

Background: The role of SREBP-1 in regulating carbohydrate metabolism is unclear.
Results: Silencing SREBP-1 reduced glycogen buildup and expression of genes involved in glycogen synthesis as well as gluconeogenesis.
Conclusion: SREBP-1 is needed to regulate carbohydrate metabolism during the fed state. Thus, its depletion does not improve insulin resistance.
Significance: This report provides a novel function for SREBP-1.
Keywords: Gene Expression, Gluconeogenesis, Glucose Metabolism, Insulin Resistance, Liver Metabolism
Abstract
Sterol regulatory element-binding protein-1 (SREBP-1) is a key transcription factor that regulates genes in the de novo lipogenesis and glycolysis pathways. The levels of SREBP-1 are significantly elevated in obese patients and in animal models of obesity and type 2 diabetes, and a vast number of studies have implicated this transcription factor as a contributor to hepatic lipid accumulation and insulin resistance. However, its role in regulating carbohydrate metabolism is poorly understood. Here we have addressed whether SREBP-1 is needed for regulating glucose homeostasis. Using RNAi and a new generation of adenoviral vector, we have silenced hepatic SREBP-1 in normal and obese mice. In normal animals, SREBP-1 deficiency increased Pck1 and reduced glycogen deposition during fed conditions, providing evidence that SREBP-1 is necessary to regulate carbohydrate metabolism during the fed state. Knocking SREBP-1 down in db/db mice resulted in a significant reduction in triglyceride accumulation, as anticipated. However, mice remained hyperglycemic, which was associated with up-regulation of gluconeogenesis gene expression as well as decreased glycolysis and glycogen synthesis gene expression. Furthermore, glycogen synthase activity and glycogen accumulation were significantly reduced. In conclusion, silencing both isoforms of SREBP-1 leads to significant changes in carbohydrate metabolism and does not improve insulin resistance despite reducing steatosis in an animal model of obesity and type 2 diabetes.
Introduction
In the past couple of decades, the prevalence of non-alcoholic fatty liver disease has escalated to alarming levels. Currently, this condition affects >30% of the general population (1, 2) and is as high as 75% in patients with type 2 diabetes (3). Hepatic lipid accumulation is associated with impaired insulin signaling and lack of inhibition of hepatic glucose production (4). However, insulin maintains de novo lipogenesis, in a process known as selective insulin resistance (5). Reduced inhibition of hepatic glucose output and increased lipogenesis lead to a combination of hyperglycemia and hypertriglyceridemia.
The transcription factor SREBP-1c6 regulates de novo lipogenesis in the liver in response to increases in insulin. SREBPs are transcription factors of the basic helix-loop-helix leucine zipper family that are synthesized as precursors and bound to the endoplasmic reticulum membrane (6). In the presence of the appropriate signals, SREBPs transition to the Golgi, where they are cleaved, releasing the mature form, which translocates to the nucleus and activates target gene expression (6). SREBP-1a and SREBP-1c are isoforms of the same gene and transcriptionally up-regulate glycolytic and lipogenic enzymes such as l-pyruvate kinase (Pklr), fatty acid synthase (Fasn), stearoyl-CoA desaturase 1 (Scd1), mitochondrial glycerol-3-phosphate acyltransferase 1 (Gpam), and acetyl-CoA carboxylase (Acac) (6). In human obese patients, increased levels of SREBP-1c correlate with hepatic steatosis and insulin resistance (7). Secondary to the hyperinsulinemia, SREBP-1c activity is higher in the liver of ob/ob and db/db mice, mouse models of obesity and type 2 diabetes, underscoring the role of this transcription factor as a contributor to hepatic steatosis and insulin resistance (8). These data suggest that strategies to reduce SREBP-1 activity have therapeutic potential to reduce hepatic lipid accumulation and improve insulin sensitivity to block gluconeogenesis and hepatic glucose output. However, ob/ob mice lacking SREBP-1 do not exhibit improved glucose levels despite a significant decrease in hepatic lipid accumulation (9). Here we have used helper-dependent adenoviral vectors to acutely silence SREBP-1 in the liver, to test the hypothesis that SREBP-1 regulates carbohydrate metabolism, in addition to hepatic de novo lipogenesis. Our data suggest that SREBP-1 is necessary to regulate carbohydrate metabolism during fed conditions. Thus, its depletion is not beneficial as a strategy to improve hepatic glucose output in animal models with hepatic insulin resistance.
EXPERIMENTAL PROCEDURES
Helper-dependent Adenoviral Vector Production
Helper-dependent adenoviral vectors were generated using a Cre-loxP system developed by Merck Laboratories and Microbix (Toronto, Canada) (10, 11). These vectors are the most advanced type of adenoviral vector; they are devoid of viral coding sequences and retain only the inverted terminal repeats and packaging signal (Fig. 1A). The lack of viral genes eliminates the possibility of inducing immune responses and toxic effects, therefore leading to transgene expression for years, in mice and nonhuman primates (12, 13). Helper-dependent adenoviral vectors have identical tropism to first generation adenoviral vectors and predominantly transduce the liver (14, 15). The construction of helper-dependent adenoviral vectors expressing a shRNA to silence SREBP-1 or a shRNA scrambled sequence (gAd.shSREBP1 and gAd.shSCR, respectively), as well as the efficacy of silencing SREBP-1 in the liver, have been described previously (16). Vectors were stored at −80 °C in 10 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 150 mm NaCl, 10% glycerol. Total particle counts were determined spectrophotometrically, as described (16).
FIGURE 1.
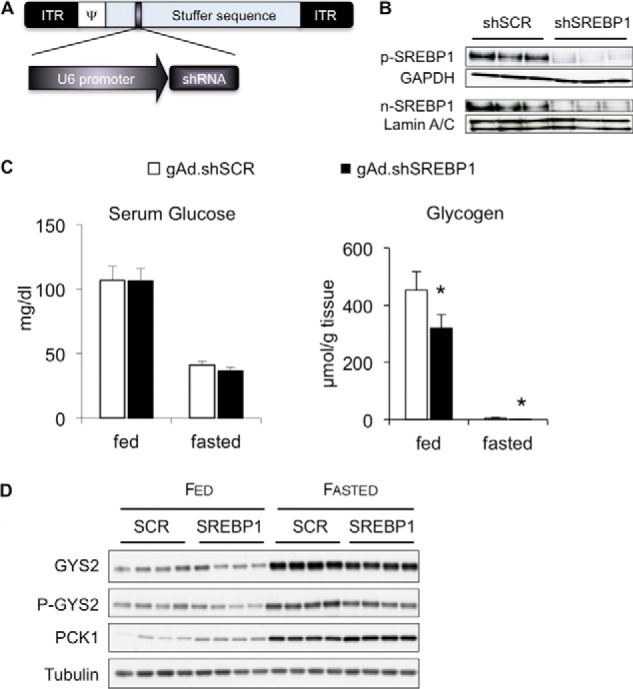
Impact of SREBP-1 knock-down on carbohydrate metabolism in normal mice. Groups of five C57BKS mice were given 1 × 1011 viral particles of gAd.shSCR or gAd.shSREBP1 and euthanized 1 week later under fed or 24-h fasted conditions. A, helper-dependent adenovirus vector used to express shRNA. Hairpin RNA expression was driven by the U6 promoter. ITR, inverted terminal repeat; Ψ, packaging signal. B, SREBP-1 precursor (p-SREBP) or nuclear (n-SREBP) protein levels. C, blood glucose unaffected by SREBP-1 silencing. Glycogen levels were significantly lower under fed and fasted conditions in mice treated with gAd.shSREBP1; *, p < 0.05; n = 5–6; error bars, S.D. D, immunoblots to detect GYS2, P-GYS2, PCK1, and tubulin.
Animals
SREBP-1 Silencing in Normal Mice
Eight- to 9-week-old C57BLKS/J mice were given 1 × 1011 viral particles and euthanized 7 days after adenovirus vector administration under ad libitum fed conditions, or 24-h fasted conditions.
SREBP-1 Silencing in db/db Mice
Male 8-week-old C57BLKS/J and db/db mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and acclimated to our facilities for a week before adenovirus administration. Animal care guidelines set forth by the Indiana University School of Medicine were followed. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). Mice were kept in a BSL2 facility and had free access to a standard chow diet and water. Mice were given 1 × 1011 viral particles by tail vein injection and euthanized 3 weeks later under fed or 24-h fasted conditions. Tissues were collected and snap frozen in liquid nitrogen or fixed in 10% buffered formalin for histology analysis.
Metabolic Tracer Study
Three weeks after gAd.shSREBP1 or gAd.shSCR adenoviral vector administration, db/db mice were given 2H2O intraperitoneally (21.4 μl/g of body weight; this dose enriches body water content to 3%) and fasted for 4 h with free access to water. Animals were euthanized, and serum and liver were collected and sent to the Mouse Metabolic Phenotyping Center at Case Western Reserve University for analysis.
Glucose Tolerance and Insulin Tolerance Tests
Adenovirus vectors were administered to db/db or C57BLKS/J mice as described above. The glucose tolerance test was performed by intraperitoneal administration of a bolus of glucose (1 mg/g body weight), after an overnight fast. Blood glucose was monitored every 30 min for 2 h. For the insulin tolerance test, mice were fasted for 5 h and given insulin intraperitoneally (1 unit/kg, Human Regular Insulin; Eli Lilly). Blood glucose was monitored every 30 min for 2 h.
Liver Lipids
Liver lipid analyses were carried out using ∼100 mg of frozen liver tissue. Total liver phospholipid, triglyceride, free fatty acid, and cholesterol were analyzed by TLC and gas chromatography at the Mouse Metabolic Phenotyping Center at Vanderbilt University. In the metabolic tracer analysis, total palmitate, de novo palmitate, malonyl-CoA, and BHB-CoA, were analyzed by the Mouse Metabolic Phenotyping Center at Case Western Reserve University School of Medicine, as described previously (17–20). For BHB-CoA, data are expressed as pmol/mg tissue relative to the internal standard [2H9]pentanoyl-CoA (19).
Serum and Tissue Biochemistries
Insulin levels were analyzed at the Vanderbilt Hormone Assay and Analytical Services Core. Blood glucose was measured every 5 days with an Ascencia Elite®XL meter (Bayer, Tarrytown, NY), from a drop collected from the tail vein. Total triglyceride in serum was measured by a triglyceride determination kit (Sigma-Aldrich), free fatty acids with the NEFA-HR(2) kit (Wako Chemicals, Richmond, VA), and β-hydroxybutyrate with a β-hydroxybutyrate determination kit from Pointe Scientific (Canton, MI). All reactions were carried out in duplicate on uncoated, flat-bottomed 96-well plate, following the manufacturer's instructions. Total liver glycogen content in tissues was determined by measuring amyloglucosidase-released glucose from glycogen as described previously (21). Glycogen synthase fractional activity with low (0.24 mm) or high (10 mm) (L/H) glucose 6-phosphate was assessed in total homogenates by monitoring the incorporation of glucose from UDP-[U-14C]glucose, as described (22).
Carnitine Palmitoyltransferase 1A (CPT1A) Activity and IC50 Assays
CPT activity and malonyl-CoA sensitivity were determined using the radiochemical forward assay as described in detail earlier (23). This assay is optimized for measuring malonyl-CoA-sensitive CPT activity. The assay is carried out with 10 μl of a 2% liver homogenate (in physiological saline) in the absence (total CPT activity) and presence of 200 μm malonyl-CoA (malonyl-CoA-insensitive activity) at 37 °C. The difference in velocity represents CPT1A activity. The malonyl-CoA sensitivity was assessed by measuring the activity in the presence of 8.0 μm malonyl-CoA that under the experimental conditions corresponds to the approximate IC50 value for malonyl-CoA in mouse liver homogenates. All values were corrected by subtracting the malonyl-CoA-insensitive values. The remaining activity upon incubation with 8 μm malonyl-CoA is plotted.
Western Blotting
To generate liver whole cell protein extracts, 100 mg of frozen liver was homogenized in lysis buffer with protease and phosphatase inhibitors (50 mm HEPES, pH 7.5, 2 mm EGTA, 2 mm EDTA, 150 mm NaCl, 10 mm NaF, 10% glycerol, 0.1% Nonidet P-40, 20 mm β-glycerolphosphate, 2 mm sodium pyrophosphate, 1 mm sodium vanadate, and 1-chloro-3-tosylamido-7-amino-2-heptanone (TLCK), 0.1 mm protease inhibitors, 2 mm benzamidine, 0.5 mm PMSF, and 10 μg/ml leupeptin). Liver extracts were centrifuged at 13,000 rpm, the fat layer carefully aspirated, and the supernatant collected for use in Western blotting. NE-PER (Thermo Scientific) extraction reagents and protease inhibitor cocktails (Thermo Scientific) were used to isolate liver nuclear and cytoplasmic proteins according to the manufacturer's recommendations. Proteins were separated by PAGE and transferred to 0.2-mm PVDF membranes (Bio-Rad). Membranes were blocked with 5% dry milk-TBST or 5% BSA-TBST for 1–2 h and incubated with the following antibodies: SREBP1 and tubulin-α (Thermo Scientific); lamin A/C, acetyl-CoA carboxylase (ACAC), phospho-ACAC, phospho-glycogen synthaseS641, and glycogen synthase (Cell Signaling); Topo II (Abcam); CPT1A and CPT2 (kind gifts from Dr. Carina Prip-Buus (INSERM, U1016, Institut Cochin, Paris, France)). Blots were developed with Pierce ECL blotting substrate (Thermo Scientific) and exposed to ECL film (GE Healthcare).
Real-time RT-PCR
Total RNA was isolated from frozen liver samples (Qiagen). Quantitative RT-PCR was carried out in an ABI 7500 instrument (Applied Biosystems), using 50 ng of RNA and the QuantiTect SYBR Green RT-PCR Kit (Applied Biosystems). Primer pairs were purchased from Qiagen or designed to bind different exons of the gene and amplify fragments of ∼200 bp, and were first confirmed to yield a single band of the expected size by agarose gel electrophoresis, as well as a negative result in wells containing sample without reverse transcriptase. Primer sequences are available upon request. The relative quantification of each gene was normalized with cyclophilin A.
Statistics
Numerical values represent mean ± S.D. p values were calculated using unpaired two-tailed Student's t tests. A p value of <0.05 was considered statistically significant.
RESULTS
Silencing SREBP-1 Increases Expression of Gluconeogenesis Genes and Reduces Glycogen Accumulation
To study the effects of silencing SREBP-1 on carbohydrate metabolism during the transition between the fed and fasted state, helper-dependent adenoviral vectors expressing an shRNA to target SREBP-1 (gAd.shSREBP1) or a scrambled shRNA (gAd.shSCR) were injected into the tail vein of normal mice (Fig. 1, A and B). No alterations in blood glucose levels were observed between the two treatments (Fig. 1C). However, glycogen accumulation was reduced by 30% under fed conditions (Fig. 1C). In addition, normal mice that received gAd.shSREBP1 had lower levels of glycogen synthase 2 (GYS2) protein (Fig. 1D). Furthermore, SREBP-1 deficiency increased expression of the gluconeogenesis enzyme phosphoenolpyruvate carboxykinase (PCK1) (Fig. 1D). These data indicate that SREBP-1 is needed to appropriately regulate glycogen synthesis and to block Pck1 expression.
SREBP-1 Knockdown Reduces Steatosis in an Animal Model of Hepatic Insulin Resistance
SREBP-1 activity is higher in the liver of db/db mice, an animal model of obesity with hepatic steatosis and severe insulin resistance (8). Thus, depleting SREBP-1 was expected to diminish lipogenesis and improve steatosis and consequently, improve insulin resistance. As predicted, silencing SREBP-1 resulted in down-regulation of hepatic de novo lipogenesis genes Fasn, Scd1, and Gpam (Fig. 2A). Interestingly, Acacb mRNA and protein expression were dramatically blunted, whereas Acaca mRNA, protein, and protein phosphorylation were not changed relative to the db/db control group (Fig. 2, A and B). These data indicate that SREBP-1 is an important regulator of ACACB and that SREBP-1 is not critical to maintain high ACACA levels in db/db mice.
FIGURE 2.
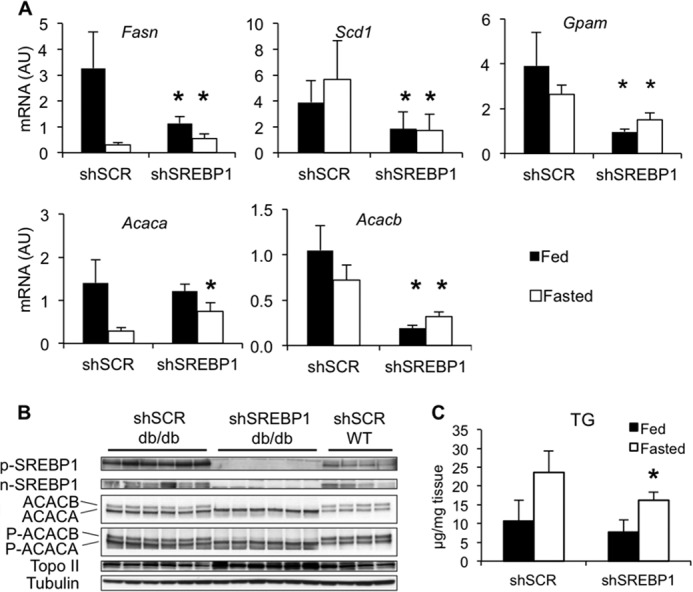
SREBP1 silencing in db/db mice. Groups of eight db/db mice were given 1 × 1011 viral particles of gAd.shSCR or gAd.shSREBP1 through the tail vein, and euthanized 3 weeks later under fed (ad libitum) or 24-h fasted conditions. Groups of four normal C57BLKS mice were used as controls and received the same dose of gAd.shSCR adenovirus. A, effect of silencing SREBP1 on lipogenic enzyme expression. Data are presented as mean ± S.D. (error bars). *, p < 0.05 versus control; n = 7–8. AU, arbitrary units. B, immunoblot analysis for SREBP-1 precursor (p-SREBP) or nuclear (n-SREBP) protein, ACAC, and phosphorylated ACAC (P-ACAC), 3 weeks after gAd.shSCR or gAd.shSREBP1 administration. ACAC and P-ACAC panels show ACACA as a bottom 265-kDa band and ACACB as a top 280-kDa band. C, hepatic triglyceride levels in db/db mice; *, p < 0.05 versus control; n = 8.
Silencing SREBP-1 resulted in lower liver triglyceride levels in the db/db gAd.shSREBP1 group compared with the gAd.shSCR under fasting conditions (Fig. 2C). Using 2H2O as a metabolic tracer, de novo liver palmitate and total liver palmitate were found to be reduced 70 and 30%, respectively, confirming that down-regulation of enzymes in the lipogenesis pathway resulted in reduced levels of glucose conversion into lipid (Fig. 3A). The decline in de novo lipogenesis (as well as in ACAC levels) was expected to decrease malonyl-CoA, an allosteric inhibitor of CPT1A. CPT1A is the enzyme that transfers an acyl group from CoA to carnitine, which is then translocated into mitochondria for oxidation. Lower malonyl-CoA levels were expected to increase CPT1A activity and subsequently, enhance fatty acid oxidation (FAO). As anticipated, malonyl-CoA levels were depleted to near half the levels observed in control animals (Fig. 3A). We confirmed that CPT1A and CPT2 protein levels were not changed between gAd.shSCR and gAd.shSREBP1-treated mice, under fed or 24-h fasting conditions (Fig. 3B), and no difference was observed in CPT1A IC50 values for malonyl-CoA between the two groups, suggesting similar capacity to be inhibited by this metabolite (Fig. 3B). Nevertheless, no evidence of increased FAO was observed in the gAd.shSREBP1-treated group, based on serum β-hydroxybutyrate and liver BHB-CoA levels (Fig. 3, A and C). Overall, these data suggest that SREBP-1 deficiency had a positive impact on hepatic steatosis, most likely as a result of reduced lipogenesis rather than increased FAO.
FIGURE 3.
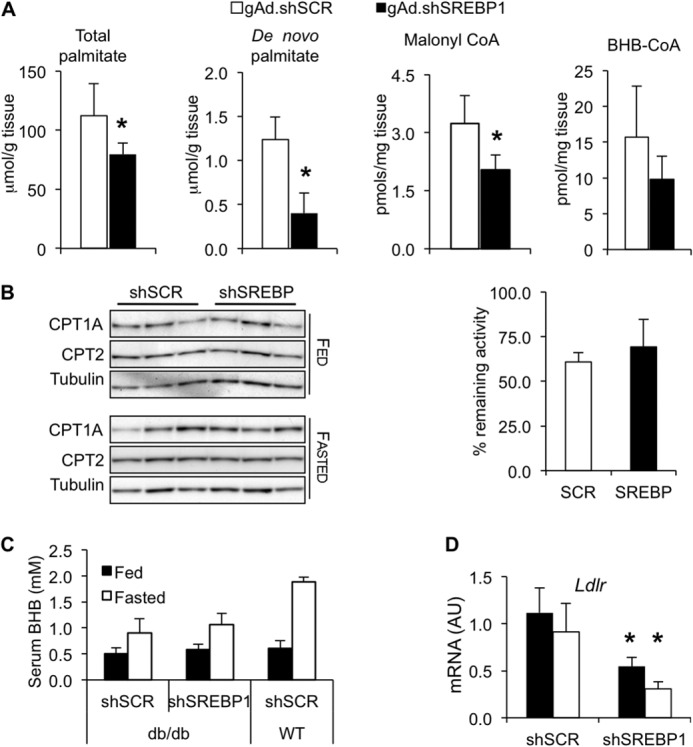
SREBP-1 silencing decreases de novo lipogenesis. A, groups of 8 db/db mice were administered 1 × 1011 viral particles of gAd.shSCR or gAd.shSREBP1. Three weeks later, 2H2O was administered intraperitoneally. Mice were fasted for 4 h and euthanized. Total palmitate, de novo synthesis of palmitate, malonyl-CoA, and BHB-CoA were analyzed (p < 0.05 versus control; n = 8). B, mice were treated as described in Fig. 2. CPT1A and CPT2 protein levels under fed and fasted conditions and CPT1A sensitivity to malonyl-CoA in livers of mice fasted for 24 h were determined. n = 5. C, mice were treated as described in Fig. 2. BHB serum levels are shown; n = 4–5 WT, n = 8 db/db. D, LDL receptor (Ldlr) mRNA expression in db/db mice; n = 7–8. AU, arbitrary units.
Serum triglyceride levels were found mildly elevated in SREBP-1-depleted mice compared with diabetic controls under fasting conditions (Table 1), and this correlated with a significant reduction of LDL receptor mRNA (Fig. 3D), indicating decreased uptake of lipoproteins by the liver. The importance of SREBP-1a as regulator of the low density lipoprotein receptor and its control of lipoprotein uptake/degradation in the liver has been highlighted in transgenic mice overexpressing SREBP-1a in this tissue, which display markedly increased levels of LDL receptor (24). Whereas null Ldlr mice accumulate triglyceride and cholesterol in serum, mice overexpressing SREBP-1a do not accumulate these metabolites in plasma because they are degraded through the action of LDL receptors (24). Furthermore, transgenic mice overexpressing SREBP-1a or SREBP-1c in liver have significantly higher levels of triglycerides in liver and lower levels in serum (25). Likewise, adenovirus-mediated SREBP-1c overexpression in liver induced a 2-fold increase in hepatic triglyceride levels and up-regulation of the LDL-receptor, leading to reduced serum levels of triglycerides (26).
TABLE 1.
Effect of SREBP-1 silencing on weight and serum chemistries
Mice were treated as described in the legend of Fig. 2. *, p < 0.05 db/db gAd.shSREBP1 versus gAd.shSCR; **, p < 0.05 compared with WT. n = 8 db/db, n = 4–5 WT.
|
db/db |
WT gAd.shSCR | ||
|---|---|---|---|
| gAd.shSCR | gAd.shSREBP | ||
| Total body weight (g) | 44.7 ± 1.92** | 40.3 ± 3.65*,** | 22.7 ± 1.32 |
| Feda | |||
| Liver weight (g) | 2.63 ± 0.22 | 1.83 ± 0.18* | 1.10 ± 0.06 |
| Serum FFAs (mm) | 1.55 ± 0.27** | 2.23 ± 0.28*,** | 0.96 ± 0.06 |
| Serum TG (mg/ml) | 1.17 ± 0.27** | 1.41 ± 0.44** | 0.75 ± 0.06 |
| Fasted | |||
| Liver weight (g) | 2.18 ± 0.20 | 1.46 ± 0.16* | 0.86 ± 0.05 |
| Serum FFAs (mm) | 1.32 ± 0.22 | 2.05 ± 0.14*,** | 1.25 ± 0.1 |
| Serum TG (mg/ml) | 0.63 ± 0.08 | 1.01 ± 0.11*,** | 0.74 ± 0.1 |
a FFA, free fatty acid; TG, triglyceride.
Increased Expression of Gluconeogenesis Genes and Lower Glycogen Accumulation in SREBP-1-depleted db/db Mice
Despite lower liver steatosis, acutely silencing SREBP-1 in db/db mice had no impact on glucose and insulin levels (Fig. 4A). Furthermore, mice were slightly more hyperglycemic upon a 24-h fast, suggesting a defect in the regulation of glucose homeostasis. To determine the impact of silencing hepatic SREBP-1 on glucose tolerance, glucose and insulin tolerance tests were performed. Consistent with the lack of glucose reduction, no differences between the gAd.shSREBP1- and gAd.shSCR-treated groups were observed (Fig. 4B). Thus, hepatic SREBP-1 silencing has no major impact on whole body glucose tolerance.
FIGURE 4.
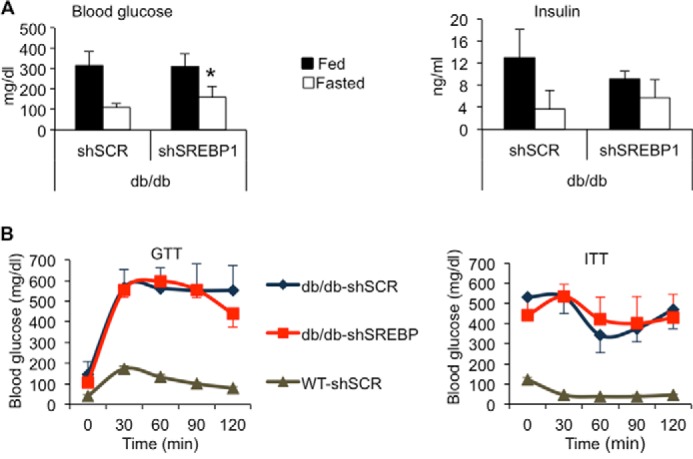
SREBP-1 silencing does not improve whole body glucose or insulin tolerance. Mice were treated as described in Fig. 2. A, glucose and insulin levels in serum; n = 5–8. B, glucose tolerance test (GTT) conducted after overnight fast and intraperitoneal administration of a glucose bolus (1 mg/g body weight). An insulin tolerance test (ITT) was carried out after fasting animals for 5 h and intraperitoneal administration of human regular insulin (1 unit/kg); n = 7 db/db, n = 5 WT.
To investigate the basis of the lack of improvement in glucose homeostasis, glycogen accumulation was measured. Remarkably, SREBP-1 silencing resulted in ∼50% lower glycogen levels under fed conditions, compared with db/db control mice, as previously seen in normal mice (Fig. 5A). In addition, glycogen synthase 2 (GYS2) mRNA, glycogen synthase activity, and protein levels were decreased, relative to diabetic control mice (Fig. 5, B–D). These observations were recapitulated in another animal model of obesity and type 2 diabetes, the ob/ob mouse (data not shown).
FIGURE 5.

SREBP-1 silencing alters gluconeogenesis and glycogen synthesis. db/db mice were treated as described in Fig. 2. A, hepatic glycogen content in db/db mice; n = 8. B, glycogen synthase 2 mRNA levels; n = 7–8. AU, arbitrary units. C, immunoblot analysis of total and phosphorylated glycogen synthase. D, ratio of the glycogen synthase activity measured with low (L) over high (H) glucose 6-phosphate, under fed and fasted conditions; n = 3; *, p < 0.05 versus control. E, Gck, Pklr, Pck1, G6pc mRNA expression; n = 7–8. F, PCK1 protein levels. Error bars, S.D.
To further understand the role of SREBP-1 at regulating carbohydrate metabolism, we analyzed expression of limiting enzymes in the glycolysis and gluconeogenesis pathways, including glucokinase (Gck), liver-pyruvate kinase (Pklr), phosphoenolpyruvate carboxykinase (Pck1), and glucose-6-phosphatase (G6pc). SREBP-1 overexpression has been shown to induce glucokinase gene expression (26). Thus, lack of SREBP-1 was expected to reduce its levels, which could have affected glycogen synthesis. Gck levels were not altered upon SREBP-1 silencing (Fig. 5E). However, Pklr gene expression was severely blunted compared with diabetic control animals, indicating reduced activity of the glycolysis pathway (Fig. 5E). In contrast, gluconeogenesis genes Pck1 and G6pc were significantly higher in the gAd.shSREBP1-treated group. PCK1 mRNA levels were >2-fold higher, both under fed and fasting conditions, and protein levels were also increased (Fig. 5, E and F). Overall, our data suggest that increased PCK1 levels, reduced PKLR activity, and reduced glycogen storage capacity may have accounted for lack of improved glycemia in the gAd.shSREBP1-treated db/db animals (Fig. 4A).
DISCUSSION
Hepatic fat accumulation is strongly associated with insulin resistance and inappropriate inhibition of hepatic glucose output. A large amount of literature has linked high SREBP-1 activity with insulin resistance and type 2 diabetes. A variety of gene ablation as well as insulin-sensitizing drug studies have shown concomitant reductions in SREBP-1 and insulin resistance. For example, it has been suggested that the beneficial effects of resveratrol and other polyphenols on hepatic steatosis and insulin sensitivity is mediated by AMPK, which phosphorylates and inactivates SREBP-1 (27). Recently, it has been proposed that multiple nuclear receptors, including ERα, CAR, LRH-1 TRβ, and FXR/SHP, exert their antidiabetic effects through down-regulation of SREBP-1c (28), placing this transcription factor at the crossroads between steatosis and hepatic insulin resistance. Nevertheless, these studies are in contrast with data generated in leptin-deficient (ob/ob) SREBP-1 whole body knock-out mice (9). Lepob/ob × Srebp-1−/− mice remain hyperglycemic despite a 50% decrease in hepatic lipid accumulation (9). Given that this study did not provide a mechanism for the lack of improvement in insulin resistance, we hypothesized that SREBP-1 may play a role in other aspects of liver metabolism. Thus, we knocked down SREBP-1 in normal mice and in db/db mice to clarify the role of this transcription factor on carbohydrate metabolism as well as investigate the therapeutic value of silencing SREBP-1 in the liver of a mouse model with established steatosis and severe insulin resistance. This is the first comprehensive in vivo study addressing both carbohydrate and lipid metabolism and the only adult model of hepatic SREBP-1 deficiency in normal and obese mice.
The results of this study have underscored the requirement for SREBP-1 in glycogen synthesis in the fed state. SREBP-1 deficiency in db/db mice decreased glycogen levels as well as glycogen synthase mRNA, protein, and enzyme activity during fed conditions, when glycogen synthesis takes place. These observations were also observed in normal mice, implying that SREBP-1 is needed to regulate glycogen metabolism appropriately. The effects on glycogen were independent from glucokinase gene expression, which was unaffected by SREBP-1 silencing. Glucokinase gene expression has been shown to be up-regulated in response to SREBP-1c overexpression (26), although other studies have shown that increases in SREBP-1c were neither necessary nor sufficient to induce glucokinase (29, 30). Our study confirms that in vivo, SREBP-1 is dispensable for expression of this gene.
In addition to alterations in glycogen synthesis, up-regulated Pck1 and G6Pase gene expression was observed under fed and fasted conditions. In vitro studies have previously demonstrated that SREBP-1 negatively regulates Pck1 gene expression by binding to two sterol regulatory elements present in the promoter of the gene (31). SREBP-1 has also been shown to inhibit Pck1 gene transcription by interacting with HNF4α and interfering with the recruitment of the transcription co-activator PGC-1α to the Pck1 promoter (32). Here we show that in vivo, SREBP-1 deficiency results in lack of appropriate inhibition of Pck1 and G6pase gene expression. Finally, Pklr was found severely diminished. Overall, this study provides evidence that SREBP-1 controls carbohydrate metabolism during fed conditions by promoting glycogen synthesis, enhancing glycolysis, and inhibiting gluconeogenic gene expression.
The importance of the de novo lipogenesis pathway as a contributor of hepatic steatosis has been underscored in a study using metabolic tracers in human patients with non-alcoholic fatty liver disease (33). In these individuals, as much as 26% of triglycerides are derived from this pathway (33). In addition, de novo lipogenesis is elevated in the fasted state and does not increase postprandially, indicating that one of the underlying causes of hepatic steatosis in non-alcoholic fatty liver disease might be the inability of the liver to regulate this pathway appropriately (33). As expected, SREBP-1 silencing resulted in decreased expression of de novo lipogenesis genes, including Fasn, Acacb, Gpam, and Scd1. Consistent with the decrease in gene expression, de novo synthesis of palmitate was 70% lower in livers of gAd.shSREBP1-treated animals relative to control db/db mice, and liver triglycerides were significantly decreased under fasting conditions. This indicates that, as observed in humans with non-alcoholic fatty liver disease (33), enhanced de novo lipogenesis in the fasted state is one of the underlying causes of hepatic lipid accumulation in db/db mice and targeting SREBP-1 lowered steatosis.
ACACB co-localizes spatially with CPT1A on the mitochondrial membrane (34), and it has been suggested that the malonyl-CoA produced from this isoform inhibits CPT1A activity and FAO, whereas the malonyl-CoA produced by ACACA is used to generate palmitate (34). Nevertheless, liver-specific Acaca-deficient mice compensate for lack of ACACA by increasing ACACB expression and maintain physiological levels of de novo lipogenesis and FAO, suggesting that compartmentalization of malonyl-CoA produced by each ACAC isoform is not stringent (35). Increased activities of transcription factors SREBP-1 and carbohydrate-responsive element-binding protein (ChREBP) and have been observed in db/db and ob/ob mice, animal models of obesity and type 2 diabetes. Silencing ChREBP in liver of ob/ob mice resulted in a significant reduction in ACACA and ACACB, de novo lipogenesis and triglyceride levels, as well as increased serum levels of β-hydroxybutyrate, which was attributed to higher FAO rates (36). In contrast, SREBP-1 silencing in db/db mice largely reduced ACACB, but had no effect on ACACA, the predominant isoform in liver. Serum β-hydroxybutyrate levels were not elevated under fed or 24-h fasting conditions. Furthermore, the levels of liver BHB-CoA, an intermediate of fatty acid oxidation (20, 37), were not significantly different between the two groups. Given that SREBP-1 silencing did not reduce ACACA, these data suggest that both isoforms need to be diminished to reduce malonyl-CoA to levels that allow increasing CPT1A activity. Thus, depleting SREBP-1 in db/db mice reduced lipogenesis, but FAO was not enhanced.
A growing number of studies are reporting a dissociation between hepatic steatosis and insulin resistance (38–40). Overexpression of diacylglycerol O-acyltransferase 2 (DGAT2) in the liver increased triglyceride, ceramide, and diacylglycerol without alterations on insulin or glucose tolerance (38). Gpam deficiency in ob/ob mice diminished SREBP-1 levels with a concomitant decrease in hepatic triglyceride accumulation, but no improvement in hepatic or peripheral insulin sensitivity (40). Furthermore, fasting glucose was exacerbated, whereas the absence of Gpam in lean mice had no effect on glucose, similar to what we observed in gAd.shSREBP1-treated db/db and lean mice, respectively. SREBP-1 silencing is another example of dissociation between hepatic steatosis and glucose tolerance. Our data provide evidence of additional roles for SREBP-1 at regulating metabolism, besides its established function at facilitating lipogenesis. Thus, SREBP-1 coordinates changes in gene expression physiologically important during the fed state: enhancing de novo lipogenesis and glycogen synthesis, facilitating glycolysis, and inhibiting gluconeogenesis.
In conclusion, this work provides evidence of novel roles for SREBP-1, a critical transcription factor that has been extensively associated with hepatic steatosis and insulin resistance. Our data indicate that SREBP-1 depletion has profound effects on expression of genes needed for carbohydrate metabolism. Hence, the benefits of reducing de novo lipogenesis in db/db mice were offset by the negative impact on gluconeogenesis and glycogen synthesis.
Acknowledgments
We thank Drs. Robert Harris and Wenhong Cao for critical reading of the manuscript, Dr. Henri Brunengraber for advice with metabolic tracer analysis, and Dr. Carina Prip-Buus for providing the antibody against CPT1A and CPT2. We thank the Mouse Metabolic Phenotyping Center (MMPC) at Vanderbilt University for lipid analyses, and the MMPC at Case Western Reserve University for the metabolic tracer analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK078595 through the NIDDK. This work was also supported by American Diabetes Association Grant 1-08-RA-135. The Mouse Metabolic Phenotyping Centers at Vanderbilt University and Case Western Reserve University are supported by National Institutes of Health Grants DK59637 and U24-DK76174, respectively.
- SREBP
- sterol regulatory element-binding protein
- ACAC
- acetyl-CoA carboxylase
- BHB
- β-hydroxybutyrate
- CPT
- carnitine palmitoyltransferase
- FAO
- fatty acid oxidation
- PCK
- phosphoenolpyruvate carboxykinase.
REFERENCES
- 1. Browning J. D., Szczepaniak L. S., Dobbins R., Nuremberg P., Horton J. D., Cohen J. C., Grundy S. M., Hobbs H. H. (2004) Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40, 1387–1395 [DOI] [PubMed] [Google Scholar]
- 2. Szczepaniak L. S., Nurenberg P., Leonard D., Browning J. D., Reingold J. S., Grundy S., Hobbs H. H., Dobbins R. L. (2005) Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 288, E462–468 [DOI] [PubMed] [Google Scholar]
- 3. Richard J., Lingvay I. (2011) Hepatic steatosis and type 2 diabetes: current and future treatment considerations. Expert Rev. Cardiovasc. Ther. 9, 321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seppälä-Lindroos A., Vehkavaara S., Häkkinen A. M., Goto T., Westerbacka J., Sovijärvi A., Halavaara J., Yki-Järvinen H. (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 87, 3023–3028 [DOI] [PubMed] [Google Scholar]
- 5. Brown M. S., Goldstein J. L. (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96 [DOI] [PubMed] [Google Scholar]
- 6. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pettinelli P., Del Pozo T., Araya J., Rodrigo R., Araya A. V., Smok G., Csendes A., Gutierrez L., Rojas J., Korn O., Maluenda F., Diaz J. C., Rencoret G., Braghetto I., Castillo J., Poniachik J., Videla L. A. (2009) Enhancement in liver SREBP-1c/PPAR-α ratio and steatosis in obese patients: correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta 1792, 1080–1086 [DOI] [PubMed] [Google Scholar]
- 8. Shimomura I., Bashmakov Y., Horton J. D. (1999) Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 274, 30028–30032 [DOI] [PubMed] [Google Scholar]
- 9. Yahagi N., Shimano H., Hasty A. H., Matsuzaka T., Ide T., Yoshikawa T., Amemiya-Kudo M., Tomita S., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., Osuga J., Harada K., Gotoda T., Nagai R., Ishibashi S., Yamada N. (2002) Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lepob/Lepob mice. J. Biol. Chem. 277, 19353–19357 [DOI] [PubMed] [Google Scholar]
- 10. Parks R. J., Chen L., Anton M., Sankar U., Rudnicki M. A., Graham F. L. (1996) A helper-dependent adenovirus vector system: removal of helper virus by cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U.S.A. 93, 13565–13570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sandig V., Youil R., Bett A. J., Franlin L. L., Oshima M., Maione D., Wang F., Metzker M. L., Savino R., Caskey C. T. (2000) Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morral N., O'Neal W., Rice K., Leland M., Kaplan J., Piedra P. A., Zhou H., Parks R. J., Velji R., Aguilar-Córdova E., Wadsworth S., Graham F. L., Kochanek S., Carey K. D., Beaudet A. L. (1999) Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. U.S.A. 96, 12816–12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim I. H., Józkowicz A., Piedra P. A., Oka K., Chan L. (2001) Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. U.S.A. 98, 13282–13287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morral N., Parks R. J., Zhou H., Langston C., Schiedner G., Quinones J., Graham F. L., Kochanek S., Beaudet A. L. (1998) High doses of a helper-dependent adenoviral vector yield supraphysiological levels of α1-antitrypsin with negligible toxicity. Hum. Gene Ther. 9, 2709–2716 [DOI] [PubMed] [Google Scholar]
- 15. Schiedner G., Morral N., Parks R. J., Wu Y., Koopmans S. C., Langston C., Graham F. L., Beaudet A. L., Kochanek S. (1998) Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 18, 180–183 [DOI] [PubMed] [Google Scholar]
- 16. Ruiz R., Witting S. R., Saxena R., Morral N. (2009) Robust hepatic gene silencing for functional studies using helper-dependent adenovirus vectors. Hum. Gene Ther. 20, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brunengraber D. Z., McCabe B. J., Kasumov T., Alexander J. C., Chandramouli V., Previs S. F. (2003) Influence of diet on the modeling of adipose tissue triglycerides during growth. Am. J. Physiol. Endocrinol. Metab. 285, E917–925 [DOI] [PubMed] [Google Scholar]
- 18. Bederman I. R., Foy S., Chandramouli V., Alexander J. C., Previs S. F. (2009) Triglyceride synthesis in epididymal adipose tissue: contribution of glucose and non-glucose carbon sources. J. Biol. Chem. 284, 6101–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris S. R., Zhang G. F., Sadhukhan S., Murphy A. M., Tomcik K. A., Vazquez E. J., Anderson V. E., Tochtrop G. P., Brunengraber H. (2011) Metabolism of levulinate in perfused rat livers and live rats: conversion to the drug of abuse 4-hydroxypentanoate. J. Biol. Chem. 286, 5895–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Q., Tomcik K., Zhang S., Puchowicz M. A., Zhang G. F. (2012) Dietary regulation of catabolic disposal of 4-hydroxynonenal analogs in rat liver. Free Rad. Biol. Med. 52, 1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki Y., Lanner C., Kim J. H., Vilardo P. G., Zhang H., Yang J., Cooper L. D., Steele M., Kennedy A., Bock C. B., Scrimgeour A., Lawrence J. C., Jr., DePaoli-Roach A. A. (2001) Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol. Cell. Biol. 21, 2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guinovart J. J., Salavert A., Massagué J., Ciudad C. J., Salsas E., Itarte E. (1979) Glycogen synthase: a new activity ratio assay expressing a high sensitivity to the phosphorylation state. FEBS Lett. 106, 284–288 [DOI] [PubMed] [Google Scholar]
- 23. Hoppel C. L., Kerner J., Turkaly P., Turkaly J., Tandler B. (1998) The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J. Biol. Chem. 273, 23495–23503 [DOI] [PubMed] [Google Scholar]
- 24. Horton J. D., Shimano H., Hamilton R. L., Brown M. S., Goldstein J. L. (1999) Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J. Clin. Invest. 103, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimano H., Horton J. D., Shimomura I., Hammer R. E., Brown M. S., Goldstein J. L. (1997) Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Invest. 99, 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bécard D., Hainault I., Azzout-Marniche D., Bertry-Coussot L., Ferré P., Foufelle F. (2001) Adenovirus-mediated overexpression of sterol regulatory element binding protein-1c mimics insulin effects on hepatic gene expression and glucose homeostasis in diabetic mice. Diabetes 50, 2425–2430 [DOI] [PubMed] [Google Scholar]
- 27. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., Cohen R. A., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore D. D. (2012) Nuclear receptors reverse McGarry's vicious cycle to insulin resistance. Cell Metab. 15, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hansmannel F., Mordier S., Iynedjian P. B. (2006) Insulin induction of glucokinase and fatty acid synthase in hepatocytes: analysis of the roles of sterol-regulatory-element-binding protein-1c and liver X receptor. Biochem. J. 399, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stoeckman A. K., Towle H. C. (2002) The role of SREBP-1c in nutritional regulation of lipogenic enzyme gene expression. J. Biol. Chem. 277, 27029–27035 [DOI] [PubMed] [Google Scholar]
- 31. Chakravarty K., Wu S. Y., Chiang C. M., Samols D., Hanson R. W. (2004) SREBP-1c and Sp1 interact to regulate transcription of the gene for phosphoenolpyruvate carboxykinase (GTP) in the liver. J. Biol. Chem. 279, 15385–15395 [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto T., Shimano H., Nakagawa Y., Ide T., Yahagi N., Matsuzaka T., Nakakuki M., Takahashi A., Suzuki H., Sone H., Toyoshima H., Sato R., Yamada N. (2004) SREBP-1 interacts with hepatocyte nuclear factor-4α and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J. Biol. Chem. 279, 12027–12035 [DOI] [PubMed] [Google Scholar]
- 33. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abu-Elheiga L., Matzuk M. M., Abo-Hashema K. A., Wakil S. J. (2001) Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291, 2613–2616 [DOI] [PubMed] [Google Scholar]
- 35. Harada N., Oda Z., Hara Y., Fujinami K., Okawa M., Ohbuchi K., Yonemoto M., Ikeda Y., Ohwaki K., Aragane K., Tamai Y., Kusunoki J. (2007) Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol. Cell. Biol. 27, 1881–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dentin R., Benhamed F., Hainault I., Fauveau V., Foufelle F., Dyck J. R., Girard J., Postic C. (2006) Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55, 2159–2170 [DOI] [PubMed] [Google Scholar]
- 37. Deng S., Zhang G. F., Kasumov T., Roe C. R., Brunengraber H. (2009) Interrelations between C4 ketogenesis, C5 ketogenesis, and anaplerosis in the perfused rat liver. J. Biol. Chem. 284, 27799–27807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monetti M., Levin M. C., Watt M. J., Sajan M. P., Marmor S., Hubbard B. K., Stevens R. D., Bain J. R., Newgard C. B., Farese R. V., Sr., Hevener A. L., Farese R. V., Jr. (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 6, 69–78 [DOI] [PubMed] [Google Scholar]
- 39. Minehira K., Young S. G., Villanueva C. J., Yetukuri L., Oresic M., Hellerstein M. K., Farese R. V., Jr., Horton J. D., Preitner F., Thorens B., Tappy L. (2008) Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J. Lipid Res. 49, 2038–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wendel A. A., Li L. O., Li Y., Cline G. W., Shulman G. I., Coleman R. A. (2010) Glycerol-3-phosphate acyltransferase 1 deficiency in ob/ob mice diminishes hepatic steatosis but does not protect against insulin resistance or obesity. Diabetes 59, 1321–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]