

Background: Force in fluid flow regulates von Willebrand factor (VWF) A1 domain binding to glycoprotein Ibα (GPIbα).
Results: X-ray crystal structures of high affinity A1-GPIbα complexes and mutations reveal interactions involving central leucine-rich repeats of GPIbα.
Conclusion: Structural changes are on a pathway to a force-induced super high affinity state.
Significance: A1-GPIbα complexes provide insight into mechanochemistry of bleeding disorders.
Keywords: Directed Evolution, Immunology, Protein Folding, Structural Biology, von Willebrand Factor, Glycoprotein Ib, Yeast Display
Abstract
Activation by elongational flow of von Willebrand factor (VWF) is critical for primary hemostasis. Mutations causing type 2B von Willebrand disease (VWD), platelet-type VWD (PT-VWD), and tensile force each increase affinity of the VWF A1 domain and platelet glycoprotein Ibα (GPIbα) for one another; however, the structural basis for these observations remains elusive. Directed evolution was used to discover a further gain-of-function mutation in A1 that shifts the long range disulfide bond by one residue. We solved multiple crystal structures of this mutant A1 and A1 containing two VWD mutations complexed with GPIbα containing two PT-VWD mutations. We observed a gained interaction between A1 and the central leucine-rich repeats (LRRs) of GPIbα, previously shown to be important at high shear stress, and verified its importance mutationally. These findings suggest that structural changes, including central GPIbα LRR-A1 contact, contribute to VWF affinity regulation. Among the mutant complexes, variation in contacts and poor complementarity between the GPIbα β-finger and the region of A1 harboring VWD mutations lead us to hypothesize that the structures are on a pathway to, but have not yet reached, a force-induced super high affinity state.
Introduction
von Willebrand factor (VWF)3 is a plasma glycoprotein with monomers that are 2,050 residues in length and are linked head-to-head and tail-to-tail into ultralong concatemers. VWF senses exposure of the subendothelium and changes in flow at sites of bleeding and is required for hemostasis in the rapid flow of the arteriolar circulation (1–5). VWF binds through its A1 domain to the GPIbα subunit of the GPIb-GPIX complex on platelets. No interaction between VWF and GPIbα is seen in stasis or in low flow; however, above a threshold flow, they bind and mediate in vivo platelet plug formation and in vitro aggregation of platelets in stirred cuvettes and rolling of platelets on VWF immobilized on the walls of flow chambers (6). Multiple mechanisms are thought to contribute to enhancement of VWF adhesiveness by flow. One mechanism by which flow can activate VWF is induction of transition from a bird's nest to an elongated conformation, leading to better exposure of the VWF monomers for multimeric binding to platelets (7).
Conformational change within the A1 domain of VWF and platelet glycoprotein Ibα is the second postulated mechanism for regulating their interaction (8). In flow, the hydrodynamic force exerted on VWF multimers results in tensile force exerted throughout the multimer at the connections between neighboring domains (e.g. on the N and C termini of the A1 domain). The force exerted near the middle of a long VWF multimer free in shear flow has been estimated to reach 10 pN, and it would be much higher for VWF in elongational flow or bound to the vessel wall at a site of hemorrhage (5, 9). Named after the three A domains in VWF, VWA domains (or VWA folds) are widely distributed and also appear in integrins as the inserted domains in their α and β subunits (i.e. the integrin αI and βI domains). As a VWA domain, A1 has largely alternating β-strands and α-helices that assemble into a Rossman-like fold with a central β-sheet surrounded by amphipathic α-helices (10). In integrin VWA (αI and βI) domains, a 10-Å axial displacement of the C-terminal α-helix is linked to communication of allostery to a neighboring domain and to a change in conformation at the metal ion-dependent adhesion site where ligand is bound (11). However, the A domains in VWF contain no bound metal ion, and GPIbα binds at a completely different site on A1 to the edge of the central β-sheet rather than to the turns between β-strands and α-helices at the C-terminal end of the β-sheet (12, 13).
Mutations in VWF are the most frequent cause of heritable bleeding disorders and provide important insights into function. Interesting gain-of-function mutations in von Willebrand disease (VWD) type 2B localize to the region of A1 in or adjacent to the ordered N- and C-terminal segments, distal to the binding site for platelet GPIbα. Therefore, it has been suggested that these mutations might release A1 from a lower affinity conformation (10, 12). Notably, the sites of these mutations are near the termini where elongational force in VWF multimers is exerted on A1.
Gain-of-function mutations in platelet type VWD (PT-VWD) locate in GPIbα and have a phenotype similar to VWD type 2B mutations in VWF A1. GPIbα has an N-terminal leucine-rich repeat (LRR) domain with eight LRRs and N- and C-terminal segments that cap the LRRs (10). A partially ordered β-switch region in the C-terminal cap of GPIbα becomes ordered upon binding to A1 and forms a β-hairpin with two β-strands that add onto the central VWA β-sheet (12). GPIbα VWD mutations all map to the β-switch and are thought to favor the β-strand conformation found in the bound state over a disordered or α-helical state found in the unbound state (12–14).
Crystal structures of the A1 and GPIbα complex show two discontinuous binding interfaces, whether the components are wild-type or contain gain-of-function A1-R1306Q and GPIb-M239V mutations (12, 13). Differences were seen between wild-type and gain-of-function complexes in the N and C termini and α1-β2 loop of A1. However, no compelling gained interactions were seen in the gain-of-function mutant complex that could explain its higher affinity, and it has remained unclear how the affinity of A1 and GPIbα for one another could be regulated by hydrodynamic flow. Thus, in contrast to integrin I domains, an allosteric mechanism for regulating affinity of A1 and GPIbα for one another remains unclear.
Two types of evidence for force-induced conformational change in the A1-GPIbα complex motivated the current study. Chimeras between human and canine GPIbα showed that the LRRs between the two discontinuous contacts with A1 were unimportant at low shear but became important at high shear (15, 16). Measurements on a receptor and ligand in a single molecule (ReaLiSM) construct in which A1 was connected at its C terminus through a polypeptide linker to the N terminus of GPIbα revealed reversible unbinding and rebinding (8). Two distinct pathways of force-dependent dissociation were found. The kinetics of force-dependent dissociation (koff) of the two pathways were well fit by biophysical models that relate koff to koff in the absence of force (koff0) and the exponential of tensile (elongational) force. A complex with faster koff0 predominated below a force of 10 pN, and a complex with a slower koff0 and lesser exponentiation by force predominated above 10 pN, suggesting two conformational states of the receptor-ligand complex (8).
Here, we use crystallographic structures and mutations to address the principles that regulate affinity of A1 and GPIbα for one another. Moving beyond the use of one gain-of-function mutation in A1 and one in GPIbα (12), we use a total of four VWD gain-of-function mutations, two each in A1 and GPIbα. Using directed evolution in yeast, we discover further gain-of-function mutations in A1. Shifting the long range disulfide bond in A1 by one residue in sequence causes a large increase in affinity. We describe the structure of this mutant alone and complexed with GPIbα containing two VWD mutations. In our high affinity structures, a gained interaction is found in the region implicated in GPIbα chimeras as important at high shear stress (15) and is found to be important in mutational experiments. Structural analysis of the region harboring gain-of-function mutations in A1 suggests clashing rather than complementary interactions with the GPIbα β-finger and leads us to suggest that our structures are on a pathway to, but have not yet reached, a hypothetical force-induced super high affinity state.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The cDNA sequence encoding VWF A1 (Glu1264–Leu1469) was cloned into the pET21a vector with an initiation Met codon and expressed in BL21(DE3) cells (17). Gain-of-function mutations were introduced using QuikChange (Stratagene). Refolding was performed essentially as described (17). Refolded A1 was purified with nickel-nitrilotriacetic acid beads (Qiagen), followed by a heparin-Sepharose affinity column (GE Healthcare), and a S75 gel filtration column (GE Healthcare). The cDNA for human platelet GPIbα (His1–Arg290) was mutated using QuikChange (Stratagene). For crystallization, two N-linked glycosylation sites were removed with N21R and N159R mutations. GPIbα mutants were cloned into an ET-6 vector, which resembles ET-1 (18), except it contains only a C-terminal His6 tag. Stable transfectants were established in HEK293T cells. Protein was expressed in 293 Freestyle medium (Invitrogen) and initially purified using an anti-GPIbα IgG-Sepharose column. Monoclonal antibody 12E4 (19) was coupled to cyanogen bromide-activated Sepharose 4B FF (GE Healthcare) using the manufacturer's protocols. Culture medium supernatant was flowed over 12E4-Sepharose and eluted with 0.1 m glycine, pH 2.4, and immediately neutralized with one-quarter volume of 1 m Tris, pH 8.5. GPIbα was further purified on a S200 gel filtration column (GE Healthcare).
Yeast Surface Display
The cDNA sequence encoding the above A1 fragment was cloned into the pCTCON2 yeast surface display plasmid in frame with Aga2-HA tag and with the C-terminal c-Myc tag. A1 random libraries were created using nucleotide analog PCR (20) and transformed into Saccharomyces cerevisiae strain EBY100 by electroporation (21). Library complexity was greater than 107 unique clones. Expression was induced using synthetic galactose medium with casamino acids (SGCAA), and 109 yeasts were sorted by magnetic activated cell sorting (LS Columns, Miltenyi Biotech) for functional expression using 200 nm biotinylated GPIbα prepared with EZ-Link NHS-PEG4-Biotin (Pierce) and coated onto anti-biotin magnetic beads (Miltenyi Biotech). The functional library was then regrown and sorted twice by fluorescence-activated cell sorting (FACS) with 50 nm biotinylated GPIbα detected with streptavadin-phycoerythrin (Invitrogen); and with a 1:100 dilution of 9E10 anti-c-Myc antibody (Covance) detected with FITC-labeled anti-mouse IgG (Invitrogen). The sorting gate was selected for improved apparent affinity. Last, the library was sorted for decreased off-rate by competition with 10 μm non-labeled GPIbα for 4 h before sorting for cells retaining fluorescence. Fourteen recovered clones were sequenced.
Flow cytometry of yeast was performed on a FACScan (BD Biosciences). 105 yeasts were harvested, washed three times in room temperature PBS + 1% BSA (PBSB), and incubated with 50 nm biotinylated GPIbα and anti-c-Myc 9E10 IgG (1:200) at room temperature for 1 h. Cells were then centrifuged and washed three times in ice-cold PBSB before incubation with secondary reagents (1:200 dilution of streptavidin-phycoerythrin and 1:100 dilution of FITC-labeled goat anti-mouse IgG) for 20 min at 4 °C. 104 yeasts were analyzed for mean fluorescence intensity for both GPIbα and c-Myc. The specific fluorescence intensity was calculated by subtracting autofluorescence from uninduced controls. Because the fluorescence signal is affected by both affinity and surface expression, we calculate the adjusted specific fluorescence intensity by dividing the specific fluorescence intensity from the GPIbα signal by the specific fluorescence intensity from c-Myc and normalizing to WT (WT adjusted specific fluorescence intensity = 1). Single mutations of A1 expressed using yeast display were generated using QuikChange mutagenesis.
Radioligand Binding Experiments
Purified high affinity GPIbα containing PT-VWD mutations G233V and M239V (GPIbα/VWD2) was radioiodinated using the IODO-GEN method (Pierce). 250 μg of purified protein was iodinated with 1 mCi of Na125I radionuclide (PerkinElmer Life Sciences) for 20 min at room temperature. The reaction was quenched with 2 mg/ml l-tyrosine for 5 min. After the addition of 5 mm potassium iodide, the radiolabeled GPIbα/VWD2 was purified on a PD-10 column (GE Healthcare) equilibrated with PBS. Radioligand concentration was determined with bicinchoninic acid assay (Pierce) with BSA as a standard. Iodination was >80% efficient. 105 yeast cells displaying A1 were incubated with increasing concentrations of iodinated GPIbα/VWD2 in PBS with 1% BSA for 2 h. Samples were then centrifuged through oil (67% n-octyl-phthalate, 33% n-butyl-phthalate) (22). The samples were frozen on dry ice, and the cell pellet at the tip was cut and analyzed in a γ counter (PerkinElmer Life Sciences). Similar procedures were used for competition assays with 5–10 nm 125I-GPIbα/VWD2 as the probe.
Binding Analysis
Binding data were analyzed by non-linear regression using software Prism version 5.03 (GraphPad). Saturation binding data were collected alongside a negative control to determine the nonspecific binding. Both sets of data were globally fit to a one-site binding model extended by a nonspecific term using Equation 1,
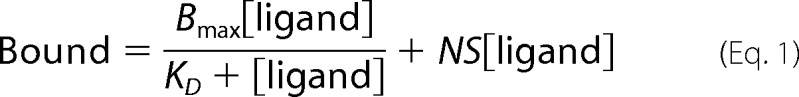 |
Specific competition binding data were fit to a one-site model with Equations 2 and 3, where KD,probe was the affinity of 125I-GPIbα/VWD2 for that particular A1 mutant, [probe] = 5 or 10 nm depending on the experiment, and B0 is the specific binding of the probe in the absence of the nonlabeled GPIbα.
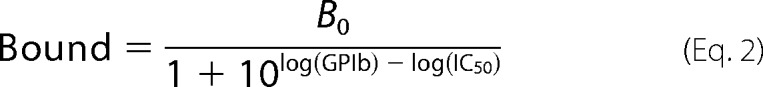 |
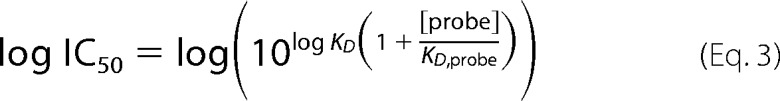 |
Protein Crystallization and Data Collection
Proteins were in 10 mm Tris, pH 7.4, and 150 mm sodium chloride. For complexes, equimolar amounts of A1 and GPIbα were mixed. Crystals of A1 with Y1271C and C1272R mutations (A1/SS) appeared in drops containing 25% PEG 3350 and 0.2 m calcium acetate. These crystals were crushed and used for seeding. Crystals grown in 8 mg/ml protein, 15% PEG 3350, and 0.2 m calcium acetate were harvested and soaked in the same buffer containing 20% glycerol for cryoprotection. Crystals of A1/SS-GPIbα/VWD2 complex appeared in drops with 20% PEG 4000, 0.16 m ammonium sulfate, 0.08 m sodium acetate, pH 4.6, and 20% glycerol. These crystals were crushed and used for seeding crystal growth in 8 mg/ml complex, 15% PEG 4000, 0.16 m ammonium sulfate, 0.08 m sodium acetate, pH 4.6, and 20% glycerol. Because these crystals were formed in buffer containing 20% glycerol, no additional cryoprotection was used. Crystals of A1 with two VWD mutations R1306Q and I1309V (A1/VWD2) in complex with GPIbα/VWD2 appeared in 12% PEG 8000, 0.1 m sodium cacodylate, pH 6.5, 0.1 m calcium acetate, and 0.4 mg/ml ristocetin (Sigma-Aldrich). These crystals were crushed and used for seeding. Crystals grew in 10 mg/ml complex, 14% PEG 8000, 0.2 m calcium acetate, 0.1 m sodium cacodylate, pH 6.5, and 0.4 mg/ml ristocetin. Next, we soaked these crystals in 14% PEG 8000, 0.2 m calcium acetate, 0.1 sodium cacodylate, pH 6.5, and 4 mg/ml ristocetin. Data for A1/SS-GPIbα/VWD2 were collected at 100 K at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline. Data for A1/SS and A1/VWD2-GPIbα/VWD2 were collected at 100°K at the General Medicine/Cancer (GM/CA)-CAT 23-ID beamline. Both beamlines are at the Advanced Photon Source, Argonne National Laboratory.
Structure Determination and Refinement
All data were processed with HKL-2000 or XDS. The structures were solved by molecular replacement using MOLREP (23). 1SQ0 served as the model for A1/VWD2-GPIbα/VWD2; A1/VWD2-GPIbα/VWD2 served as the model for A1/SS-GPIbα/VWD2; and 1AUQ served as the search model for A1/SS. Structure refinement was performed using COOT (24) and PHENIX (25). The resolution for A1/SS-GPIbα/VWD2 was found to extend to 2.8 Å using cross-correlation (26); we could not similarly extend resolution of the data sets for the other two crystals because their higher resolution reflections went beyond the edge of the detector. Validation and Ramachandran statistics used MOLPROBITY (27). APBS was used to calculate electrostatic surfaces (28).
RESULTS
Directed Evolution of A1 Affinity
The A1 domain of VWF was fused to the Aga2 protein in S. cerevisiae and expressed on the yeast surface (29). A library of greater than 107 random mutants was selected for high affinity for GPIbα using several steps of isolation with magnetic activated cell sorting and FACS. Fourteen mutants were selected, most of which contained more than one mutation (Table 1). Remarkably, about half contain mutations of residues previously found to be mutated in VWD (Table 1).
TABLE 1.
A1 clones with high affinity for GPIbα selected by yeast surface display
Many of the isolates contained more than one mutation. Mutations of residues known to be mutated in VWD are in boldface type, and those with the exact substitutions in VWD are additionally underlined.
| Clone | A1 mutation(s) |
|---|---|
| 1 | L1267S/I1309T |
| 2 | I1309F |
| 3 | Y1271C/C1458R |
| 4 | C1272R |
| 5 | V1279A /S1310F |
| 6 | V1314A |
| 7 | R1287G /V1398I/I1410T/E1429K |
| 8 | F1270S/M1393I/K1406R |
| 9 | Y1271C/Q1388R/L1460F |
| 10 | S1378P |
| 11 | L1267S/D1269G/Y1271C/D1302A/I1309V/V1409T/K1430R/D1451G |
| 12 | R1274G/H1322R/K1332R |
| 13 | I1309T/R1426C |
| 14 | F1270S/W1313P/V1360I/Q1391R/P1465L |
The highest affinity single mutant found was Y1271C (Table 2). Mutant Y1271S was markedly lower in affinity although still higher than wild type. This finding, together with the observation that the Y1271C mutation introduces a cysteine adjacent to Cys1272, which forms a long range disulfide bond to Cys1458, suggested that a novel disulfide bond introduced by the mutation might be responsible for the increase in affinity. To test this possibility, we prepared Y1271C/C1272R, which could form an alternative, long range Cys1271 disulfide bond to Cys1458, and Y1271C/C1458R, which could form a vicinal disulfide between Cys1271 and Cys1272. The latter showed GPIbα binding similar to wild type (Table 2). In contrast, Y1271C/C1272R showed a 10-fold increase in fluorescent GPIbα binding compared with the 5-fold increase in Y1271C (Table 2). These results are consistent with a mixture of Cys1271–Cys1458 and Cys1272–Cys1458 long range disulfide bonds in Y1271C and only a Cys1271–Cys1458 long range disulfide in Y1271C/C1272R. Formation of a disulfide bond in Y1271C/C1272R was confirmed by its differing migration in reducing and non-reducing SDS-PAGE (data not shown). We conclude that moving the long range disulfide bond in A1 so that it connects to residue 1271 rather than 1272 markedly increases affinity.
TABLE 2.
Binding of VWF-A1 mutants to GPIbα
| Mutation | ASFIa for WT GPIbα | KD for WT GPIbαb | KD for GPIbα G233V/M239V (VWD2)b |
|---|---|---|---|
| nm | nm | ||
| WT | 1 | 2470 (1920–3170) | 109 (70–148) |
| Y1271C | 4.87 | 487 (403–589) | 81.2 (44–119) |
| Y1271S | 1.69 | NDc | ND |
| C1272R | 1.30 | ND | ND |
| Y1271C/C1272R (A1/SS) | 9.82 | 245 (170–357) | 9.0 (2.6–15.3) |
| Y1271C/C1458R | 0.95 | ND | ND |
| C1272R/C1458R | 1.00 | ND | ND |
| R1306Q | 1.71 | 1010 (848–1210) | 83 (32–135) |
| I1309V | 3.41 | 737 (651–834) | 95 (42–148) |
| R1306Q/I1309V (A1/VWD2) | 9.06 | 245 (214–281) | 9.7 (3.7–15.7) |
a ASFI, adjusted specific fluorescence intensity; the ratio of specific GPIbα fluorescence/c-Myc tag fluorescence normalized to WT.
b KD values were determined by radiometric titration with 125I-labeled GPIbα/VWD2 and competition with unlabeled GPIbα or GPIbα/VWD2. Error is reported as 95% confidence interval.
c ND, not determined.
We quantitated affinity with saturation binding of 125I-G233V/M239V-GPIbα (GPIbα/VWD2). The G233V and M239V GPIbα PT-VWD mutations have previously been found to be additive or synergistic in VWF functional assays (30, 31). Competition was used to determine the affinity of A1 mutants for wild-type GPIbα and GPIbα/VWD2 (Table 2). Individual A1 VWD gain-of-function R1306Q and I1309V mutants showed a severalfold increase in affinity, and the double R1306Q/I1309V mutant (A1/VWD2) showed a 10-fold increase in affinity (17). Notably, the novel Y1271C/C1272R mutant (A1/SS) also showed a 10-fold increase in affinity (Table 2). The KD values for A1/VWD2 and A1/SS were each close to 250 nm for wild-type GPIbα and 10 nm for GPIbαVWD2 (Table 2).
Overall Crystal Structures
To investigate the structural basis for increased affinity, we determined crystal structures of GPIbα/VWD2 complexed with A1/VWD2 (2.08 Å resolution, 1 complex/asymmetric unit) (Fig. 1A), GPIbα/VWD2 complexed with A1/SS (2.8 Å resolution, 4 complexes/asymmetric unit), and for comparison, A1/SS alone (2.2 Å resolution, 2 molecules/asymmetric unit) (Table 3). Furthermore, we compare our structures with a complex with R1306Q A1 and M239V GPIbα mutations (A1/VWD1- GPIbα/VWD1, 3.1 Å, 1 complex/asymmetric unit) (12) and wild-type A1 complexed with wild-type GPIbα (A1/WT-GPIbα/WT, 2.6 Å, 1 complex/asymmetric unit) (13). A number of structures of A1 and GPIbα, either alone or complexed with other proteins, are available for comparison (Table 4).
FIGURE 1.

The VWF A1-GPIbα complex. A, overall structure in a schematic diagram. A1 (cyan) has a central β-sheet that interacts with GPIbα (pink) by forming an extended β-sheet with the GPIbα β-switch. A single disulfide bond in A1 and three disulfide bonds in GPIbα are shown in yellow stick form. VWD type 2B mutations locate near the A1 termini distant from the GPIbα interface and are shown as Cα spheres in red (R1306Q and I1309V type 2B mutations used in this study), silver (other 2B mutations), and yellow (Y1271C and C1272R mutations used here). PT-VWD mutations locate in the GPIbα β-switch region (green Cα spheres for the G233V and M239V mutations used here). GPIbα LRR2 to -4, shown not to bind to A1 in previous crystal structures but to be important for ristocetin and shear-induced binding to VWF (15), are shown in gray. B and C, previous and current A1-GPIbα complexes superimposed on A1 and shown as Cα ribbons. Magenta, A1/VWD2-GPIbα/VWD2; orange, A1/SS-GPIbα/VWD2, chains E and F; yellow, chains G and H; cyan, A1/VWD1-GPIbα/VWD1 (12), PDB code 1M10; silver, A1/WT-GPIbα/WT (44), PDB code 1SQ0. C, the same superposition, rotated 90° to show the A1-binding face of GPIbα with A1 removed. D and E, sequences with secondary structure overlined, GPIbα regions marked, and decadal residues dotted. Regions present in constructs but disordered in crystals are in italic type. PT-VWD mutations used here and known VWD type 2B mutations are marked with asterisks.
TABLE 3.
X-ray diffraction and refinement statistics
| A1/VWD2-GPIbα/VWD2 | A1/SS | A1/SS-GPIbα/VWD2 | |
|---|---|---|---|
| Data | |||
| Space group | P61 | P21 | P21 |
| Examples/asymmetric unit | 1 | 2 | 4 |
| Cell dimensions | |||
| a, b, c (Å) | 89.1, 89.1, 124.5 | 41.3, 50.1, 96.9 | 96.5, 103.8, 119.9 |
| α, β, γ (degrees) | 90, 90, 120 | 90, 92.7, 90 | 90, 90.1, 90 |
| Wavelength (Å) | 1.03322 | 1.03322 | 1.00000 |
| Resolution (Å) | 48.4-2.08 (2.19-2.08)a | 50.0-2.2 (2.24-2.20)a | 48.3-2.80 (2.87-2.80)a |
| Observations/unique | 251,729/66,245 | 288,382/20,133 | 188,956/56,526 |
| I/s overall | 8.9 (1.7)a | 13.2 (2.0)a | 2.56 (0.3)a |
| CC½b | NAc | NA | 94.9 (15.6)a |
| Completeness | 99.9 (99.5)a | 99.5 (98.0)a | 96.6 (80.9)a |
| Rmerged | 8.9 (73.9)a | 14.8 (53.6)a | 58.8 (418)a |
| Structure refinement | |||
| Rwork (%) e | 17.6 | 18.6 | 25.3 |
| Rfree (%)f | 19.7 | 23.5 | 28.6 |
| Protein/solvent atoms | 3721/204 | 3234/175 | 14634/66 |
| Root mean square deviation bond length (Å) | 0.003 | 0.004 | 0.002 |
| Root mean square deviation bond angle (degrees) | 0.70 | 0.66 | 0.47 |
| Ramachandran favored/allowed/outliers (%) | 95.9/3.9/0.2 | 97.0/3.0/0 | 93.2/6.5/0.3 |
| Protein Data Bank codeg | 4C2A | 4C29 | 4C2B |
a Values in parentheses are for the highest resolution shell.
b Pearson's correlation coefficient between average intensities of random half-data sets of the measurements for each unique reflection (26).
c NA, not applicable; higher resolution reflections went beyond the edge of the detector, preventing extension of resolution using the CC½ criterion.
d Rmerge = Σi,h|I(i,h) − 〈I(h))〉|/Σi,h|I(i,h)|, where I(i,h) and 〈I(h)〉 are the ith and mean measurement of intensity of reflection h.
e Rwork = Σh(‖Fobs(h)| − |Fcalc(h)‖)/Σ|Fobs(h)|, where Fobs(h) and Fcalc(h) are the observed and calculated structure factors, respectively.
f Rfree is the R-factor for a selected subset of the reflections that are not included in refinement calculations.
g Experimental data has been deposited with the indicated deposition ID codes.
TABLE 4.
Structures of A1 and GPIbα examined
| Codea | A1 | A1 chains | GPIbα | GPIbα chains | Å | Additional molecules | Source/Reference |
|---|---|---|---|---|---|---|---|
| A1/VWD2-GPIb/VWD2 | R1306Q/I1309V | A | G233V/M239V | B | 2.08 | This work | |
| A1/SS-GPIb/VWD2 | Y1271C/C1272R | A,C,E,G | G233V/M239V | B,D,F,H | 2.8 | This work | |
| 1M10 | R1306Q | A | M239V | B | 3.1 | Ref. 12 | |
| 1SQ0 | WT | A | WT | B | 2.6 | Ref. 13 | |
| 1U0N | WT | A | WT | D | 2.95 | Botrocetin | Ref. 33 |
| A1/SS | Y1271C/C1272R | A, B | 2.2 | This work | |||
| 1AUQ | WT | A | 2.3 | Ref. 10 | |||
| 1OAK | WT | A | 2.2 | Fab | Ref. 43 | ||
| 1U0O | WT mouse | C | 2.7 | Botrocetin | Ref. 33 | ||
| 1IJK | I1309V | A | 2.6 | Botrocetin | Ref. 35 | ||
| 1IJB | I1309V | A | 1.8 | Fab | Ref. 35 | ||
| 1FNS | I1309V | A | 2.0 | Fab | Ref. 34 | ||
| 1UEX | WT | C | 2.85 | Biticetin | Ref. 50 | ||
| 3HXO | WT | A | 2.4 | Aptamer | Ref. 51 | ||
| 3HXQ | WT | A | 2.69 | Aptamer | Ref. 51 | ||
| 1GWB | WT | A,B | 2.8 | Ref. 49 | |||
| 1M0Z | M239V | A,B | 1.85 | Ref. 12 | |||
| 1OOK | WT | G | 2.3 | α-Thrombin | Ref. 45 | ||
| 1P8V | WT | A | 2.6 | α-Thrombin | Ref. 44 | ||
| 1QYY | WT | A,G | 2.8 | Ref. 46 | |||
| 1P9A | WT | G | 1.7 | Ref. 45 | |||
| 3P72 | WT | A | 1.9 | Peptide | Ref. 47 | ||
| 3PMH | WT | G | 3.2 | α-Thrombin | Ref. 48 |
a Abbreviation or PBD accession code.
The primary interaction between A1 and GPIbα occurs between the β-switch region of GPIbα and the central β-sheet of the A1 domain (Fig. 1A). LRR proteins contain caps over their N- and C-terminal LRR (32). The β-switch of GPIbα contains two β-strands and extends from the C-cap, which also contains α-helices and two disulfide bonds (Fig. 1, A and D). More C-terminal is an anionic region containing sulfated tyrosines that is not ordered in any complex crystal structure to date (Fig. 1D). The β-switch joins the central A1 β-sheet to form an extended β-sheet (Fig. 1A). PT-VWD mutations locate in the β-switch (Fig. 1A). The green spheres in Fig. 1A mark Cα atom positions of the VWD G233V and M239V mutations in GPIbα/VWD2. In the N-cap of GPIbα, a disulfide-stabilized β-finger forms a second site of interaction with A1 (Fig. 1, A and B). Sites of VWD mutations in A1 (spheres, Fig. 1A) are located not at the A1-GPIbα interface but near the N and C termini of A1. Red and yellow spheres in Fig. 1A mark Cα atom positions of the mutated residues in A1/VWD2 and A1/SS, respectively.
Complex structures solved in this work and previously published complex structures (12, 13) are shown superimposed on the A1 domain in Fig. 1B. Large conformational changes in the A1 domain are not observed for any structure. Structural differences in A1 are restricted to the N and C termini and to the α1-β2 loop (Fig. 1, B and E). Differences in the orientation of GPIbα relative to A1 are visualized by rotating the superimposed structures shown in Fig. 1B so that A1 faces the reader and then removing A1 to show the concave A1-binding face of GPIbα (Fig. 1C). In the WT complex, GPIbα is shifted laterally relative to complexes with high affinity mutants (i.e. to the right in Fig. 1C). Further differences between the structures are noted in the β-finger of GPIbα (Fig. 1, B and C). In contrast, there is little difference in position of the β-switch when superposition is performed on A1, in agreement with the constraints imposed by forming hydrogen bonds between the β-switch and the β-sheet in A1, and forming a large interface.
The β-Switch
The β-switch is the major binding interface between A1 and GPIbα and also the site of PT-VWD mutations. In complex with A1, the β-switch consists of two antiparallel β-strands linked by a β-hairpin turn (12, 13) (Figs. 1A and 2A). The β-hairpin joins the central A1 β-sheet, with switch residues 237–239 forming antiparallel hydrogen bonds to A1 β-strand 3 (Fig. 2, A–C). WT and all mutant structures show identical backbone hydrogen bonds between A1 and β-switch and between the two β-switch β-strands (Fig. 2, A–C) (data not shown). The GPIbα/VWD1 (M239V) and GPIbα/WT complexes show different β-hairpin turn geometries near turn residue Val234 (Fig. 2, B and C) (13); however, it is difficult to relate this to the Met to Val substitution at residue 239, which is four positions away in the β-ladder from the β-turn. In contrast, the G233V substitution in GPIbα/VWD2 is adjacent to the β-turn (Fig. 2A). To make room for the Val233 side chain adjacent to the Val236 side chain, the backbone at Gln232 and Val233 rotates and, together with a hydrogen bond between the Gln232 side chain and Val234 backbone, causes a markedly different β-turn conformation (Fig. 2, A–C).
FIGURE 2.

The β-switch. A–C, diagrams show Cα traces with key side chains and the backbone in β-sheet regions in stick representations with carbons in cyan (A1) and pink (GPIbα), nitrogens in blue, and oxygens in red. Hydrogen bonds in the switch region and the Lys132–Trp230 π-cation bond are shown as black dashes. The structures shown are indicated in each panel. D, flexibility in the β-switch in the absence of A1. GPIbα structures in the absence of A1 are superimposed on GPIbα complex structures with A1. The Cα atom positions of residues 233 and 239, mutated in VWD, are shown as small spheres. Structures are A1/VWD2-GPIbα/VWD2 (magenta); A1/WT-GPIbα/WT (44), PDB code 1SQ0 (cyan); GPIbα/VWD1 (12), PDB code 1M0Z chain B (purple); GPIbα/WT (45), PDB code 1P9A chain A (green); GPIbα/WT (44), PDB code 1P8V (dark green); GPIbα/WT (46), PDB code 1QYY chain A (orange); GPIbα/WT PDB code 3P72 (47) (yellow); GPIbα/WT (48), PDB code 3PMH (light green); GPIbα/WT (49), PDB code 1GWB chain A (lavender).
The Val side chain added by the G233V substitution does not interact with the A1 domain (Fig. 2A). Although the M239V substitution lies adjacent to A1, it does not markedly change the buried interface (12, 13) (Fig. 2, B and C). Residues 233 and 239 each lie in regions of the β-switch that form β-strands in A1-bound but not unliganded GPIbα structures (Fig. 2D). Val is a β-branched side chain and favors β-strand conformation (12); however, PT-VWD mutations may also act by destabilizing the unbound state of GPIbα (14). The finding that Val233 does not interact with A1 supports the concept that substitutions of β-switch residues may raise affinity by shifting the conformation of the β-switch away from that present in the unbound state and toward that present in the bound state (12, 14).
We point out here that in the unbound state of GPIbα, Trp230 and preceding residues in the β-switch adopt a β-conformation in common with the bound state (Fig. 2D). A remarkable π-cation bond between Trp230 and LRR5 residue Lys132 is found in all GPIbα structures (Fig. 2, A–D) and, together with nearby aromatic and hydrophobic residues (Fig. 2D), stabilizes this region in β-conformation. Because GPIbα residues Val229 and Trp230 are in β-conformation prior to A1 binding, they can act as a template for forming β-sheet hydrogen bonds to switch residues 240–238, followed by joining to the β-sheet in A1 and zipping up the remainder of the β-switch as a β-ribbon. Aromatic residues are abundant on both the A1 and GPIbα sides of their β-sheet interface and contribute to the stability of this interface (Fig. 2, A–D).
The α1-β2 Loop
As already noted, when complex structures are superimposed on the A1 domain (Fig. 3A), the most notable differences in A1 are in the N and C termini and the α1-β2 loop (Figs. 1 (A and B) and 3 (A–C); boxed in Fig. 3A). Previous comparison of single examples in crystals of A1/WT-GPIbα/WT and A1/VWD1-GPIbα/VWD1 complexes demonstrated differences in the conformation of the A1 α1-β2 loop and suggested that this correlated with and was responsible for affinity differences (13). However, the larger number of high affinity complexes made available by this work does not support a simple correlation between α1-β2 loop conformation and affinity (a more complex relationship is described under “Discussion”). Multiple examples of the A1/SS-GPIbα/VWD2 complex show an α1-β2 loop conformation essentially identical to that found in uncomplexed A1/WT (1AUQ; Fig. 3B). Further examples of WT-like A1 α1-β2 loop conformations are found in an A1-GPIbα complex formed with botrocetin (1UON; Fig. 3B), A1 I1309V mutants (1IJK, 1IJB, and 1FNS; Fig. 3B), A1 complexes with botrocetin or biticetin (1U0O, 1IJK, and 1UEX; Fig. 3B), and A1 complexes with Fabs or an aptamer (IU0O, 1IJB, 1FNS, 3HXO, and 3HXQ; Fig. 3B). In contrast to the A1/SS-GPIbα/VWD2 complex (and the A1/WT- GPIbα/WT complex with botrocetin, which shows “sliding” of the A1-GPIbα interface (33)), all other complexes show shifts of the α1-β2 loop from uncomplexed A1 WT (Fig. 3C). The α1-β2 loop conformational change is shown in Fig. 3, D–I, by a solid line connecting Gln1311 (Cα sphere) to its position in uncomplexed A1WT. In A1/VWD1-GPIbα/VWD1, the α1-β2 loop is shifted slightly away from the GPIbα interface (Fig. 3, C and G); in the A1/VWD2-GPIbα/VWD2 complex, the loop is shifted further (Fig. 3, C and H); and in the A1/WT-GPIbα/WT complex, the loop is shifted most and in a different direction toward the A1 termini (Fig. 3, C and I). The greater shift of the α1-β2 loop in the WT complex relates to the different overall orientation between A1 and GPIbα (Fig. 1, B and C). Variation in α1-β2 loop conformation also relates to variation in GPIbα β-finger orientation among complexes (Figs. 1 (B and C) and 3 (C and F–I)). This variation appears to result from avoidance of clashes rather than from maintenance of specific interactions because contacts between A1 and GPIbα β-finger residues vary markedly among the complexes (Fig. 3, F–I).
FIGURE 3.

Structural alterations near the A1 termini and α1-β2 loop and GPIbα β-finger upon complex formation. A, overview of the complex in a schematic diagram. B and C, details of the region boxed in A, shown for isolated A1 domains and an A1-GPIbα complex with little α1-β2 loop perturbation (B) and for all A1-GPIbα complexes (C). Keys in A–C show the color of each structure. Superposition is on A1. D–I, stereoviews of alterations around the A1 α1-β2 loop and long range disulfide upon complex formation. D and E, examples of uncomplexed molecules; F–I, complexes. All superposition is on the A1 domain; additionally, in D–E, isolated GPIbα is shown in the same orientation as in complexes. A1 has a silver Cα trace and cyan side chains; GPIbα has a wheat Cα trace and magenta side chains; and nitrogen and oxygen are blue and red, respectively. In F–I, solid black lines show movements of Cα atoms of selected residues from the uncomplexed structure in D to the indicated complex structure. Dashes in D and E show backbone-backbone or backbone-water hydrogen bonds discussed under “Results,” and small red spheres in E and H show waters at an opening to the hydrophobic core of the A1 domain. Structures are as indicated in the panels. Additionally, D shows PDB codes 1AUQ (A1/WT) and 1GWB (GPIbα/WT) chain B; E shows the A1/SS structure and 3P72 (GPIbα/WT). The latter two GPIbα structures show examples of large divergence of β-finger orientation. Table 4 has references for structures.
The A1 Termini
Superposition of the uncomplexed A1 domains listed in Table 4 reveals a structurally conserved pattern of hydrogen bonds that stabilize the conformation of their termini (Fig. 4A). The N-terminal end is stabilized at Cys1272, both through its long range disulfide bond and a backbone hydrogen bond to residue 1308 (Fig. 4A). The C-terminal end is stabilized by a network of backbone-backbone and backbone-side chain hydrogen bonds from Cys1458 to Pro1462 (Fig. 4A). VWD type 2B mutations occur in regions that neighbor the N and C termini and, with the exception of Trp1313, are distal from GPIbα (pink side chains; Fig. 4, A–F). The C-terminal network of hydrogen bonds in uncomplexed A1/WT (Fig. 4A) is conserved in the A1/WT-GPIbα/WT complex, except for minor alterations due to the Arg1274 rotamer (Fig. 4B). Many of these hydrogen bonds and consistently the Pro1462–Leu1275 backbone hydrogen bond are also preserved in the mutant, high affinity A1-GPIbα complexes (Fig. 4, A–E), and uncomplexed A1/SS (Fig. 4F).
FIGURE 4.

Alteration of the termini and VWD mutation-proximal region of A1 by mutations and complex formation with GPIbα shown in stereoview. Cα traces are supplemented with key side chains and hydrogen bond-forming backbone regions in stick representations. A1 side chains mutated in VWD (whether wild-type or mutated in the structures shown) have pink carbons. Nitrogen, oxygen, and sulfur are blue, red, and gold, respectively. Black dashes show key hydrogen bonds. Structures are named in A–F; A1/WT in A is PDB code 1AUQ.
Upon binding GPIbα, more changes are evident near the A1 N terminus than C terminus. For example, the N-proximal 2.8 Å backbone hydrogen bond between Cys1272 and residue 1308 in uncomplexed WT A1 (Fig. 4A) is not seen in the WT complex (Fig. 4B), mutant complexes (Fig. 4, C–E), or A1/SS (Fig. 4F).
It is difficult to deconvolute those A1 N terminus-proximal and GPIbα-proximal changes that are due to GPIbα binding and those that are due to affinity-enhancing mutations; nonetheless, it is possible to deduce from the structures the effects of the mutations on their immediate A1 environment. In A1/WT, the Arg1306 side chain hydrogen-bonds to the backbone of residue 1302 (Fig. 4A). In the complexes with A1/VWD1 and A1/VWD2, the mutated Gln1306 side chain points in a different direction than the WT Arg1306 side chain (Fig. 4, C and E). At least in the higher resolution VWD2 complex, Gln1306 forms two hydrogen bonds to the Asp1451 and Arg1450 side chains (Fig. 4C); Arg1450 is a known site of mutation in VWD.
The I1309V mutation present in A1/VWD2 complexes is one of the most subtle VWD mutations; previous uncomplexed A1/I1309V structures showed little change in backbone or even side chain rotamer compared with wild type (34, 35). The I1309V mutation, along with R1306Q, is in the A1 α1-β2 loop. The A1/VWD2-GPIbα/VWD2 complex reveals the behavior of the I1309V mutation in a complex (Fig. 3H). The smaller size of the Val1309 side chain compared with Ile allows it to dive deeper into the hydrophobic core and hence closer to Met1304 in the α1-helix and Val1316 in the β2-strand (Fig. 3H), compared with Ile1309 in the A1/VWD1-GPIbα/VWD1 and A1/SS-GPIbα/VWD2 complexes (Fig. 3, F and G). However, the markedly different shape of the α1-β2 loop in the A1/WT-GPIbα/WT complex allows its Ile1309 to dive almost as deep as Val1309 in the A1/VWD2-GPIbα/VWD2 complex (Fig. 3I). Lack of a clear structural phenotype for Val1309 in a complex structure is consistent with hypothesized further structural change to a super high affinity state (see “Discussion”).
Shifting the long range disulfide in the Y1271C/C1272R A1/SS mutant imparts greater freedom to the N terminus of A1. Cys1271 takes the place of Cys1272, with no significant change in the backbone and Cβ position of Cys1458 and only 1-Å differences in positions of the Cα and Cβ atoms of Cys1271 compared with Cys1272 (Fig. 3, D and E).
The comparisons to A1/SS reveal the Cys1272/Cys1458 disulfide as an important transducer of A1 conformational change upon complex formation with GPIbα. Upon complex formation, there is no significant change in the backbone position of Cys1458 in A1/SS (Fig. 3, E and F), whereas a 2-Å shift occurs in A1/WT, A1/VWD1, and A1/VWD2 (Fig. 3, G–I). The shift at Cys1458 is enforced by a shift in a similar direction at Cys1272 of >2 Å (Fig. 3, G–I). In turn, the shift at Cys1272 appears to be enforced by a subtle adjustment in the α1-β2 loop backbone at residues Leu1307 and Arg1308; the Leu1307 side chain would clash with the Cys1272 side chain unless Cys1272 moved (Fig. 3, G–I). In the A1/SS-GPIbα/VWD2 complex, Cys1271 moves away from Leu1307 similarly to Cys1272; however, this movement is accommodated by the extra backbone residue at 1272 in the A1/SS-GPIbα/VWD complex and does not require movement of Cys1458 (Fig. 3F).
The Electrostatic Interface
The calculated pI values for GPIbα (residues 1–265) and A1 (residues 1267–1465) are 5.5 and 9.2, respectively. Furthermore, their interaction interface is highly electrostatic (12) (Fig. 5). As described above, the orientation differs between the WT complex (Fig. 5A) and the high affinity mutant complexes, exemplified by A1/VWD2-GPIbα/VWD2 (Fig. 5B). The change in orientation shifts a cluster of six basic residues in A1 more into an acidic concave surface in GPIbα in the high affinity complexes (note the change in position of the non-overlapping portion of the A1 backbone in Fig. 5) and increases charge complementarity. This shift is also observed for A1/SS-GPIbα/VWD2 (Fig. 5C).
FIGURE 5.

Electrostatic complementarity and interaction of A1 Lys1371 with GPIbα. A–C, A1-GPIbα complexes are shown in identical orientations, with A1 in green Cα trace with key side chains in stick representations and GPIbα as a solvent-accessible surface colored by electrostatics from −7 eV (red) to 0 eV (white) to 7 eV (blue). D–G, the interaction between A1 K1371 and GPIbα. GPIbα (silver carbons to left) and A1 (green carbons to right) are shown as Cα traces with relevant side chains in stick representations. Hydrogen bonds to Lys1371 are shown as black dashes. 2Fo − Fc electron density contoured at 1σ is shown as orange mesh within 1.8 Å of Cα, and other atoms of the side chains are shown in stick representations.
The curvature of LRR proteins varies depending on the structure and number of residues in the portion of each repeat on the convex surface (32). Furthermore, there is some inherent flexibility in a given LRR protein. The curvature of an LRR protein may be characterized by its radius of curvature or the number of degrees subtended by each LRR (32). Flexion in GPIbα is more conveniently measured as the distance between LRR1 and LRR8 (Table 5). LRR1–LRR8 distances of 31 and 30.3 Å in the two examples of unliganded GPIbα in the 1M0Z structure correspond to total rotations of 85.9 and 94.0° about a central axis, respectively (32). Among six high affinity complexes, this distance is 31.06 ± 0.16 Å (mean ± S.D.) (Table 5). This distance is lower in the wild-type complex and the wild-type complex with bound botrocetin, at 30.48 ± 0.02 Å (mean ± difference from mean), showing that GPIbα in the two wild-type complexes is more concave. Among 11 independent examples of GPIbα in isolation or in complex with peptide or α-thrombin (Table 5), the distances show much greater variation of 30.48 ± 0.48 Å. The tighter distributions of distances among the high affinity and among the wild-type complexes and the differences between the two distributions suggest that their binding modes constrain GPIbα curvature. The greater stretch (lesser concavity) of GPIbα in the high affinity complexes enables A1 to come closer to the concave, acidic surface of GPIbα, increasing electrostatic attraction.
TABLE 5.
GPIbα curvature measured as LRR1 to LRR8 distance
The distance between LRR1 and LRR8 in GPIbα is measured as a surrogate for curvature (curvature increases as distance decreases) (32). All distances are given in Å.
| Structure | LRR1–LRR8 Cα atom distances |
Average Cα distance | ||
|---|---|---|---|---|
| 35–199 | 36–200 | 37–201 | ||
| Å | Å | |||
| A1/VWD2-GPIbα/VWD2 | 29.9 | 32.3 | 30.7 | 31.0 |
| A1/VWD1-GPIbα/VWD1 | 29.9 | 32.1 | 30.7 | 30.9 |
| A1/SS-GPIbα/VWD2, B | 30.3 | 32.8 | 30.9 | 31.3 |
| A1/SS-GPIbα/VWD2, D | 30.1 | 32.5 | 30.7 | 31.1 |
| A1/SS-GPIbα/VWD2, F | 30.1 | 32.4 | 30.8 | 31.1 |
| A1/SS-GPIbα/VWD2, H | 29.9 | 32.4 | 30.5 | 30.9 |
| A1/WT-GPIbα/WT | 29.4 | 31.9 | 30.2 | 30.5 |
| A1/WT-GPIbα/WT-botrocetin | 29.5 | 31.7 | 30.2 | 30.5 |
| 1M0Z, B | 29.1 | 31.8 | 30 | 30.3 |
| 1M0Z, A | 30 | 32.4 | 30.7 | 31.0 |
| 1OOK | 29.6 | 31.9 | 30.1 | 30.5 |
| 1P8V | 30.2 | 32.5 | 30.7 | 31.1 |
| 1P9A | 29 | 31.6 | 29.8 | 30.1 |
| 1QYY, A | 28.8 | 31.4 | 29.6 | 29.9 |
| 1QYY, B | 28.5 | 31.1 | 29.6 | 29.7 |
| 3P72 | 29.5 | 32.1 | 30.3 | 30.6 |
| 3PMH | 29.5 | 31.8 | 30.3 | 30.5 |
| 1GWB, B | 29.4 | 32.2 | 30.3 | 30.6 |
| 1GWB, A | 29.6 | 32.1 | 30.2 | 30.6 |
| Average in high affinity complexes ± S.D. | 31.06 ± 0.16 | |||
| Average in the absence of A1 association ± S.D. | 30.48 ± 0.43 | |||
| Average in wild type A1 complexes ± difference from mean | 30.48 ± 0.02 | |||
An Interaction with LRR4 and LRR5
The closer approach of A1 to the less concave surface of GPIbα in high affinity complexes compared with the low affinity wild-type complex brings the Cα position of A1 Lys1371 closer to acidic patch residues GPIbα Glu128 and Glu151 and nearby polar residues Thr103 and Gln127 (Fig. 5, D–G). No hydrogen bond across the interface of Lys1371 is present in the wild-type complex (Fig. 5D). The density of the Lys1371 side chain is weak in the previous A1/VWD1-GPIbα/VWD1 3.1 Å complex crystal structure, and no hydrogen bond of Lys1371 across the interface was modeled (Fig. 5E). However, Lys1371 density is well defined in the 2.08 Å A1/VWD2-GPIbα/VWD2 structure and demonstrates that the Lys1371 side chain hydrogen-bonds across the interface to Thr103 and Gln127 in GPIbα LRR4 and LRR5, respectively (Fig. 5F). The 2.8 Å A1/SS-GPIbα/VWD2 crystal provides four independent examples in the crystal lattice of complex formation. Although there is some variation in A1 Lys1371 side chain orientation, all four examples show hydrogen bonding across the interface to one or more of GPIbα residues 81, 103, 127, and 128. Density is best defined in chains G and H, which show hydrogen bonding of A1 Lys1371 to GPIbα residues 81 and 103 (Fig. 5G). This is the first reported A1 interaction within LRR3–LRR5, which separate the two previously reported discontinuous interfaces in GPIbα with A1.
To test the importance of the A1 Lys1371 hydrogen bond to GPIbα Thr103 and Gln127 without altering overall charge, we mutated Thr103 and Gln127 to Ala. 125I-GPIbα/VWD2 binding to yeast displaying A1/SS (Fig. 6A) or A1/VWD2 (Fig. 6B) was competed with GPIbα/WT or GPIbα/VWD2 with or without additional alanine mutations. The T103A and Q127A mutations each substantially decreased affinity (Fig. 6, A and B). The Q127A mutation showed the greatest effect, consistent with the close proximity of Gln127 to nearby acidic residues Glu128 and Glu151.
FIGURE 6.
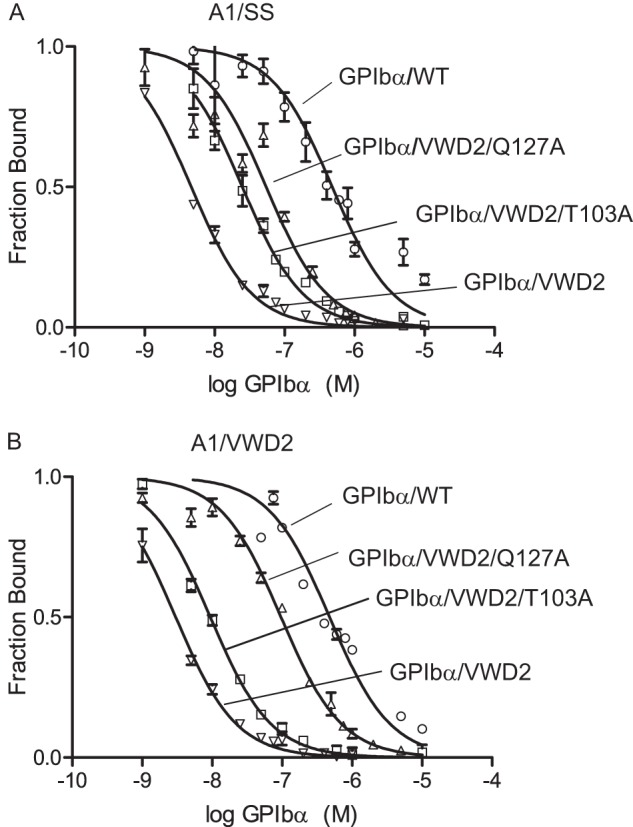
Mutational importance of GPIbα middle LRRs in binding A1 in high affinity complexes. A and B, 125I-GPIbα/VWD2 binding to A1/SS (A) or A1/VWD2 (B) displayed on yeast was competed with unlabeled GPIbα proteins as labeled. The Ki values (95% confidence interval) measured in A are as follows: GPIbα/WT = 317 nm (270–365), GPIbα/VWD2 = 1.98 nm (1.78–2.21), GPIbα/VWD2-Q127A = 63.5 nm (57.0–70.8), and GPIbα/VWD2-T103A = 6.2 nm (5.60–6.87). The Ki values (95% confidence interval) measured in B are as follows: GPIbα/WT = 307.5 nm (241–393), GPIbα/VWD2 = 2.97 nm (2.70–3.26), GPIbα/VWD2-Q127A = 35.0 nm (27.6–44.4), and GPIbα/VWD2-T103A = 16.0 nm (14.3–18.0). Error bars, S.D.
DISCUSSION
Binding of the A1 domain of VWF to GPIbα on platelets is important in arteriolar hemostasis and has the most interesting property of being regulated by hydrodynamic flow. Naturally occurring mutations in the A1 domain in VWD and in GPIbα in PT-VWD have been identified that enhance A1 and GPIbα interaction and cause disease by gain of function (3–5). Here, we have used directed evolution in yeast to identify further gain-of-function mutations. Moving the long range disulfide bond to encompass one more residue with the Y1271C/C1272R mutation (A1/SS) increased affinity for GPIbα by 10-fold, similarly to the combined effect of two independent VWD mutations, R1306Q and I1309V (A1/VWD2).
We determined multiple crystal structures that extend the resolution of complexes containing VWD mutations from 3.1 to 2.08 Å and extend structural knowledge to complexes with different mutations and with markedly higher affinity. Furthermore, we now have a set of three high affinity mutant complexes (not counting independent lattice environments) in contrast to the single previous example for comparison with the wild-type complex. GPIbα with the double G233V/M239V mutation is much more active than either single mutation alone in binding to VWF (30, 36). This is in agreement with measurement here of 25-fold higher affinity of G233V/M239V than WT and previous measurements of 3-fold higher affinity of M239V than WT (12). The R1306Q/I1309V double mutant (A1/VWD2) showed 10-fold higher affinity than WT, as did the A1/SS mutant, whereas single R1306Q and I1309V substitutions increased affinity severalfold, in agreement with a report of a 2.5-fold increase for the R1306Q mutation (12). Altogether, our complexes of A1/SS or A1/VWD2 with GPIbα/VWD2 show a 250-fold increase in affinity relative to WT, compared with the 7.5-fold increase in affinity of the A1/VWD1-GPIbα/VWD1 complex used previously for crystallization (12).
Despite the markedly higher affinity of the two types of double mutant complexes crystallized here, all mutant complexes show remarkably similar orientations between A1 and GPIbα. This high affinity orientation differs from that seen in the wild-type complex, both in terms of having less concavity in GPIbα and allowing LRR4 and LRR5 to approach closer to A1 and in allowing lateral sliding of A1 over the concave GPIbα surface with better electrostatic complementarity. A caveat is that we only have one example thus far of the WT complex in crystals.
The closer approach to A1 at LRR4 and LRR5 in the high affinity complexes is consistent with previous evidence implicating this region in shear-regulated increased adhesiveness of GPIbα for VWF (15). LRR3 to -5 were previously shown to be required for ristocetin-stimulated binding of VWF to platelets (16). Ristocetin is a glycopeptide antibiotic that adventitiously activates VWF binding and is used to measure VWF function in the diagnosis of VWD. VWD type 2B mutations, two of which were used here in the A1/VWD2 mutant, reduce the concentration of ristocetin required for VWF to bind platelets (37). It is widely believed that ristocetin mimics the effect of shear on VWF (38). Ristocetin has been shown to bind to residues in the N and C terminus of A1 (39). The observations on the importance of LRR3 to -5 in ristocetin-stimulated binding were made by substituting canine for human sequence in this region of GPIbα, followed by expression in CHO cells co-transfected with the associating GPIbβ and GPIX subunits (16). Subsequently, the same chimeras were examined for shear-dependent interaction of transfectants with VWF. Remarkably, as shear was increased, having human rather than canine LRR3 to -5 was increasingly important (15).
For the first time, we observed a structural interaction in this region, between Thr103 and Gln127 of GPIbα and Lys1371 of A1. Furthermore, we demonstrated by mutation that Thr103 and Gln127 are important for high affinity for A1. We examined conservation of these residues in humans, canines, mice, rats, pigs, cows, and chickens. Lys1371 is conserved as Lys or Arg in VWF in all of these species. Gln127 is similarly conserved as Gln or Glu in GPIbα in the same species; however, canine is the exception with a His residue, possibly explaining the poor performance of human-canine chimeric GPIbα in ristocetin-stimulated adhesion and in adhesion at high shear to VWF (15, 16).
The previous functional assays using canine-human GPIbα chimeras, together with a closer approach in LRR of high affinity complexes, and our mutational studies suggest that the high affinity complexes are on a pathway toward the high affinity state induced by shear flow. An increase in shear flow to supraphysiologic levels, as well as transition of shear flow to elongational flow at sites of vasoconstriction in hemostasis, will induce an overall change in VWF concatemer shape from bird's nest to extended and therefore expose multiple A1 domains in VWF for multimeric binding to GPIbα on platelets (5, 7). The A1 domain is flexibly connected to the neighboring D′D3 assembly and to the A2 domain by highly flexible, O-glycosylated mucin-like linkers (40). After elongation of VWF, tensile force applied by hydrodynamic flow to VWF concatemers will be communicated through the flexible linkers between domains (9) to the structured portions of the A1 domain that lie N- and C-terminal of its long range disulfide bond. These are the regions in which many VWD mutations are found and in which rearrangements occur in A1 in both high affinity and WT complexes, relative to uncomplexed A1. The hydrodynamic force exerted on the A1 N and C termini could potentially have much larger effects on A1 structure than seen here as a consequence of mutation and complex formation. Moreover, this force would increase after platelet binding to the A1 domain in VWF, as a consequence of the additional hydrodynamic force on the platelet.
Such forces might induce large scale conformational rearrangements in A1, as are seen in the VWA folds present in integrin αI and βI domains. The presence of long range disulfide bonds does not prevent conformational change in VWA domains, as illustrated by the 15-Å movement of the long range disulfide in the VWA domain of a sporozoite adhesin (41). In integrin I domains, a force exerted in a similar C-terminal direction on the C-terminal α-helix induces axial pistoning of this helix that leads to a large increase in affinity for ligand (11). Integrin I domains and the A1 domain differ greatly in the location of their ligand-binding sites, and any hypothetical large scale rearrangements in A1 would necessarily affect affinity through a different mechanism than in integrins. Above, we have argued that high affinity mutant complexes studied here and previously are on-pathway to a super high affinity A1-GPIbα state induced by force; however, so as not to discourage further research in this area, it is important to operate under the hypothesis that the force-induced super high affinity state of the A1-GPIbα complex has not yet been seen by crystallography.
There are several reasons to think that a high affinity, force-extended state of A1-GPIbα has not been seen in the current study. The PT-VWD mutations in GPIbα used here are thought to increase the propensity for β-strand formation in the β-switch (12) and, like other PT-VWD mutations, may also destabilize the unliganded conformation of the β-switch relative to the ligand-bound conformation (14). We hypothesize that the β-switch has the same conformation in complexes in the presence and absence of force; thus, PT-VWD mutations would not shift the equilibrium between these types of complexes. VWD mutations in A1 locate in or proximal to regions where force would be exerted physiologically and where shape shifting might occur; these mutations are thus hypothesized to shift the equilibrium to the high affinity state. In integrins, co-crystallization with ligand is sufficient to induce the high affinity state, and this state is even sometimes glimpsed fortuitously in a crystal lattice-dependent manner (11). However, single molecule experiments suggest that A1-GPIbα binding is not sufficient to induce a high affinity, extended state and that this state requires a force over 10 pN to be exerted on the N terminus of A1 and the C terminus of GPIbα and across the receptor-ligand complex (8). It is currently not possible to mimic such a force in a crystallization experiment. Our VWD R1306Q and I1309V mutations may have lowered the energy to reach such a high affinity state; however, in contrast to one report (42), we find in single molecule experiments with R1306Q and M239V mutations that two states are still observed.4 Ristocetin selectively strengthens the state seen in high force in single molecule experiments (8). The best-diffracting A1/VWD2-GPIbα/VWD2 crystal selected for refinement here was crystallized in the presence of 0.4 mg/ml ristocetin and was further soaked with 4 mg/ml ristocetin. However, we found no electron density for a bound ristocetin, suggesting either that the state we have crystallized is not that stabilized by ristocetin or that lattice contacts prevented ristocetin binding.
Overall, comparisons among complexes and with uncomplexed A1 suggest that strain in VWD mutation-proximal regions occurs in both wild-type and mutant A1-GPIbα complexes. The long range disulfide bond moves at the Cys1272 or Cys1271 backbone in all complexes and at the Cys1458 backbone except in the Cys1271 mutant A1/SS complex. The hydrophobic cavity between the wild-type disulfide and the core of the A1 domain is sealed by a backbone hydrogen bond between Arg1308 and Cys1272 in wild-type A1 (Fig. 3D). The cavity becomes exposed to water in the A1/SS mutant, in which a water replaces residue 1272 in hydrogen bonding to the Arg1308 backbone (Fig. 3E). The movement of the disulfide upon binding GPIbα appears to be transmitted from N-terminal-proximal portions of A1 that contact GPIbα near its β-finger (Fig. 3, F–I). The dislocation of the disulfide opens up a cavity at the position vacated by Cys1272. In the high resolution A1/VWD2-GPIbα/VWD2 complex, water also gains access to the interface between the disulfide and A1 core (Fig. 3H). Similarly, a cavity formed by the I1309V mutation is occupied by a water (43). Burial of waters in hydrophobic cavities is energetically unfavorable and thus a symptom of strain.
Movement of the long range disulfide appears to result from movement of contacting residue Leu1307 at the end of the α1-helix (Fig. 3, D–I). The α1-β2 loop neighbors Leu1307, contains the R1306Q and I1309V VWD mutations studied here in addition to Trp1313, and has been extensively discussed previously with respect to a role in affinity regulation (44). The α1-β2 loop occupies multiple conformations in the different structures. Compared with uncomplexed A1, the position of the Cα atom of residue Q1311 in the α1-β2 loop varies only slightly in A1/SS-GPIbα/VWD (0.7 Å), more in A1/VWD1-GPIbα/VWD1 (1.7 Å), greatly in A1/VWD2-GPIbα/VWD2 (4 Å), and severely with an overall different loop conformation in A1/WT-GPIbα/WT (7.5 Å) (Fig. 3, D and F–I). The β-finger is variable in conformation in isolated GPIbα structures (Fig. 3, D and E) and is sometimes disordered (see structures listed in Table 4).
Remarkably, upon A1-GPIbα binding, Leu1307, Ile1309 in the α1/β2 loop, and Cys1272 (or Cys1271) in the long range disulfide in A1 move in a similar direction away from GPIbα, despite the many different conformations of the contact sites in the α1-β2 loop and β-finger (Fig. 3, G–I). These results are consistent with a clash, rather than a favorable binding interaction, in the α1-β2 loop and β-finger region. Clashes can be avoided by different types of structural rearrangements, whereas favorable interactions generally result in a single type of binding interface. The overall binding interaction between A1 and GPIbα appears to be largely driven by β-sheet formation and burial of hydrophobic and aromatic groups around the β-switch and electrostatic complementarity at the A1 interface with the concave LRR surface of GPIbα. None of the binding interfaces between the α1-β2 loop and β-finger region show notable burial of hydrophobic residues or favorable hydrogen bond or electrostatic complementarity (Fig. 3, F–I).
In summary, the structures described here markedly advance our understanding of high affinity A1-GPIbα complexes. Strain appears to be induced in the α1-β2 loop, long range disulfide, and VWD mutation-containing regions of A1 upon binding at the interface between the A1 α1-β2 loop and GPIbα β-finger. We hypothesize that a large conformational change in these regions could dissipate this strain and reshape A1 to form a complementary interface possibly extending across the concave GPIbα surface. For these reasons, we wish to keep alive the hypothesis that in structures crystallized to date, the A1-GPIbα complex has not yet reached a hypothesized VWD mutation-facilitated, force-extended, high affinity conformation.
Acknowledgments
We thank A. Koksal and J. Zhu for discussion and crystallographic data acquisition. We thank K. D. Wittrup and K. von Hoorelbeke for reagents and K. Ketman (IDI Flow Cytometry Core) for help with FACS.
This work was supported, in whole or in part, by National Institutes of Health Grant HL-103526 and Postdoctoral Fellowship NIH-1F32HL-099167 (to M. A. B.). This work was also supported by American Heart Association Postdoctoral Fellowship AHA-10POST4170043 (to M. A. B.).
The atomic coordinates and structure factors (codes 4C2A, 4C2B, and 4C29) have been deposited in the Protein Data Bank (http://wwpdb.org/).
J. Kim, N. Hudson, and T. A. Springer, unpublished observations.
- VWF
- von Willebrand factor
- GPIbα
- glycoprotein Ibα
- VWD
- von Willebrand disease
- PT-VWD
- platelet-type VWD
- LRR
- leucine-rich repeat
- pN
- piconewtons
- PDB
- Protein Data Bank.
REFERENCES
- 1. Sadler J. E. (2005) New concepts in von Willebrand disease. Annu. Rev. Med. 56, 173–191 [DOI] [PubMed] [Google Scholar]
- 2. Wagner D. D. (1990) Cell biology of von Willebrand factor. Annu. Rev. Cell Biol. 6, 217–246 [DOI] [PubMed] [Google Scholar]
- 3. Sadler J. E. (1998) Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67, 395–424 [DOI] [PubMed] [Google Scholar]
- 4. Ruggeri Z. M., Mendolicchio G. L. (2007) Adhesion mechanisms in platelet function. Circ. Res. 100, 1673–1685 [DOI] [PubMed] [Google Scholar]
- 5. Springer T. A. (2011) Biology and physics of von Willebrand factor concatamers. J. Thromb. Haemost. 9, 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savage B., Saldívar E., Ruggeri Z. M. (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84, 289–297 [DOI] [PubMed] [Google Scholar]
- 7. Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J., Zhang C. Z., Zhang X., Springer T. A. (2010) A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature 466, 992–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X., Halvorsen K., Zhang C. Z., Wong W. P., Springer T. A. (2009) Mechanoenzymatic cleavage of the ultralarge vascular protein, von Willebrand Factor. Science 324, 1330–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Emsley J., Cruz M., Handin R., Liddington R. (1998) Crystal structure of the von Willebrand factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 273, 10396–10401 [DOI] [PubMed] [Google Scholar]
- 11. Luo B.-H., Carman C. V., Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., Gros P. (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 13. Dumas J. J., Kumar R., McDonagh T., Sullivan F., Stahl M. L., Somers W. S., Mosyak L. (2004) Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibα complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J. Biol. Chem. 279, 23327–23334 [DOI] [PubMed] [Google Scholar]
- 14. Othman M., Kaur H., Emsley J. (2013) Platelet-type von Willebrand disease. New insights into the molecular pathophysiology of a unique platelet defect. Semin. Thromb. Hemost. 39, 663–673 [DOI] [PubMed] [Google Scholar]
- 15. Shen Y., Cranmer S. L., Aprico A., Whisstock J. C., Jackson S. P., Berndt M. C., Andrews R. K. (2006) Leucine-rich repeats 2–4 (Leu60–Glu128) of platelet glycoprotein Ibα regulate shear-dependent cell adhesion to von Willebrand factor. J. Biol. Chem. 281, 26419–26423 [DOI] [PubMed] [Google Scholar]
- 16. Shen Y., Romo G. M., Dong J. F., Schade A., McIntire L. V., Kenny D., Whisstock J. C., Berndt M. C., López J. A., Andrews R. K. (2000) Requirement of leucine-rich repeats of glycoprotein (GP) Ibα for shear-dependent and static binding of von Willebrand factor to the platelet membrane GP Ib-IX-V complex. Blood 95, 903–910 [PubMed] [Google Scholar]
- 17. Miura S., Li C. Q., Cao Z., Wang H., Wardell M. R., Sadler J. E. (2000) Interaction of von Willebrand factor domain A1 with platelet glycoprotein Iba-(1–289). Slow intrinsic binding kinetics mediate rapid platelet adhesion. J. Biol. Chem. 275, 7539–7546 [DOI] [PubMed] [Google Scholar]
- 18. Mi L. Z., Grey M. J., Nishida N., Walz T., Lu C., Springer T. A. (2008) Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry 47, 10314–10323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cauwenberghs N., Vanhoorelbeke K., Vauterin S., Westra D. F., Romo G., Huizinga E. G., Lopez J. A., Berndt M. C., Harsfalvi J., Deckmyn H. (2001) Epitope mapping of inhibitory antibodies against platelet glycoprotein Ibα reveals interaction between the leucine-rich repeat N-terminal and C-terminal flanking domains of glycoprotein Ibα. Blood 98, 652–660 [DOI] [PubMed] [Google Scholar]
- 20. Zaccolo M., Williams D. M., Brown D. M., Gherardi E. (1996) An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255, 589–603 [DOI] [PubMed] [Google Scholar]
- 21. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 22. Schürpf T., Springer T. A. (2011) Regulation of integrin affinity on cell surfaces. EMBO J. 30, 4712–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity. All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boder E. T., Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 [DOI] [PubMed] [Google Scholar]
- 30. Tait A. S., Cranmer S. L., Jackson S. P., Dawes I. W., Chong B. H. (2001) Phenotype changes resulting in high-affinity binding of von Willebrand factor to recombinant glycoprotein Ib-IX. Analysis of the platelet-type von Willebrand disease mutations. Blood 98, 1812–1818 [DOI] [PubMed] [Google Scholar]
- 31. Flood V. H., Friedman K. D., Gill J. C., Morateck P. A., Wren J. S., Scott J. P., Montgomery R. R. (2009) Limitations of the ristocetin cofactor assay in measurement of VWF function. J. Thromb. Haemost. 7, 1832–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Enkhbayar P., Kamiya M., Osaki M., Matsumoto T., Matsushima N. (2004) Structural principles of leucine-rich repeat (LRR) proteins. Proteins 54, 394–403 [DOI] [PubMed] [Google Scholar]
- 33. Fukuda K., Doggett T., Laurenzi I. J., Liddington R. C., Diacovo T. G. (2005) The snake venom protein botrocetin acts as a biological brace to promote dysfunctional platelet aggregation. Nat. Struct. Mol. Biol. 12, 152–159 [DOI] [PubMed] [Google Scholar]
- 34. Celikel R., Ruggeri Z. M., Varughese K. I. (2000) von Willebrand factor conformation and adhesive function is modulated by an internalized water molecule. Nat. Struct. Biol. 7, 881–884 [DOI] [PubMed] [Google Scholar]
- 35. Fukuda K., Doggett T. A., Bankston L. A., Cruz M. A., Diacovo T. G., Liddington R. C. (2002) Structural basis of von Willebrand factor activation by the snake toxin botrocetin. Structure 10, 943–950 [DOI] [PubMed] [Google Scholar]
- 36. Flood V. H., Gill J. C., Morateck P. A., Christopherson P. A., Friedman K. D., Haberichter S. L., Branchford B. R., Hoffmann R. G., Abshire T. C., Di Paola J. A., Hoots W. K., Leissinger C., Lusher J. M., Ragni M. V., Shapiro A. D., Montgomery R. R. (2010) Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood 116, 280–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong J. F., Berndt M. C., Schade A., McIntire L. V., Andrews R. K., López J. A. (2001) Ristocetin-dependent, but not botrocetin-dependent, binding of von Willebrand factor to the platelet glycoprotein Ib-IX-V complex correlates with shear-dependent interactions. Blood 97, 162–168 [DOI] [PubMed] [Google Scholar]
- 38. Ginsburg D., Sadler J. E. (1993) von Willebrand disease. A database of point mutations, insertions, and deletions. For the Consortium on von Willebrand Factor Mutations and Polymorphisms, and the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 69, 177–184 [PubMed] [Google Scholar]
- 39. De Luca M., Facey D. A., Favaloro E. J., Hertzberg M. S., Whisstock J. C., McNally T., Andrews R. K., Berndt M. C. (2000) Structure and function of the von Willebrand factor A1 domain. Analysis with monoclonal antibodies reveals distinct binding sites involved in recognition of the platelet membrane glycoprotein Ib-IX-V complex and ristocetin-dependent activation. Blood 95, 164–172 [PubMed] [Google Scholar]
- 40. Zhou Y. F., Eng E. T., Nishida N., Lu C., Walz T., Springer T. A. (2011) A pH-regulated dimeric bouquet in the structure of von Willebrand factor. EMBO J. 30, 4098–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song G., Koksal A. C., Lu C., Springer T. A. (2012) Shape change in the receptor for gliding motility in plasmodium sporozoites. Proc. Natl. Acad. Sci. U.S.A. 109, 21420–21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yago T., Lou J., Wu T., Yang J., Miner J. J., Coburn L., López J. A., Cruz M. A., Dong J. F., McIntire L. V., McEver R. P., Zhu C. (2008) Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 118, 3195–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Celikel R., Varughese K. I., Madhusudan, Yoshioka A., Ware J., Ruggeri Z. M. (1998) Crystal structure of the von Willebrand factor A1 domain in complex with the function blocking NMC-4 Fab. Nat. Struct. Biol. 5, 189–194 [DOI] [PubMed] [Google Scholar]
- 44. Dumas J. J., Kumar R., Seehra J., Somers W. S., Mosyak L. (2003) Crystal structure of the GpIbα-thrombin complex essential for platelet aggregation. Science 301, 222–226 [DOI] [PubMed] [Google Scholar]
- 45. Celikel R., McClintock R. A., Roberts J. R., Mendolicchio G. L., Ware J., Varughese K. I., Ruggeri Z. M. (2003) Modulation of α-thrombin function by distinct interactions with platelet glycoprotein Ibα. Science 301, 218–221 [DOI] [PubMed] [Google Scholar]
- 46. Varughese K. I., Ruggeri Z. M., Celikel R. (2004) Platinum-induced space-group transformation in crystals of the platelet glycoprotein Ibα N-terminal domain. Acta Crystallogr. D Biol. Crystallogr. 60, 405–411 [DOI] [PubMed] [Google Scholar]
- 47. McEwan P. A., Andrews R. K., Emsley J. (2009) Glycoprotein Ibα inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism. Blood 114, 4883–4885 [DOI] [PubMed] [Google Scholar]
- 48. Zarpellon A., Celikel R., Roberts J. R., McClintock R. A., Mendolicchio G. L., Moore K. L., Jing H., Varughese K. I., Ruggeri Z. M. (2011) Binding of α-thrombin to surface-anchored platelet glycoprotein Ibα sulfotyrosines through a two-site mechanism involving exosite I. Proc. Natl. Acad. Sci. U.S.A. 108, 8628–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Uff S., Clemetson J. M., Harrison T., Clemetson K. J., Emsley J. (2002) Crystal structure of the platelet glycoprotein Ibα N-terminal domain reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 277, 35657–35663 [DOI] [PubMed] [Google Scholar]
- 50. Maita N., Nishio K., Nishimoto E., Matsui T., Shikamoto Y., Morita T., Sadler J. E., Mizuno H. (2003) Crystal structure of von Willebrand factor A1 domain complexed with snake venom, bitiscetin. Insight into glycoprotein Iba binding mechanism induced by snake venom proteins. J. Biol. Chem. 278, 37777–37781 [DOI] [PubMed] [Google Scholar]
- 51. Huang R. H., Fremont D. H., Diener J. L., Schaub R. G., Sadler J. E. (2009) A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von Willebrand factor domain A1. Structure 17, 1476–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]