

Background: Apolipoprotein (apo)A-IV is involved in lipid and glucose metabolism, but its full-length structure is not known.
Results: Stable isotope-assisted cross-linking combined with molecular modeling produced new models of the full-length protein.
Conclusion: At least three hydrophobic residues participate in a unique clasp mechanism that regulates apoA-IV function.
Significance: We report the most detailed models of lipid-free apoA-IV to date and demonstrate their utility in terms of functional predictions.
Keywords: Apolipoproteins, Lipid-binding Protein, Mass Spectrometry (MS), Molecular Dynamics, Protein Cross-linking, Protein Structure, Apolipoprotein A-IV, Lipid-free, Small Angle X-ray Scattering
Abstract
Apolipoprotein (apo)A-IV plays important roles in dietary lipid and glucose metabolism, and knowledge of its structure is required to fully understand the molecular basis of these functions. However, typical of the entire class of exchangeable apolipoproteins, its dynamic nature and affinity for lipid has posed challenges to traditional high resolution structural approaches. We previously reported an x-ray crystal structure of a dimeric truncation mutant of apoA-IV, which showed a unique helix-swapping molecular interface. Unfortunately, the structures of the N and C termini that are important for lipid binding were not visualized. To build a more complete model, we used chemical cross-linking to derive distance constraints across the full-length protein. The approach was enhanced with stable isotope labeling to overcome ambiguities in determining molecular span of the cross-links given the remarkable similarities in the monomeric and dimeric apoA-IV structures. Using 51 distance constraints, we created a starting model for full-length monomeric apoA-IV and then subjected it to two modeling approaches: (i) molecular dynamics simulations and (ii) fitting to small angle x-ray scattering data. This resulted in the most detailed models yet for lipid-free monomeric or dimeric apoA-IV. Importantly, these models were of sufficient detail to direct the experimental identification of new functional residues that participate in a “clasp” mechanism to modulate apoA-IV lipid affinity. The isotope-assisted cross-linking approach should prove useful for further study of this family of apolipoproteins in both the lipid-free and -bound states.
Introduction
Human apolipoprotein (apo)A-IV4 is a lipid-binding protein synthesized by the small intestine and secreted into the gastric lymphatics primarily associated with chylomicrons. Upon entering the plasma compartment, it rapidly dissociates from chylomicrons and distributes between high density lipoprotein particles (roughly 30–50%) and a lipid-free state (1). Unlike the other exchangeable apolipoproteins, apoA-IV message and protein increase markedly during lipid absorption in the gut (2, 3). Although its specific function has not been fully settled, it has been suggested that apoA-IV co-evolved with apoB mRNA editing in mammals to play a role in chylomicron assembly (4), perhaps acting as a “barostat” to regulate interfacial pressures on nascent chylomicrons (5). Other evidence suggests that apoA-IV modulates chylomicron size by affecting its rate of passage through the cellular secretory pathway (6) or by facilitating triglyceride packing into the nascent particle (7). Additional proposed functions include cholesterol transport in high density lipoprotein (8–11), an inhibitor of lipoprotein oxidation (12), a regulator of food intake (13), and a mediator of gastric emptying (14, 15). More recently, apoA-IV has been implicated in the control of glucose homeostasis (16). Its presence enhanced glucose-stimulated release of insulin from pancreatic islet cells, implicating it as a target for the treatment of diabetes.
To better define the role of apoA-IV in these varied processes, there has been significant interest in determining its structure. Furthermore, given its evolutionary relationship to other disease-relevant players like apoA-I and apoE (17), structural understanding of apoA-IV should translate to the entire class of exchangeable apolipoproteins. All three proteins, apoA-IV being the largest at 376 amino acids (46 kDa), are dominated by 22-amino acid repeats, which are predicted to form amphipathic α helices (18) and likely impart the ability to emulsify lipids into stable lipoproteins. ApoA-IV contains 12 such repeats, most of which are punctuated by proline residues (18), tightly clustered between residues 40 and 332. Like apoA-I, the N terminus of apoA-IV (residues 1–39) is encoded by a different exon than the remainder of the protein (19). Unlike apoA-I, the C terminus of apoA-IV contains a unique glutamine-rich sequence (residues 354–367) with unknown function (20). In the absence of lipid, human apoA-IV exists as a mixture of monomers (25%) and homodimers (75%) (21).
We have reported an x-ray crystal structure of the central domain of human apoA-IV (apoA-IV64–335) in the dimeric form that shows a unique helix swapping interface (22). This interface was remarkably similar to that found in the crystal structure of a dimeric C-terminal truncated form of apoA-I (23). The nature of the interaction allowed us to propose that apoA-IV can monomerize when this shared helical domain doubles back onto the originating molecule, forming a four-helix bundle. Unfortunately, our structure did not shed light on the N- and C-terminal residues. This is important because we have previously identified residues in these domains that can strongly affect the ability of the protein to interact with lipid. We proposed that these sites interact to form a “clasp” that regulates the unfolding of the protein in response to lipid (24, 25). More recent work using small angle x-ray scattering (SAXS) indicated that these terminal domains cluster at one end of the helical bundle, but their detailed three-dimensional arrangement remains unresolved (26).
Chemical cross-linking represents a powerful method for determining three-dimensional distance constraints for protein molecules as they exist in solution. We have used this technique extensively to study the structure of apoA-I (27, 28), apoA-II (29), and even apoA-IV (25). However, the technique is limited when studying the structure of homodimeric proteins because it cannot distinguish between intramolecular and intermolecular cross-links. This is particularly important because the apoA-IV64–335 structure predicts that the same cross-links can be generated both intra- and intermolecularly. In this work, we have overcome this technical challenge by combining isotopic labeling with chemical cross-linking to extend our model of monomeric apoA-IV. Using the cross-linking distance constraints, we created a starting model and then subjected it to two modeling approaches: (i) molecular dynamics simulations and (ii) fitting to SAXS data. These data-driven models are the most detailed yet achieved for lipid-free human apoA-IV.
EXPERIMENTAL PROCEDURES
ApoA-IV Expression and Endogenous Stable Isotope Labeling
Recombinant human apoA-IV was expressed and purified as described previously in BL21 Escherichia coli (30). The construct contained the mature sequence with an additional glycine at the N terminus after the histidine tag was removed by the tobacco etch virus protease. Purity was routinely >95% as determined by SDS-PAGE and mass spectrometry. For the 15N-labeled version, a modified expression system was used. Briefly, 1 ml of bacterial cells containing the expression vector was pelleted and resuspended in rich minimal medium containing vitamins, salts, glucose, and 15N-lableled ammonium chloride with no other nitrogen source. This was seeded into a 1-liter culture of the same media and grown overnight. From here, the expression and purification of the labeled protein was similar to that of the unlabeled proteins.
Separation of Stable Monomeric and Dimeric Forms of ApoA-IV
Recombinant 14N and 15N apoA-IV in PBS at pH 7.8 were individually brought to 3 m guandine HCl with dry reagent to assure complete dissociation of any dimeric species. The two solutions were then mixed at a 1:1 molar ratio to achieve a final concentration of >1 mg/ml at 37 °C for 1 h to assure complete mixing of the isotopically labeled forms. After removal of the guanidine HCl by dialysis, the proteins were applied to a Superdex® 200 16/60 (GE Healthcare) gel filtration column equilibrated in PBS and run at 4 °C. Fractions corresponding to monomeric and dimeric apoA-IV were pooled, concentrated by ultrafiltration, and stored at 4 °C until used. We previously showed that isolated monomeric and dimeric species were stable for longer than a week at 4 °C. Protein concentrations were determined by the Markwell modified Lowry assay (31).
Chemical Cross-linking
The monomeric and dimeric samples were cross-linked with bis(sulfosuccinimidyl) suberate as previously reported (25). Freshly solubilized bis(sulfosuccinimidyl) suberate (spacer arm of 11.4 Å) was added to the protein solutions at molar ratios of cross-linker to protein that varied from 10:1 to 50:1. The samples were incubated at 4 °C for periods of 2 or 12 h, with no differences in total cross-linking noted between the two incubation times. After quenching with an excess of Tris-HCl, the cross-linked proteins were exhaustively digested with trypsin at a 1:20 mass ratio of trypsin to apoA-IV for 2 h at 37 °C followed by a second spike of trypsin and another 2-h incubation. The resulting peptides were then lyophilized to dryness and stored at −80 °C until analyzed by MS.
Mass Spectrometry
Nano-LC-MS/MS analyses were performed on a TripleTOF® 5600+ (AB Sciex, Toronto, Canada) coupled to an Eksigent (Dublin, CA) NanoLC-Ultra® nanoflow system. Dried samples were reconstituted in formic acid/H2O 0.1/99.9 (v/v), and 5 μl (containing 1–3 μg of digest) was loaded onto C18 IntegraFritTM trap column (outer diameter of 360 μm, inner diameter of 100, and packed bed of 25 μm) from New Objective, Inc. (Woburn, MA) at 2 μl/min in FA/H2O 0.1/99.9 (v/v) for 15 min to desalt and concentrate the samples. For the chromatographic separation, the trap column was switched to align with the analytical column, Acclaim® PepMap100 (inner diameter of 75 μm, length of 15 cm, C18 particle size of 3 μm, and pore size of 100 Å) from Dionex-Thermo Fisher Scientific (Sunnyvale, CA). The protein was eluted at 300 nl/min using a varying mobile phase gradient from 95% phase A (FA/H2O 0.1/99.9, v/v) to 40% phase B (FA/acetonitrile 0.1/99.9, v/v) for 35 min (1% per min) and then from 40% B to 85% B in 5 min with re-equilibration. The effluent from the nanoLC was introduced in to the mass spectrometer using a NANOSpray® III source (AB Sciex, Toronto, Canada). The instrument was operated in positive ion mode for 65 min, where each cycle consisted of one TOF-MS scan (0.25-s accumulation time, in a 350–1500 m/z window) followed by 30 information-dependent acquisition mode MS/MS scans on the most intense candidate ions selected from initially performed TOF-MS scan during each cycle. Each product ion scan was operated under vender-specified high sensitivity mode with an accumulation time of 0.075 s and CE of 43 with an 8-unit scan range. The .wiff files were converted to Mascot generic files using a file converting algorithm embedded in PeakView® v1.2.0.3 software (AB Sciex).
Mass Spectral Data Analysis
The MS data, in the form of a Mascot generic file, was uploaded into Cross-ID. Given the sequence of apoA-IV, the cross-linker used, the isotopic possibilities, and the enzyme used for peptide generation, the program first identifies all possible cross-link interactions based on a user defined window of error for each mass detected in the experiment (20 ppm for this instrument). Once candidate identifications are determined, the program then compares theoretical MS/MS fragmentation patterns to the actual data, generating a probability score that reflects the confidence of the identification. These identifications are then visually verified by experienced personnel before the cross-link is declared valid. Valid identifications required the presence of co-eluting 14N and 15N versions of the same species and appeared in two independent analyses of purified monomers and dimers.
Generation of Initial Model for Molecular Dynamics Studies
An initial full-length model of apoA-IV was generated using the Phyre2 server (32). Phyre2 server searches for sequence homology within the Protein Data Bank database and generates models based on previously solved structures. The initial structure generated by Phyre2 was based on the apoA-IV64–335 structure, and the secondary structure of this model consisted primarily of helices. This was then further refined manually using PyMOL to manipulate residues to conform to the distance constraints from cross-linking experiments. In conforming to these constraints, proline, glycine, or serine residues were used to initiate helical breaks or bends, similar to what is observed in the apoA-IV64–335 structure. Additionally, hydrophobic residues were buried into the core of the helical bundle, again similar to what is observed in the apoA-IV64–335 structure.
All Atom Molecular Dynamics Simulations
All atom MD simulations were performed using NAMD 2.9 (33) as described by Jones et al. (34). Using Visual Molecular Dynamics (35), each system was solvated with the solvate plug-in and ionized and charge-neutralized with NaCl to 0.15 m with the ionize plug-in. The TIP3P model was used for water (36), and the CHARMM 22 (37, 38) force fields were used for protein.
The all atom simulations began with the starting structure of full-length monomeric apoA-IV described above. After heating to 310 K, the system was simulated for 15 ns. In another experiment, the system was subjected to a 15-ns MD-simulated annealing (MDSA) protocol as described by Jones et al. (34), involving a quick heating to 500 K that was maintained for 5 ns, followed by a cool down from 500 to 310 K over 5 ns, followed by 5 ns at 310 K. As a measure of the convergence of our simulations, a root mean square deviation was performed over the course of each simulation.
To obtain contact maps over the trajectories of the simulations, the number of times that α-carbon atoms were within 25 Å of each other was counted over the course of that simulation, combined, and then shown as a contour plot of different colors indicating percentages of counts (except for a count of 0, which has no color) relative to the maximum count over all pairs of residues, respectively, using the graphics software GNUPLOT.
Model Comparisons to SAXS Data
To generate multiple models for SAXS comparison, the starting model or the MD generated model described above was processed with the AllosMod-FoXS web server (39, 40) using MODELLER (41) and constrained with the identified cross-links that link the N/C termini (20.0 ± 8.0Å from C-α to C-α). The simulation was allowed to sample all intermediate conformations consistent with the input structure using a temperature scan. A total of 30 runs were conducted generating 101 possible structures for each run, yielding both the single best fit model and the multiple ensemble search results (42).
Liposome Clearance Assay
ApoA-IV or its mutants were added to dimyristoyl phosphatidylcholine (DMPC) liposomes at a 2:1 specified mole lipid: mole protein ratio, and the absorbance of the solution at 325 nm was recorded every 30 s for 20 min. Samples were measured in triplicate, and the values were averaged. The data were expressed as a normalized optical density calculated by dividing the sample optical density by the initial optical density. All measurements were made on an Amersham Biosciences Ultraspec 4000 UV-visible spectrophotometer within a temperature-controlled cuvette at 24.5 °C.
RESULTS
Overall Strategy
We previously reported an x-ray crystal structure of the central region of human apoA-IV in the dimeric state (22). However, despite continuing work aimed at crystallizing the entire protein, the N- and C-terminal regions have not yet been visualized. In the meantime, we reasoned that chemical cross-linking could provide distance constraints for building a new model for full-length apoA-IV in both its monomeric and dimeric forms. However, when applied to homodimeric proteins, a drawback of the technique is uncertainty with regard to cross-link molecular span. The structures of truncated forms of apoA-IV and apoA-I clearly show that the monomeric and dimeric forms of both proteins are highly similar with respect to the protein contacts made. Thus, one can observe cross-linking of the same two peptides in both monomer and dimer forms, but these linkages occur intramolecularly in the former and intermolecularly in the latter.
To unambiguously distinguish between intra- and intermolecular cross-links, we produced versions of apoA-IV containing either naturally occurring nitrogen (14N) or its heavier isotope (15N). Thus, for every nitrogen atom in a peptide (one for each amino acid in the peptide backbone and more in some side chains), the mass increases by 1 Da in the labeled protein. These mass differences are easily detected by modern mass spectrometers and can be exploited to distinguish between intramolecular and intermolecular cross-linked peptides. Differentially labeled species were mixed 1:1 under denaturing conditions and then allowed to refold/reassociate, creating a mixture of isotopically labeled proteins that are either purely 14N or 15N, or a mixed 14N/15N dimer. Fig. 1 shows the example of the crystal structure of dimeric apoA-IV64–335 with 14N-apoA-IV (green) and 15N-apoA-IV (red). For an intermolecular cross-link between two peptides (A and B), there are four possible mass combinations: 14A to 14B, 15A to 15B, and the mixed forms 14A to 15B and 14B to 15A. Thus, intermolecular cross-links will exhibit four mass peaks of equal intensity. On the other hand, intramolecular cross-links only have two possibilities: 14A to 14B and 15A to 15B. Thus, cross-link span can be easily and unambiguously determined.
FIGURE 1.
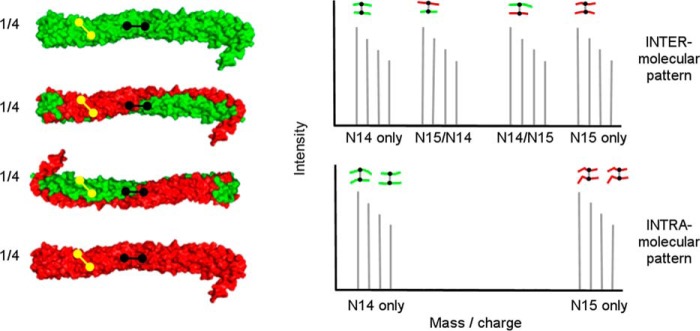
Principle behind isotope-assisted chemical cross-linking for determination of intra- versus intermolecular cross-links in oligomerized proteins. Recombinant proteins produced with either (i) 14N amino acids (green) or (ii) 15N amino acids (red) are thoroughly mixed at a 1:1 mass ratio under denaturing conditions and then allowed to reassociate, resulting in the four combinations shown on the left. The proteins are then cross-linked and fully digested with trypsin. For an intermolecular cross-link (shown in black) between two peptides (A and B), there are four possible mass combinations: 14A to 14B, 15A to 15B, and the mixed forms 14A to 15B and 14B to 15A. Thus, intermolecular cross-links will exhibit four mass peaks of approximately equal intensity as shown in the mass spectrum on the right. On the other hand, intramolecular cross-links (shown in yellow) only have two possibilities: 14A to 14B and 15A to 15B, allowing for an unambiguous determination of the cross-link molecular span.
Cross-linking Monomeric and Dimeric ApoA-IV
The 14N and 15N versions of the full-length apoA-IV were generated using our bacterial expression system (43). Fig. 2a shows that in solution at 1 mg/ml, both forms distributed into monomers (lower bands) and dimers (upper bands) at a ∼1:2 ratio, respectively. Fig. 2b shows mass spectra for a typical peptide from each preparation. The expected 9-Da mass increase was apparent in the 15N-labeled peptide. The lack of significant peaks lower than 413.221 Da shows nearly complete 15N labeling of the protein. The similarity of the MS/MS spectra indicates that both peptides are otherwise identical.
FIGURE 2.

Characterization of lipid-free 14N and 15N apoA-IV. a, native PAGE analysis of apoA-IV at 1 mg/ml: 14N apoA-IV (lane 1), 15N apoA-IV (lane 2), and a 1:1 mixture of the two (lane 3). The gel was stained with Coomassie Blue. b, mass spectra of M2H+ ion resulting from the peptide LKEEIGK. The top spectrum was derived from 14N apoA-IV, and the bottom spectrum from 15N apoA-IV, illustrating the mass difference resulting from isotopic labeling. The MS/MS spectra for each ion are shown in the insets indicating a similar fragmentation pattern for each.
Fig. 3 shows a native gel analysis of a 1:1 molar mixture of 14N and 15N apoA-IV. At equilibrium, the mixture exhibited the characteristic distribution of dimer and monomer shown in Fig. 2. These were separated by gel filtration chromatography to yield isolated monomer and dimer species, with minimal cross-contamination. We have previously demonstrated that the isolated species can be maintained for weeks at 4 °C without redistribution (22).
FIGURE 3.
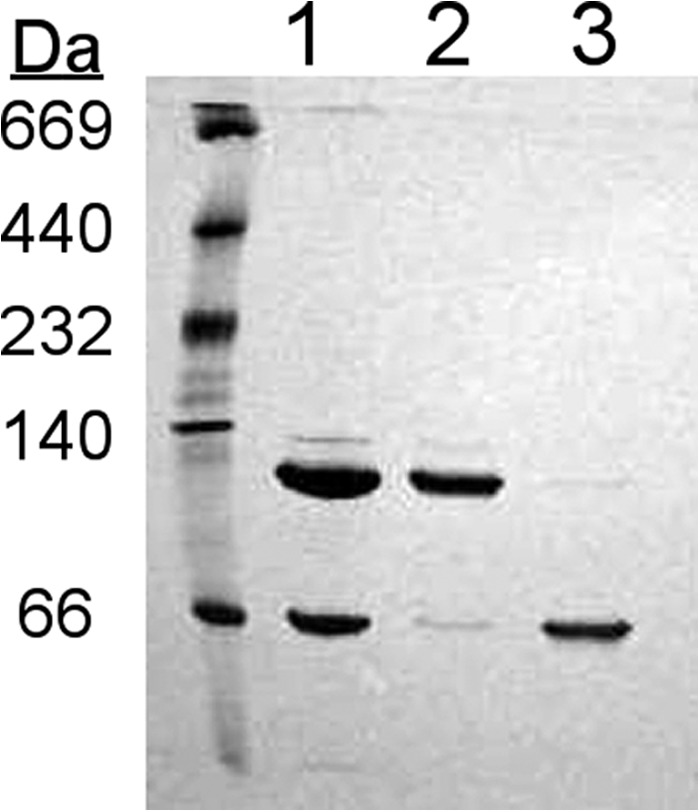
Separation of lipid-free apoA-IV monomers and dimers by gel filtration chromatography. 14N apoA-IV and 15N apoA-IV were mixed and renatured as described under “Experimental Procedures.” The proteins were then subjected to gel filtration chromatography, and fractions corresponding to the stable monomeric and dimeric species were pooled. Shown is a native PAGE analysis of the sample prior to separation (lane 1) and the purified dimeric (lane 2) and monomeric (lane 3) samples. The gel was stained with Coomassie Blue.
The separated species were then individually cross-linked, trypsinized, and analyzed by MS as described under “Experimental Procedures.” We identified 51 peptide pairs that contained an intact cross-link in both the monomer and dimer samples. The MS signatures of these species fell into two categories exemplified in Fig. 4. The majority contained 14N and 15N masses only (Fig. 4a), which are indicative of intramolecular linkages (Fig. 1). Eleven cross-linked pairs, always appearing in dimeric samples, exhibited the additional mixed forms, all with similar intensities (Fig. 4b, green spectrum), clearly indicating intermolecular span. Table 1 lists the peptides containing an intra-peptide cross-link, i.e., cross-links that occurred between two lysine residues located on the same tryptic peptide. By definition, these links are intramolecular. Table 2 lists the cross-links that were interpeptide. These can be intramolecular, occurring between distant peptides within the same protein molecule, or intermolecular. Inspection of the tables reveals several important observations. First, almost every cross-link that appeared in the monomeric samples also appeared in the dimeric samples. There were only two that were unique to the dimer and one unique to the monomer. Second, there were several instances where the same two peptides were cross-linked intramolecularly in the monomer but were intermolecular in the dimer (for example, Lys59–Lys330 in Table 2, with spectra shown in Fig. 4b).
FIGURE 4.
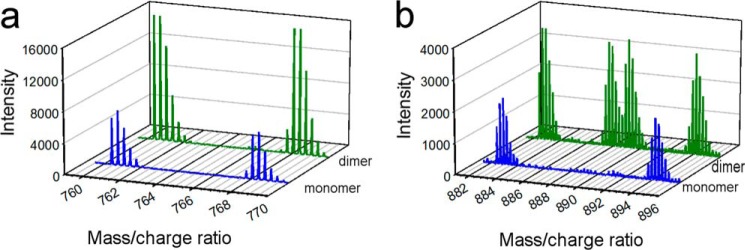
Example MS spectra of intra-molecular and intermolecular cross-links identified in monomeric and dimeric apoA-IV. a shows the mass spectra for the M3H+ ion resulting from the cross-link 182–193 × 228–234 in the monomeric (blue) and dimeric (green) samples indicating a clear intramolecular span in both. b shows the same for the M4H+ ion resulting from the cross-link 94–110 × 191–202 showing that the cross-link is intermolecular in the dimer sample but intramolecular in the monomeric sample.
TABLE 1.
Identified intrapeptide BS3 cross-links in isolated apoA-IV monomeric and dimeric samples derived from mixed 14N- and 15N-labeled proteins
| Cross-link | Peptides involveda | Modificationsb | Samplec | 14N massd | 15N mass |
|---|---|---|---|---|---|
| Da | Da | ||||
| Lys73–Lys77 | 71LAKDSEKLK79 | XL | M | 1168.67 | 1180.64 |
| D | 1168.67 | 1180.64 | |||
| Lys226–Lys233 | 228KNAEELKAR235 | XL | M | 1195.65 | 1210.61 |
| D | 1195.66 | 1210.61 | |||
| Lys77–Lys79 | 74DSEKLKEEIGK84 | XL | M | 1412.74 | 1426.70 |
| D | 1412.74 | 1426.70 | |||
| Lys169–Lys178 | 168AKIDQNVEELKGR180 | XL | M | 1636.88 | 1656.82 |
| D | 1636.87 | 1656.81 | |||
| Lys79–Lys84 | 78LKEEIGKELEELR90 | XL | M | 1722.94 | 1740.89 |
| D | 1722.95 | 1740.89 | |||
| Lys73–(Lys77,Lys79) | 71LAKDSEKLKEEIGK84 | XL | M | 1724.95 | 1742.90 |
| D | 1724.95 | 1742.91 | |||
| Lys339–Lys341 | 331VNSFFSTFKEKESQDK341 | XL | M | 2057.99 | 2078.93 |
| D | 2058.00 | 2078.95 | |||
| Lys79–Lys84 | 74DSEKLKEEIGKELEELR90 | XL | M | 2182.14 | 2205.08 |
| D | 2182.14 | 2205.07 | |||
| Lys330–Lys341 | 329DKVNSFFSTFKEKESQDK346 | XL | M | 2301.12 | 2325.05 |
| D | 2301.12 | 2325.05 | |||
| Lys73–Lys84 | 71LAKDSEKLKEEIGKELEELR90 | XL | M | 2494.35 | 2521.28 |
| D | 2494.35 | 2521.30 | |||
| Lys226–Lys227 | 214LNHQLEGLTFQMKKNAEELK233 | XL | M | 2508.30 | 2537.25 |
| D | 2508.30 | 2537.22 | |||
| Lys189–Lys191 | 181LTPYADEFKVKIDQTVEELR200 | XL | M | 2531.32 | 2557.24 |
| D | 2531.33 | 2557.25 | |||
| Lys308–Lys325 | 307QKLGPHAGDVEGHLSFLEKDLR328 | XL | M | 2583.34 | 2615.25 |
| D | 2583.35 | 2615.25 | |||
| Lys189–Lys191 | 181LTPYADEFKVKIDQTVEELRR201 | XL | M | 2687.42 | 2717.33 |
| D | 2687.42 | 2717.34 | |||
| Lys58–Lys59 | 46LGEVNTYAGDLQKKLVPFATELHER70 | XL | M | 2965.56 | 2999.46 |
| D | 2965.56 | 2999.47 | |||
| Lys213–Lys228 | 202SLAPYAQDTQEKLNHQLEGLTFQMKK229 | XL | M | 3155.60 | 3191.49 |
| D | 3155.62 | 3191.50 | |||
| Lys167–Lys169 | 150ENADSLQASLRPHADELKAKIDQNVEELK201 | XL | M | 3398.73 | 3439.60 |
| D | 3398.74 | 3439.62 |
a Lysines involved in cross-links are in bold.
b Chemical modifications: XL = 1 complete cross-link (+138.06808 Da) and H = 1 hydrolyzed cross-linker (+156.07864 Da).
c Monomers (M) and dimers (D) were separated prior to cross-linking by gel filtration. See “Experimental Procedures.”
d Experimentally derived monoisotopic mass for each peptide with each isotope and the combinations.
TABLE 2.
Identified interpeptide BS3 cross-links in isolated apoA-IV monomeric and dimeric samples derived from mixed 14N- and 15N-labeled proteins
| Cross-link | Peptides involveda | Modificationsb | Samplec | Peptide massd |
Span | |||
|---|---|---|---|---|---|---|---|---|
| 14N | 14N/15N | 15N/14N | 15N | |||||
| Da | ||||||||
| Lys73–Lys79 | 71LAKDSEK77 | XL | M | 1742.96 | 1760.92 | Intra | ||
| 78LKEEIGK84 | D | 1742.96 | 1760.91 | Intra | ||||
| Lys73–Lys341 | 71LAKDSEK77 | XL | M | 1789.88 | 1808.84 | Intra | ||
| 340EKESQDK346 | D | 1789.89 | NRe | 1798.86 | 1808.83 | Inter | ||
| Lys77–Lys84 | 74DSEKLK79 | XL | M | 2200.15 | 2223.10 | Intra | ||
| 80EEIGKELEELR90 | D | 2200.15 | 2223.08 | Intra | ||||
| Lys73–Lys330 | 71LAKDSEK77 | XL | M | 2246.15 | 2269.09 | Intra | ||
| 329DKVNSFFSTFK339 | D | 2246.15 | 2260.14 | 2255.12 | 2269.08 | Inter | ||
| Lys189–Lys227 | 181LTPYADEFKVK191 | XL | M | 2278.21 | 2301.14 | Intra | ||
| 227KNAEELK233 | D | 2278.21 | 2301.15 | Intra | ||||
| Lys79–Lys339 | 78LKEEIGK84 | XL | M | 2286.22 | 2309.13 | Intra | ||
| 331VNSFFSTFKEK341 | D | 2286.22 | 2300.176 | 2295.21 | 2309.15 | Inter | ||
| Lys330–Lys341 | 329DKVNSFFSTFK339 | XL | M | 2319.13 | 2343.06 | Intra | ||
| 340EKESQDK346 | D | 2319.15 | 2343.08 | Intra | ||||
| Lys178–Lys233 | 170IDQNVEELKGR180 | XL | M | 2367.26 | 2397.17 | Intra | ||
| 228NAEELKAR235 | D | 2367.24 | 2397.15 | Intra | ||||
| Lys189–Lys233 | 181LTPYADEFKVK191 | XL | M | 2377.25 | 2403.17 | Intra | ||
| 228NAEELKAR235 | D | 2377.25 | 2403.18 | Intra | ||||
| Lys191–Lys233 | 190VKIDQTVEELR200 | XL | M | 2396.29 | 2425.20 | Intra | ||
| 228NAEELKAR235 | D | 2396.30 | 2425.21 | Intra | ||||
| Lys59–Lys341 | 59KLVPFATELHER70 | XL | M | 2439.26 | 2467.18 | Intra | ||
| 340EKESQDK346 | D | 2439.27 | 2448.22 | 2457.211 | 2467.18 | Inter | ||
| Lys189–(Lys227,Lys233) | 181LTPYADEFKVK191 | XL | M | 2505.34 | 2533.26 | Intra | ||
| 227KNAEELKAR235 | D | 2505.35 | 2533.26 | Intra | ||||
| Lys73–(Lys79,Lys84) | 71LAKDSEK77 | XL | M | 2512.36 | 2539.28 | Intra | ||
| 78LKEEIGKELEELR90 | D | 2512.36 | 2539.29 | Intra | ||||
| Lys191–Lys233 | 190VKIDQTVEELRR201 | XL | M | 2552.39 | 2585.30 | Intra | ||
| 228NAEELKAR235 | D | 2552.39 | 2585.30 | Intra | ||||
| (Lys169,Lys178)–Lys233 | 168AKIDQNVEELKGR180 | XL,H | M | 2722.47 | 2755.37 | Intra | ||
| 228NAEELKAR235 | D | 2722.48 | 2755.38 | Intra | ||||
| Lys59–Lys330 | 59KLVPFATELHER70 | XL | M | 2895.53 | 2927.47 | Intra | ||
| 329DKVNSFFSTFK339 | D | 2895.53 | 2909.52 | NR | 2927.43 | Inter | ||
| Lys79–Lys339 | 78LKEEIGK84 | XL | M | 2913.53 | 2945.44 | Intra | ||
| 326DLRDKVNSFFSTFKEK341 | D | 2913.54 | 2945.44 | Intra | ||||
| Lys84–Lys330 | 80EEIGKELEELRAR92 | XL | M | 3027.51 | 3061.44 | Intra | ||
| 329DKVNSFFSTFK339 | D | 3027.53 | 3061.44 | Intra | ||||
| (Lys169,Lys178)–Lys189 | 168AKIDQNVEELKGR180 | XL,H | M | 3102.68 | 3135.59 | Intra | ||
| 181LTPYADEFKVK191 | D | 3102.69 | 3135.59 | Intra | ||||
| Lys227–Lys296 | 227KNAEELK233 | XL | M | 3360.70 | 3399.59 | Intra | ||
| 287VEPYGENFNKALVQQMEQLR306 | D | 3360.71 | - | 3399.61 | Intra | |||
| Lys103–Lys191 | 93LLPHANEVSQKIGDNLR109 | XL | M | 3369.82 | 3411.69 | Intra | ||
| 190VKIDQTVEELR200 | D | 3369.83 | 3385.787 | 3395.757 | 3411.71 | Inter | ||
| Lys31–Lys73 | 24EAVEHLQKSELTQQLNALFQDK45 | XL | M | 3495.83 | 3535.71 | Intra | ||
| 71LAKDSEK77 | D | 3495.81 | 3535.69 | Intra | ||||
| Lys227–Lys296 | 227KNAEELK233 | XL | M | 3516.80 | 3559.66 | Intra | ||
| 286RVEPYGENFNKALVQQMEQLR306 | D | 3516.80 | 3559.68 | Intra | ||||
| Lys103–Lys191 | 93LLPHANEVSQKIGDNLR109 | XL | M | 3525.93 | 3571.80 | Intra | ||
| 190VKIDQTVEELRR200 | D | 3525.93 | 3545.872 | 3551.860 | 3571.80 | Inter | ||
| Lys189–Lys325 | 181LTPYADEFKVK191 | XL | M | 3636.88 | 3677.78 | Intra | ||
| 309LGPHAGDVEGHLSFLEKDLR328 | D | 3636.88 | 3677.75 | Intra | ||||
| (Lys189,Lys191)–Lys227 | 181LTPYADEFKVKIDQTVEELRR201 | XL,H | M | 3673.98 | 3712.88 | Intra | ||
| 227KNAEELK233 | D | 3673.99 | 3712.89 | Intra | ||||
| Lys233–Lys267 | 228NAEELKAR235 | XL | M | 3821.91 | 3869.79 | Intra | ||
| 260GNTEGLQKSLAELGGHLDQQVEEFR284 | D | f | ||||||
| Lys227–Lys325 | 214LNHQLEGLTFQMKK227 | XL | M | f | ||||
| 309LGPHAGDVEGHLSFLEKDLR328 | D | 4013.09 | 4061.94 | Intra | ||||
| Lys193–Lys296 | 190VKIDQTVEELR200 | XL | M | 4015.09 | 4063.95 | Intra | ||
| 286RVEPYGENFNKALVQQMEQLR306 | D | 4015.11 | 4063.98 | Intra | ||||
| N-terminus-Lys339 | −1-GEVSADQVATVMWDYFSQLSNNAK23 | XL | M | 4130.01 | 4173.88 | Intra | ||
| 331VNSFFSTFKEK341 | D | 4130.02 | 4143.98 | 4159.93 | 4173.90 | Inter | ||
| (Lys79,Lys84)–Lys213 | 78LKEEIGKELEELR90 | XL | M | 4612.41 | 4664.28 | Intra | ||
| 202SLAPYAQDTQEKLNHQLEGLTFQMK226 | D | 4612.38 | 4630.35 | 4646.33 | 4664.27 | Inter | ||
| (Lys79,Lys84)–Lys213 | 78LKEEIGKELEELR90 | XL,H | M | 4768.47 | 4820.35 | Intra | ||
| 202SLAPYAQDTQEKLNHQLEGLTFQMK226 | D | 4768.50 | 4786.46 | 4801.37 | 4820.35 | Inter | ||
| Lys267–Lys267 | 260GNTEGLQKSLAELGGHLDQQVEEFR284 | XL | M | f | ||||
| 260GNTEGLQKSLAELGGHLDQQVEEFR284 | D | 5646.88 | 5681.74 | 5681.74 | 5716.65 | Inter | ||
a Lysines involved in cross-links are in bold.
b Chemical modifications: XL = 1 complete cross-link (+138.06808 Da) and H = 1 hydrolyzed cross-linker (+156.07864 Da).
c Monomers (M) and dimers (D) were separated prior to cross-linking by gel filtration. See “Experimental Procedures.”
d Experimentally derived monoisotopic mass for each peptide with each isotope and the combinations.
e NR, not resolved. The mass was clearly present, but an accurate molecular weight could not be determined because of interference from other ions.
f Not detected. These ions were detectable in one sample (i.e., monomer or dimer) but not in the other.
Cross-link Consistency with the ApoA-IV64–335 Crystal Structure
The cross-linking technique analyzes proteins in solution, whereas x-ray crystallography reports on proteins in a crystalline state. We first compared the cross-linked Lys residues listed in Tables 1 and 2 with predictions from the apoA-IV64–335 crystal structure. Fig. 5 is a contact plot rendered from the crystal structure which shows the proximity of each residue with respect to all other residues in a two-dimensional format. The colored areas show where residues indicated on the axes come within 20 Å of each other (i.e., close enough to be cross-linked). Plotting the experimentally derived cross-links showed that all landed within the colored regions and were thus consistent with the crystal structure, even when far apart in the sequence. The observation of these cross-links on a previously validated homo-dimeric crystal structure shows that we can clearly differentiate intra- and intermolecular contacts.
FIGURE 5.
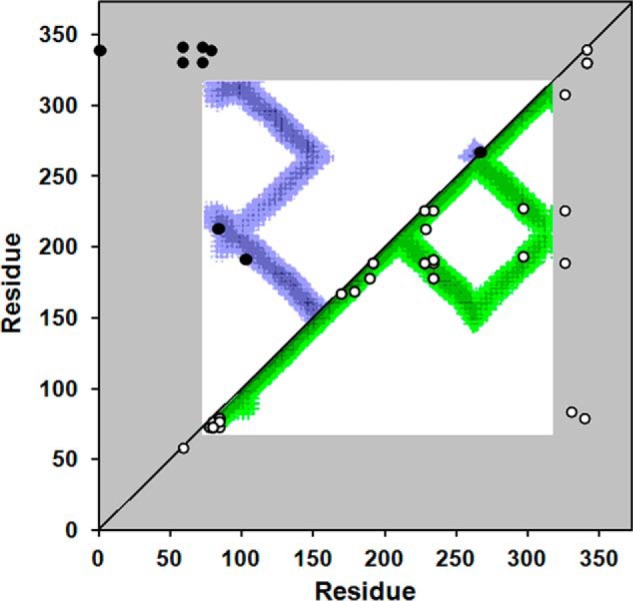
Cross-linking data from Tables 1 and 2 superimposed on a molecular contact plot generated from the crystal structure of dimeric apoA-IV64–335. The x and y axes of the plot indicate the residue number of apoA-IV from 1–376. The diagonal line bisects the figure; the top left is used to evaluate intermolecular interactions, whereas the bottom right shows intramolecular interactions. Using the crystal structure of human apoA-IV64–335 (22), we produced a contact map for each amino acid. The regions of the darkest shade of purple represent proximity of 1–10 Å, lighter gradations represent 11–15, 16–20, and 21–24 Å, respectively. White indicates greater than 24 Å. For example, residue 100 of one molecule (x axis) is within 11–15 Å of residue 200 (y axis) of the other molecule in the dimeric crystal structure as indicated by the purple color at their intersection on the graph. On the other hand, residue 100 is quite far away from residue 250 of the other molecule (white where they intersect). Because the crystal structure only provides structural data between residue 74 and 314 (in white), areas where the contact plot could not be generated are shown in gray. The cross-links between Lys residues shown in Tables 1 and 2 are superimposed on the figure (intermolecular links in black in the upper left, intramolecular links in white on the lower right). In cases where Lys assignments in cross-links were ambiguous, the Lys residues with the shortest separation distance were plotted.
Based on the dimeric apoA-IV64–335 crystal structure and later supported by SAXS (26), we proposed that apoA-IV could monomerize by keeping the shared helix to itself rather than swapping it with another molecule (22). The helix, bent at its midpoint, was posited to fold back onto the molecule making intramolecular interactions that are identical to the intermolecular interactions made in the dimer. One of the cross-links in Table 2 allowed direct evaluation of this prediction. Fig. 6 shows the cross-link between Lys103 and Lys191. Table 2 and Fig. 4b indicate that this cross-link is intramolecular in the monomer but intermolecular in the dimer. The insets in Fig. 6 show that this linkage can easily occur (∼12 Å) in both models. In fact, this was the only model that we could envision that satisfies the data. Furthermore, this concept easily explains the remarkable similarity in the overall cross-linking patterns between the dimeric and monomeric samples. We interpreted this data to be strongly supportive of the proposed monomer model and elected to pursue further modeling of full-length apoA-IV in the context of the monomer. After creating a starting model based on available data, we took two separate modeling approaches: (i) molecular dynamics simulation and (ii) constrained simulation using our previously generated SAX data for the full-length protein (26).
FIGURE 6.
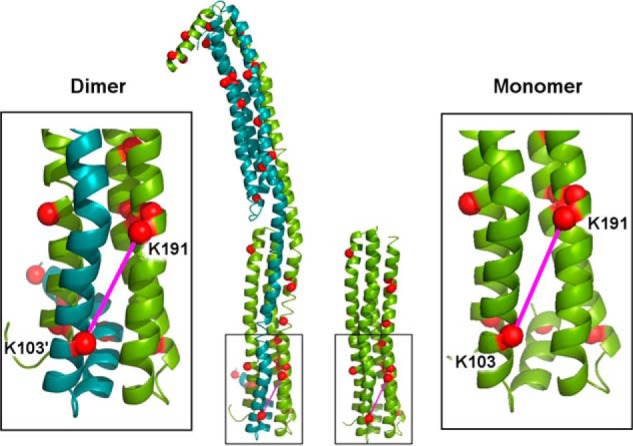
Representative cross-link depicting an intra- versus intermolecular contact in the monomer versus dimer of apoA-IV. The dimeric crystal structure for apoA-IV64–335 is shown on the left with molecule A in green and molecule B in teal. The α-carbons of Lys residues are shown as red spheres. The long, “swapped” helices that reciprocate across the dimer interface can be seen on the right side of the dimer. In our proposed model of the monomeric structure (right), the long helix in molecule A has a hinge introduced at its midpoint and folds back onto the molecule, making exactly the same interactions as the swapped helix from molecule B makes in the dimer. The insets show the cross-link between Lys191 and Lys103 (pink line), which was determined to be intermolecular in the dimer but intramolecular in the monomer (the mass spectra for a representative ion for this cross-link is shown in Fig. 4b).
Generation of a Starting Model for the Full-length ApoA-IV Monomer
Once we obtained the cross-linking distance constraints within regions that were not visible in the crystal structure of apoA-IV64–335, we set out to generate a model of full-length apoA-IV that would be suitable as a starting point for in silico modeling. Using homology modeling techniques, coupled with secondary structure analysis, we created a preliminary model of full-length monomeric apoA-IV in which unknown regions of the protein were placed as directed by the cross-linking data (see “Experimental Procedures”). Subsequently, we energy-minimized the structure to generate the “starting” model.
Modeling Approach 1: Molecular Dynamics Simulation
We subjected the starting model to two 15-ns MD simulations: one at 310 K and a second MDSA simulation (5 ns at 500 K, 5 ns of cool down, and 5 ns at 310 K). These two methods are useful for comparing the stability of the initial model at physiological temperature with more aggressive simulation using the temperature jump in the MDSA approach. Root mean square deviation plots for both simulations showed good convergence after 10 ns (not shown).
The structure resulting from the 15-ns simulation is shown in Fig. 7 with its contact plot (with cross-linking data overlaid) and sequences predicted to be helical. The model is dominated by a four-helix bundle composed of the same residues that form the reciprocating four-helix bundle in the apoA-IV64–335 crystal structure. The N and C termini are clustered at one end of the bundle in a significantly less helical, globular conformation. The calculated α-helicity of the model for the last 5 ns is 67%. The experimentally determined α-helical content of lipid-free apoA-IV varies considerably in the literature: 35% (44), 40% (30), 43% (45), 54% (46), 60% (47), and 68% (48). Thus, this model is on the high end of the range. The contact plot in Fig. 7a differs from Fig. 5 in that the distances plotted are over the entire 15 ns of the simulation rather than distances determined from a static structure. When superimposed on the plot, it was clear that all cross-links were plausible in the model. Because the cross-links were not set as a constraint during the simulations, this analysis indicates that the general organization of our starting model held up during the simulation. However, we caution that MD simulations may reflect localized energy minima that may not fully reflect the native solution structure of the protein.
FIGURE 7.

Simulated monomeric full-length apoA-IV structures generated with non-MDSA molecular dynamics. a, contact plot derived from the 15-ns simulation. The plot is laid out similarly to Fig. 5. Only the lower right half of the plot contains data because this monomeric sample only has intramolecular contacts. b, cartoon models showing the α-carbons of the C-terminal residue in yellow spacefill and the N-terminal residue in red spacefill. Graphics were generated using PyMOL. c, sequence of full-length human apoA-IV showing amino acids involved in helical domains (underlined) in the simulated structure.
The results of the simulated annealing (MDSA) simulation are not shown. The model was overall similar with a four-helical bundle and globular domain, although the bundle was less helical and slightly more curved. The N terminus folded back across the helical bundle and was in closer proximity to the C terminus versus the non-MDSA model. The helicity was ∼66%. The cross-links were generally compatible with the model with the exception of Lys189–Lys325, which was not plausible in this model.
Modeling Approach 2: Comparing the Monomer Models to SAXS Data
We have previously used SAXS to determine the molecular shape of apoA-IV64–335 (26). The molecular envelope of the dimer exhibited an excellent match with that predicted from the crystal structure. In that work, we also derived SAXS envelopes for isolated full-length monomer and dimer. We first compared the starting model to the full-length SAXS profile. Fig. 8a shows that it was in good agreement with the SAXS profile (χ = 1.51) and significantly better than a model that only includes the helical bundle (χ = 5.80). We then used the AllosMod-FoXS web server (39, 40) to generate alternate conformations of the starting model to determine whether we could better represent the SAXS profile. The starting model was constrained with the experimentally observed cross-links as described under “Experimental Procedures.” Fig. 8 (a and b) shows the single best fit from the analysis, which is in excellent agreement with the experimental SAXS profile (χ = 0.93). A minimal ensemble search (42) of the AllosMod-FoXS generated models revealed that a combination of two models resulted in a slight improvement to the fit of the SAXS profile (χ = 0.57). However, the models were divergent in their N/C termini, suggesting that these segments can sample multiple conformations (Fig. 8c).
FIGURE 8.

AllosMod-FoXS modeling of the apoA-IV monomer using SAXS data. a, comparison of apoA-IV x-ray scattering profile (black line) to truncation of the N/C termini from the single best fit model (cyan line), initial model (orange line), full-length single best fit (red line), and minimal ensemble search (MES, green line) theoretical profiles determined by AllosMod-FoXS. Residuals are shown below with chi distribution values in parentheses. b, AllosMod-FoXS single best fit molecular model shown in cartoon. c, C-α root mean square deviation (RMSD) values for each residue determined using SuperPose 1.0 (55) of the molecular models derived from minimal ensemble search within the AllosMod-FoXS pipeline. Note that significant C-α deviations between models are within the N/C termini (amino acids 1–96 and 312–376) and not the four-helical bundle (amino acids 97–376). d, superposition of the single best fit model onto the GASBOR ab intio molecular envelope. e, flexibility plots of the experimental SAXS profile of the apoA-IV monomer. The graphics were generated using PyMOL.
The single best fit molecular model is shown in Fig. 8b and shows that the general overall shape is maintained, whereas parts of the N/C termini are less compact and extended away from the bundle. Overall, the orientation satisfies the experimentally determined cross-linking profiles and SAXS scattering profile. Furthermore, we generated a GASBOR ab initio molecular envelope of the apoA-IV monomer, which contained extra density adjacent to the four-helical bundle (26). Our newly derived single best fit model strongly indicates that the extra density derives from the N/C termini (Fig. 8d). Nonetheless, alternate/multiple conformations may exist that could also satisfy these parameters.
Although the single best fit is consistent with the experimental data, we took a closer look at the experimental SAXS profile. Further analysis confirmed that monomeric apoA-IV exhibits some flexibility. This is demonstrated by the lack of a distinct plateau in the Porod-Debye plot (q4 versus q4·I(q)), whereas there is a clear plateau in the q3 versus q3·I(q) plot (Fig. 8e). A plateau in this plot is suggestive of intrinsic flexibility in the protein (49, 50). This should not be mistaken for an unstructured protein because the q2 versus q2·I(q) plot displays a distinct decrease and not a plateau (Fig. 8e) (49, 50).
For completeness, we also analyzed the MD model (non-MDSA) with respect to the SAXS data (not shown). The simulated model was in good agreement with the SAXS envelope (χ = 2.38) though not as strong as the starting model. The AllosMod-FoXS analysis yielded an improved single best fit model (χ = 0.97). Again, the models were divergent in the N and C termini (Fig. 8c), consistent with significant conformational flexibility in these regions.
Extrapolation of the Simulated Monomer Model to the Dimeric Form
Based on the cross-linking data in Tables 1 and 2 and our previous work, we reasoned that the structure of the globular domain was similar in the monomer and dimer. In the monomer, this domain consists of the N and C termini from the same molecule. In the dimer, there are two of these domains, each composed of termini from different molecules. Therefore, we generated a symmetry related full-length dimer with swapped chains by superimposing the monomer MD results onto the apoA-IV64–335 crystal structure dimer. Unfortunately, MD simulations of this model were not successful because of significant instability at the crossover in the middle of the model. Thus, we were unable to develop a consensus model and corresponding contact plot. However, by simple extrapolation, it is clear that the cross-links occurring between the N- and C-regions in the monomer can also occur intermolecularly in the dimer. An example is shown in Fig. 9. This conclusion also applies to the single best fit models derived from the SAXS analysis (not shown).
FIGURE 9.
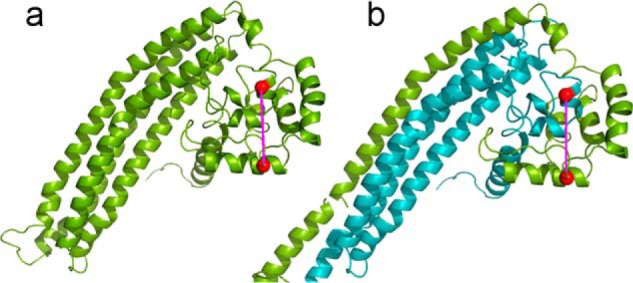
Representative cross-link depicting an intra- versus intermolecular contact between the N- and C-terminal residues in the monomer versus dimer of apoA-IV. a, the monomeric non-MDSA simulated model for full-length apoA-IV is shown in green. The α-carbons of Lys residues 59 and 330 are shown as red spheres and are connected in an intramolecular cross-link as indicated in Table 2. b, the non-MDSA model was used to generate a preliminary model of the dimeric form of full-length apoA-IV as shown with molecule A in green and molecule B in teal. The same Lys residues are shown participating in an intermolecular cross-link.
Functional Predictions from the Models
From the modeling approaches described above, it is evident that the N/C-terminal domains interact and likely fold back to interact with the helical bundle. Further inspection of the bundle revealed that a phenylalanine (Phe223) was positioned near hydrophobic residues known to be involved in triggering the lipid affinity of apoA-IV (24) (see “Discussion” for further details). Speculating that this residue may comprise part of the clasp mechanism, we mutated it to an alanine and then evaluated the effect on the ability of apoA-IV to reorganize DMPC liposomes into lipoproteins. Fig. 10 shows that apoA-I cleared the lipid avidly as demonstrated previously (51), whereas WT apoA-IV did so at a more moderate rate (47). On the other hand, a mutant of apoA-IV (apoA-IV F334A) in which the clasp interaction has been disrupted (24) cleared the lipid nearly as efficiently as apoA-I. Interestingly, apoA-IV(F223A) was significantly more effective than WT apoA-IV and nearly as effective as apoA-IV(F334A), indicating that this mutation may indeed affect the lipid binding clasp interaction. We observed the same effect when Phe223 was mutated to His (not shown). By contrast, other point mutations in the region, such as Phe294, failed to exert significant effects on lipid solubilization (Fig. 10).
FIGURE 10.

Ability of apoA-IV mutants to reorganize DMPC multilamellar liposomes. DMPC liposomes were incubated with at a 1:2 mass ratio of lipid to protein at 24.5 °C for the time indicated. The conversion of the light scattering liposomes to nonscattering lipoproteins was monitored at 325 nm. DMPC liposomes without added protein were included as a negative control, and apoA-I was included as a positive control because of its avid ability to clear lipid. The traces are representative from three separate experimental runs.
DISCUSSION
The remarkable structural plasticity of apolipoproteins is evident in the different forms they adopt. They can exist lipid-free (or poor) in solution where they undergo a concentration-dependent self-association that bears a remarkable similarity to chemical detergents. Upon interaction with lipid, significant structural rearrangements are induced to generate and stabilize lipoprotein particles. These properties have made them notoriously difficult to study by traditional high resolution techniques such as nuclear magnetic resonance and x-ray crystallography. Although moderate success has been achieved with truncated mutants, our structural understanding of these proteins as they exist in lipoproteins and how they transition to this state remains limited. This has required the clever integration of a wide range of complementary structural techniques to understand the structure of the full-length proteins.
Power of Isotope-assisted Cross-linking for Determining Spatial Constraints in Oligomeric Proteins
The recent crystal structures of dimeric apoA-I (23) and apoA-IV (22) fragments have highlighted the remarkable similarities in molecular contacts among the various oligomeric states of apolipoproteins. This may hint that their lipid-bound forms also exhibit similar reciprocating contacts. Therefore, it is critical to develop structural techniques that can distinguish between intra- and intermolecular contacts, even if they occur between exactly the same protein regions.
In earlier work, we proposed a crude way to estimate the molecular span of cross-links called the “oligomer isolation strategy.” This involved setting the cross-linking conditions so that linked monomers and dimers could be separated by gel filtration chromatography and independently analyzed by MS (52). In theory, the dimeric species should contain all intra- and intermolecular cross-links, whereas the monomeric species have only intramolecular links. Those unique to the dimer were interpreted to be intermolecular. This approach was adopted by other laboratories (53, 54) as well. However, this concept suffers from three limitations. First, it is not always possible to completely separate monomers and dimers in the analysis, resulting in uncertainty assigning molecular span (52). Second, it is not possible to distinguish cross-link span in trimers or higher, even if they can be individually isolated. Third, and most importantly, the crystal structures of dimeric apoA-I (23) and apoA-IV (22) show that the same cross-link can appear in both the dimers and monomers but have different molecular spans in each.
The dual isotope labeling approach reported here addresses all these issues. Although we separated monomers and dimers in this study, this is not a requirement. Contributions of mixed systems should be interpretable by comparing the relative intensities of the mixed isotope cross-links compared with the pure 14N and 15N versions. A mixed system of equal monomer and dimer should produce a pattern in which the 14N and 15N peptide ion intensities are X and the mixed peptide ion intensities are 0.5X. A similar argument can be made for evaluating oligomers of higher order than dimer. For example, if all four ion intensities are equal, then all three molecules in a trimer can be interpreted as making intermolecular contacts. However, if the mixed species are some fraction of the 14N and 15N species, one of the molecules may be making different contacts than the other two. Therefore, we anticipate that this technique will be highly useful for studying the structure of oligomeric species of other proteins such as apoA-I as well as the lipid-bound forms of all apolipoproteins. However, because this technique requires purified protein from two different sources, it is most applicable to systems that can (re)assemble postproduction.
Structure of ApoA-IV
We made four major conclusions from the current study. First, the cross-linking pattern from soluble protein in buffer closely matched the predictions of the apoA-IV65–335 crystal structure. This, combined with our previous SAXS work (26), is strong evidence that the crystal structure reflects soluble apoA-IV and the crystallization conditions did not significantly perturb the structure. Second, because the cross-linking was performed on full-length apoA-IV and the crystal structure on a truncation mutant, the addition of the N and C termini does not dramatically affect the structure of the central helical bundle of the protein, again arguing for the relevance of the crystal structure to full-length soluble apoA-IV. Third, the molecular span information from the cross-links strongly support our proposed structure for monomeric apoA-IV. Finally, using the cross-linking/SAXS data and molecular dynamics, we created two new models for monomeric apoA-IV. In both, the hydrophobic components of each termini interact to form a globular domain that appears to sit atop the four-helix bundle.
The best test of a protein structural model is whether it can be used as a basis for making functional predictions. Our previous work has provided significant evidence that sequences near the N terminus (particularly residue 12) and C terminus (residue 334 and possibly residue 335) interact to form a clasp that prevents apoA-IV from unfolding in response to lipid (24, 25, 47). Unfortunately, these residues were not visible in the apoA-IV65–335 crystal structure. Upon inspection of the new full-length models in Figs. 7 and 8, we found that both residues are in proximity in both models (α-carbons within 12.3 Å). Furthermore, a phenylalanine (Phe223) was also clustered near these hydrophobic clasp residues. This residue had previously caught our attention because of its unusual exposure on the surface of the four-helix bundle in the crystal structure. The models suggested that Phe223 may comprise part of the clasp mechanism, possibly acting to keep the globular domain associated with the folded helical bundle. Consistent with this, mutating Phe223 had similar stimulatory effects on apoA-IV lipid binding as Phe334. However, manipulation of another phenylalanine in the same general area failed to alter lipid binding. This may indicate a three-way interaction between the N terminus (residue 12), the C-terminal region (Phe334), and the helical bundle (Phe223) that plays a major role in regulating apoA-IV lipid binding (Fig. 11). When the loosely folded globular domain interacts with lipid, there may be a conformational cascade that results in its dissociation from the helical bundle, allowing it to open and solubilize lipid bilayers more avidly. Based on the similarity of the structures, this process should occur similarly in the monomer or the dimer.
FIGURE 11.
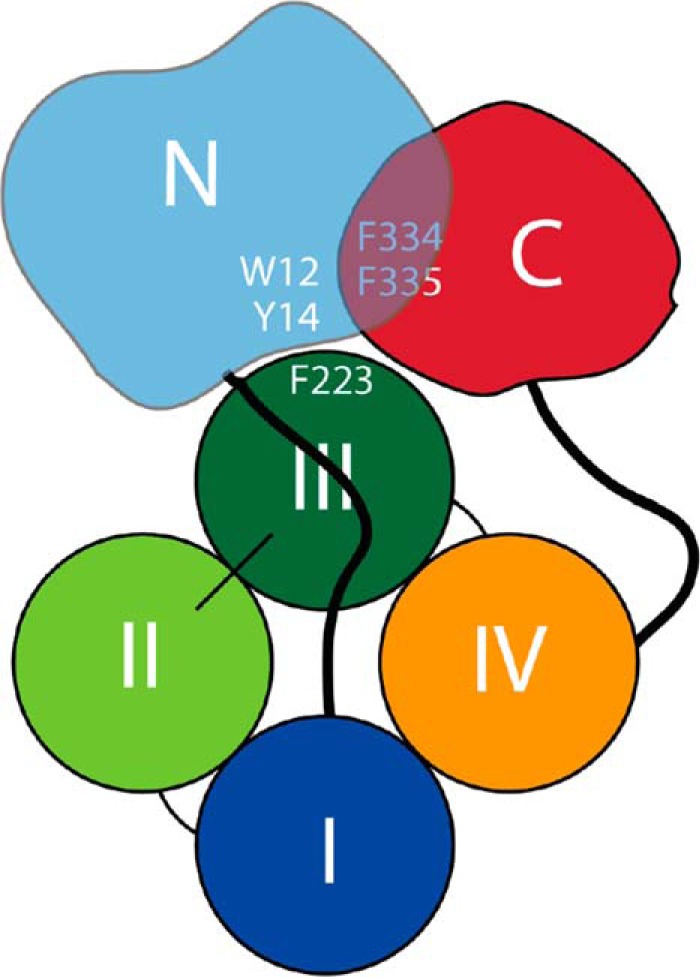
Schematic representation of the domain interaction and participating residues within monomeric apoA-IV. The N-terminal residues (approximately 1–96) are shown in light blue with Trp12 and Tyr14, both previously shown to be required for effective apoA-IV lipid binding, highlighted. The central four-helix bundle domain (approximately residues 97–311) is shown with each helix in a different color and Phe223 (shown in Fig. 10 to be a modulator of lipid binding) highlighted. The C-terminal domain (approximately residues 312–376) is shown in red with Phe334 and Phe335, both previously shown to be critical for apoA-IV lipid binding (47), highlighted.
Another interesting issue relates to the impact of the globular domain on the transition between monomer and dimer. The degree to which the two termini are intertwined may be envisioned to impact the kinetics of the interconversion. Our new models indicate that the N and C termini are more or less independently folded, although each may depend on the other for stabilizing interactions. Thus, the dimer to monomer transition may not require a complex conformational change in this domain. This is supported by our previous studies showing that both the WT apoA-IV and apoA-IV65–335 (lacking the N and C termini) have similar dimer to monomer interconversion rates (22). If the N and C termini interacted in a more integrated or tight fashion, the transition should be slower when the termini are present. However, we caution that the structural resolution of these domains needs to be improved before more definitive conclusions can be made.
As additional data become available, the quality of full-length human apolipoprotein models is consistently improving. The currently reported apoA-IV models are consistent with (i) our partial crystal structure, (ii) SAXS molecular shape data, and (iii) upwards of 50 chemical cross-links. In addition, they are consistent with secondary structure estimates made by circular dichroism. Nevertheless, there remains some uncertainty in the structure of the globular domain. This area is likely dynamic enough that it may not exist in any one structure at a given time. Indeed, this is consistent with our data showing that these domains are the first to be digested under limited proteolysis (30). Although crystallography and nuclear magnetic resonance methods will continue to be pursued, the dynamic nature of these apolipoproteins may preclude complete structural visualization by these methods. Thus, it is important to continue to develop alternative methodologies, such as the isotope assisted cross-linking technique, to meet this goal.
Acknowledgments
We thank the University of Alabama at Birmingham's Academic Computing and Technology resource and the Department of Mechanical Engineering for use of the Cheaha that they jointly maintain.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 GM098458 (to W. S. D. and T. B. T.) and HL67093 (to W. S. D.).
- apo
- apolipoprotein
- DMPC
- dimyristoyl phosphatidylcholine
- MD
- molecular dynamics simulation
- MDSA
- molecular dynamics-simulated annealing
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1. Green P. H., Glickman R. M., Riley J. W., Quinet E. (1980) Human apolipoprotein A-IV. Intestinal origin and distribution in plasma. J. Clin. Invest. 65, 911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayashi H., Nutting D. F., Fujimoto K., Cardelli J. A., Black D., Tso P. (1990) Transport of lipid and apolipoproteins A-I and A-IV in intestinal lymph of the rat. J. Lipid Res. 31, 1613–1625 [PubMed] [Google Scholar]
- 3. Kratz M., Wahrburg U., von Eckardstein A., Ezeh B., Assmann G., Kronenberg F. (2003) Dietary mono- and polyunsaturated fatty acids similarly increase plasma apolipoprotein A-IV concentrations in healthy men and women. J. Nutr. 133, 1821–1825 [DOI] [PubMed] [Google Scholar]
- 4. Weinberg R. B., Anderson R. A., Cook V. R., Emmanuel F., Denefle P., Hermann M., Steinmetz A. (2000) Structure and interfacial properties of chicken apolipoprotein A-IV. J. Lipid Res. 41, 1410–1418 [PubMed] [Google Scholar]
- 5. Weinberg R. B., Cook V. R., DeLozier J. A., Shelness G. S. (2000) Dynamic interfacial properties of human apolipoproteins A-IV and B-17 at the air/water and oil/water interface. J. Lipid Res. 41, 1419–1427 [PubMed] [Google Scholar]
- 6. Gallagher J. W., Weinberg R. B., Shelness G. S. (2004) apoA-IV tagged with the ER retention signal KDEL perturbs the intracellular trafficking and secretion of apoB. J. Lipid Res. 45, 1826–1834 [DOI] [PubMed] [Google Scholar]
- 7. Lu S., Yao Y., Cheng X., Mitchell S., Leng S., Meng S., Gallagher J. W., Shelness G. S., Morris G. S., Mahan J., Frase S., Mansbach C. M., Weinberg R. B., Black D. D. (2006) Overexpression of apolipoprotein A-IV enhances lipid secretion in IPEC-1 cells by increasing chylomicron size. J. Biol. Chem. 281, 3473–3483 [DOI] [PubMed] [Google Scholar]
- 8. Emmanuel F., Steinmetz A., Rosseneu M., Brasseur R., Gosselet N., Attenot F., Cuiné S., Séguret S., Latta M., Fruchart J. C. (1994) Identification of specific amphipathic α-helical sequence of human apolipoprotein A-IV involved in lecithin:cholesterol acyltransferase activation. J. Biol. Chem. 269, 29883–29890 [PubMed] [Google Scholar]
- 9. Duverger N., Ghalim N., Theret N., Fruchart J. C., Castro G. (1994) Lipoproteins containing apolipoprotein A-IV. Composition and relation to cholesterol esterification. Biochim. Biophys. Acta 1211, 23–28 [DOI] [PubMed] [Google Scholar]
- 10. Main L. A., Ohnishi T., Yokoyama S. (1996) Activation of human plasma cholesteryl ester transfer protein by human apolipoprotein A-IV. Biochim. Biophys. Acta 1300, 17–24 [DOI] [PubMed] [Google Scholar]
- 11. Remaley A. T., Stonik J. A., Demosky S. J., Neufeld E. B., Bocharov A. V., Vishnyakova T. G., Eggerman T. L., Patterson A. P., Duverger N. J., Santamarina-Fojo S., Brewer H. B., Jr. (2001) Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 280, 818–823 [DOI] [PubMed] [Google Scholar]
- 12. Qin X., Swertfeger D. K., Zheng S., Hui D. Y., Tso P. (1998) Apolipoprotein AIV. A potent endogenous inhibitor of lipid oxidation. Am. J. Physiol. 274, H1836–H1840 [DOI] [PubMed] [Google Scholar]
- 13. Fujimoto K., Cardelli J. A., Tso P. (1992) Increased apolipoprotein A-IV in rat mesenteric lymph after lipid meal acts as a physiological signal for satiation. Am. J. Physiol. 262, G1002–G1006 [DOI] [PubMed] [Google Scholar]
- 14. Okumura T., Fukagawa K., Tso P., Taylor I. L., Pappas T. N. (1996) Apolipoprotein A-IV acts in the brain to inhibit gastric emptying in the rat. Am. J. Physiol. 270, G49–G53 [DOI] [PubMed] [Google Scholar]
- 15. Raybould H. E., Meyer J. H., Tabrizi Y., Liddle R. A., Tso P. (1998) Inhibition of gastric emptying in response to intestinal lipid is dependent on chylomicron formation. Am. J. Physiol. 274, R1834–R1838 [DOI] [PubMed] [Google Scholar]
- 16. Wang F., Kohan A. B., Kindel T. L., Corbin K. L., Nunemaker C. S., Obici S., Woods S. C., Davidson W. S., Tso P. (2012) Apolipoprotein A-IV improves glucose homeostasis by enhancing insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 109, 9641–9646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karathanasis S. K., Oettgen P., Haddad I. A., Antonarakis S. E. (1986) Structure, evolution, and polymorphisms of the human apolipoprotein A4 gene (APOA4). Proc. Natl. Acad. Sci. U.S.A. 83, 8457–8461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boguski M. S., Elshourbagy N., Taylor J. M., Gordon J. I. (1985) Comparative analysis of repeated sequences in rat apolipoproteins A-I, A-IV, and E. Proc. Natl. Acad. Sci. U.S.A. 82, 992–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elshourbagy N. A., Walker D. W., Paik Y. K., Boguski M. S., Freeman M., Gordon J. I., Taylor J. M. (1987) Structure and expression of the human apolipoprotein A-IV gene. J. Biol. Chem. 262, 7973–7981 [PubMed] [Google Scholar]
- 20. Weinberg R. B. (1994) Identification of functional domains in the plasma apolipoproteins by analysis of inter-species sequence variability. J. Lipid Res. 35, 2212–2222 [PubMed] [Google Scholar]
- 21. Weinberg R. B., Spector M. S. (1985) The self-association of human apolipoprotein A-IV. Evidence for an in vivo circulating dimeric form. J. Biol. Chem. 260, 14279–14286 [PubMed] [Google Scholar]
- 22. Deng X., Morris J., Dressmen J., Tubb M. R., Tso P., Jerome W. G., Davidson W. S., Thompson T. B. (2012) The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure 20, 767–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mei X., Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tubb M. R., Silva D., Gangani R. A., Pearson K. J., Tso P., Liu M., Davidson W. S. (2007) Modulation of apolipoprotein A-IV lipid binding by an interaction between the N and C termini. J. Biol. Chem. 282, 28385–28394 [DOI] [PubMed] [Google Scholar]
- 25. Tubb M. R., Silva R. A., Fang J., Tso P., Davidson W. S. (2008) A three-dimensional homology model of lipid-free apolipoprotein A-IV using cross-linking and mass spectrometry. J. Biol. Chem. 283, 17314–17323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deng X., Morris J., Chaton C., Schröder G. F., Davidson W. S., Thompson T. B. (2013) Small-angle x-ray scattering of apolipoprotein A-IV reveals the importance of its termini for structural stability. J. Biol. Chem. 288, 4854–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silva R. A., Hilliard G. M., Fang J., Macha S., Davidson W. S. (2005) A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry 44, 2759–2769 [DOI] [PubMed] [Google Scholar]
- 28. Huang R., Silva R. A., Jerome W. G., Kontush A., Chapman M. J., Curtiss L. K., Hodges T. J., Davidson W. S. (2011) Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat. Struct. Mol. Biol. 18, 416–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Silva R. A., Schneeweis L. A., Krishnan S. C., Zhang X., Axelsen P. H., Davidson W. S. (2007) The structure of apolipoprotein A-II in discoidal high density lipoproteins. J. Biol. Chem. 282, 9713–9721 [DOI] [PubMed] [Google Scholar]
- 30. Pearson K., Saito H., Woods S. C., Lund-Katz S., Tso P., Phillips M. C., Davidson W. S. (2004) Structure of human apolipoprotein A-IV. A distinct domain architecture among exchangeable apolipoproteins with potential functional implications. Biochemistry 43, 10719–10729 [DOI] [PubMed] [Google Scholar]
- 31. Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E. (1978) A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 87, 206–210 [DOI] [PubMed] [Google Scholar]
- 32. Nema V., Pal S. K. (2013) Exploration of freely available web-interfaces for comparative homology modelling of microbial proteins. Bioinformation 9, 796–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones M. K., Catte A., Patterson J. C., Gu F., Chen J., Li L., Segrest J. P. (2009) Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics. Biophys. J. 96, 354–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 36. Jorgensen W. L., Chandrasekahar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 37. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) CHARMM. A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 [Google Scholar]
- 38. MacKerell A. D., Bashford D., Bellott, Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wirkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 39. Weinkam P., Pons J., Sali A. (2012) Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U.S.A. 109, 4875–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneidman-Duhovny D., Hammel M., Sali A. (2010) FoXS. A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 42. Pelikan M., Hura G. L., Hammel M. (2009) Structure and flexibility within proteins as identified through small angle x-ray scattering. Gen. Physiol. Biophys. 28, 174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tubb M. R., Smith L. E., Davidson W. S. (2009) Purification of recombinant apolipoproteins A-I and A-IV and efficient affinity tag cleavage by tobacco etch virus protease. J. Lipid Res. 50, 1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dvorin E., Mantulin W. W., Rohde M. F., Gotto A. M., Jr., Pownall H. J., Sherrill B. C. (1985) Conformational properties of human and rat apolipoprotein A-IV. J. Lipid Res. 26, 38–46 [PubMed] [Google Scholar]
- 45. Weinberg R. B., Jordan M. K. (1990) Effects of phospholipid on the structure of human apolipoprotein A-IV. J. Biol. Chem. 265, 8081–8086 [PubMed] [Google Scholar]
- 46. Weinberg R. B., Spector M. S. (1985) Structural properties and lipid binding of human apolipoprotein A-IV. J. Biol. Chem. 260, 4914–4921 [PubMed] [Google Scholar]
- 47. Tubb M. R., Silva R. A., Pearson K. J., Tso P., Liu M., Davidson W. S. (2007) Modulation of apolipoprotein A-IV lipid binding by an interaction between the N and C termini. J. Biol. Chem. 282, 28385–28394 [DOI] [PubMed] [Google Scholar]
- 48. Jonas A., Steinmetz A., Churgay L. (1993) The number of amphipathic α-helical segments of apolipoproteins A-I, E, and A-IV determines the size and functional properties of their reconstituted lipoprotein particles. J. Biol. Chem. 268, 1596–1602 [PubMed] [Google Scholar]
- 49. Rambo R. P., Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hammel M. (2012) Validation of macromolecular flexibility in solution by small-angle x-ray scattering (SAXS). Eur. Biophys. J. 41, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Panagotopulos S. E., Witting S. R., Horace E. M., Hui D. Y., Maiorano J. N., Davidson W. S. (2002) The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J. Biol. Chem. 277, 39477–39484 [DOI] [PubMed] [Google Scholar]
- 52. Silva R. A., Hilliard G. M., Li L., Segrest J. P., Davidson W. S. (2005) A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry 44, 8600–8607 [DOI] [PubMed] [Google Scholar]
- 53. Bhat S., Sorci-Thomas M. G., Alexander E. T., Samuel M. P., Thomas M. J. (2005) Intermolecular contact between globular N-terminal fold and C-terminal domain of ApoA-I stabilizes its lipid-bound conformation. Studies employing chemical cross-linking and mass spectrometry. J. Biol. Chem. 280, 33015–33025 [DOI] [PubMed] [Google Scholar]
- 54. Wu Z., Gogonea V., Lee X., May R. P., Pipich V., Wagner M. A., Undurti A., Tallant T. C., Baleanu-Gogonea C., Charlton F., Ioffe A., DiDonato J. A., Rye K. A., Hazen S. L. (2011) The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J. Biol. Chem. 286, 12495–12508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maiti R., Van Domselaar G. H., Zhang H., Wishart D. S. (2004) SuperPose. A simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594 [DOI] [PMC free article] [PubMed] [Google Scholar]