

Background: Replication factor C contains four active AAA+ ATPase sites.
Results: Mutation of any one ATP binding site affects clamp loader activities.
Conclusion: Although ATP sites may fill sequentially, ATP binding to all four sites is needed for RFC activity.
Significance: This provides insight into the ATP-dependent conformational changes in RFC that drive the clamp loading reaction.
Keywords: ATP, DNA Enzymes, DNA Replication, Fluorescence, Nucleic Acid Enzymology, AAA+ ATPase, Proliferating Cell Nuclear Antigen, Replication Factor C, Clamp Loader
Abstract
Clamp loaders belong to a family of proteins known as ATPases associated with various cellular activities (AAA+). These proteins utilize the energy from ATP binding and hydrolysis to perform cellular functions. The clamp loader is required to load the clamp onto DNA for use by DNA polymerases to increase processivity. ATP binding and hydrolysis are coordinated by several key residues, including a conserved Lys located within the Walker A motif (or P-loop). This residue is required for each subunit to bind ATP. The specific function of each ATP molecule bound to the Saccharomyces cerevisiae clamp loader is unknown. A series of point mutants, each lacking a single Walker A Lys residue, was generated to study the effects of abolishing ATP binding in individual clamp loader subunits. A variety of biochemical assays were used to analyze the function of ATP binding during discrete steps of the clamp loading reaction. All mutants reduced clamp binding/opening to different degrees. Decreased clamp binding activity was generally correlated with decreases in the population of open clamps, suggesting that differences in the binding affinities of Walker A mutants stem from differences in stabilization of proliferating cell nuclear antigen in an open conformation. Walker A mutations had a smaller effect on DNA binding than clamp binding/opening. Our data do not support a model in which each ATP site functions independently to regulate a different step in the clamp loading cycle to coordinate these steps. Instead, the ATP sites work in unison to promote conformational changes in the clamp loader that drive clamp loading.
Introduction
AAA+ ATPases are a family of enzymes that utilize the energy from ATP binding and hydrolysis to perform a variety of cellular functions ranging from proteolysis to organelle assembly (for reviews, see Refs. 1–3). These molecular motors also play vital functions within DNA replication and repair processes (4). Efficient, faithful DNA replication and repair are essential for cell survival. Fidelity of DNA synthesis is achieved by the polymerase itself, whereas efficiency is greatly increased by a variety of accessory proteins. Increased processivity is conferred by a protein known as proliferating cell nuclear antigen (PCNA).3 This toroid-shaped protein encircles DNA and serves to tether the polymerase to the template DNA, reducing the number of binding events that occur during replication (5). This protein is stable as a closed ring in solution (6) and requires a clamp loader protein complex known as replication factor C (RFC) to load it onto DNA (see Fig. 1A). The clamp loading reaction is complex, requiring many conformational changes and binding events. Simplistically, the clamp loader, in the presence of ATP, binds to the clamp and opens it. This allows for DNA binding and formation of a ternary complex of RFC-PCNA-DNA. Formation of this ternary complex triggers ATP hydrolysis, closing of the clamp, and dissociation of the clamp loader, leaving the clamp loaded onto DNA.
FIGURE 1.
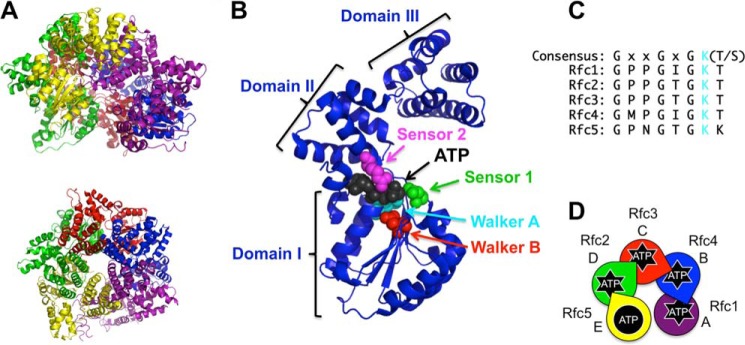
RFC structure and ATP binding sites. A, schematic diagram of S. cerevisiae RFC (Protein Data Bank code 1SXJ) shown from a side (top) and top (bottom) view (7). α-Helices are shown as coils, and β-strands are shown as arrows. RFC subunits are colored as follows: Rfc1, purple; Rfc4, blue; Rfc3, red; Rfc2, green; and Rfc5, yellow. This color scheme is used throughout the figures. B, ribbon diagram of the Rfc4 subunit showing the three dominant structural domains and the amino acid residues within the ATP binding pocket that coordinate ATP binding and hydrolysis. The highlighted residues are: Walker A (cyan; Lys-55), Walker B (red; Asp-114), sensor 1 (green; Asn-145), and sensor 2 (magenta; Arg-203). C, alignment of the Walker A (P-loop) sequence for each of the RFC subunits. The conserved Lys residue (mutated to Ala to generate point mutants in this study) is shown in cyan. D, schematic representation of the RFC subunit arrangement using S. cerevisiae nomenclature for each subunit along with a species-independent A–E designation. The color scheme matches the structure in A. Subunits that can bind and hydrolyze ATP have “starbursts.” Arginine fingers are shown as wedgelike protrusions. This schematic is included in each figure with data color-coded in the same way (e.g. data for the Rfc1 mutant are shown in purple).
The subunits of the clamp loader complex belong to the AAA+ family of ATPases and contain three structural domains: domains I and II contribute to ATP binding, and the C-terminal domain III supports oligomerization (Fig. 1B) (7). Several key residues within the ATP binding pocket serve to coordinate ATP binding and hydrolysis, including sensors 1 and 2, the Walker A and B motifs, and the trans-acting arginine finger (see Fig. 1B) (8–10). The Walker A motif (consensus sequence GXXGXGKT where “X” is any amino acid) has a conserved Lys residue that has been shown to be important for ATP binding (see Fig. 1C). Several studies have looked at the function of this residue by making a series of point mutations in Saccharomyces cerevisiae (11), human (12), and Archaeoglobus fulgidus (13) RFC complexes as well as the bacterial clamp loader (14, 15), but elucidation of the function of this residue within each individual step of the clamp loading reaction utilizing steady-state and pre-steady-state analyses has not been performed.
Each of the genes encoding a subunit of RFC is unique, allowing for site-specific mutation in a single subunit within the complex. In this study, a series of point mutations were introduced wherein the conserved Lys residue was mutated to Ala in each RFC subunit with active ATPase activity to generate four mutant RFC complexes (see Fig. 1D). A series of biochemical assays was used to measure the activity of the mutant complexes at various steps within the clamp loading reaction, ranging from clamp binding to formation of a ternary complex (RFC-PCNA-DNA). This systematic biochemical analysis provides a framework wherein the function of ATP binding to specific subunits can be evaluated for individual steps within the reaction.
EXPERIMENTAL PROCEDURES
Nucleotides and Oligonucleotides
Concentrations of ATP (GE Healthcare) diluted with 30 mm HEPES, pH 7.5 and ATPγS (Roche Diagnostics) diluted with water were determined by measuring the absorbance at 259 nm and using the extinction coefficient 15,400 m−1 cm−1. Synthetic oligonucleotides were obtained from Integrated DNA Technologies and purified by 10% (60-mer) or 12% (30-mer) denaturing polyacrylamide gel electrophoresis. The sequences of the 60-nucleotide template and the complementary 30-nucleotide primer used in the equilibrium fluorescence binding assays were: 5′-TTC AGG TCA GAA GGG TTC TAT CTC TGT TGG CCA GAA (C6dT)GT CCC TTT TAT TAC TGG TCG TGT-3′ and 5′-ACA CGA CCA GTA ATA AAA GGG ACA TT-3′, respectively, where C6dT is a T with a C6 amino linker that was covalently labeled with 7-diethylaminocoumarin-3-carboxylic acid succinimidyl ester (DCC) (Invitrogen) as described previously (16, 17). The sequences of the 60-nucleotide template and the complementary 30-nucleotide primer used in anisotropy assays were: 5′-(5AmMC6)TTC AGG TCA GAA GGG TTC TAT CTC TGT TGG CCA GAA TGT CCC TTT TAT TAC TGG TCG TGT-3′ and 5′-ACA CGA CCA GTA ATA AAA GGG ACA TTC TGG-3′, respectively, where 5AmMC6 is a T with a C6 amino linker that was covalently labeled with X-rhodamine (RhX) (Invitrogen) as described previously (16–18). Primed templates were annealed by incubating the primer with the template in 30 mm HEPES, pH 7.5 and 150 mm NaCl at 85 °C for 5 min and then allowing the solution to slowly cool to room temperature. For all assays, the molar ratios of fluorescently labeled template to unlabeled primer were 1:1.2.
Buffers and Reagents
RFC storage buffer is 30 mm HEPES, pH 7.5, 0.5 mm EDTA, 300 mm sodium chloride (NaCl), 2 mm dithiothreitol (DTT), 10% glycerol, and 750 mm maltose. PCNA storage buffer is 30 mm HEPES, pH 7.5, 0.5 mm EDTA, 150 mm NaCl, 2 mm DTT, and 10% glycerol. Assay buffer is 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10% glycerol, 10 mm magnesium chloride (MgCl2), and 750 mm maltose.
Proteins
The DNA constructs for expressing Δ283 RFC (wild type and the Rfc2, Rfc3, and Rfc4 mutants) and WT PCNA were generated as described previously (19–21). A fragment of the Rfc1 DNA sequence from the wild-type expression vector (between restriction sites AatII and MluI) was ligated into a pETBlue-2 vector. Site-directed mutagenesis was performed to introduce the K359A mutation into Rfc1 using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The following primer (and complementary strand) was used: Rfc1 K359A, 5′-CGG TCC TCC TGG TAT TGG GGC AAC AAC TGC TGC-3′. Sequencing to verify the mutation was performed at the Interdisciplinary Center for Biotechnology Research (University of Florida). The Rfc1 fragment (between restriction sites AatII and MluI) containing the mutation was then ligated into the parent vector (19) for expression. PCNA was expressed, purified (22–24), and labeled (22) as described previously. RFC (19, 21, 25) and the RFC Walker A mutants (26) were expressed and purified as described previously with minor modifications. Purity was assessed by SDS-PAGE (see Fig. 2). The molecular masses for each subunit are given in the figure legend. Approximately 5 mg of purified protein was obtained from 1 liter of expression medium for all complexes except the Rfc1 mutant. This complex generally yielded about 1 mg/liter of expression medium. This difference in protein yield has been reported previously (11).
FIGURE 2.
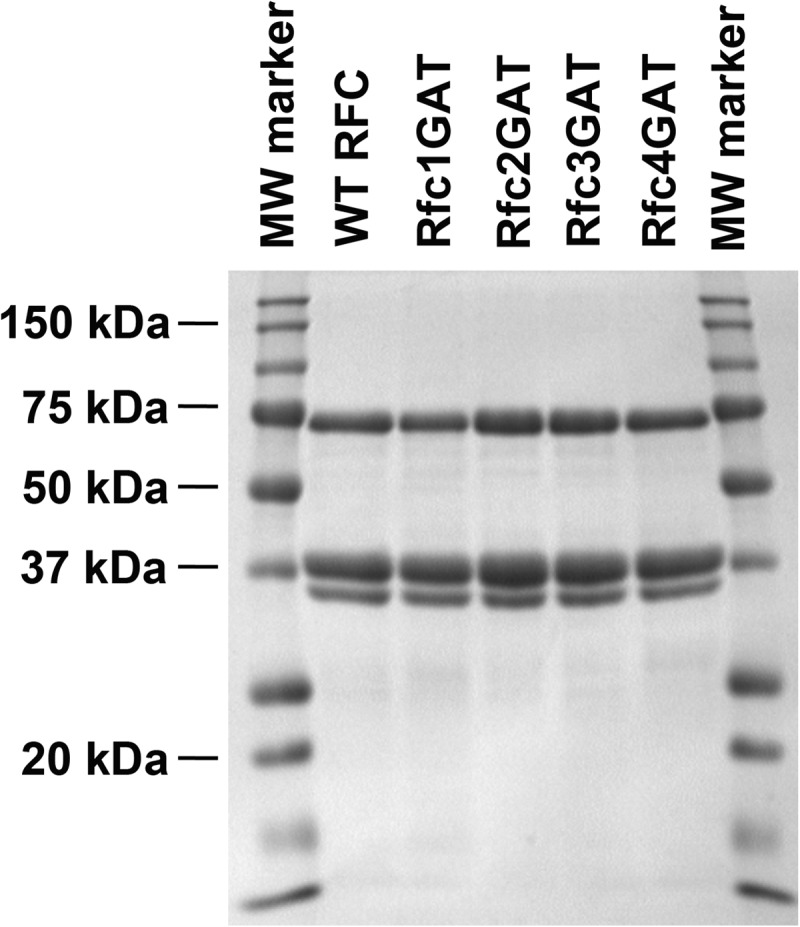
SDS-PAGE analysis of RFC complexes. SDS-PAGE of the final purified protein complexes stained with Coomassie Brilliant Blue is shown for WT RFC and each of the mutants. Subunit molecular masses are as follows: Δ283 Rfc1, 65 kDa; Rfc2, 40 kDa; Rfc3, 38 kDa; Rfc4, 36 kDa; and Rfc5, 40 kDa. Because of their similarity in size, the bands corresponding to the Rfc2, Rfc3, and Rfc5 subunits overlap.
Equilibrium Fluorescence Clamp Binding Assays
Steady-state clamp binding measurements were made on a QuantaMaster QM1 spectrofluorometer (Photon Technology International) at room temperature using a quartz cuvette with a 3 × 3-mm light path (Hellma 105.251-QS). MDCC was excited at 420 nm, and emission spectra were recorded from 450 to 500 nm using a 4-nm band pass after reagents were added sequentially to the cuvette. First, assay buffer with ATP (when present) was added for background signal, then PCNA-MDCC was added for unbound PCNA signal, and finally RFC was added for bound PCNA signal. Final concentrations were: 0.5 mm ATP (when present), 10 nm PCNA-MDCC, and 0–1600 nm clamp loader. Storage buffer was added instead of RFC to generate the 0 nm RFC point. After subtracting the buffer background, the intensity of bound PCNA was divided by the peak intensity (467 nm) of unbound PCNA. Each bound PCNA value at 467 nm was then divided by the 0 nm RFC point, setting this point to 1, to account for fluorescence change due to dilution. All other points are relative to this value. Kd,app values were calculated using Equation 1 where PCNA is the total concentration of PCNA, RFC is the total concentration of RFC, Imax is the maximum intensity, and Imin is the minimum intensity (22).
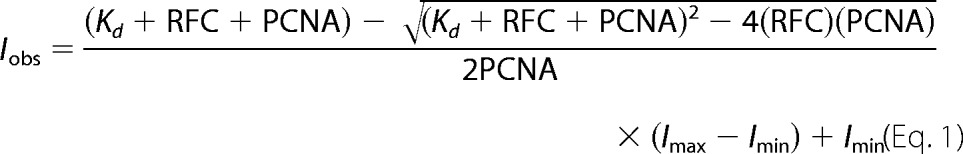 |
The average values and standard deviations for three independent experiments are reported, and the average relative intensities versus RFC concentration and standard deviations were graphed and fit using Kaleidagraph (22).
Equilibrium Fluorescence Clamp Opening Assays
Steady-state clamp opening assays were performed and analyzed in the same manner as the clamp binding assays except that Alexa Fluor 488 (AF488) was excited at 495 nm, emission spectra were recorded from 505 to 555 nm using a 2.5-nm band pass, and clamp loader was titrated from 0 to 400 nm. The peak intensity for unbound PCNA was 517 nm. The average values and standard deviations for three independent experiments are reported, and the average relative intensities versus RFC concentration and standard deviations were graphed and fit using Kaleidagraph (22).
Equilibrium ATP Titration
Steady-state clamp opening assays were performed as described above but with varying ATP concentrations. Final concentrations were: 0–8 mm ATP, 10 nm PCNA-AF4882, and 200 nm clamp loader (where present). Storage buffer was added instead of RFC to generate a 0 nm RFC point. After subtracting the buffer background, the intensity of bound PCNA was divided by the peak intensity (517 nm) of unbound PCNA. Each bound PCNA value at 517 nm was then divided by the 0 nm RFC point to determine the -fold change in binding. The average relative intensities for three independent experiments versus ATP concentration and standard deviations were graphed and fit using Kaleidagraph.
Pre-steady-state Clamp Binding Assays
Pre-steady-state binding assays were performed using an Applied PhotoPhysics SX20MV stopped-flow apparatus at 20 °C with a 3.72-nm band pass. MDCC fluorescence was monitored using a 455-nm cut-on filter while exciting at 420 nm. Single mix reactions were performed by mixing equal volumes (60 μl) of a solution of RFC and 0.5 mm ATP (when present) with a solution of PCNA-MDCC and 0.5 mm ATP (when present) in assay buffer immediately before they entered the cuvette. Data were collected for a total of 4 s at 0.4-ms intervals, and four or more kinetic traces were averaged. The time courses were corrected for background by subtracting the average signal for buffer. The corrected time courses were relativized to the average signal for free clamp intensity, and the data from the first 1 s were fit to single exponential rises (Equation 2) using Kaleidagraph.
Final concentrations were: 0.5 mm ATP (where present), 20 nm PCNA-MDCC, and 200 nm RFC. The average values and standard deviations for three independent experiments are reported, and a representative data set is shown.
Pre-steady-state Clamp Opening Assays
Pre-steady-state clamp opening assays were performed in the same manner as the clamp binding assays except that AF488 was excited at 495 nm and fluorescence was monitored using a 515-nm cut-on filter. Data were collected for a total of 8 s at 0.8-ms intervals, and four or more kinetic traces were averaged. The time courses were corrected for background by subtracting the average signal for buffer. The corrected time courses were relativized to the average signal for free clamp intensity, and the data from the first 4 s were fit to single exponential rises (Equation 2) using Kaleidagraph (22). Final concentrations were: 0.5 mm ATP (where present), 20 nm PCNA-AF4882, and 200 nm RFC. The average values and standard deviations for three independent experiments are reported, and a representative data set is shown.
Equilibrium Fluorescence DNA Binding Assays
Steady-state DNA binding measurements were made on a QuantaMaster QM1 spectrofluorometer (Photon Technology International) at room temperature using a quartz cuvette with a 3 × 3-mm light path (Hellma 105.251-QS). DCC was excited at 440 nm, and emission spectra were recorded from 460 to 500 nm using a 4-nm band pass after reagents were added sequentially to the cuvette. First, assay buffer with ATPγS (when present) was added for background signal, then DNA-DCC was added for unbound DNA signal, and finally RFC was added for bound DNA signal. Final concentrations were: 0.5 mm ATP (when present), 10 nm DNA-DCC, and 0–2530 nm clamp loader (see Fig. 6 legend for details). Storage buffer was added instead of RFC to generate the 0 nm RFC point. After subtracting the buffer background, the intensity of bound DNA was divided by the peak intensity of unbound DNA (470 nm). Each bound DNA value at 470 nm was then divided by the 0 nm RFC point, setting this point to 1, to account for fluorescence change due to dilution. All other points are relative to this value. Kd values were calculated using Equation 3 where DNA is the total concentration of DNA, RFC is the total concentration of RFC, Imax is the maximum intensity, and Imin is the minimum intensity (17, 27).
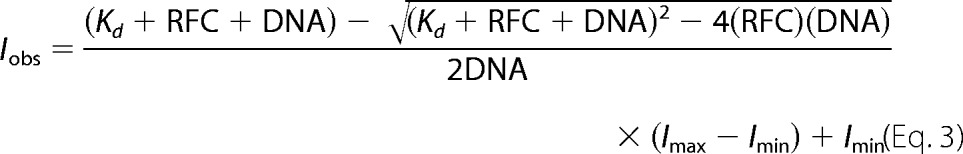 |
The average values and standard deviations for three independent experiments are reported, and the average relative intensities versus RFC concentration and standard deviations were graphed and fit using Kaleidagraph (17, 27).
FIGURE 6.
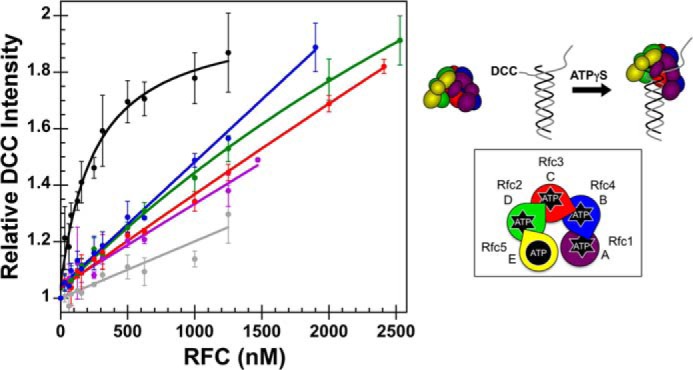
Fluorescence-based DNA binding. A schematic representation of the RFC subunit arrangement and a schematic of the assay are shown to the right of the data. Equilibrium DNA binding is shown for WT RFC (black), WT RFC without ATPγS (gray), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4 GAT (blue). DNA-DCC fluorescence was measured in solutions containing 10 nm DNA-DCC, 0.5 mm ATPγS, and 0–2530 nm clamp loader in assay buffer. The average relative DCC intensities and standard deviations from three independent experiments are plotted and fit to Equation 3. Dissociation constants (Kd values) were calculated for each experiment with WT RFC, and the average values and standard deviations are given in the text. Final assay buffer contained 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 10% glycerol, and 750 mm maltose. Error bars represent S.D.
Equilibrium Anisotropy DNA Binding Assays
Steady-state anisotropy DNA binding measurements were made on a QuantaMaster QM1 spectrofluorometer (Photon Technology International) with polarizers at room temperature using a quartz cuvette with a 3 × 3-mm light path (Hellma 105.251-QS). RhX was excited with vertically and horizontally polarized light using an 8-nm band pass, vertical and horizontal emissions were monitored at 605 nm for 30 s, and the average value was calculated. Anisotropy values were calculated using a G factor, determined under the same experimental conditions, to account for polarization bias using Equation 4.
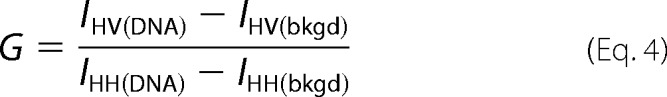 |
To generate each point, polarized emissions were monitored after reagents were added sequentially to the cuvette. First, assay buffer with ATPγS (when present) was added for background signal, then DNA-RhX was added for unbound DNA signal, then RFC was added for RFC-bound DNA signal (DNA-RFC), and finally PCNA was added for RFC-PCNA-bound DNA signal (DNA-RFC-PCNA). Final concentrations were: 0.5 mm ATPγS (when present), 25 nm DNA-RhX, 0–2 μm clamp loader, and 2 μm PCNA (see Fig. 7 legend for details). Storage buffer was added instead of RFC to generate the 0 nm RFC point. Anisotropy values were calculated for DNA-RFC and DNA-RFC-PCNA using Equation 5 where IVV and IVH are either the DNA-RFC values or DNA-RFC-PCNA values.
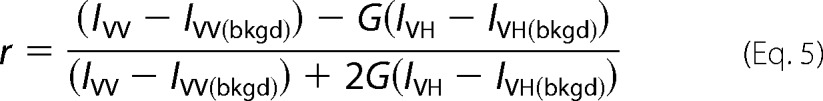 |
Kd values for the two bound states, DNA-RFC and DNA-RFC-PCNA, were calculated using Equation 3 where DNA is the total concentration of DNA; RFC is the DNA-RFC or DNA-RFC-PCNA anisotropy value, respectively; Imax is the maximum intensity; and Imin is the minimum intensity. The average values and standard deviations for three independent experiments are reported, and the average anisotropy values versus RFC concentration and standard deviations were graphed and fit using Kaleidagraph (18, 26, 27).
FIGURE 7.

Anisotropy-based DNA binding. A schematic representation of the RFC subunit arrangement and a schematic of the assay are shown above the data. Equilibrium DNA binding is shown for WT RFC (black) (A), WT RFC without ATPγS (light gray) (B), Rfc1GAT (purple) (C), Rfc2GAT (green) (D), Rfc3GAT (red) (E), and Rfc4 GAT (blue) (F). In each panel, the data for free DNA is shown in black, and the data for clamp loader binding in the presence of PCNA is shown in dark gray. DNA-RhX anisotropy was measured in solutions containing 25 nm DNA-RhX, 2 μm WT PCNA, 0.5 mm ATPγS, and 0–2 μm clamp loader in assay buffer. The average anisotropy and standard deviations from three independent experiments are plotted and fit to Equation 2 for clamp loader-DNA binding and clamp loader-DNA-PCNA binding. Dissociation constants (Kd values) were calculated for each experiment, and the average values and standard deviations are given in Table 3. Final assay buffer contained 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 10% glycerol, and 750 mm maltose. Error bars represent S.D.
RESULTS
RFC Mutants
Mutations were made at individual ATP active sites within RFC to investigate the contribution of ATP binding in each subunit to discrete steps in the clamp loading reaction cycle. RFC is a heteropentamer (Fig. 1A) of subunits that belong to the AAA+ ATPase family of molecular motors (for reviews, see Refs. 4, 28, and 29). A conserved Lys residue is found in the Walker A motif of all five RFC subunits (Fig. 1C) and is required for ATP binding (9, 30, 31). Four of the five RFC subunits, Rfc1, Rfc2, Rfc3, and Rfc4 (Fig. 1D), are able to bind and hydrolyze ATP (11, 32, 33). Rfc5 contains mutations to the canonical Walker B motif and lacks a trans-acting Arg finger and thus is not an active ATPase (7, 29, 32, 34). As a separate gene encodes each subunit, four separate clamp loader mutants were created, each harboring a single mutation: Rfc1, K359A; Rfc2, K71A; Rfc3, K59A; and Rfc4, K55A. These mutants and WT RFC were expressed and purified (Fig. 2) as described previously (26). Throughout the text, the mutants are named by the subunit containing the mutation and the letters “GAT” to denote conversion of the conserved Lys to Ala in the Walker A “GKT” sequence (for example, a mutation in the Rfc1 subunit would be named Rfc1GAT).
Clamp Binding
Equilibrium clamp binding assays were performed to determine whether mutation of the conserved Lys residue in the ATP binding pocket had any effect on the ability of the clamp loader to bind to the PCNA clamp. This assay, described previously (22), takes advantage of the environmental sensitivity of MDCC to monitor clamp binding. PCNA is covalently labeled with MDCC on the surface to which the clamp loader binds. Upon clamp loader binding, an increase in MDCC fluorescence occurs. To measure equilibrium binding quantitatively, RFC was added to PCNA-MDCC (10 nm) and ATP (0.5 mm), and MDCC fluorescence was measured before and after RFC addition to determine the increase in fluorescence. A schematic of the assay and the RFC subunit arrangement (data are color-coded to match this schematic) are shown above the data. The increase in MDCC fluorescence was measured as a function of clamp loader concentration (Fig. 3A) and apparent dissociation constants (Kd,app values) for the clamp loader-clamp interaction were calculated using Equation 1 (Table 1). PCNA binding was reduced for all of the RFC Walker A mutants but to differing degrees. The Rfc1 mutation gave the smallest increase in Kd,app (decrease in binding) of about 7-fold, and the Rfc3 mutation gave the greatest increase in Kd,app of about 100-fold. The Rfc2 and Rfc4 mutants bound PCNA with similar affinity with Kd,app values about 10-fold greater than that of WT RFC. In comparison, the Kd,app value for the single site Rfc3GAT mutant is only about 4-fold lower than that for WT RFC binding PCNA in the complete absence of ATP. The difference in Kd,app values, about 2 orders of magnitude, for WT RFC in the presence and absence of ATP is similar to the difference measured for the Escherichia coli β-clamp and γ-complex clamp loader (27). A smaller difference in these Kd,app values for ATP-dependent and -independent RFC-PCNA binding, about 10-fold, has been reported previously (35). Inclusion of maltose in the assay buffer, as we have done here, increases the stability of RFC mutant complexes and reduces nonspecific binding interactions between RFC and PCNA (26).
FIGURE 3.

Binding equilibria and observed rates for RFC complexes binding PCNA. A schematic representation of the RFC subunit arrangement and schematics of the assays are shown above the data. A, equilibrium clamp binding is shown for WT RFC (black), WT RFC without ATP (gray), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4 GAT (blue). PCNA-MDCC fluorescence was measured in solutions containing 10 nm PCNA-MDCC and 0–1600 nm clamp loader in assay buffer. The average relative MDCC intensities and standard deviations from three independent experiments are plotted and fit to Equation 1. Dissociation constants (Kd,app values) were calculated for each experiment, and the average values and standard deviations are given in Table 1. B, pre-steady-state clamp binding is shown for WT RFC (black), WT RFC without ATP (gray), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4GAT (blue). The increase in MDCC intensity was measured as a function of time when a solution of clamp loader and 0.5 mm ATP (when present) in assay buffer was rapidly mixed with a solution of PCNA-MDCC and 0.5 mm ATP (when present) in assay buffer. Reactions contained 20 nm PCNA-MDCC and 200 nm clamp loader. Solid lines are the result of an empirical fit of the data to a single exponential rise. Observed rates were calculated for three independent experiments, and the average values and standard deviations are given in Table 2. Final assay buffer for both experiments contained 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 10% glycerol, 750 mm maltose, and 0.5 mm ATP (where present). Error bars represent S.D.
TABLE 1.
Equilibrium binding and opening of PCNA by WT RFC and Walker A mutants
Reactions contained 10 nm clamp, 0–1600 nm (binding) or 0–400 nm (opening) clamp loader, and 0.5 mm ATP. ND, not determined.
| Clamp loader | PCNA-MDCC binding Kd | PCNA-AF4882 opening |
|
|---|---|---|---|
| Kd | Imax − Imin | ||
| nm | nm | ||
| WT | 1.2 ± 0.9 | 2.4 ± 2 | 0.86 ± 0.1 |
| Rfc1GAT | 9.2 ± 3 | 15 ± 1 | 0.67 ± 0.01 |
| Rfc2GAT | 20 ± 2 | 60 ± 30 | 0.13 ± 0.04 |
| Rfc3GAT | 130 ± 90 | ND | 0.072 ± 0.04 |
| Rfc4GAT | 29 ± 9 | 60 ± 20 | 0.11 ± 0.1 |
| WT no ATP | 540 ± 160 | ND | 0.048 ± 0.02 |
As clamp binding activity was reduced for each of the mutants when compared with WT, it is also possible that the mutations cause a change in the kinetics of clamp binding that is not observable under equilibrium conditions. Pre-steady-state clamp binding assays used the same PCNA-MDCC substrate as the equilibrium clamp binding assays, and a schematic of the assay and the RFC subunit arrangement (data are color-coded to match this schematic) are shown above the data. MDCC fluorescence was monitored in real time when a solution of clamp loader with 0.5 mm ATP in assay buffer was rapidly mixed with a solution of PCNA-MDCC with 0.5 mm ATP in assay buffer. Reactions contained 20 nm PCNA-MDCC and 200 nm clamp loader. The data from the first 1 s of the time courses were fit to a single exponential rise (Equation 2) to calculate observed rates (kobs) (Table 2), and the lines through each kinetic trace are the result of these fits (Fig. 3B). Relative intensities at the end points reflect the equilibrium fraction of PCNA bound. In contrast to equilibrium dissociation constants, the Walker A mutations had small effects on observed binding rates. For example, the observed rate for binding was only 3-fold lower for the Rfc3 mutant than for WT, whereas the Kd,app was 100-fold greater. Note that these observed rates reflect the rate of approach to equilibrium, which is a function of both the on- and off-rates. Given that the observed rates are similar but Kd values are different, one of the rates dominates the observed rate, and this rate is not affected by the mutations.
TABLE 2.
Observed rates for PCNA binding and opening by WT RFC and Walker A mutants
Reactions contained 20 nm labeled clamp, 200 nm clamp loader, and 0.5 mm ATP. ND, not determined.
| Clamp loader | PCNA-MDCC binding kobs | PCNA-AF4882 opening kobs |
|---|---|---|
| s−1 | s−1 | |
| WT | 8.3 ± 0.4 | 0.63 ± 0.04 |
| Rfc1GAT | 4.3 ± 0.3 | 0.36 ± 0.09 |
| Rfc2GAT | 4.5 ± 0.2 | 0.57 ± 0.2 |
| Rfc3GAT | 2.7 ± 0.2 | 0.70 ± 0.51 |
| Rfc4GAT | 3.0 ± 0.1 | 2.1 ± 1.8 |
| WT no ATP | 8.5 ± 0.7 | ND |
Clamp Opening
PCNA opening is a two-step reaction in which RFC binds PCNA before PCNA opens (22). Because mutation of the conserved Lys residues in the ATP binding pocket had differing effects on the clamp binding activity of the mutants, equilibrium clamp opening assays were performed to determine whether these mutations also affected the clamp opening activity of RFC. This assay, described previously (22), takes advantage of the fact that two fluorophores in close proximity will self-quench. PCNA is covalently labeled with AF488 on two Cys residues, one on each side of the dimer interface. Upon clamp loader binding, an increase in AF488 fluorescence is observed due to clamp opening and relief of AF488 quench. AF488 fluorescence was measured as a function of clamp loader concentration when RFC was added to PCNA-AF4882, and the relative fluorescence increase following RFC addition is plotted in Fig. 4A. A schematic of the assay and the RFC subunit arrangement (data are color-coded to match this schematic) are shown above the data. This experiment provides two pieces of information about the PCNA opening reaction. The relative fluorescence intensity at saturating concentrations of RFC provides information about the relative population of clamp loader-clamp complexes in an open conformation, and the RFC concentration dependence of the fluorescence increase provides information about the binding affinity. Intensity values (Imax) at saturating RFC concentrations and dissociation constants (Kd values) for the clamp loader-clamp interaction were calculated using Equation 1 (Table 1). The Rfc1 mutant showed the smallest defects in clamp opening with only a 1.5-fold decrease in the population of open clamps relative to WT and a 7-fold increase in Kd value. For the remaining mutants, the defects in clamp opening were more severe, 7–13-fold decreases in the relative population of open clamps compared with WT RFC. For Rfc2GATand Rfc4GAT, the small increases in fluorescence led to large errors in calculated Kd values; however, the trend was the same as in the binding assays. Kd values for Rfc2GAT and Rfc4GAT measured in the opening assay were about the same and were greater than that for Rfc1GAT. The fraction of open clamps was too small to accurately determine Kd values for WT RFC in the absence of ATP and for Rfc3GAT. For WT RFC and Rfc1GAT where the change in intensities was large, Kd values measured in opening assays were in good agreement with Kd values measured in binding assays.
FIGURE 4.

Clamp opening by RFC complexes under equilibrium conditions and in real time. A schematic representation of the RFC subunit arrangement and schematics of the assays are shown above the data. A, equilibrium clamp opening is shown for WT RFC (black), WT RFC without ATP (gray), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4 GAT (blue). PCNA-AF4882 fluorescence was measured in solutions containing 10 nm PCNA-AF4882 and 0–400 nm clamp loader in assay buffer. The average relative AF488 intensities and standard deviations from three independent experiments are plotted and fit to Equation 1. Dissociation constants (Kd values) were calculated for each experiment, and the average values and standard deviations are given in Table 1. B, pre-steady-state clamp opening is shown for WT RFC (black), WT RFC without ATP (gray), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4GAT (blue). The increase in AF488 intensity was measured as a function of time when a solution of clamp loader and 0.5 mm ATP (where present) in assay buffer was rapidly mixed with a solution of PCNA-AF4882 and 0.5 mm ATP (where present) in assay buffer. Reactions contained 20 nm PCNA-AF4882 and 200 nm clamp loader. Solid lines are the result of an empirical fit of the data to a single exponential rise. Observed rates were calculated for three independent experiments, and the average values and standard deviations are given in Table 2. Final assay buffer for both experiments contained 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 10% glycerol, 750 mm maltose, and 0.5 mm ATP (where present). Error bars represent S.D.
Rates of clamp opening were measured for WT RFC and mutants that gave a large enough signal change using the same PCNA-AF4882 substrate as in the equilibrium clamp opening assays. AF488 fluorescence was monitored in real time when a solution of clamp loader and 0.5 mm ATP (where present) in assay buffer was rapidly mixed with a solution of PCNA-AF4882 and 0.5 mm ATP (where present) in assay buffer. The data from the first 4 s of the time courses were fit to a single exponential rise (Equation 2) to calculate observed rates (kobs) (Table 2), and the lines through each kinetic trace are the result of this fit (Fig. 4B). Observed changes in intensity were similar to those measured under equilibrium conditions at the same concentrations. Rates of clamp opening were comparable with that of WT for the Rfc1 and Rfc2 mutants, suggesting that the 7–30-fold increase in Kd value observed for these mutants may be due to faster rates of clamp closing/dissociation. When compared with clamp binding, the observed rate of clamp opening is 10-fold slower for WT, indicating that clamp opening is rate-limiting at these concentrations, which is consistent with previous kinetic studies (22, 36). The observed opening rate is also about 10-fold slower for Rfc1GAT and Rfc2GAT (Table 2). Because of the small amplitudes, rates for Rfc3GAT and Rfc4GAT could not be measured reproducibly.
Equilibrium ATP Titration
As ATP binding is required for clamp loader-clamp binding/opening (37, 38), it is possible that higher concentrations of ATP could rescue the clamp binding/opening defects observed for the RFC Walker A mutants. To address this question, equilibrium PCNA opening assays were performed with increasing concentrations of ATP. Final conditions were: 0–8 mm ATP, 10 nm PCNA-AF4882, and 200 nm clamp loader. The average AF488 change in intensity and standard deviations from three independent experiments are plotted as a function of ATP concentration (Fig. 5). As expected, no PCNA opening was observed for WT or mutant clamp loaders in the absence of ATP. At ATP concentrations ranging from 0.25 to 4 mm, opening was only observed for WT RFC and the Rfc1 mutant, and the fraction of open clamp loader-clamp complexes was lower for Rfc1GAT at each ATP concentration when compared with WT. At the highest ATP concentration tested (8 mm), a reduction in clamp opening was observed for both the WT RFC and Rfc1 mutant. Thus, increasing the concentration of ATP up to 4 mm did not rescue clamp opening activities of the mutants.
FIGURE 5.
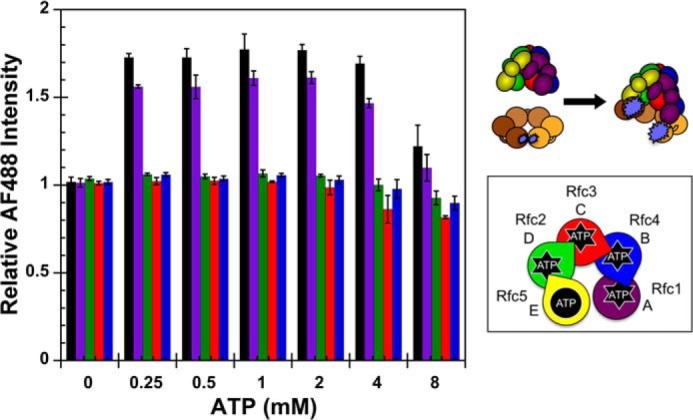
Equilibrium ATP titration. A schematic representation of the RFC subunit arrangement and a schematic of the assay are shown to the right of the data. Equilibrium clamp opening as a function of ATP concentration is shown for WT RFC (black), Rfc1GAT (purple), Rfc2GAT (green), Rfc3GAT (red), and Rfc4GAT (blue). PCNA-AF4882 fluorescence was measured in solutions containing 10 nm PCNA-AF4882 and 200 nm clamp loader in assay buffer. The average relative AF488 intensities and standard deviations from three independent experiments are plotted against ATP concentration. Final assay buffer contained 30 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 10% glycerol, and 750 mm maltose. Error bars represent S.D.
Fluorescent DNA Binding
Although most of these mutants exhibited strong defects in ATP-dependent clamp binding and/or clamp opening, it is possible that they retain the ability to interact with primer-template DNA. To test this hypothesis, primer-template labeled with DCC was used (17, 27). The DCC fluorophore is covalently attached to the template near the primer-template junction (3 nucleotides from the primer 3′-end). An increase in DCC fluorescence occurs when RFC binds to DNA-DCC (17, 27). Because of differences in the contacts RFC makes with the DNA in the presence of PCNA, little change in fluorescence occurs, and therefore, binding of RFC-PCNA to DNA-DCC was not assayed. Final conditions in assay buffer were: 0.5 mm ATPγS, 10 nm DNA-DCC, and 0–2560 nm clamp loader. DCC fluorescence was measured as a function of clamp loader concentration (Fig. 6). The fluorescence increased hyperbolically for WT RFC, approaching a saturating value, whereas the fluorescence increased linearly for each of the mutants, never approaching a maximum. Several possibilities may account for this difference. 1) The mutants may have a weaker affinity for DNA; 2) the mutants may make different contacts with the DNA than WT, resulting in a higher quantum yield for the bound state; or 3) the mutants could have a higher affinity for blunt ends or single-stranded DNA than for the primer-template junction. This fluorescence assay is specific for binding to primer-template junctions in that binding to other sites will not affect DCC fluorescence. A dissociation constant (Kd value) for the clamp loader-DNA interaction of 230 ± 60 nm was calculated for WT RFC using Equation 3. This value is similar to that observed for E. coli γ complex DNA binding using a similar assay (27).
Anisotropy DNA Binding
To address some of the questions raised by the fluorescence-based DNA binding assay, an anisotropy-based assay was used. For this assay, DNA is labeled with X-rhodamine at the free 5′-end, and changes in the rotational motion of the fluorophore were quantified by measuring polarized emission when exciting with polarized light (16, 18, 26). This assay has advantages over the fluorescence-based DCC assay in that 1) DNA binding to any site on the small primer-template DNA substrate will produce a change in anisotropy, and 2) clamp loader binding in the presence of PCNA can be measured. An increase in anisotropy or decrease in fluorophore rotational motion is observed upon clamp loader or clamp loader-clamp binding to the DNA. Final conditions in assay buffer were: 0.5 mm ATPγS, 25 nm DNA-RhX, 2 μm WT PCNA, and 0–2 μm clamp loader. Polarized emission was measured as a function of clamp loader concentration (Fig. 7), and anisotropy values were calculated using Equations 4 and 5. For all the RFC mutants, the anisotropy increased with increasing RFC concentration and approached a saturating value at high concentrations of RFC. Dissociation constants (Kd values) for the DNA-RFC and DNA-RFC-PCNA interactions were calculated using Equation 3 (Table 3). RFC-DNA dissociation constants for the mutants are similar to that of WT with the largest difference giving a 2-fold increase in Kd, whereas binding of WT RFC in the absence of ATPγS is 7-fold weaker. PCNA appeared to stimulate DNA binding for WT RFC but not for the mutants. This result is consistent with PCNA binding/opening results for the Rfc2, Rfc3, and Rfc4 mutants as they have a higher affinity for PCNA than DNA and are unable to support an open clamp structure. Thus, PCNA binding may actually inhibit DNA binding. The one caveat to these results is that PCNA alone (0 nm RFC) increases the anisotropy of RhX on DNA, suggesting that PCNA alone at this concentration (2 μm) interacts with DNA. These results agree well with previous studies of Rfc1 Δ272 complexes containing a Walker A Lys to Glu mutation (rather than the Lys to Ala used in this study) (11).
TABLE 3.
Anisotropy-based equilibrium DNA binding
Reactions contained 25 nm DNA-RhX, 2 μm WT PCNA, 0–2 μm clamp loader, and 0.5 mm ATPγS.
| Clamp loader | RFC only Kd | RFC + PCNA Kd |
|---|---|---|
| nm | nm | |
| WT | 120 ± 20 | 60 ± 20 |
| Rfc1GAT | 230 ± 20 | 440 ± 40 |
| Rfc2GAT | 140 ± 50 | 340 ± 120 |
| Rfc3GAT | 130 ± 50 | 300 ± 170 |
| Rfc4GAT | 230 ± 30 | 570 ± 150 |
| WT no ATPγS | 800 ± 150 | 4700 ± 4900 |
DISCUSSION
RFC, a member of the AAA+ ATPase family of proteins, uses the energy from ATP binding and hydrolysis to load the sliding clamp, PCNA, onto DNA for use by polymerases and various other proteins within the cell (for reviews, see Refs. 29 and 39–42). The five core AAA+ subunits will be designated A–E (Fig. 1D) to compare yeast RFC with other clamp loaders. Within each AAA+ subunit, conserved amino acid residues directly support ATP binding and hydrolysis. In addition, trans-acting Arg residues, “Arg fingers,” located in the subunits adjacent to the ATP binding sites play a key role in responding to ATP binding and hydrolysis events to support conformational changes in the complex that drive the mechanical clamp loading reactions. In this work, functions of the Lys residue in the conserved Walker A (or P-loop) sequence motifs of the ATP binding sites of S. cerevisiae Rfc1–4 (sites A–D) were investigated. Because Rfc5 (E site) lacks a trans-acting Arg finger and contains mutations in the Walker B motif that are key for hydrolysis, this mutant was not investigated. Our study extends previous work measuring the effects of Walker A mutations on clamp loading and DNA synthesis in vitro and in vivo (11, 12, 14, 15, 43, 44) by analyzing the effect of single site Walker A mutations on individual interactions and steps in the clamp loading reaction cycle.
ATP binding to clamp loaders supports formation of a ternary clamp loader-clamp-DNA complex, and ATP hydrolysis promotes closure and release of the clamp on DNA. From a mechanistic point of view, sequential binding and hydrolysis of ATP at individual sites would provide an attractive means of temporal coordination of steps in the clamp loading reaction cycle such that each binding/hydrolysis event would be coupled to a distinct step. RFC binds ATP in a stepwise manner, leaving open the possibility for this hypothetical model. Two (or three) sites in RFC fill prior to clamp and DNA binding, and subsequent binding of the clamp and DNA each promote binding of an additional ATP molecule (13, 32, 45). Similarly, structural data suggest that hydrolysis of ATP at a single site can promote clamp closure. When an ATP analog, ADP-BeF3, was bound to sites B, C, and D of the bacteriophage T4 clamp loader, an open clamp loader-clamp complex was captured in crystals, but when site B is bound to ADP, the clamp is closed (46). To test the possibility that ATP binding to a single site might be coupled to a discrete step or interaction in the clamp loading reaction, single ATP sites in RFC were mutated, and the mutant complexes were characterized biochemically, focusing on individual ATP-dependent steps leading up to formation of a ternary complex.
Given the topology of the proteins and DNA, the first step in a productive clamp loading reaction is likely RFC-PCNA binding. Our results show that none of the ATP sites is completely dispensable for PCNA binding/opening reactions, although mutation of Rfc1 (A site) has the smallest effect on the activity. Similarly, mutation of yeast Rfc1 also has small effects on ATP hydrolysis and clamp loading (11). In contrast, mutation of the A site in human RFC and an archaeal RFC decreased both clamp loading and processive DNA synthesis to the same degree as mutation of other sites (12, 13, 44). One possible reason for the difference in the human and yeast proteins is that the yeast Rfc1 was truncated to remove the N-terminal DNA ligase homology domain that is not required for clamp loading activity (47–49). ATP binding could affect the conformation of the protein to prevent the N-terminal domain from interfering with clamp loading, and this activity would not be required in assays with the truncated protein. This truncation does not necessarily explain why ATP binding to the A subunit in archaeal RFC is important, suggesting that there may be some other interesting mechanistic differences between RFC from yeast when compared with archaea or humans.
RFC binds PCNA in a two-step reaction (Fig. 8) forming a closed RFC-PCNA complex prior to an open complex (22, 36). Our PCNA-MDCC binding assay measures the total population of bound clamps, both open and closed, and therefore, experimentally determined binding constants will also be sensitive to the population of open clamps. Our working model to explain the differences in binding affinities of RFC mutants for PCNA is that the binding constant for the initial step forming a closed RFC-PCNA complex is the same, or nearly so, for WT RFC and Walker A mutants, but the opening equilibrium differs by a greater degree. This initial binding step may have a Kd value similar to the value for WT RFC binding PCNA in the absence of ATP. As observed in the absence of ATP, mutation of the four Arg fingers in RFC nearly eliminates PCNA opening (50). The crystal structure of a closed RFC-PCNA complex containing these Arg finger mutations shows that contacts between each subunit and PCNA decrease going around the ring such that the A subunit has the most extensive interactions, but the D and E subunits do not contact the surface of PCNA at all (7). We propose that the initial closed RFC-PCNA complexes for WT RFC and the Walker A mutants resemble this structure with more limited interactions between RFC subunits and PCNA (Fig. 8). When the clamp opens, more extensive contacts between each subunit and the surface of the clamp are expected as seen in the structure of the bacteriophage T4 clamp loader bound to the clamp and DNA (51). These extensive interactions stabilize the open conformation of PCNA (52). The single site Walker A mutations likely affect the conformation of RFC in a way that distorts the pitch of the spiral geometry that RFC subunits adopt, resulting in weaker RFC-PCNA interactions. This would ultimately decrease the stability of the open complex relative to the WT complex and shift the equilibrium between open and closed complexes to favor the closed form. Thus, ATP binding to all of the subunits is required for this optimal spiral geometry in the clamp loader that is necessary to stabilize the open conformation of the clamp.
FIGURE 8.

Model for ATP-dependent conformational changes in forming a ternary clamp loader-clamp-DNA complex. The clamp loading reaction is a multistep reaction that requires the clamp loader to bind and open the clamp, place the open ring on DNA, and release the closed ring on DNA. Our results support a model in which the multiple ATP sites in RFC function together at specific steps rather than functioning independently to promote changes at different steps. ATP binding promotes conformational changes in RFC, producing a spiral configuration of the complex optimized for binding the surface of an open clamp and a DNA helix. RFC initially binds PCNA in a closed conformation, which is followed by clamp opening. When ATP is bound to all sites in RFC, isomerization to the open complex is favored, and the extensive RFC-PCNA interactions in this conformation stabilize the open clamp and contribute to high affinity binding. Conversely, when ATP binding is blocked at one site, the geometry of RFC subunits is distorted, destabilizing the open RFC-PCNA complex and shifting the equilibrium toward a closed complex with reduced RFC-PCNA interactions.
Mutation of the ATP site in the Rfc3 subunit (C site) has the greatest effect on PCNA binding/opening, reducing PCNA binding by a factor of about 100 compared with WT RFC, indicating that ATP binding to this single site plays a key role. Although this RFC mutant forms predominantly closed complexes, the site missing ATP (C site) differs from the site bound to ADP (B site) in the closed bacteriophage T4 complex, suggesting that there is not a single conserved site that regulates clamp opening and closing. Mutation of the Arg finger in the D subunit of yeast RFC, which responds to ATP binding at site C, also had the largest effect on PCNA opening in comparison with mutation of other sites (50). As pointed out by Sakato et al. (50), it is interesting to note that this subunit is located in the center of the complex (Fig. 1D), suggesting that it plays a pivotal role in coordinating ATP-dependent conformational changes in the RFC complex that are required for high affinity clamp binding. In terms of our model, we propose that mutation of site C at the center of the complex makes a “kink” in the spiral geometry such that the open complex is destabilized relative to WT RFC. Similarly, mutations at sites B and D also affect this overall spiral geometry, but because they are not centrally located in the complex, mutation of these sites gives a smaller 20–25-fold decrease in binding.
Clamp opening assays support our model that defects in PCNA binding may be largely due to defects in the abilities of the Walker A mutants to stabilize open clamps. The opening assay provides not only a Kd of binding but also a measure of the relative populations of open clamps for WT RFC and the mutants. The relative populations of open clamps correlate in general with the relative binding affinities of the Walker A mutants; i.e. the greater the population of open clamps, the greater the binding affinity. The increase in AF488 intensity, which directly reflects the population of open clamps, is smallest for the Rfc3 mutant and largest for the Rfc1 mutant. Although we can globally fit our data to a model in which the initial equilibrium constant for binding is similar for all mutants but the equilibrium constant for opening varies to a greater degree, we cannot obtain a unique solution to this model. To rigorously analyze our data to test the idea that binding defects of the Walker A mutants are largely due to destabilization of the open complex, a value for the quantum yield of the open state of PCNA is required. This quantum yield cannot be deduced from fluorescence values at saturating WT RFC concentrations because we do not know what fraction of the clamps are open. We are working on methods to measure the absolute population of open clamps to test this model.
Mutation of Walker A sites or the complete absence of ATP had larger effects on RFC interactions with PCNA than with DNA. Here, DNA binding was measured in the absence of PCNA so that differences in the abilities of RFC mutants to bind/open PCNA did not influence DNA binding activity of RFC. DNA binding does not appear to be as tightly coupled to ATP binding as clamp binding is. The Kd value for RFC binding DNA in the absence of ATP was only about 7-fold greater than in the presence of ATP, whereas the difference in Kd values for PCNA binding was over 100-fold. One possible caveat to this interpretation is that ATPγS was used in DNA binding assays to block DNA-stimulated ATP hydrolysis, and it is possible that ATPγS does not support DNA binding as well as ATP. Clamp opening is not as robust in assays with ATPγS as with ATP (34). Not only were effects on DNA binding smaller than on clamp binding, but the individual mutations also affected DNA binding differently than clamp binding. The Rfc1 (A site) mutant was the most active of the mutants in clamp binding but the least active in DNA binding. The DNA binding chamber of RFC is lined with positively charged residues and residues that hydrogen bond to the DNA duplex (7, 20). Perhaps this environment generally supports DNA binding such that ATP-dependent conformational changes in RFC do not increase RFC-DNA interactions as much as RFC-PCNA interactions.
CONCLUSION
A series of multiple discrete steps, driven by ATP binding and hydrolysis, is required for clamp loaders to assemble clamps on DNA. Although our results show that mutations to individual ATP binding sites affect clamp loader function to different degrees, our data do not support a model in which ATP binding to a single site promotes a specific step in the clamp loading reaction cycle. Instead, our data combined with structural data suggest that the ATP sites work in unison to promote conformational changes within the entire complex that increase the affinity of the clamp loader for the clamp and DNA. The ATP sites may fill sequentially (45), but all sites must be filled to efficiently drive ATP-dependent steps in the clamp loading reaction. As illustrated in Fig. 8, binding of ATP to clamp loaders promotes a spiral geometry of the clamp loader-clamp complex that matches the surface of an open clamp and the pitch of the DNA helix (46, 53). Mutation of a single ATP binding site likely creates a kink in this spiral geometry that destabilizes the open complex by reducing RFC-PCNA interactions, favoring the closed complex. Thus, ATP binding and hydrolysis events at multiple sites work together as an on/off switch to promote formation of a ternary clamp loader-clamp-DNA complex and release of the clamp on DNA, respectively. AAA+ proteins, including the origin recognition complex Cdc6 and the minichromosome maintenance helicase, which play essential roles in DNA replication, are required for many cellular processes that generally involve relocating or remodeling macromolecules. The results of this work have implications for the role of ATP binding at individual sites in other AAA+ proteins where reaction mechanisms have not yet been established and discrete steps in the reaction cycles are difficult to measure quantitatively.
This work was supported, in whole or in part, by National Institutes of Health Grants GM082849 (to L. B. B.), T32 CA009126-32 (to J. N. H. through the University of Florida Shands Cancer Center), and GM38839 (to M. O. D.).
- PCNA
- proliferating cell nuclear antigen
- RFC
- replication factor C
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- DCC
- 7-diethylaminocoumarin-3-carboxylic acid succinimidyl ester
- RhX
- X-rhodamine
- MDCC
- 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin
- PCNA-MDCC
- PCNA labeled with MDCC
- AF488
- Alexa Fluor 488
- PCNA-AF4882
- PCNA labeled with Alexa Fluor 488
- Imax
- maximum intensity
- Imin
- minimum intensity.
REFERENCES
- 1. Hanson P. I., Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 2. Snider J., Houry W. A. (2008) AAA+ proteins: diversity in function, similarity in structure. Biochem. Soc. Trans. 36, 72–77 [DOI] [PubMed] [Google Scholar]
- 3. Dougan D. A., Mogk A., Zeth K., Turgay K., Bukau B. (2002) AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 529, 6–10 [DOI] [PubMed] [Google Scholar]
- 4. Duderstadt K. E., Berger J. M. (2008) AAA+ ATPases in the initiation of DNA replication. Crit. Rev. Biochem. Mol. Biol. 43, 163–187 [DOI] [PubMed] [Google Scholar]
- 5. Krishna T. S., Kong X. P., Gary S., Burgers P. M., Kuriyan J. (1994) Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 6. Yao N., Turner J., Kelman Z., Stukenberg P. T., Dean F., Shechter D., Pan Z. Q., Hurwitz J., O'Donnell M. (1996) Clamp loading, unloading and intrinsic stability of the PCNA, β and gp45 sliding clamps of human, E. coli and T4 replicases. Genes Cells 1, 101–113 [DOI] [PubMed] [Google Scholar]
- 7. Bowman G. D., O'Donnell M., Kuriyan J. (2004) Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429, 724–730 [DOI] [PubMed] [Google Scholar]
- 8. Davey M. J., Jeruzalmi D., Kuriyan J., O'Donnell M. (2002) Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 3, 826–835 [DOI] [PubMed] [Google Scholar]
- 9. Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neuwald A. F., Aravind L., Spouge J. L., Koonin E. V. (1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 11. Schmidt S. L., Gomes X. V., Burgers P. M. (2001) ATP utilization by yeast replication factor C. III. The ATP-binding domains of Rfc2, Rfc3, and Rfc4 are essential for DNA recognition and clamp loading. J. Biol. Chem. 276, 34784–34791 [DOI] [PubMed] [Google Scholar]
- 12. Cai J., Yao N., Gibbs E., Finkelstein J., Phillips B., O'Donnell M., Hurwitz J. (1998) ATP hydrolysis catalyzed by human replication factor C requires participation of multiple subunits. Proc. Natl. Acad. Sci. U.S.A. 95, 11607–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seybert A., Wigley D. B. (2004) Distinct roles for ATP binding and hydrolysis at individual subunits of an archaeal clamp loader. EMBO J. 23, 1360–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wieczorek A., Downey C. D., Dallmann H. G., McHenry C. S. (2010) Only one ATP-binding DnaX subunit is required for initiation complex formation by the Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem. 285, 29049–29053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Downey C. D., Crooke E., McHenry C. S. (2011) Polymerase chaperoning and multiple ATPase sites enable the E. coli DNA polymerase III holoenzyme to rapidly form initiation complexes. J. Mol. Biol. 412, 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bloom L. B., Turner J., Kelman Z., Beechem J. M., O'Donnell M., Goodman M. F. (1996) Dynamics of loading the beta sliding clamp of DNA polymerase III onto DNA. J. Biol. Chem. 271, 30699–30708 [DOI] [PubMed] [Google Scholar]
- 17. Anderson S. G., Williams C. R., O'donnell M., Bloom L. B. (2007) A function for the ψ subunit in loading the Escherichia coli DNA polymerase sliding clamp. J. Biol. Chem. 282, 7035–7045 [DOI] [PubMed] [Google Scholar]
- 18. Anderson S. G., Thompson J. A., Paschall C. O., O'Donnell M., Bloom L. B. (2009) Temporal correlation of DNA binding, ATP hydrolysis, and clamp release in the clamp loading reaction catalyzed by the Escherichia coli gamma complex. Biochemistry 48, 8516–8527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finkelstein J., Antony E., Hingorani M. M., O'Donnell M. (2003) Overproduction and analysis of eukaryotic multiprotein complexes in Escherichia coli using a dual-vector strategy. Anal. Biochem. 319, 78–87 [DOI] [PubMed] [Google Scholar]
- 20. Yao N. Y., Johnson A., Bowman G. D., Kuriyan J., O'Donnell M. (2006) Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J. Biol. Chem. 281, 17528–17539 [DOI] [PubMed] [Google Scholar]
- 21. Johnson A., Yao N. Y., Bowman G. D., Kuriyan J., O'Donnell M. (2006) The replication factor C clamp loader requires arginine finger sensors to drive DNA binding and proliferating cell nuclear antigen loading. J. Biol. Chem. 281, 35531–35543 [DOI] [PubMed] [Google Scholar]
- 22. Thompson J. A., Marzahn M. R., O'Donnell M., Bloom L. B. (2012) Replication factor C is a more effective proliferating cell nuclear antigen (PCNA) opener than the checkpoint clamp loader, Rad24-RFC. J. Biol. Chem. 287, 2203–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ayyagari R., Impellizzeri K. J., Yoder B. L., Gary S. L., Burgers P. M. (1995) A mutational analysis of the yeast proliferating cell nuclear antigen indicates distinct roles in DNA replication and DNA repair. Mol. Cell. Biol. 15, 4420–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bauer G. A., Burgers P. M. (1988) The yeast analog of mammalian cyclin/proliferating-cell nuclear antigen interacts with mammalian DNA polymerase δ. Proc. Natl. Acad. Sci. U.S.A. 85, 7506–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yao N., Coryell L., Zhang D., Georgescu R. E., Finkelstein J., Coman M. M., Hingorani M. M., O'Donnell M. (2003) Replication factor C clamp loader subunit arrangement within the circular pentamer and its attachment points to proliferating cell nuclear antigen. J. Biol. Chem. 278, 50744–50753 [DOI] [PubMed] [Google Scholar]
- 26. Marzahn M. R., Bloom L. B. (2012) Improved solubility of replication factor C (RFC) Walker A mutants. Protein Expr. Purif. 83, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thompson J. A., Paschall C. O., O'Donnell M., Bloom L. B. (2009) A slow ATP-induced conformational change limits the rate of DNA binding but not the rate of β clamp binding by the Escherichia coli γ complex clamp loader. J. Biol. Chem. 284, 32147–32157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. White S. R., Lauring B. (2007) AAA+ ATPases: achieving diversity of function with conserved machinery. Traffic 8, 1657–1667 [DOI] [PubMed] [Google Scholar]
- 29. Majka J., Burgers P. M. (2004) The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 78, 227–260 [DOI] [PubMed] [Google Scholar]
- 30. Cronet P., Bellsolell L., Sander C., Coll M., Serrano L. (1995) Investigating the structural determinants of the p21-like triphosphate and Mg2+ binding site. J. Mol. Biol. 249, 654–664 [DOI] [PubMed] [Google Scholar]
- 31. Story R. M., Steitz T. A. (1992) Structure of the recA protein-ADP complex. Nature 355, 374–376 [DOI] [PubMed] [Google Scholar]
- 32. Chen S., Levin M. K., Sakato M., Zhou Y., Hingorani M. M. (2009) Mechanism of ATP-driven PCNA clamp loading by S. cerevisiae RFC. J. Mol. Biol. 388, 431–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakato M., Zhou Y., Hingorani M. M. (2012) ATP binding and hydrolysis-driven rate-determining events in the RFC-catalyzed PCNA clamp loading reaction. J. Mol. Biol. 416, 176–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chiraniya A., Finkelstein J., O'Donnell M., Bloom L. B. (2013) A novel function for the conserved glutamate residue in the Walker B motif of replication factor C. Genes 4, 134–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomes X. V., Burgers P. M. (2001) ATP utilization by yeast replication factor C. I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem. 276, 34768–34775 [DOI] [PubMed] [Google Scholar]
- 36. Zhuang Z., Yoder B. L., Burgers P. M., Benkovic S. J. (2006) The structure of a ring-opened proliferating cell nuclear antigen-replication factor C complex revealed by fluorescence energy transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hingorani M. M., O'Donnell M. (1998) ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 273, 24550–24563 [DOI] [PubMed] [Google Scholar]
- 38. Turner J., Hingorani M. M., Kelman Z., O'Donnell M. (1999) The internal workings of a DNA polymerase clamp-loading machine. EMBO J. 18, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Indiani C., O'Donnell M. (2006) The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 7, 751–761 [DOI] [PubMed] [Google Scholar]
- 40. Grabowski B., Kelman Z. (2003) Archeal DNA replication: eukaryal proteins in a bacterial context. Annu. Rev. Microbiol. 57, 487–516 [DOI] [PubMed] [Google Scholar]
- 41. Hübscher U. (2009) DNA replication fork proteins. Methods Mol. Biol. 521, 19–33 [DOI] [PubMed] [Google Scholar]
- 42. McHenry C. S. (2011) DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80, 403–436 [DOI] [PubMed] [Google Scholar]
- 43. Schmidt S. L., Pautz A. L., Burgers P. M. (2001) ATP utilization by yeast replication factor C. IV. RFC ATP-binding mutants show defects in DNA replication, DNA repair, and checkpoint regulation. J. Biol. Chem. 276, 34792–34800 [DOI] [PubMed] [Google Scholar]
- 44. Podust V. N., Tiwari N., Ott R., Fanning E. (1998) Functional interactions among the subunits of replication factor C potentiate and modulate its ATPase activity. J. Biol. Chem. 273, 12935–12942 [DOI] [PubMed] [Google Scholar]
- 45. Gomes X. V., Schmidt S. L., Burgers P. M. (2001) ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA. J. Biol. Chem. 276, 34776–34783 [DOI] [PubMed] [Google Scholar]
- 46. Kelch B. A., Makino D. L., O'Donnell M., Kuriyan J. (2012) Clamp loader ATPases and the evolution of DNA replication machinery. BMC Biol. 10, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uhlmann F., Cai J., Gibbs E., O'Donnell M., Hurwitz J. (1997) Deletion analysis of the large subunit p140 in human replication factor C reveals regions required for complex formation and replication activities. J. Biol. Chem. 272, 10058–10064 [DOI] [PubMed] [Google Scholar]
- 48. Podust V. N., Tiwari N., Stephan S., Fanning E. (1998) Replication factor C disengages from proliferating cell nuclear antigen (PCNA) upon sliding clamp formation, and PCNA itself tethers DNA polymerase delta to DNA. J. Biol. Chem. 273, 31992–31999 [DOI] [PubMed] [Google Scholar]
- 49. Gomes X. V., Gary S. L., Burgers P. M. (2000) Overproduction in Escherichia coli and characterization of yeast replication factor C lacking the ligase homology domain. J. Biol. Chem. 275, 14541–14549 [DOI] [PubMed] [Google Scholar]
- 50. Sakato M., O'Donnell M., Hingorani M. M. (2012) A central swivel point in the RFC clamp loader controls PCNA opening and loading on DNA. J. Mol. Biol. 416, 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kelch B. A., Makino D. L., O'Donnell M., Kuriyan J. (2011) How a DNA polymerase clamp loader opens a sliding clamp. Science 334, 1675–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tainer J. A., McCammon J. A., Ivanov I. (2010) Recognition of the ring-opened state of proliferating cell nuclear antigen by replication factor C promotes eukaryotic clamp-loading. J. Am. Chem. Soc. 132, 7372–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simonetta K. R., Kazmirski S. L., Goedken E. R., Cantor A. J., Kelch B. A., McNally R., Seyedin S. N., Makino D. L., O'Donnell M., Kuriyan J. (2009) The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 137, 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]