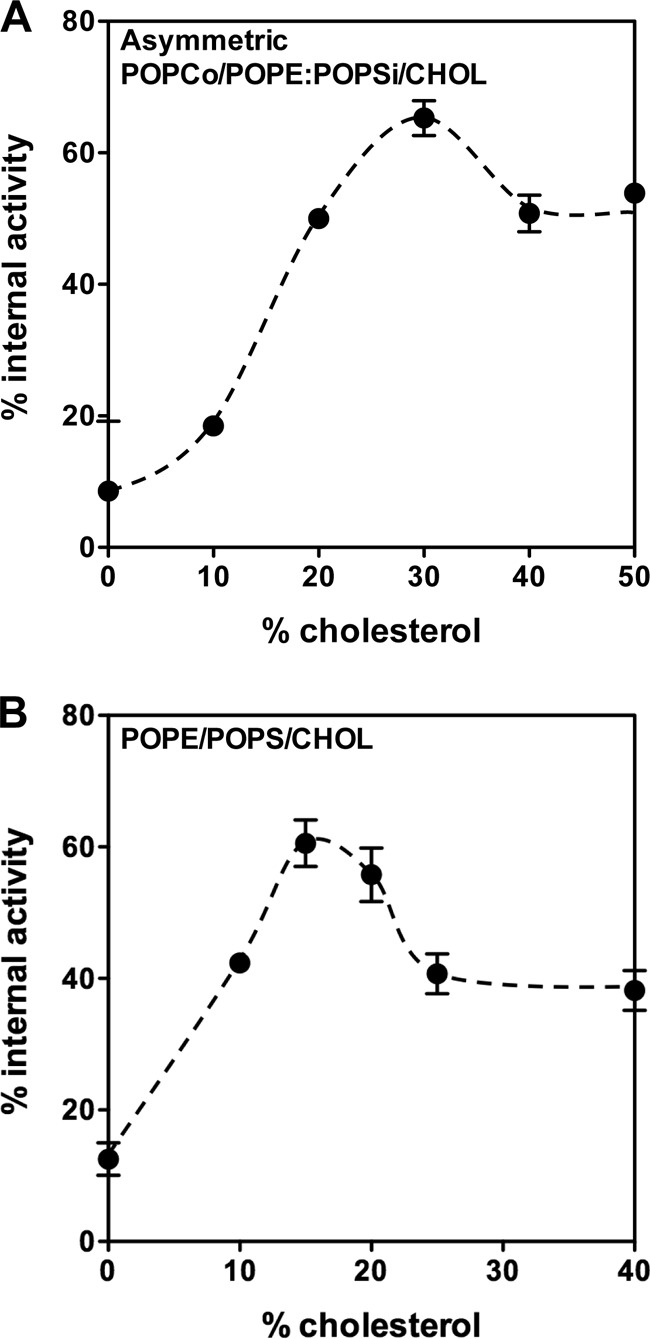
TM segment formation assayed using biotinylated PFO E204C. The percentage of biotin reacting with internally trapped BOD-SA is shown for PFO mixed with asymmetric POPCo/POPE:POPSi/cholesterol vesicles (A) or with symmetric POPE/POPS/cholesterol vesicles (B). Samples contained 50 nm biotinylated PFOE204C, 0.1 μg/ml BOD-SA, and ∼100 μm lipid in PBS, pH 5.1. At cholesterol concentrations below that at which pores form, the percentage of internal reactivity gives the percentage of TMH1 segments in a TM configuration. Average (mean) values and range from duplicates are shown.
The reactivity of biotinylated protein with BOD-SA was measured in the vesicle compositions in which the inserted intermediate forms, varying cholesterol concentration. Fig. 4 shows the percentage of internal reactivity of PFO biotinylated on residue 204 as a function of cholesterol concentration (percentage of internal reactivity equals [biotin exposed to internal solution/sum of biotin exposed to external solution and to vesicle lumen] × 100%). In the absence of cholesterol, PFO was on the outside of the vesicles, and as expected, the percentage of internal reactivity was close to zero (the small amount of apparent reactivity with vesicle-trapped BOD-SA may reflect a small amount of BOD-SA in the external solution). With increasing amounts of cholesterol, the percentage of internal reactivity increased. The maximum value corresponded to 65–70% of biotin translocated to the vesicle lumen. This maximum was observed at 30 mol % cholesterol in POPCo/POPE:POPSi/cholesterol vesicles (Fig. 4A) and 15 mol % in POPE/POPS/cholesterol vesicles (Fig. 4B). For both of these lipid compositions, these are the concentrations of cholesterol at which Fig. 1 shows that the formation of the inserted intermediate conformation is most predominant (i.e., cholesterol concentrations just below that at which there is extensive pore formation). When the cholesterol concentration was further increased, so that the pore forming conformation was present, the percentage of internal reactivity decreased to 40–50%. A value close to 50% is expected under these conditions because pores formed by PFO are large enough to allow proteins to easily pass through (36). Thus when pores are present, BOD-SA should be able to react with biotin no matter what side of the membrane the biotin was exposed. Based on the combination of these experiments, we conclude that the inserted intermediate spans the bilayer and so has formed TM segments.
The observation that the percentage of internal reactivity never reaches 100% may simply reflect the presence of a mixture of conformations, such that there is incomplete formation of the inserted intermediate state, or may be due to partial blockage of translocation by biotinylation, or may be due to a small amount of BOD-SA leakage into vesicles. Values not exactly equal to 50% in the presence of pores may indicate a small fraction of vesicles containing PFO that is not in the pore-forming state.
The Inserted Intermediate Is Not a Kinetically Trapped Dead-end State
An interesting question is whether the inserted intermediate state has the potential to be a kinetic intermediate in the insertion process rather than simply a structural intermediate. To determine when the inserted intermediate forms, the interaction of PFO with POPE/POPS/cholesterol vesicles was followed as a function of time, using low temperature (15 °C) to slow down insertion and pore formation. The time dependence of PFO behavior showed that membrane binding preceded formation of the deeply inserted state and that the deeply inserted state appeared to form slightly ahead of pore formation (Fig. 5). Thus, it is possible that the inserted intermediate is on the pathway to pore formation.
FIGURE 5.
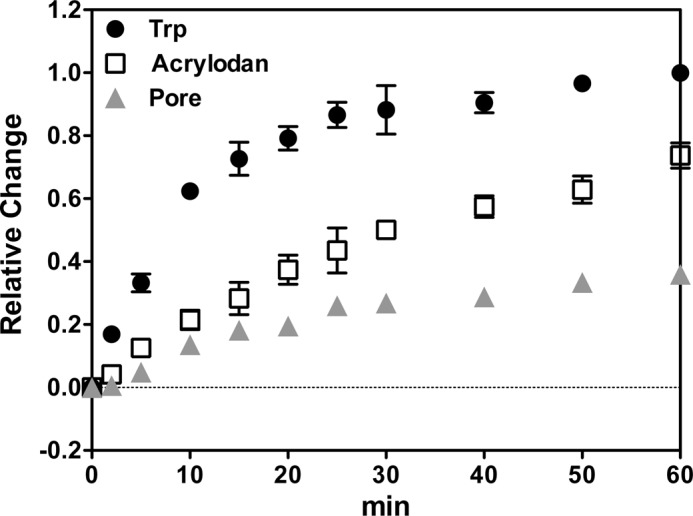
Time dependence of PFO interaction with LUVs composed of 1:1 POPE/POPS with 25 mol % cholesterol at 15 °C. Samples contained 100 μm lipids with 537 μm entrapped biocytin and 95 nm PFO A215C for Trp measurement, or 25 nm acrylodan-labeled PFO A215C for acrylodan measurement, or 95 nm PFO A215C plus 10 nm BOD-SA for pore formation measurement. Trp, acrylodan, and BODIPY fluorescence was measured as a function of time for up to 60 min at 15 °C. Zero time is the time of addition of PFO. After 60 min of incubation at 15 °C, samples were gradually heated up to 37 °C and kept at 37 °C for 15 min. Normalized values are shown with the fluorescence intensity at zero time assigned as 0 and values at 37 °C assigned as 1. Average (mean) values and ranges from duplicates are shown. Notice that because the acrylodan samples used the lowest protein concentration, insertion may have been slowed down, so that the differences between the time of insertion and that pore formation may be even greater than shown here.
A related question is whether the formation of the inserted intermediate is irreversible. To test this hypothesis, cholesterol was extracted from the asymmetric membranes with MβCD. MβCD treatment has been shown to reverse the interaction of PFO with cholesterol (23). The behavior of PFO in asymmetric vesicles was compared with that of the Y181A PFO mutant, which forms a noninserted prepore state (Fig. 6). The cholesterol interaction and binding of prepore PFO to vesicles was mostly lost upon extraction of cholesterol, showing that binding to the vesicle surface is a reversible process. The membrane binding of the inserted intermediate formed by WT PFO was almost as reversible as that of the prepore mutant (Fig. 6A). However, the pore-forming conformation remained very largely membrane-bound after cholesterol extraction, consistent with the observation that the pore-forming TM β-barrel is so stable that it cannot be destroyed by incubation in SDS. In agreement with these results, both for the prepore and the inserted intermediate, MβCD also largely reversed the increase in Trp fluorescence that is observed when PFO binds to membranes, while again having little effect upon the pore-forming state (Fig. 6B). Finally, addition of MβCD to acrylodan-labeled PFO induced a partial red shift in emission λmax for the inserted intermediate, but not for the pore-forming state (Fig. 6C). This assay exaggerates the extent of residual membrane insertion after MβCD treatment, because acrylodan fluorescence from membrane bound PFO is much stronger than that of PFO in solution and so tends to dominate the observed λmax, which is a weighted average of that in solution and in membranes. Additional evidence that the inserted intermediate does not represent an irreversible dead end comes from the observation that it can be converted to the pore-forming conformation, as assayed by pore formation, when cholesterol concentration in membranes containing the inserted intermediate is increased by addition of cholesterol-loaded MβCD (data not shown).
FIGURE 6.
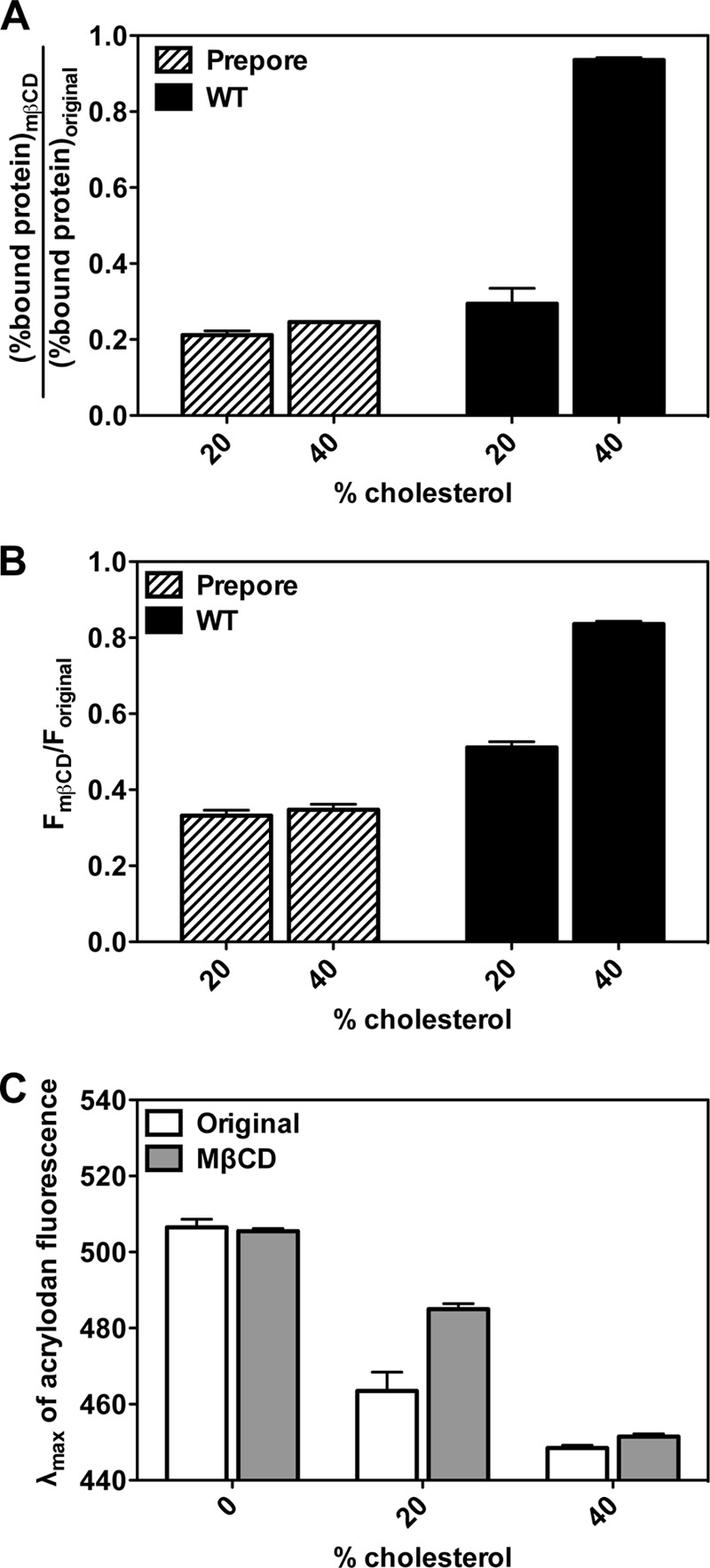
The effect of removal of membrane cholesterol by MβCD upon the interaction of PFO with asymmetric vesicles. A, association of BODIPY-labeled prepore PFO mutant (PFO Y181A/A215C) (striped bars) and WT PFO (PFO A215C) (black bars) with POPCo/POPE:POPSi/cholesterol vesicles containing 20 or 40 mol % cholesterol with or without MβCD. “Original samples” contained 1 ml of asymmetric vesicles (∼100 μm total lipid) and 50 nm BODIPY-PFO, whereas “MβCD samples” contained 1 ml of asymmetric vesicles (∼100 μm total lipid), 50 nm BODIPY-PFO, and 10 mm MβCD in PBS, pH 5.1. After centrifugation to pellet the vesicles and bound protein, BODIPY fluorescence was measured in both the supernatant and the pellet. B, Trp fluorescence of prepore PFO mutant (striped bars) and WT PFO (black bars) interacting with POPCo/POPE:POPSi/cholesterol asymmetric vesicles containing 20 or 40 mol % cholesterol with or without MβCD. Original samples contained 1 ml of asymmetric vesicles (∼100 μm total lipid) and 50 nm PFO, whereas MβCD samples contained 1 ml of asymmetric vesicles (∼100 μm total lipid), 50 nm PFO, and 10 mm MβCD in PBS, pH 5.1. C, λmax of acrylodan-labeled WT PFO interacting with POPCo/POPE:POPSi/cholesterol asymmetric vesicles containing 0, 20, or 40 mol % cholesterol with or without MβCD. Original samples contained 1 ml of asymmetric vesicles (∼100 μm total lipid) and 25 nm acrylodan-PFO, whereas MβCD samples contained 1 ml of asymmetric vesicles (∼100 μm total lipid), 25 nm acrylodan-PFO, and 10 mm MβCD in PBS, pH 5.1. Average (mean) values and S.D. from triplicates are shown.
We conclude that the inserted intermediate state and noninserted prepore states can be in equilibrium with PFO in solution, but reversibility is at least much reduced after pore formation. Combined with the structural properties of the different states and the kinetic sequence of events, this suggests possible pathways for the conformational changes that occur during pore formation (see “Discussion”).
DISCUSSION
Identification of a Stable Deeply Inserted Intermediate PFO Conformation
The mechanism of pore formation by PFO and other cholesterol-dependent cytolysins involves several events, including membrane binding, oligomerization, TM insertion, and formation of the TM β-barrel walled aqueous pore. These events occur in a series of steps, triggered when cholesterol is present above a threshold concentration, and mutants stuck at various intermediate (prepore) conformations have been identified (28, 29, 37, 38). Prepore states involve intermediate conformations that are often defined by whether the ring complex is SDS-sensitive or -resistant. Ramachandran et al. (37) have shown that the disruption of an intermolecular π-stacking interaction between Tyr-181 and Phe-318 results in the formation of an SDS-sensitive prepore complex. Hotze et al. (28) engineered a disulfide between TMH1 and domain 2 that prevented the extension of TMH1 but allowed PFO to form a SDS-resistant prepore complex. Whether these mutated prepore states are completely identical to those formed by WT PFO prior to pore formation is not clear.
Our results suggest the presence of a novel state that is structurally intermediate between a noninserted prepore and the pore-forming conformation. This type of membrane-inserted state has been thought to form only transiently, but we find that it is quite stable over a wide range of cholesterol concentrations in POPE/POPS/cholesterol and POPCo/POPE:POPSi/cholesterol vesicles. Although the inserted intermediate has TM segments and the behavior of residues 214 and 215 suggests that β-strands similar to the pore-forming conformation are present, its structure cannot yet be fully specified. One possibility is that it has TM β-sheets with polar surfaces pressed against one another in place of a pore. This could suggest that it forms a very small oligomer lacking a pore. This type of structure has been seen in the bacterial OmpA protein, which has an eight-strand β-barrel lacking a pore (39). Alternately, a larger, but highly strained β-barrel with a collapsed pore might form. This could explain the relative ease of reversing formation of this state by removal of cholesterol. In addition, although it seems unlikely, we cannot rule out the possibility that TMH1 forms TM segments, whereas TM hairpin 2 does not. The details of the structure of the inserted intermediate are important questions for future studies. However, this does not lessen the main conclusion from our study, that PFO structure and behavior is strongly influenced by lipid asymmetry.
Possible Pathways for Assembly of Pore-forming Conformation of PFO
The behavior of PFO in asymmetric vesicles suggests the possibility of novel pathways for the assembly of the pore-forming conformation of PFO. In this regard, the key observation is that a TM-inserted state that has not yet formed a pore was thought to only exist as a highly unstable, transient conformation. This study shows that it can be stable and thus experimentally accessible. The most plausible assembly pathway is one in which PFO undergoes a series of conformational changes to states that share increasingly more structural features with the pore-forming state. As shown in Fig. 7 this would mean that the assembly pathway involves conversion of the noninserted prepore to the inserted intermediate, which then converts to the pore conformation (arrow c followed by arrow b). However, because the prepore and inserted intermediate states form reversibly, they might not be identical to the true intermediate states on the pathway leading to pore formation. Instead, either or both could both be slightly off-pathway intermediates, which convert to a closely related conformation just prior to assembly of the pore-forming state.
FIGURE 7.
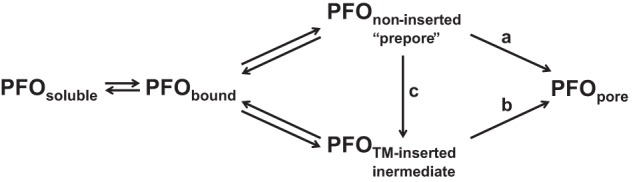
Schematic diagram of the possible steps in PFO pore formation. Pathways a, b, and c represent different possible steps in the assembly process.
Furthermore, it is possible that the assembly of the pore avoids direct involvement of either the prepore or inserted intermediate. For example, the assembly process might involve conversion of the noninserted prepore to the pore conformation by passing through a highly transient inserted state distinct from the inserted intermediate (arrow a). Alternately, the assembly process might involve conversion of the inserted intermediate to the pore conformation (arrow b), via a pathway in which the precursor to the inserted intermediate is a transient noninserted oligomer that differs significantly from the stable prepore conformation. These possibilities are not mutually exclusive. Different pathways might be followed under different physiological situations (e.g., at the plasma membrane and in endocytic vesicles).
It is also possible that the inserted intermediate has functional significance. Pore formation causes mammalian cells to mount an innate immune response to repair toxin-induced damage (40). If PFO formed many pores in inappropriate cell membranes containing lower amounts of cholesterol, it could trigger this response. Forming the inserted intermediate would allow for efficient membrane insertion while avoiding unnecessary pore formation in inappropriate membranes.
Implications and Future Applications of Asymmetric Vesicles for Protein-Lipid Studies
Asymmetric vesicles should be a powerful system for more detailed examination of lipid-protein interactions than heretofore possible. Although most membrane proteins are helical, there is no reason that the interactions of helical protein residues should not also respond to lipid asymmetry. The amino acids of plasma membrane proteins in, and adjacent to, the inner and outer leaflets are not identical in terms of both charge and size (41), and so should have different interactions with inner and outer leaflet lipids.
It is also interesting that lipid asymmetry might be important for two immunologically crucial mammalian pore-inducing proteins that form TM β-barrels, complement proteins, and perforin. These proteins have membrane-inserting domains analogous to that in cholesterol-dependent cytolysins. Although complement and perforin do not require cholesterol to bind to membranes, their binding may be influenced by phosphatidylserine, which appears in the outer leaflet upon loss of plasma membrane asymmetry (42–44). Thus, it will be interesting to see whether their membrane insertion and pore-forming pathways are influenced by phosphatidylserine asymmetry.
Studies of β-barrel pore-forming proteins in asymmetric vesicles may have important biomedical implications. Cholesterol-dependent cytolysins are important virulence factors in infection, and mammalian pore-forming proteins have important roles in combatting infection (11). Understanding the interaction of such proteins with membranes and how their pore-forming ability is controlled by membranes may provide important clues to designing agents that interfere or modulate pore-forming abilities in a biomedically useful fashion.
Acknowledgment
We thank Caroline Lee for characterization of the effect of lipid composition upon PFO binding to membranes.
This work was supported by National Science Foundation Grants DMR 1104367 and MCB 1019986.
- TM
- transmembrane
- PFO
- perfringolysin O
- POPC
- 1-palmitoyl-2-oleoyl-phosphatidylcholine
- POPE
- 1-palmitoyl-2-oleoyl-phosphatidylethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-phosphatidyl-l-serine
- Rho-DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)
- HPαCD
- (2-hydroxypropyl)-α-cyclodextrin
- MβCD
- methyl-β-cyclodextrin
- acrylodan
- 6-acryloyl-2-dimethylaminonaphthalene
- BODIPY-FL-IA
- N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl iodoacetamide
- BOD-SA
- streptavidin BODIPY-FL conjugate
- LUV
- large unilamellar vesicle(s)
- 10-DN
- 10-doxylnonadecane
- TMH
- TM hairpin.
REFERENCES
- 1. Devaux P. F. (1991) Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30, 1163–1173 [DOI] [PubMed] [Google Scholar]
- 2. Bretscher M. S. (1972) Asymmetrical lipid bilayer structure for biological membranes. Nat. New Biol. 236, 11–12 [DOI] [PubMed] [Google Scholar]
- 3. von Heijne G., Gavel Y. (1988) Topogenic signals in integral membrane proteins. European journal of biochemistry / FEBS 174, 671–678 [DOI] [PubMed] [Google Scholar]
- 4. Shahidullah K., Krishnakumar S. S., London E. (2010) The effect of hydrophilic substitutions and anionic lipids upon the transverse positioning of the transmembrane helix of the ErbB2 (neu) protein incorporated into model membrane vesicles. J. Mol. Biol. 396, 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharpe H. J., Stevens T. J., Munro S. (2010) A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tweten R. K., Parker M. W., Johnson A. E. (2001) The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 257, 15–33 [DOI] [PubMed] [Google Scholar]
- 7. Giddings K. S., Johnson A. E., Tweten R. K. (2003) Redefining cholesterol's role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. U.S.A. 100, 11315–11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cockeran R., Anderson R., Feldman C. (2003) Pneumolysin in the immunopathogenesis and treatment of pneumococcal disease. Expert Rev. Anti. Infect. Ther. 1, 231–239 [DOI] [PubMed] [Google Scholar]
- 9. Braun J. S., Hoffmann O., Schickhaus M., Freyer D., Dagand E., Bermpohl D., Mitchell T. J., Bechmann I., Weber J. R. (2007) Pneumolysin causes neuronal cell death through mitochondrial damage. Infect. Immun. 75, 4245–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rood J. I. (1998) Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52, 333–360 [DOI] [PubMed] [Google Scholar]
- 11. Rosado C. J., Kondos S., Bull T. E., Kuiper M. J., Law R. H., Buckle A. M., Voskoboinik I., Bird P. I., Trapani J. A., Whisstock J. C., Dunstone M. A. (2008) The MACPF/CDC family of pore-forming toxins. Cell. Microbiol. 10, 1765–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng H. T., London E. (2011) Preparation and properties of asymmetric large unilamellar vesicles. Interleaflet coupling in asymmetric vesicles is dependent on temperature but not curvature. Biophys. J. 100, 2671–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H. T., Megha, London E. (2009) Preparation and properties of asymmetric vesicles that mimic cell membranes. Effect upon lipid raft formation and transmembrane helix orientation. J. Biol. Chem. 284, 6079–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schick P. K., Kurica K. B., Chacko G. K. (1976) Location of phosphatidylethanolamine and phosphatidylserine in the human platelet plasma membrane. J. Clin. Invest. 57, 1221–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marinetti G. V., Love R. (1976) Differential reaction of cell membrane phospholipids and proteins with chemical probes. Chem. Phys. Lipids 16, 239–254 [DOI] [PubMed] [Google Scholar]
- 16. Lin Q., London E. (2014) Preparation of artificial plasma membrane mimicking vesicles with lipid asymmetry. PLoS One, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin Q., London E. (2013) Altering hydrophobic sequence lengths shows that hydrophobic mismatch controls affinity for ordered lipid domains (rafts) in the multitransmembrane strand protein perfringolysin O. J. Biol. Chem. 288, 1340–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson L. D., Johnson A. E., London E. (2008) How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH. Insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 283, 4632–4642 [DOI] [PubMed] [Google Scholar]
- 19. Nakamura M., Sekino N., Iwamoto M., Ohno-Iwashita Y. (1995) Interaction of θ-toxin (perfringolysin-O), a cholesterol-binding cytolysin, with liposomal membranes. Change in the aromatic side-chains upon binding and insertion. Biochemistry 34, 6513–6520 [DOI] [PubMed] [Google Scholar]
- 20. Ramachandran R., Tweten R. K., Johnson A. E. (2005) The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc. Natl. Acad. Sci. U.S.A. 102, 7139–7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J., Rosconi M. P., London E. (2006) Topography of the hydrophilic helices of membrane-inserted diphtheria toxin T domain. TH1-TH3 as a hydrophilic tether. Biochemistry 45, 8124–8134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosconi M. P., Zhao G., London E. (2004) Analyzing topography of membrane-inserted diphtheria toxin T domain using BODIPY-streptavidin. At low pH, helices 8 and 9 form a transmembrane hairpin but helices 5–7 form stable nonclassical inserted segments on the cis side of the bilayer. Biochemistry 43, 9127–9139 [DOI] [PubMed] [Google Scholar]
- 23. Heuck A. P., Savva C. G., Holzenburg A., Johnson A. E. (2007) Conformational changes that effect oligomerization and initiate pore formation are triggered throughout perfringolysin O upon binding to cholesterol. J. Biol. Chem. 282, 22629–22637 [DOI] [PubMed] [Google Scholar]
- 24. Moe P. C., Heuck A. P. (2010) Phospholipid hydrolysis caused by Clostridium perfringens α-toxin facilitates the targeting of perfringolysin O to membrane bilayers. Biochemistry 49, 9498–9507 [DOI] [PubMed] [Google Scholar]
- 25. Flanagan J. J., Tweten R. K., Johnson A. E., Heuck A. P. (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shepard L. A., Heuck A. P., Hamman B. D., Rossjohn J., Parker M. W., Ryan K. R., Johnson A. E., Tweten R. K. (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O. An α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37, 14563–14574 [DOI] [PubMed] [Google Scholar]
- 27. Shatursky O., Heuck A. P., Shepard L. A., Rossjohn J., Parker M. W., Johnson A. E., Tweten R. K. (1999) The mechanism of membrane insertion for a cholesterol-dependent cytolysin. A novel paradigm for pore-forming toxins. Cell 99, 293–299 [DOI] [PubMed] [Google Scholar]
- 28. Hotze E. M., Wilson-Kubalek E. M., Rossjohn J., Parker M. W., Johnson A. E., Tweten R. K. (2001) Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane β-sheet from a prepore intermediate. J. Biol. Chem. 276, 8261–8268 [DOI] [PubMed] [Google Scholar]
- 29. Hotze E. M., Heuck A. P., Czajkowsky D. M., Shao Z., Johnson A. E., Tweten R. K. (2002) Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem. 277, 11597–11605 [DOI] [PubMed] [Google Scholar]
- 30. Nicol F., Nir S., Szoka F. C., Jr. (1999) Orientation of the pore-forming peptide GALA in POPC vesicles determined by a BODIPY-avidin/biotin binding assay. Biophys. J. 76, 2121–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nir S., Nicol F., Szoka F. C., Jr. (1999) Surface aggregation and membrane penetration by peptides. Relation to pore formation and fusion. Mol. Membr. Biol. 16, 95–101 [DOI] [PubMed] [Google Scholar]
- 32. Shepard L. A., Shatursky O., Johnson A. E., Tweten R. K. (2000) The mechanism of pore assembly for a cholesterol-dependent cytolysin. Formation of a large prepore complex precedes the insertion of the transmembrane β-hairpins. Biochemistry 39, 10284–10293 [DOI] [PubMed] [Google Scholar]
- 33. O'Brien D. K., Melville S. B. (2004) Effects of Clostridium perfringens α-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect. Immun. 72, 5204–5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Portnoy D. A., Tweten R. K., Kehoe M., Bielecki J. (1992) Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect. Immun. 60, 2710–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Caputo G. A., London E. (2003) Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic α-helix locations within membranes. Biochemistry 42, 3265–3274 [DOI] [PubMed] [Google Scholar]
- 36. Heuck A. P., Tweten R. K., Johnson A. E. (2003) Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J. Biol. Chem. 278, 31218–31225 [DOI] [PubMed] [Google Scholar]
- 37. Ramachandran R., Tweten R. K., Johnson A. E. (2004) Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit β-strand alignment. Nat. Struct. Mol. Biol. 11, 697–705 [DOI] [PubMed] [Google Scholar]
- 38. Dowd K. J., Tweten R. K. (2012) The cholesterol-dependent cytolysin signature motif. A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 8, e1002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schulz G. E. (2002) The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta 1565, 308–317 [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez M. R., Bischofberger M., Pernot L., van der Goot F. G., Frêche B. (2008) Bacterial pore-forming toxins. The (w)hole story? Cell. Mol. Life Sci. 65, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munro S. (1995) An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 14, 4695–4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Païdassi H., Tacnet-Delorme P., Garlatti V., Darnault C., Ghebrehiwet B., Gaboriaud C., Arlaud G. J., Frachet P. (2008) C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J. Immunol. 180, 2329–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Païdassi H., Tacnet-Delorme P., Verneret M., Gaboriaud C., Houen G., Duus K., Ling W. L., Arlaud G. J., Frachet P. (2011) Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J. Mol. Biol. 408, 277–290 [DOI] [PubMed] [Google Scholar]
- 44. Martin G., Sabido O., Durand P., Levy R. (2005) Phosphatidylserine externalization in human sperm induced by calcium ionophore A23187. Relationship with apoptosis, membrane scrambling and the acrosome reaction. Hum. Reprod. 20, 3459–3468 [DOI] [PubMed] [Google Scholar]
- 45. Solovyova A. S., Nöllmann M., Mitchell T. J., Byron O. (2004) The solution structure and oligomerization behavior of two bacterial toxins. Pneumolysin and perfringolysin O. Biophys. J. 87, 540–552 [DOI] [PMC free article] [PubMed] [Google Scholar]