

Background: Protein-protein interactions often involve intrinsically disordered protein domains.
Results: The binding interface between two disordered protein domains is suboptimal, or frustrated, with regard to the energetics.
Conclusion: The frustration likely results from the promiscuous binding behavior of these disordered domains.
Significance: Suboptimal binding interfaces may be common among intrinsically disordered proteins with multiple binding partners.
Keywords: Intrinsically Disordered Proteins, Kinetics, Protein Domains, Protein Engineering, Protein-Protein Interactions
Abstract
Intrinsically disordered proteins are very common in the eukaryotic proteome, and many of them are associated with diseases. Disordered proteins usually undergo a coupled binding and folding reaction and often interact with many different binding partners. Using double mutant cycles, we mapped the energy landscape of the binding interface for two interacting disordered domains and found it to be largely suboptimal in terms of interaction free energies, despite relatively high affinity. These data depict a frustrated energy landscape for interactions involving intrinsically disordered proteins, which is likely a result of their functional promiscuity.
Introduction
Intrinsically disordered proteins (IDPs)3 are abundant in the eukaryotic proteome and participate in various cellular processes such as signaling, cell cycle control, transcription, and translation (1). Due to their frequent occurrence and the fact that many IDPs are involved in diseases such as cancer and neurodegenerative disorders such as Parkinson and Alzheimer (2), the relationship between disorder and (dys)function has been the subject of intense research in the last decade. A recurrently cited reason for the prevalence of IDPs is that disorder enables a combination of low affinity and high specificity and that it also facilitates binding to several different targets (1). However, this binding malleability could also lead to interactions with targets for which IDPs were not intended to bind and hence lower their specificity. This apparent contradiction leads to the following question. How optimized are the interactions involving IDPs? To address this issue, we have analyzed in detail the largely hydrophobic interaction surface between the molten globule nuclear coactivator-binding domain (NCBD) of CREB-binding protein and the fully disordered activation domain from the p160 transcription coactivator for thyroid hormone and retinoid receptors (ACTR) (3–6).
We have previously used ACTR/NCBD as a model system to investigate the mechanisms of coupled binding and folding (7–9). The association between NCBD and ACTR is fast, with a transition state that contains only a few native hydrophobic interactions, followed by a cooperative formation of native contacts after the rate-limiting barrier, resembling the nucleation condensation mechanism in protein folding (10).
In this work, we used protein engineering, namely the double mutant cycle (11), in combination with kinetic experiments to map in detail the binding interface between ACTR and NCBD. We found that the binding interface contains energetic frustration and propose that this is a necessary result of the promiscuity of IDPs such as ACTR and NCBD.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human NCBD and ACTR were expressed and purified as described previously (7, 8).
Binding Kinetics
Kinetic measurements of the interactions between mutant NCBD and mutant ACTR were performed using an upgraded SX-17MV stopped-flow spectrometer (Applied Photophysics, Leatherhead, United Kingdom). Stopped-flow binding data for single mutant interactions with the wild type are from Dogan et al. (8). The change in Trp fluorescence was monitored, with excitation at 280 nm and a 320-nm long-pass emission filter. Experiments were done at 277 K in 20 mm sodium phosphate (pH 7.4) and 150 mm NaCl. Apparent association rate constants of binding (konapp) were obtained by measuring kobs values for binding at different concentrations of ACTR (0.5–25 μm) and 1 μm NCBD. The data (kobs versus [ACTR]) were fitted to the general equation for a reversible association of two molecules (12). Displacement experiments were performed to determine apparent dissociation rate constants of binding (koffapp) by mixing a preformed ACTR-NCBD complex with an excess of NCBDWT (7, 8). Kd values were obtained by taking the ratio of koffapp to konapp. Calculation of ΔΔΔGc was performed as described previously (13).
Isothermal Titration Calorimetry Experiments
The thermodynamics of ACTR/NCBD association was characterized using an iTC200 isothermal titration calorimeter (GE Healthcare). Proteins were dialyzed prior to isothermal titration calorimetry measurements, which were performed in 20 mm sodium phosphate (pH 7.4) and 150 mm NaCl and at 278 K. NCBD (150–365 μm) was titrated into an ACTR solution with concentrations ranging from 15 to 30 μm. Each titration series consisted of an initial 0.4- or 0.5-μl injection, followed by 18 subsequent 2-μl injections. Data were corrected for the heats of dilution and fitted to a one-to-one binding model using the Origin software provided with the iTC200 instrument.
RESULTS
We mapped the energetics of the binding interface between ACTR and NCBD using site-directed mutagenesis and binding kinetics. In a previous study (8), we made 10 deletion mutations at hydrophobic positions in both ACTR and NCBD and performed a Φ value analysis for binding. Here, we used the same 20 mutants to measure the coupling free energies of binding (ΔΔΔGc) between different positions in ACTR-NCBD using the double mutant cycle analysis (11). The side chains subjected to mutation are in the interface of ACTR and NCBD as defined by the NMR structure (4). Thus, our measured ΔΔΔGc values reflect the energetic cross-talk in the binding interface.
Stopped-flow fluorometry was used to determine the binding dissociation constants (Kd) as the ratio between koff and kon for each pair of mutants. These Kd values are identical within error to Kd values determined by isothermal titration calorimetry (Fig. 1). ΔΔΔGc values were obtained from Kd values as described in detail previously (13, 14). Briefly, the Kd values of four bimolecular ACTR-NCBD complexes were measured in each cycle (7, 8): the wild-type complex, single mutants X and Y, and the double mutant (in which both X and Y mutations are present), as illustrated in Fig. 2. If the sum of the differences in the free energy of binding between the wild-type and single mutant complexes (ΔΔGmutant−X + ΔΔGmutant−Y) is identical to ΔΔGmutant−XY when both mutations are present, then the free energies are additive, and positions X and Y are not energetically coupled to each other. However, if ΔΔΔGc does not equal zero, then a cross-talk in terms of free energy between X and Y exists.
FIGURE 1.

Comparison of equilibrium dissociation binding constants (Kd) obtained by stopped-flow fluorometry (NCBDY2108W/ACTRWT (a) and NCBDY2108W/ACTRL1049A (b)) and isothermal titration calorimetry (NCBDY2108W/ACTRWT (c) and NCBDY2108W/ACTRL1049A (d)). Observed rate constants (kobs) are plotted against the concentration of ACTR, and from the fitting (12), the apparent association rate constant of binding (konapp) is obtained. The insets show a trace in a displacement experiment in which a preformed NCBDY2108W/ACTR solution was mixed with an excess of NCBDWT to obtain the apparent dissociation rate constant (koffapp). Stopped-flow data shown in a are from Dogan et al. (7), and data shown in b are from Dogan et al. (8). The two different methods produced the same Kd.
FIGURE 2.
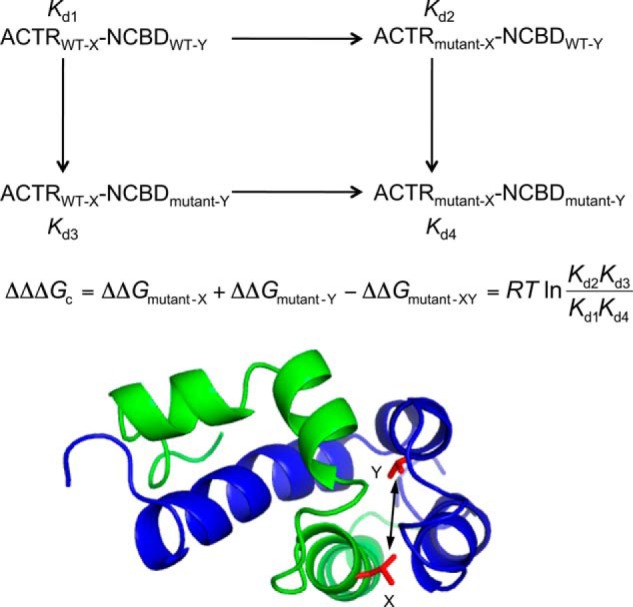
In the double mutant cycle, the dissociation constant (Kd) of four complexes are measured to calculate the coupling free energy (ΔΔΔGc) between positions X and Y (shown in red) in ACTR (green) and NCBD (blue), respectively.
If the wild-type protein-protein interaction is highly optimized in terms of all intra- and intermolecular interactions, it is likely that it becomes less optimized by the first mutation in the cycle. This means that the first mutation weakens the effect of the second mutation, and ΔΔΔGc becomes positive. Conversely, if the first mutation boosts the effect of the second mutation, then ΔΔΔGc becomes negative, which is a sign of a less optimized wild-type complex.
We were able to measure the interaction free energy for 63 pairs of amino acid side chains situated in the interface between ACTR and NCBD in the complex (Table 1). ΔΔΔGc values for several other pairs were not possible to obtain by the stopped-flow technique due to elevated observed rate constants (kobs), which in turn result from severe destabilization of the ACTR-NCBD complex upon double mutation. Nevertheless, the obtained values were well distributed, thus ensuring a good coverage of the whole ACTR-NCBD binding surface. The coupling free energies for a few positions are, to some extent, distance-dependent. However, when all of the coupling free energies are plotted against the distance between the positions, no clear correlation is observed (Fig. 3).
TABLE 1.
Coupling free energies (ΔΔΔGc) between residues in NCBD and ACTR
| NCBD mutant | ACTR mutant | ΔΔΔGc | Error |
|---|---|---|---|
| kcal/mol | kcal/mol | ||
| I2062V | L1049A | 0.09 | 0.03 |
| I2062V | L1048A | −0.09 | 0.03 |
| I2062V | I1067V | −0.11 | 0.02 |
| I2062V | V1077A | −0.12 | 0.03 |
| I2062V | L1055A | −0.16 | 0.06 |
| I2062V | I1073V | 0.02 | 0.05 |
| L2067A | L1048A | 0.44 | 0.04 |
| L2067A | L1049A | 0.40 | 0.04 |
| L2067A | L1055A | −0.58 | 0.07 |
| L2067Aa | I1067Va | −0.32 | 0.06 |
| L2067A | V1077A | −0.09 | 0.04 |
| L2067Aa | I1073Va | −0.13 | 0.10 |
| L2067A | A1061G | −0.17 | 0.06 |
| L2070A | L1055A | −0.82 | 0.10 |
| L2070A | L1048A | −0.23 | 0.09 |
| L2070A | L1049A | 0.52 | 0.06 |
| L2070A | I1067V | −0.33 | 0.11 |
| L2070Aa | I1073Va | −0.29 | 0.08 |
| L2070Aa | A1061Ga | −0.94 | 0.11 |
| L2074A | L1055A | −0.46 | 0.10 |
| L2074A | I1067V | −0.24 | 0.09 |
| L2074A | L1048A | −0.15 | 0.04 |
| L2074A | L1049A | 0.49 | 0.05 |
| L2074A | A1061G | −0.77 | 0.11 |
| L2074Aa | I1073Va | −0.28 | 0.08 |
| V2086A | I1073V | −0.13 | 0.05 |
| V2086A | I1067V | −0.50 | 0.04 |
| V2086A | V1077A | −0.07 | 0.04 |
| V2086A | L1055A | −0.32 | 0.06 |
| V2086Aa | L1048Aa | −0.36 | 0.06 |
| V2086A | L1049A | −0.08 | 0.09 |
| V2086A | A1061G | −0.27 | 0.06 |
| L2087A | I1067V | 0.65 | 0.12 |
| L2087A | V1077A | 0.48 | 0.09 |
| L2087A | L1048A | 1.01 | 0.13 |
| L2087A | A1061G | 0.41 | 0.14 |
| L2087A | I1073V | 0.75 | 0.20 |
| L2087A | L1055A | 0.64 | 0.11 |
| L2096Aa | V1077Aa | −0.17 | 0.11 |
| L2096Aa | L1055Aa | −0.12 | 0.26 |
| L2096Aa | L1048Aa | 0.01 | 0.26 |
| L2096Aa | A1061Ga | −0.72 | 0.21 |
| A2099Ga | L1048Aa | −0.28 | 0.07 |
| A2099G | L1049A | 0.15 | 0.04 |
| A2099G | L1055A | −0.25 | 0.05 |
| A2099Ga | I1067Va | −0.07 | 0.08 |
| A2099Ga | I1073Va | −0.15 | 0.11 |
| A2099G | A1061G | −0.21 | 0.07 |
| A2099G | V1077A | −0.12 | 0.03 |
| I2101V | L1055A | −0.14 | 0.05 |
| I2101V | V1077A | −0.02 | 0.03 |
| I2101V | I1067V | −0.10 | 0.04 |
| I2101V | L1048A | −0.14 | 0.03 |
| I2101Va | L1049Aa | −0.37 | 0.06 |
| I2101V | I1073V | −0.42 | 0.05 |
| I2101V | A1061G | −0.05 | 0.07 |
| V2109A | V1077A | −0.15 | 0.04 |
| V2109A | I1073V | −0.05 | 0.04 |
| V2109A | I1067V | 0.09 | 0.02 |
| V2109A | L1055A | −0.07 | 0.05 |
| V2109A | L1049A | −0.13 | 0.04 |
| V2109A | L1048A | 0.02 | 0.03 |
| V2109Aa | L1056Aa | −0.13 | 0.13 |
a koffapp for the double mutant complex was determined from fitting kobs versus [ACTR] to the general equation for a reversible association of two molecules (12). Otherwise, koffapp was determined using displacement experiments as described under “Experimental Procedures.”
FIGURE 3.
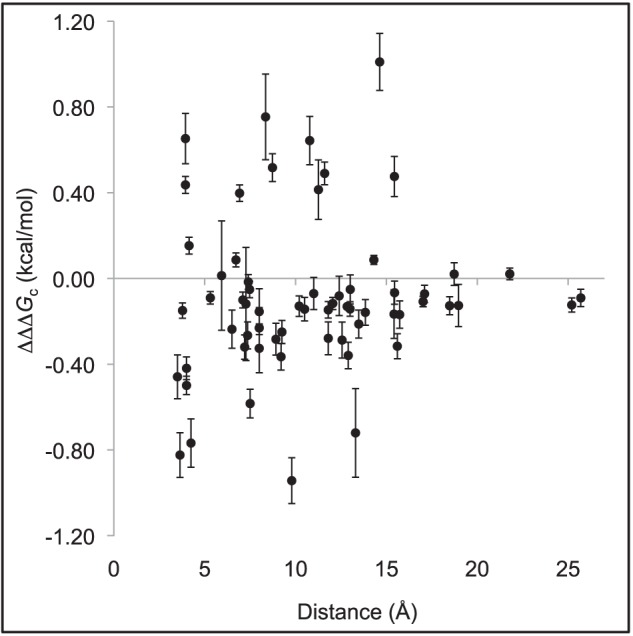
Coupling free energies (ΔΔΔGc; Table 1) between pairs of residues plotted against the shortest distance between them using the first model in the NMR structure of the ACTR-NCBD complex (Protein Data Bank code 1KBH) (4).
DISCUSSION
Despite a recent avalanche of articles on IDPs, there is still a paucity of quantitative data on the interactions in the interface of disordered proteins. Such knowledge is needed for a general understanding of IDP interactions but may also help drug design. We have here investigated in detail the interaction energetics in the interface formed in a coupled binding and folding reaction of two disordered protein domains, ACTR and NCBD, and found it to be rather distinct from those of ordered domains.
The coupling free energies determined for ACTR and NCBD reflect the network of noncovalent bonds holding the two domains together in the complex. Note that our interpretation of ΔΔΔGc values is not dependent on mechanism. Nevertheless, for the following reasons, it is likely that they reflect effects in the binding interface: (i) the nature of the mutations (conservative deletion mutations of hydrophobic residues), (ii) their location (in the interface), and (iii) the effect on koff rather than kon (8), which means that the short-range interdomain hydrophobic interactions break before the rate-limiting step of dissociation.
To visualize the spatial distribution of the interaction energies, we mapped the ΔΔΔGc values onto the structure of the ACTR-NCBD complex (Fig. 4). Surprisingly, ∼80% of the ΔΔΔGc values adopt negative values (Fig. 3 and Table 1), which means that the effect of having both mutations present at the same time is more unfavorable compared with the theoretical combination of single mutations. One interpretation of these negative ΔΔΔGc values is that the binding interface is not fully optimized for the ACTR/NCBD interaction. Previously determined coupling free energies in the context of folded and stable proteins generally displayed positive values (13, 15–19), although for some systems, both positive and negative ΔΔΔGc values have been reported (20). A positive ΔΔΔGc means that the first mutation diminishes the effect of the second mutation, and along the same line of reasoning as above, the interpretation is that the interactions of these ordered proteins are highly specific and optimized throughout the binding interface and even the whole protein domain (Fig. 5a) (13).
FIGURE 4.
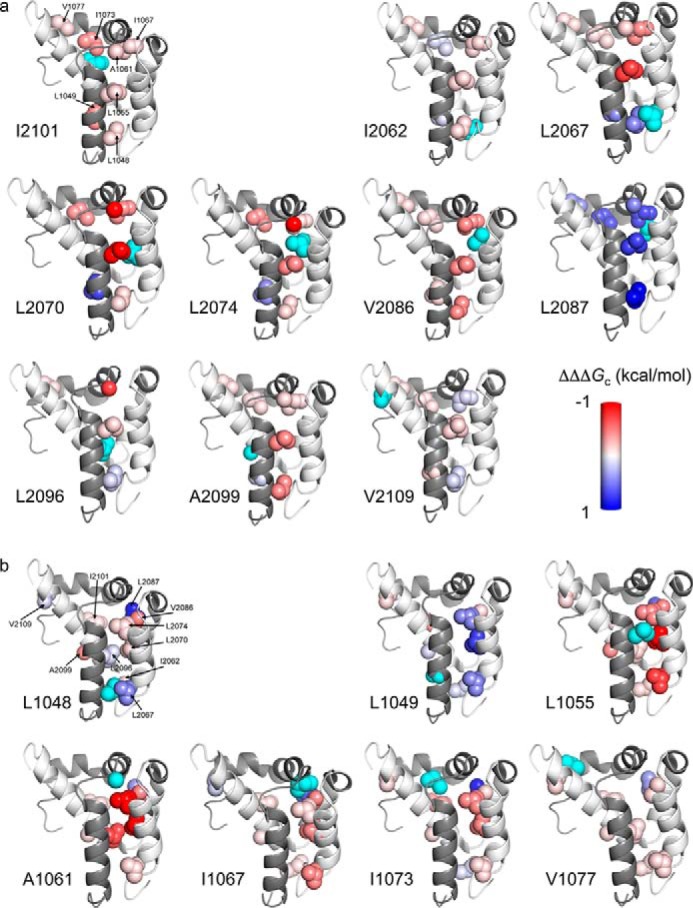
Coupling free energies mapped onto the structure of the bimolecular complex between ACTR and NCBD (Protein Data Bank code 1KBH) (4). Residues are color-coded by ΔΔΔGc values between a certain position colored cyan, as indicated by the label next to each image, in NCBD to different residues in ACTR (a) and in ACTR to different residues in NCBD (b), with a gradient that ranged from −1 (red) to 1 (blue) kcal/mol. Images were generated using PyMOL (39).
FIGURE 5.

Energetic frustration in protein-protein interfaces for two hypothetical complexes with four intermolecular bonds. a, complex between two well folded protein domains, in which all interactions are satisfied and there is no frustration. The first mutation will perturb the interaction network such that the effect of the second mutation is reduced, leading to a positive coupling free energy. b, the interactions in a complex between promiscuous disordered domains are less likely to be optimized and instead contain strain in the form of unfavorable interactions. The first mutation will relax some of the strain and increase the strength of certain interactions. The effect of the second mutation is therefore augmented by the first mutation, leading to a negative coupling free energy. These hypothetical protein-protein interaction networks were drawn using Protein Data Bank files 1BRS and 2L14 for a and b, respectively.
The large number of negative coupling free energies in the interaction surface between ACTR and NCBD may be understood in terms of frustration. The concept of minimal frustration was initially proposed for protein folding (21) but applies also to binding. In a hypothetical fully optimized protein-protein interaction, all possible bonds are satisfied in the complex, and there is no frustration. In the free ground states, however, the respective binding interface may contain frustration, which, upon binding, is minimized due to the favorable interactions between the two proteins. In a protein-protein interface with all interactions satisfied, a perturbation of the interface by mutation may affect neighboring residues and result in a positive coupling free energy in a double mutant cycle. The first mutation introduces strain in the protein-protein complex, which lowers the effect of the second mutation. Conversely, in a suboptimal strained binding interface, a mutation might even “relax,” i.e. stabilize the protein-protein complex and make it more sensitive for the second mutation, leading to a negative coupling free energy (Fig. 5b).
Thus, protein-protein interactions are optimized by minimizing the frustration upon binding. However, IDPs with multiple binding partners, such as ACTR and NCBD, cannot minimize the frustration in the binding interface to the same degree as proteins involved in more specific interactions because their amino acid residues make different interactions with different partners and may even adopt distinct conformations. For example, NCBD adopts a completely different conformation when bound to IRF-3 compared with the ACTR-NCBD complex (22), and the C terminus of p53 has been shown to have at least four different bound conformations depending on the interacting ligand (23). We therefore suggest that, in general, protein-protein interactions involving functionally promiscuous IDPs display more frustration than those of well folded proteins with specific binding partners.
One important implication of this suggestion is that it challenges the prevalent low affinity-high specificity argument for binding of IDPs. It has been previously suggested that the interactions forming the highly hydrophobic binding interfaces by α-helical molecular recognition features (so-called α-MORFs, which undergo a disorder-to-order transition upon binding) are predominantly nonspecific (24). NCBD has been shown to bind to several widely different ligands (4, 22, 25–30). The ACTR-NCBD complex has a rather high affinity (93 nm at physiological salt) resulting from hydrophobic and electrostatic interactions but yet apparently combined with a frustrated interface. There are previous studies that have collected data from literature on the binding kinetics for stable proteins and IDPs (31, 32), and they found that IDPs tend to have higher koff values than folded proteins. It is believed that such a difference would be one of the key advantages of being disordered (33) because it means that complexes formed by IDPs can dissociate rapidly, which is important for proteins acting as hubs in signaling pathways. Thus, it is tempting to speculate that the frustrated interface is related to a high koff (26 s−1 at 25 °C for ACTR-NCBD). However, there are a couple of shortcomings with such tabulations of binding kinetics, such as data being collected at different ionic strengths, temperatures, and pH values and the use of different experimental methods. Therefore, at present moment, it is difficult to make such comparisons and draw any conclusions about the magnitudes of binding rate constants when comparing IDPs with stable and folded proteins. However, it is clear that IDPs interact with their targets with a wide range of binding affinities, from picomolar to micromolar (34–37). The most likely general scenario for IDPs is, almost by necessity, a suboptimized binding site in combination with basically any affinity depending on the relevant intracellular concentration of ligand and biological function.
Interestingly, although the majority of coupling free energies were negative, most of the positive coupling free energies were obtained for two residues: Leu-2087 in NCBD and Leu-1049 in ACTR. The positions in NCBD that displayed positive interaction energies with Leu-1049 in ACTR may form a contiguous positive pathway (Fig. 4), although the coupling between NCBDLeu-2096 and ACTRLeu-1049, which seems to be important for this pathway, is missing because we were not able to determine an accurate value of ΔΔΔGc between these two positions. Similarly, the residues in ACTR that displayed positive interaction energies with Leu-2087 in NCBD form a putative contiguous pathway (Fig. 4). Indeed, both of these hydrophobic side chains could interact with other proteins: they are partially solvent-exposed in the complex, with 50 and 30% of the surface area being accessible in the bound state for Leu-2087 in NCBD and Leu-1049 in ACTR, respectively. Such contiguous pathways were previously observed for PDZ domains (14), and they offer the possibility for a specific route for long-range signal transmission (38).
This work was supported by the Swedish Research Council (Natural and Engineering Sciences) and the Human Frontiers Young Investigator Science Program (to P. J.) and by the Magnus Bergvall Foundation (to J. D.).
- IDP
- intrinsically disordered protein
- NCBD
- nuclear coactivator-binding domain.
REFERENCES
- 1. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 2. Uversky V. N., Oldfield C. J., Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 3. Kjaergaard M., Teilum K., Poulsen F. M. (2010) Conformational selection in the molten globule state of the nuclear coactivator binding domain of CBP. Proc. Natl. Acad. Sci. U.S.A. 107, 12535–12540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demarest S. J., Martinez-Yamout M., Chung J., Chen H., Xu W., Dyson H. J., Evans R. M., Wright P. E. (2002) Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415, 549–553 [DOI] [PubMed] [Google Scholar]
- 5. Demarest S. J., Deechongkit S., Dyson H. J., Evans R. M., Wright P. E. (2004) Packing, specificity, and mutability at the binding interface between the p160 coactivator and CREB-binding protein. Protein Sci. 13, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ebert M. O., Bae S. H., Dyson H. J., Wright P. E. (2008) NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 47, 1299–1308 [DOI] [PubMed] [Google Scholar]
- 7. Dogan J., Schmidt T., Mu X., Engström Å., Jemth P. (2012) Fast association and slow transitions in the interaction between two intrinsically disordered protein domains. J. Biol. Chem. 287, 34316–34324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dogan J., Mu X., Engström Å., Jemth P. (2013) The transition state structure for coupled binding and folding of disordered protein domains. Sci. Rep. 3, 2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iešmantavičius V., Dogan J., Jemth P., Teilum K., Kjaergaard M. (2014) Helical propensity in an intrinsically disordered protein accelerates ligand binding. Angew. Chem. Int. Ed. Engl. 10.1002/anie.201307712 [DOI] [PubMed] [Google Scholar]
- 10. Itzhaki L. S., Otzen D. E., Fersht A. R. (1995) The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 254, 260–288 [DOI] [PubMed] [Google Scholar]
- 11. Carter P. J., Winter G., Wilkinson A. J., Fersht A. R. (1984) The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38, 835–840 [DOI] [PubMed] [Google Scholar]
- 12. Malatesta F. (2005) The study of bimolecular reactions under non-pseudo-first order conditions. Biophys. Chem. 116, 251–256 [DOI] [PubMed] [Google Scholar]
- 13. Gianni S., Haq S. R., Montemiglio L. C., Jürgens M. C., Engström Å., Chi C. N., Brunori M., Jemth P. (2011) Sequence-specific long range networks in PSD-95/Discs large/ZO-1 (PDZ) domains tune their binding selectivity. J. Biol. Chem. 286, 27167–27175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hultqvist G., Haq S. R., Punekar A. S., Chi C. N., Engström Å., Bach A., Strømgaard K., Selmer M., Gianni S., Jemth P. (2013) Energetic pathway sampling in a protein interaction domain. Structure 21, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 15. Schreiber G., Fersht A. R. (1995) Energetics of protein-protein interactions: analysis of the Barnase-Barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248, 478–486 [DOI] [PubMed] [Google Scholar]
- 16. Goldman E. R., Dall'Acqua W., Braden B. C., Mariuzza R. A. (1997) Analysis of binding interactions in an idiotope-antiidiotope protein-protein complex by double mutant cycles. Biochemistry 36, 49–56 [DOI] [PubMed] [Google Scholar]
- 17. Dall'Acqua W., Goldman E. R., Lin W., Teng C., Tsuchiya D., Li H., Ysern X., Braden B. C., Li Y., Smith-Gill S. J., Mariuzza R. A. (1998) A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry 37, 7981–7991 [DOI] [PubMed] [Google Scholar]
- 18. Pons J., Rajpal A., Kirsch J. F. (1999) Energetic analysis of an antigen/antibody interface: alanine scanning mutagenesis and double mutant cycles on the HyHEL-10/lysozyme interaction. Protein Sci. 8, 958–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kiel C., Serrano L., Herrmann C. (2004) A detailed thermodynamic analysis of Ras/effector complex interfaces. J. Mol. Biol. 340, 1039–1058 [DOI] [PubMed] [Google Scholar]
- 20. Reichmann D., Cohen M., Abramovich R., Dym O., Lim D., Strynadka N. C., Schreiber G. (2007) Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J. Mol. Biol. 365, 663–679 [DOI] [PubMed] [Google Scholar]
- 21. Bryngelson J. D., Wolynes P. G. (1987) Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. U.S.A. 84, 7524–7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qin B. Y., Liu C., Srinath H., Lam S. S., Correia J. J., Derynck R., Lin K. (2005) Crystal structure of IRF-3 in complex with CBP. Structure 13, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 23. Oldfield C. J., Meng J., Yang J. Y., Yang M. Q., Uversky V. N., Dunker A. K. (2008) Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9, Suppl. 1, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vacic V., Oldfield C. J., Mohan A., Radivojac P., Cortese M. S., Uversky V. N., Dunker A. K. (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee C. W., Martinez-Yamout M. A., Dyson H. J., Wright P. E. (2010) Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 49, 9964–9971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuda S., Harries J. C., Viskaduraki M., Troke P. J., Kindle K. B., Ryan C., Heery D. M. (2004) A conserved α-helical motif mediates the binding of diverse nuclear proteins to the SRC1 interaction domain of CBP. J. Biol. Chem. 279, 14055–14064 [DOI] [PubMed] [Google Scholar]
- 27. Lin C. H., Hare B. J., Wagner G., Harrison S. C., Maniatis T., Fraenkel E. (2001) A small domain of CBP/p300 binds diverse proteins: solution structure and functional studies. Mol. Cell 8, 581–590 [DOI] [PubMed] [Google Scholar]
- 28. Waters L., Yue B., Veverka V., Renshaw P., Bramham J., Matsuda S., Frenkiel T., Kelly G., Muskett F., Carr M., Heery D. M. (2006) Structural diversity in p160/CREB-binding protein coactivator complexes. J. Biol. Chem. 281, 14787–14795 [DOI] [PubMed] [Google Scholar]
- 29. Ryan C. M., Harries J. C., Kindle K. B., Collins H. M., Heery D. M. (2006) Functional interaction of CREB binding protein (CBP) with nuclear transport proteins and modulation by HDAC inhibitors. Cell Cycle 5, 2146–2152 [DOI] [PubMed] [Google Scholar]
- 30. Mas C., Lussier-Price M., Soni S., Morse T., Arseneault G., Di Lello P., Lafrance-Vanasse J., Bieker J. J., Omichinski J. G. (2011) Structural and functional characterization of an atypical activation domain in erythroid Krüppel-like factor (EKLF). Proc. Natl. Acad. Sci. U.S.A. 108, 10484–10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang Y., Liu Z. (2009) Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 393, 1143–1159 [DOI] [PubMed] [Google Scholar]
- 32. Shammas S. L., Rogers J. M., Hill S. A., Clarke J. (2012) Slow, reversible, coupled folding and binding of the spectrin tetramerization domain. Biophys. J. 103, 2203–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou H. X. (2012) Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 37, 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drobnak I., De Jonge N., Haesaerts S., Vesnaver G., Loris R., Lah J. (2013) Energetic basis of uncoupling folding from binding for an intrinsically disordered protein. J. Am. Chem. Soc. 135, 1288–1294 [DOI] [PubMed] [Google Scholar]
- 35. Sugase K., Dyson H. J., Wright P. E. (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 [DOI] [PubMed] [Google Scholar]
- 36. Rogers J. M., Steward A., Clarke J. (2013) Folding and binding of an intrinsically disordered protein: fast, but not 'diffusion-limited.' J. Am. Chem. Soc. 135, 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gianni S., Morrone A., Giri R., Brunori M. (2012) A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem. Biophys. Res. Commun. 428, 205–209 [DOI] [PubMed] [Google Scholar]
- 38. del Sol A., Tsai C. J., Ma B., Nussinov R. (2009) The origin of allosteric functional modulation: multiple pre-existing pathways. Structure 17, 1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]