

Background: Whether RIP1 directly contributes to chemotherapy response in cancer has not been determined.
Results: RIP1 knockdown resulted in miR-146a-mediated catalase reduction, ROS induction, IAP degradation, and increased cisplatin cytotoxicity.
Conclusion: RIP1 blunts the anticancer activity of cisplatin by releasing miR-146a-mediated catalase suppression.
Significance: Our results establish a chemoresistant role for RIP1, and intervention within the RIP1-mediated pathway may be exploited for chemosensitization.
Keywords: Apoptosis, Chemoresistance, Lung Cancer, Reactive Oxygen Species (ROS), RIP
Abstract
Although receptor-interacting protein 1 (RIP1) is well known as a key mediator in cell survival and death signaling, whether RIP1 directly contributes to chemotherapy response in cancer has not been determined. In this report, we found that, in human lung cancer cells, knockdown of RIP1 substantially increased cytotoxicity induced by the frontline anticancer therapeutic drug cisplatin, which has been associated with robust cellular reactive oxygen species (ROS) accumulation and enhanced apoptosis. Scavenging ROS dramatically protected RIP1 knockdown cells against cisplatin-induced cytotoxicity. Furthermore, we found that, in RIP1 knockdown cells, the expression of the hydrogen peroxide-reducing enzyme catalase was dramatically reduced, which was associated with increased miR-146a expression. Inhibition of microRNA-146a restored catalase expression, suppressed ROS induction, and protected against cytotoxicity in cisplatin-treated RIP1 knockdown cells, suggesting that RIP1 maintains catalase expression to restrain ROS levels in therapy response in cancer cells. Additionally, cisplatin significantly triggered the proteasomal degradation of cellular inhibitor of apoptosis protein 1 and 2 (c-IAP1 and c-IAP2), and X-linked inhibitor of apoptosis (XIAP) in a ROS-dependent manner, and in RIP1 knockdown cells, ectopic expression of c-IAP2 attenuated cisplatin-induced cytotoxicity. Thus, our results establish a chemoresistant role for RIP1 that maintains inhibitor of apoptosis protein (IAP) expression by release of microRNA-146a-mediated catalase suppression, where intervention within this pathway may be exploited for chemosensitization.
Introduction
Chemotherapy is one of the major means used to reduce cancer mortality and prolong patient survival. However, chemoresistance, either primary or acquired, is a major clinical challenge that diminishes the anticancer efficacy of chemotherapeutic drugs (1). Current anticancer chemotherapeutic drugs suppress cancer growth through the inhibition of cell proliferation and angiogenesis. However, the major anticancer mechanism for directly killing cancer cells is induction of apoptosis (2, 3). In unfavorable microenvironments, cancer cells employ a cell survival and death signaling network for tumor growth (4). Increasing survival and evading apoptosis are recognized as hallmarks of cancer cells (5). Thus, interventions that tip the survival and death balance toward death would sensitize anticancer chemotherapy drugs (4).
In cancer cells, chemotherapeutic drugs also induce the production of reactive oxygen species (ROS).2 As active and toxic molecules, ROS damage cellular components by oxidizing DNA, proteins, and lipids, resulting in cytotoxicity. ROS also serve as mediators in cell death signaling (6–9). ROS are mainly produced in mitochondria and are detoxified in the cell by ROS reductases such as manganese superoxide dismutase for superoxide and catalase for hydrogen peroxide (H2O2). Cancer cells may utilize these endogenous ROS scavengers to attenuate cytotoxicity induced by chemotherapeutic drugs (10). Accordingly, boosting anticancer drug-induced cellular ROS through the targeting of ROS reductases may be exploited for chemosensitization (11).
Receptor-interacting protein 1 (RIP1) is an important signaling factor in survival and death pathways. Initially, RIP1 was found to be required for TNF-α receptor 1 (TNFR1)-mediated NF-κB activation for cell survival. Subsequent studies substantiate RIP1 as a major mediator for cell survival signaling, along with other death receptors (12–15), Toll-like receptors and growth factor receptors (6, 16–19). However, recent studies have revealed a prodeath role for RIP1 under certain conditions. For example, when caspase 8 is suppressed, RIP1 mediates apoptosis or necrosis induced by TNF-α or genotoxic stresses (6, 20–22). Therefore, RIP1 merges signals induced by different stimuli for either cell survival or death. The role of RIP1 in the response to chemotherapy in cancer cells has not been elucidated, and whether RIP1 is involved in chemoresistance is still elusive.
In this report, we provide evidence that, for human lung cancer cells, RIP1 plays a role in survival. RIP1 knockdown enhanced cytotoxicity induced by the frontline therapeutic drug cisplatin, which is associated with increased miRNA-146a, reduced catalase expression, ROS induction, and degradation of antiapoptotic IAP proteins. Thus, our results establish a chemoresistant signaling pathway consisting of RIP1, miR-146a, catalase, and IAPs. Targeting of this pathway may be utilized for chemosensitization.
EXPERIMENTAL PROCEDURES
Reagents
Cisplatin (catalog no. 479306) was from Sigma-Aldrich. Antibodies against caspase 3 (catalog no. 559565), c-IAP2 (catalog no. 552782), and RIP1 (catalog no. 610458) were from BD Biosciences (San Jose, CA). c-IAP1 (catalog no. AF8181) was from R&D Systems (Minneapolis, MN). Anti-XIAP (catalog no. 2042) was purchased from Cell Signaling Technology (Danvers, MA). Anti-poly(ADP-ribose) polymerase (catalog no. ALX-210-302) and the pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-FMK, catalog no. ALX-260-020) were from Enzo Life Sciences Inc. (Ann Arbor, MI). Catalase antibody (catalog no. sc-50508) and input control GAPDH (catalog no. sc-32233) were from Santa Cruz Biotechnology (Dallas, TX), and anti-β-actin (catalog no. A1978) was from Sigma. The inhibitors Q-VD-OPh Non-O-methylate [N-(2-quinolyl)valyl-aspartyl-(2,6-difluorophenoxy)methyl ketone] (catalog no. 551476) and MG-132 (catalog no. 474790) were from EMD Millipore. Chloroquine (CQ, catalog no. C6628), cycloheximide (CHX, catalog no. C1988), and butylated hydroxyanisole (BHA, catalog no. B1253) were from Sigma-Aldrich. N-acetyl-l-cysteine (NAC, catalog no. A7250) was from Sigma-Aldrich. The ROS detection reagent 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), the Amplex Red catalase assay kit (catalog no. A22180), and the superoxide indicator dihydroethidium (catalog no. D11347) were purchased from Life Technologies. The anti-hsa-miRNA-146a-5p miScript miRNA inhibitor targeting miR-146a was from Qiagen (Germantown, MD).
Cell Culture
The lung cancer cell lines A549 and H460 were obtained from the ATCC and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cells were grown under standard incubator conditions at 37 °C with 5% CO2.
Lentivirus Infection and Establishment of Stable Cell Lines
Lentivirus vectors with shRNA against RIP1 and control vectors were purchased from Open Biosystems (Lafayette, CO). The V5-catalase expressing lentiviral vector was from the Biodesign Institute. Viruses were produced and packaged in HEK293T cells following the instructions of the manufacturer. A pLKO.1 backbone harboring the shRNA sequence CCGGAGGTCATGTTCTTTCAGCTTACTCGAGTAAGCTGAAAGAACATGACCTTTTTT (mature sequence AGGTCATGTTCTTTCAGCTTA, catalog no. RHS3979-9569092) was used to establish A549 RIP1 knockdown cell lines. H460 RIP1 knockdown cell lines were created with the pGIPZ vector and the shRNA sequence TGCTGTTGACAGTGAGCGCGCAGTTGATAATGTGCATAAATAGTGAAGCCACAGATGTATTTATGCACATTATCAACTGCTTGCCTACTGCCTCGGA (mature sequence TTATGCACATTATCAACTG, catalog no. RHS4430-98902904). Cells were infected with viruses and selected with 5 μg/ml puromycin. Positive clones were expanded and maintained in medium supplemented with 1 μg/ml puromycin.
Western Blot Analysis and Immunoprecipitation
Cells were treated as indicated in each figure legend and lysed in M2 buffer (20 mm Tris-HCl (pH 7.6), 0.5% Nonidet P-40, 250 mm NaCl, 3 mm EDTA, 3 mm EGTA, 2 mm DTT, 0.5 mm phenylmethylsulfonyl fluoride, 20 mm β-glycerophosphate, 1 mm sodium vanadate, and 1 μg/ml leupeptin). Equal amounts of protein from cell lysates were resolved on 12% or 15% SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and analyzed by Western blot analysis using various antibodies. The proteins were visualized with enhanced chemiluminescence (EMD Millipore) following the instructions of the manufacturer. The results are shown in each figure. For immunoprecipitation, cell extracts were incubated with a c-IAP1 antibody and protein A-agarose beads at 4 °C for 16 h. The beads were washed five times with M2 buffer, and precipitated proteins were detected by Western blot analysis (23).
Cytotoxicity Assay
Cell death was assessed on the basis of the release of lactate dehydrogenase with a cytotoxicity detection kit (Promega (Madison, WI)) using a protocol described previously (24). Cells were seeded in a 48-well plate 1 day before treatment and then treated as indicated in each figure legend. Experiments were repeated at least three times, and representative results are shown in each figure.
Detection of ROS
Cells were treated with cisplatin as shown in the figure legends. CM-H2DCFDA (5 μm) or dihydroethidium (5 μm) was added to the cell culture 30 min before cell harvest. ROS were measured with a fluorescence plate reader. Results were normalized to total protein concentration (25). All experiments were repeated at least three times, and representative results are shown in each figure.
Catalase Activity Detection
Cells were seeded in a 12-well plate and cultured for 24 h before collection. Cells were lysed in M2 buffer without dithiothreitol. Catalase activity was measured using the Amplex Red catalase assay kit following the instructions of the manufacturer. All the experiments were performed in triplicate.
miR-146a Expression Analysis
Total RNA was isolated from cells with TRIzol reagent (Life Technologies). Total RNA (1 μg) was reverse-transcribed with a miScript II RT kit (Qiagen). Quantitative real-time PCR was carried out with an ABI PRISM 7900HT using Power SYBR Green PCR Master Mix (Life Technologies). Experiments were normalized to SNORD44. Data were analyzed as Relative Quantitation (RQ) with respect to a calibrator sample using the 2−ΔΔCT method (25).
Fluorescence Microscopy
A549 cells with stable RIP1 knockdown were transfected overnight with pEGFP and pcDNA or EGFP and c-IAP2 expression plasmids, and then cells were treated with cisplatin (20 μm) for 48 h and examined under a fluorescence microscope. The images shown are representative of three experiments. The percentage of fluorescent cells was calculated.
Statistics
All data were expressed as mean ± S.D. and examined by Student's t test for statistical significance. p < 0.05 was considered statistically significant.
RESULTS
Down-regulation of RIP1 Sensitizes Chemotherapeutic Drug-induced Cytotoxicity
To investigate the role of RIP1 in lung cancer cell response to chemotherapy, we established stable RIP1 knockdown in A549 and H460 cells with transfection of RIP1 shRNA and examined the effect of RIP1 knockdown on drug response (Fig. 1A). Although cisplatin killed both A549 and H460 cells in a dose- and time-dependent manner, RIP1 knockdown substantially increased the cytotoxicity induced by cisplatin (Fig. 1A, data not shown). The increased cisplatin sensitivity in RIP1 knockdown cells was confirmed in vivo in a nude mouse xenograft tumor and therapy model (Fig. 1B). These results demonstrate that down-regulation of RIP1 sensitizes lung cancer cells to chemotherapy in vitro and in vivo.
FIGURE 1.

Down-regulation of RIP1 sensitizes cisplatin-induced cytotoxicity. A, A549 and H460 cells (control and RIP1 stable knockdown) were treated with increasing concentrations of cisplatin (cDDP) for 48 h. Cell death was detected by lactate dehydrogenase release assay. Data are mean ± S.D. *, p < 0.05; **, p < 0.01. Knockdown of RIP1 was confirmed by Western blot analysis, and β-actin was used as an input control. B, A549 control and RIP1 KD1 cells were injected subcutaneously into the flanks of nude mice for the development of xenograft tumors. After tumor development, mice were randomly divided into two groups and subjected to the following treatments by intraperitoneal injection once a week for 5 weeks from day 13: vehicle control, and 6 mg/kg cisplatin. Tumor sizes were measured once or twice a week using calipers and were calculated using the following formula: tumor volume = 0.5 × (length × width2). Tumors were measured from 6–60 days post-injection, and the mean tumor size is shown as mean ± S.D. KD, knockdown. C, cells were untreated or treated with cisplatin (A549, 20 μm; H460, 10 μm for 24 h), and then active caspase 3 (Casp 3) and poly(ADP-ribose) polymerase (PARP) were examined by Western blot analysis. β-Actin was detected as an input control. D, A549 cells (control and RIP1 knockdown) were pretreated with zVAD (10 μm)) for 30 min and then treated with cisplatin (A549, 20 μm; H460 10 μm) for an additional 48 h. Cell death was detected by lactate dehydrogenase assay. Columns shown are mean ± S.D. *, p < 0.05; **, p < 0.01.
RIP knockdown evidently increased apoptosis markers such as those for caspase 3 activation and poly(ADP-ribose) polymerase cleavage (Fig. 1C). Additionally, the pan-caspase inhibitors zVAD-FMK and Q-VD significantly suppressed the potentiation of cell death by RIP1 knockdown (Fig. 1D and data not shown), indicating that RIP1 knockdown potentiated cisplatin-induced apoptosis.
Cisplatin-induced Intracellular ROS Accumulation Contributes to Potentiated Cytotoxicity in RIP1 Knockdown Cells
Because chemotherapeutics induce ROS and excessive ROS are cytotoxic to cells, we examined whether ROS accumulation is involved in the sensitivity difference between control and RIP1 knockdown cells. Staining with dihydroethidium, which mainly detects superoxide anion (25, 26), was not obviously increased (data not shown). In contrast, CM-H2DCFDA, which is mainly oxidized by H2O2 and the hydroxyl radical (25, 26), detected a robust increase in cisplatin-treated A549 and H460 cells (Fig. 2, A and C), suggesting that cisplatin-induced accumulation of ROS was enhanced in RIP1 knockdown cells. Notably, the basal ROS levels were comparable between control and RIP1 knockdown cells (Fig. 2, A and C). Scavenging ROS with BHA and NAC effectively attenuated cisplatin-induced cell death in RIP1 knockdown cells (Fig. 2, B and D).These results suggest that RIP1 suppresses cisplatin-induced cytotoxicity through the inhibition of ROS accumulation.
FIGURE 2.

Cisplatin-induced intracellular ROS accumulation contributes to potentiated cytotoxicity in RIP1 knockdown cells. A and C, cells were treated with cisplatin (cDDP, 20 μm for A549, 10 μm for H460) for 12 h and incubated with CM-H2DCFDA (5 μm) for 30 min before being collected for ROS detection. Data are mean ± S.D. *, p < 0.05; **, p < 0.01. KD, knockdown. B and D, cells were pretreated with the indicated ROS scavengers (BHA, 100 μm; NAC, 3 mm) for 30 min and then treated with cisplatin (20 μm for A549; 10 μm for H460) for another 48 h. Cytotoxicity was detected by lactate dehydrogenase release assay. Data are mean ± S.D. *, p < 0.05; **, p < 0.01.
Reduced Catalase Expression and Activity Is Involved in Cisplatin-induced Cytotoxicity in RIP1 Knockdown Cells
Because ROS are detoxified by endogenous ROS reductases and because catalase eliminates H2O2 and the hydroxyl radical, we then investigated whether this scavenger is involved in cisplatin-induced ROS accumulation. The catalase protein expression level was decreased dramatically in RIP1 knockdown cells (Fig. 3A and B). Consistently, catalase activity in these cells was also decreased significantly (Fig. 3, C and D). Importantly, ectopic expression of catalase suppressed cisplatin-induced ROS accumulation and cytotoxicity (Fig. 3, E–G). Taken together, these results suggest that RIP1 suppresses ROS accumulation induced by cisplatin through maintenance of catalase expression.
FIGURE 3.

Reduced catalase expression and activity is involved in cisplatin-induced cytotoxicity in RIP1 knockdown cells. A and B, catalase expression in the indicated cells was detected by Western blot analysis, and β-actin was detected as an input control. KD, knockdown. C and D, catalase activity of A549 and H460 cells (control and RIP1 knockdown) were detected as described in the text. Data are mean ± S.D. **, p < 0.01. E, cells were infected with control and catalase viruses for 24 h. V5-catalase expression in the indicated cells was detected by Western blot analysis, and β-actin was detected as an input control. F, cells were infected with control and catalase viruses for 24 h, then treated with cisplatin (cDDP, 20 μm) for 12 h, and incubated with CM-H2DCFDA (5 μm) for 30 min before being collected for ROS detection. Data are mean ± S.D. **, p < 0.01. V5-cat, V5-catalase. G, cells were infected with control and catalase viruses for 24 h, treated with cisplatin (20 μm), or left untreated for 48 h. Cell death was detected by lactate dehydrogenase release assay. Data are mean ± S.D. *, p < 0.05; **, p < 0.01.
miR-146a Mediates Catalase Suppression in RIP1 Knockdown Cells
Suppressed catalase expression in RIP1 knockdown cells is unlikely to occur through transcriptional regulation because there were no detectable changes in catalase mRNA levels between control and RIP1 knockdown cells (Fig. 4A). Thus, we focused on posttranscriptional regulation of this protein. The stability of catalase was slightly suppressed when RIP1 was down-regulated (Fig. 4B). However, the synthesis rate of catalase was suppressed substantially in RIP1 knockdown cells (Fig. 4C), suggesting a dual regulation in the expression level of catalase involving protein synthesis and degradation. Because microRNA suppresses gene expression by inhibiting translation, we investigated the potential microRNA-mediated regulation of catalase. In a search with miRWalk, several miRNAs, including miR-146a, were found to be the best hits for potential regulation of catalase. miR-146a, but not other miRNAs, were increased significantly when RIP1 was knocked down (Fig. 4D and data not shown). Suppression of miR-146a expression with a targeted anti-miR clearly restored catalase expression in both A549 and H460 cells (Fig. 4E and data not shown). Importantly, inhibition of miR-146a in RIP1 knockdown cells effectively suppressed cisplatin-induced ROS accumulation and cytotoxicity (Fig. 4, F and G). These results suggest that RIP1 releases the miR-146a-mediated brake on catalase expression to suppress cisplatin-induced ROS accumulation.
FIGURE 4.

miR-146a mediates catalase suppression in RIP1 knockdown cells. A, A549 cells (control and RIP1 knockdown (KD)) were collected for RNA isolation with TRIzol reagent. Catalase and RIP1 mRNA levels were detected by PCR, and β-actin was detected as an input control. B, A549 cells (control and RIP1 knockdown) were treated with cycloheximide (CHX, 10 μg/ml) for the indicated time points. Catalase was detected by Western blot analysis. GAPDH was used as the input control. The intensity of the individual bands was quantified by Quantity One® software and normalized to the corresponding input control (GAPDH) bands. C, A549 (control and RIP1 knockdown) cells were pretreated with cycloheximide (10 μg/ml) for 16 h. Then, the culture medium was refreshed, and cells were treated with the proteasome inhibitor MG-132 (10 μm) and actinomycin D (Act-D,5 μg/ml) for the indicated times. Catalase and β-actin were detected by Western blot analysis. The intensity of the individual bands was quantified as described in A. D, A549 cells (control and RIP1 knockdown) were collected for RNA isolation with TRIzol reagent, and miR-146a expression was detected by quantitative real-time PCR. E, A549 RIP1 knockdown cells were transfected with a negative control (NC) or miR-146a siRNA (10 nm) for 48 h, catalase expression was detected by Western blot analysis, and GAPDH was used as an input control. F, A549 cells were transfected with the indicated siRNA (10 nm) for 24h and then treated with cisplatin (cDDP, 20 μm) for 12 h. Before collection for ROS detection, cells were incubated with CM-H2DCFDA (5 μm) for 30 min. Data are mean ± S.D. *, p < 0.05. G, A549 cells transfected with a negative control or an miR-146a miScript miRNA inhibitor for 24 h, and then the cells were left untreated or treated with cisplatin for an additional 48 h. Cell death was detected by lactate dehydrogenase assay. Data are mean ± S.D. **, p < 0.01.
RIP1 Suppresses Proteasomal Degradation of IAPs Induced by Cisplatin
Because the IAP family proteins are apoptosis inhibitors that are regulated by ROS (27), we then examined whether IAPs are involved in the cisplatin sensitivity increase in RIP1 knockdown cells. Interestingly, although cisplatin caused little change in expression of c-IAP1, c-IAP2, and XIAP in the control cells, it induced a dramatic reduction of these antiapoptotic factors in RIP1 knockdown cells (Fig. 5A). Because IAP proteins are degraded through the proteasome or lysosome pathways (27, 28), we next examined the mechanism for down-regulation of these proteins in RIP1 knockdown cells by cisplatin. The proteasome inhibitor MG-132, but not the lysosome inhibitor chloroquine, effectively restored IAP expression in cisplatin-treated RIP1 knockdown cells (Fig. 5, B and C), suggesting that RIP1 retains IAP expression mainly through suppressing cisplatin-induced proteasomal degradation.
FIGURE 5.
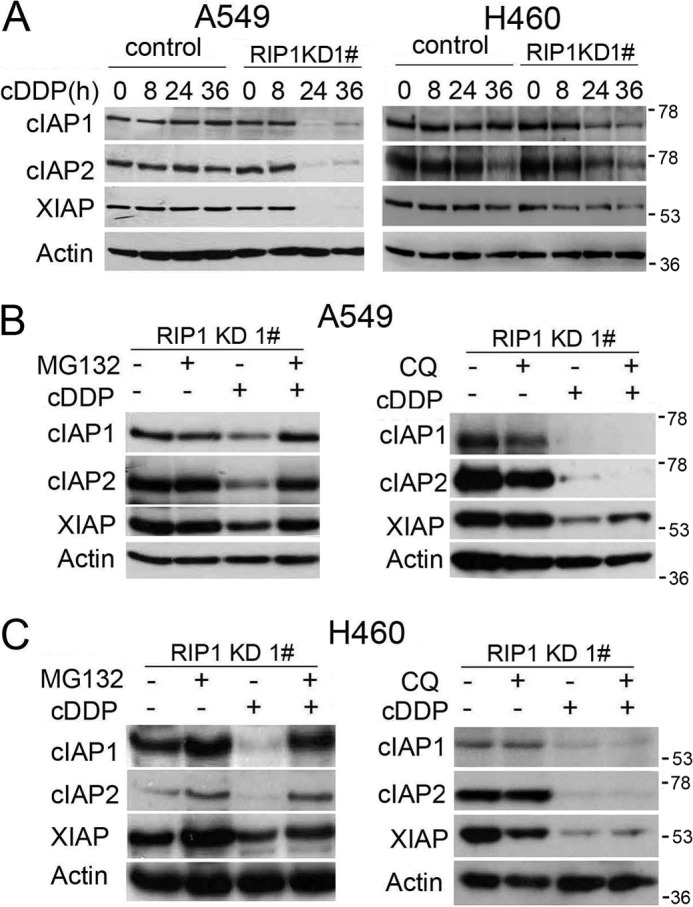
RIP1 suppresses proteasomal degradation of IAPs induced by cisplatin. A, A549 and H460 cells (control and RIP1 knockdown (KD)) were treated with cisplatin (cDDP, 20 μm for A549, 10 μm for H460) or remained untreated for the indicated times. IAPs were detected by Western blot analysis. β-Actin was used as the input control. C, cells were pretreated with the proteasome inhibitor MG-132 (10 μm) or chloroquine (CQ, 20 μm) for 30 min and then treated with cisplatin (cDDP, 20 μm for A549, 10 μm for H460) for 24 h. The indicated proteins were detected by Western blot analysis. β-Actin was used as an input control.
Cisplatin-induced Degradation of IAPs in RIP1 Knockdown Cells Is ROS-dependent
Because ROS is involved in cisplatin-induced apoptosis (Fig. 2, B and D) and IAP expression was reduced in RIP1 knockdown cells exposed to cisplatin (Fig. 5, A and B), we investigated the role of ROS in cisplatin-induced IAP degradation. The ROS scavengers BHA and NAC efficiently attenuated cisplatin-induced IAP reduction in RIP1 knockdown cells (Fig. 6, A and B). These results clearly show that down-regulation of cisplatin-induced IAP degradation was mediated by ROS, which is suppressed by RIP1. Consistent with the role of miR-146a in catalase and ROS regulation, inhibition of this microRNA efficiently suppressed cisplatin-induced IAP reduction in RIP1 knockdown cells (Fig. 6, C and D). Furthermore, cisplatin-induced c-IAP1 ubiquitination was increased in RIP1 knockdown cells (Fig. 6E). Although the MAPK inhibitors had little effect, NAC strongly suppressed cisplatin-induced c-IAP1 ubiquitination (Fig. 6F). These results suggest that suppression of ROS by RIP1 prevents cisplatin-induced IAP ubiquitination, which promotes the stability of these antiapoptotic proteins.
FIGURE 6.

Cisplatin-induced degradation of IAPs in RIP1 knockdown cells is ROS-dependent. A and B, A549 and H460 (control and RIP1 knockdown (KD)) cells were pretreated with the indicated ROS scavengers (BHA, 100 μm; NAC, 3 mm) for 30 min and then treated with cisplatin (cDDP, 20 μm for A549, 10 μm for H460) for another 24 h. The indicated proteins were detected by Western blot analysis, and β-actin and GAPDH were used as input controls. C and D, A549 and H460 (control and RIP1 knockdown) cells were transfected with a negative control (NC) or an miR-146a miScript miRNA inhibitor (10 nm) for 24 h and then treated (20 μm for A549, 10 μm for H460) for another 24 h. The indicated proteins were detected by Western blot analysis, and β-Actin was used as input control. E, A549 (control and RIP1 knockdown) cells were treated with cisplatin (20 μm) for 16 h. The indicated proteins were detected by Western blot analysis after coimmunoprecipitation (IP) with an antibody for cIAP1. β-Actin was used as input control. F, A549 RIP1 knockdown cells were pretreated with the JNK inhibitor SP600125 (SP, 10 μm), the ERK inhibitor U0126 (10 μm), the p38 inhibitor SB203580 (SB, 5 μm), and the ROS scavenger NAC (3 mm) for 30 min and then treated with cisplatin (20 μm) for 16 h. The indicated proteins were detected by Western blot analysis after coimmunoprecipitation with an antibody for cIAP1. β-Actin was detected as an input control.
Ectopic Expression of cIAP2 Protects RIP1 Knockdown Cells from Cisplatin-induced Cytotoxicity
We further examined the role of IAPs in increased cisplatin sensitivity in RIP1 knockdown cells. cIAP2 and EGFP were coexpressed in RIP1 knockdown A549 cells by transfection. EGFP was used as a marker for identification of transfected cells. An empty pCDNA vector served as a negative control. The cells were treated with cisplatin and observed under a fluorescence microscope (Fig. 7B). Although cisplatin killed most cells in the transfected control cells, c-IAP2 transfection attenuated cisplatin-induced cytotoxicity (Fig. 7, B and C). The relatively low level of rescue in cell death by c-IAP2 may be due to degradation of ectopically expressed c-IAP2 protein and the inability to restore other IAPs. Nevertheless, the results clearly suggest that down-regulation of IAPs is involved in increased cisplatin sensitivity in RIP1 knockdown cells.
FIGURE 7.

Ectopic expression of c-IAP2 protects RIP1 knockdown cells from cisplatin-induced cytotoxicity. A, cIAP2 or empty vector pcDNA were coexpressed with the readout marker EGFP inA549 RIP1 knockdown cells by overnight ectopic transfection. Transfection efficiency was confirmed by Western blot analysis with an anti-cIAP2 antibody, and GAPDH was used as an input control. B, cells were treated with cisplatin (cDDP, 20 μm) for 24 h. Photographs were taken under a fluorescence microscope. C, quantification of survival cells with EGFP fluorescence was calculated. Data are mean ± S.D. **, p < 0.01. D, model of the function of RIP1 in cancer cell response to cisplatin.
DISCUSSION
In this report, we present evidence substantiating a cell survival role for RIP1 that contributes to chemoresistance in cancer cells. In human lung cancer cells, RIP1 knockdown significantly increased apoptotic cytotoxicity induced by cisplatin. Cisplatin induced a robust cellular ROS accumulation, and scavenging ROS dramatically attenuated cisplatin-induced cytotoxicity in RIP1 knockdown cells. Increased miR-146a expression and decreased catalase expression were observed when RIP1 was suppressed. Knockdown of miR-146a restored catalase expression, suppressed ROS induction, and protected cytotoxicity in cisplatin-treated RIP1 knockdown cells. Additionally, cisplatin significantly triggered ROS-dependent proteasomal degradation of IAPs, and ectopic expression of c-IAP2 attenuated cisplatin-induced cytotoxicity in RIP1 knockdown cells. These results establish a chemoresistant role for RIP1 that occurs by maintaining IAP expression through releasing miR-146a-mediated catalase suppression that could be exploited for chemosensitization (Fig. 7D).
Because the functions of RIP1 in cell survival regulation are complex, it is important to elucidate the role of this signaling factor in the response of cancer cells, response to chemotherapeutic drugs. In this study, we clearly show a cell survival function for RIP1 in lung cancer cells. RIP1 knockdown significantly increased cisplatin-induced apoptosis and cytotoxicity in human lung cancer cells. These results are in agreement with the observations that RIP1 knockout mouse embryonic fibroblast cells are more susceptible to doxorubicin-induced cytotoxicity (29). However, a prodeath role for RIP1 has also been reported. In exposure of acute lymphoblastic leukemia cells to IAP inhibitors plus anticancer drugs such as AraC, Adriamycin, and taxol, RIP1 is required for inducing cytotoxicity (30). Similarly, suppressing RIP1 attenuated apoptosis caused by the combination of TRAIL receptor 2 antibody (lexatumumab) and IAP inhibitors in rhabdomyosarcoma cells (31). These results suggest that the role of RIP1 in the response of cancer cells to therapy is complex, which may depend on cell context or cytotoxicity inducers.
We further found that induction of cellular ROS is responsible for increased cytotoxicity in RIP1 knockdown cells. Although excessive ROS are cytotoxic, cancer cells develop detoxifying mechanisms to ensure survival and proliferation (32). Up-regulation of the antioxidant capacity to manage oxidative stress confers chemoresistance to cancer cells. Thus, suppressing the antioxidant capacity in cancer cells may be exploited to increase therapeutic efficacy (8). We found that the synthesis rate of catalase was suppressed substantially, whereas the stability of this ROS scavenging protein was decreased slightly within RIP1 knockdown cells, suggesting a dual regulation for catalase expression. This is distinct from the carcinogen benzo(a)pyrene diolepoxide-induced catalase suppression that mainly occurs through proteasomal degradation in immortalized human bronchial epithelial cells (33). These observations suggest that the regulation of catalase expression is complex and, again, may be cell type- or stimulation-specific. Our results show, for the first time, that miR-146a was increased significantly when RIP1 was suppressed and that suppression of miR-146a effectively restored catalase expression, which establishes a novel mechanism whereby RIP1 releases the miR-146a-mediated brake on catalase expression. Suppressing this RIP1-mediated catalase expression pathway may sensitize chemotherapy. Indeed, ectopic catalase expression increases radioresistance in vitro and in vivo (34), and targeting tumor cell-protective catalase has been proposed for anticancer therapy (35).
How RIP1 regulates miR-146a expression is currently elusive. RIP1- mediated pathways may be involved in controlling the expression of miR-146a. However, modulating RIP1-mediated pathways did not obviously impact miR-146a expression (data not shown). Alternatively, as a nuclear protein (12), RIP1 may act as a coactivator for miR-146a transcription. Further investigation is needed to uncover the defined mechanism of this observation.
Our results further suggest that excessive ROS triggers degradation of IAPs. As major apoptosis inhibitors, IAPs have received extensive attention for improving anticancer therapy. Smac mimics that target IAPs for degradation are potential anticancer drugs undergoing preclinical and clinical investigations (36, 37). It is well known that IAPs function upstream of RIP1 for cell survival signaling. For example, because IAPs function as E3 ubiquitin ligases, they are able to directly ubiquitinate RIP1 to mediate NF-κB activation (38, 39). IAPs also restrain RIP1 to suppress the formation of the ripoptosome that is required under certain circumstances for either apoptosis or necrosis (40). In this study, we show evidence that RIP1 works upstream, maintaining expression of the IAP proteins through suppressing proteasomal degradation to cope with genotoxic insults for cell survival. In addition, we found that cisplatin-induced c-IAP1 ubiquitination, which was dependent on ROS, was increased in RIP1 knockdown cells. These results suggest that suppression of ROS by RIP1 is involved in prevention of cisplatin-induced c-IAP1 ubiquitination and degradation. Although further work to elucidate the mechanism by which RIP1-mediated ROS suppression attenuates cisplatin-induced ubiquitination of IAPs for proteasomal degradation is warranted, our results strongly suggest that reciprocal regulation of RIP1 and IAPs play a significant role in cancer cell's resistance to chemotherapy.
In summary, our results establish, for the first time, a chemoresistant role for RIP1, which maintains the expression of IAP through the release of miR-146a-mediated catalase suppression, and intervention targeting this pathway may sensitize anticancer chemotherapy.
This work was supported in part by NIEHS, National Institutes of Health Grant R01ES017328. This work was also supported by Department of Energy Low Dose Radiation Research Program Grant DE-SC0001173 and by National Natural Science Foundation of China Grant 30973403.
- ROS
- reactive oxygen species
- miR
- microRNA
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- EGFP
- enhanced GFP
- c-IAP1
- cellular inhibitor of apoptosis protein 1
- c-IAP2
- cellular inhibitor of apoptosis protein 2
- XIAP
- X-linked inhibitor of apoptosis
- NAC
- N-acetyl-l-cysteine.
REFERENCES
- 1. Chang A. (2011) Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 71, 3–10 [DOI] [PubMed] [Google Scholar]
- 2. Maione P., Rossi A., Sacco P. C., Bareschino M. A., Schettino C., Gridelli C. (2010) Advances in chemotherapy in advanced non-small-cell lung cancer. Expert Opin. Pharmacother. 11, 2997–3007 [DOI] [PubMed] [Google Scholar]
- 3. Ulahannan S. V., Brahmer J. R. (2011) Antiangiogenic agents in combination with chemotherapy in patients with advanced non-small cell lung cancer. Cancer Invest. 29, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer. The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 5. Lin Y., Bai L., Chen W., Xu S. (2010) The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 14, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin Y., Choksi S., Shen H. M., Yang Q. F., Hur G. M., Kim Y. S., Tran J. H., Nedospasov S. A., Liu Z. G. (2004) Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279, 10822–10828 [DOI] [PubMed] [Google Scholar]
- 7. Starkov A. A. (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N.Y. Acad. Sci. 1147, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trachootham D., Alexandre J., Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms. A radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 [DOI] [PubMed] [Google Scholar]
- 9. Bai L., Xu X., Wang Q., Xu S., Ju W., Wang X., Chen W., He W., Tang H., Lin Y. (2012) A superoxide-mediated mitogen-activated protein kinase phosphatase-1 degradation and c-Jun NH(2)-terminal kinase activation pathway for luteolin-induced lung cancer cytotoxicity. Mol. Pharmacol. Drug Discov. 81, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin Y., Shi R., Wang X., Shen H. M. (2008) Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr. Cancer Drug Targets 8, 634–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Yi J. (2008) Cancer cell killing via ROS. To increase or decrease, that is the question. Cancer Biol. Ther. 7, 1875–1884 [DOI] [PubMed] [Google Scholar]
- 12. Festjens N., Vanden Berghe T., Cornelis S., Vandenabeele P. (2007) RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ. 14, 400–410 [DOI] [PubMed] [Google Scholar]
- 13. Kelliher M. A., Grimm S., Ishida Y., Kuo F., Stanger B. Z., Leder P. (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 [DOI] [PubMed] [Google Scholar]
- 14. Ting A. T., Pimentel-Muiños F. X., Seed B. (1996) RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J. 15, 6189–6196 [PMC free article] [PubMed] [Google Scholar]
- 15. Lin Y., Devin A., Cook A., Keane M. M., Kelliher M., Lipkowitz S., Liu Z. G. (2000) The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IκB kinase and c-Jun N-terminal kinase. Mol. Cell. Biol. 20, 6638–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meylan E., Tschopp J. (2005) The RIP kinases. Crucial integrators of cellular stress. Trends Biochem. Sci. 30, 151–159 [DOI] [PubMed] [Google Scholar]
- 17. Kaiser W. J., Offermann M. K. (2005) Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952 [DOI] [PubMed] [Google Scholar]
- 18. Hur G. M., Lewis J., Yang Q., Lin Y., Nakano H., Nedospasov S., Liu Z. G. (2003) The death domain kinase RIP has an essential role in DNA damage-induced NF-κB activation. Genes Dev. 17, 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J., Kanayama A., Yamamoto K., Miyamoto Y. (2012) ARD1 binding to RIP1 mediates doxorubicin-induced NF-κB activation. Biochem. Biophys. Res. Commun. 422, 291–297 [DOI] [PubMed] [Google Scholar]
- 20. Wang L., Du F., Wang X. (2008) TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 [DOI] [PubMed] [Google Scholar]
- 21. Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 [DOI] [PubMed] [Google Scholar]
- 22. Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F., Zachariou A., Lopez J., MacFarlane M., Cain K., Meier P. (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 [DOI] [PubMed] [Google Scholar]
- 23. Bai L., Chen W., Chen W., Wang X., Tang H., Lin Y. (2009) IKKβ-mediated nuclear factor-κB activation attenuates Smac mimetic-induced apoptosis in cancer cells. Mol. Cancer Ther. 8, 1636–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X., Chen W., Lin Y. (2007) Sensitization of TNF-induced cytotoxicity in lung cancer cells by concurrent suppression of the NF-κB and Akt pathways. Biochem. Biophys. Res. Commun. 355, 807–812 [DOI] [PubMed] [Google Scholar]
- 25. Ju W., Wang X., Shi H., Chen W., Belinsky S. A., Lin Y. (2007) A critical role of luteolin-induced reactive oxygen species in blockage of tumor necrosis factor-activated nuclear factor-κB pathway and sensitization of apoptosis in lung cancer cells. Mol. Pharmacol. 71, 1381–1388 [DOI] [PubMed] [Google Scholar]
- 26. Sakon S., Xue X., Takekawa M., Sasazuki T., Okazaki T., Kojima Y., Piao J. H., Yagita H., Okumura K., Doi T., Nakano H. (2003) NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 22, 3898–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang L., Wang Q., Li D., Zhou Y., Zheng X., Sun H., Yan J., Zhang L., Lin Y., Wang X. (2013) Wogonin enhances antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo through ROS-mediated downregulation of cFLIPL and IAP proteins. Apoptosis 18, 618–626 [DOI] [PubMed] [Google Scholar]
- 28. He W., Wang Q., Xu J., Xu X., Padilla M. T., Ren G., Gou X., Lin Y. (2012) Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 8, 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Y., Xia F., Hermance N., Mabb A., Simonson S., Morrissey S., Gandhi P., Munson M., Miyamoto S., Kelliher M. A. (2011) A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 31, 2774–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Löder S., Fakler M., Schoeneberger H., Cristofanon S., Leibacher J., Vanlangenakker N., Bertrand M. J., Vandenabeele P., Jeremias I., Debatin K. M., Fulda S. (2012) RIP1 is required for IAP inhibitor-mediated sensitization of childhood acute leukemia cells to chemotherapy-induced apoptosis. Leukemia 26, 1020–1029 [DOI] [PubMed] [Google Scholar]
- 31. Basit F., Humphreys R., Fulda S. (2012) RIP1 protein-dependent assembly of a cytosolic cell death complex is required for inhibitor of apoptosis (IAP) inhibitor-mediated sensitization to lexatumumab-induced apoptosis. J. Biol. Chem. 287, 38767–38777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caputo F., Vegliante R., Ghibelli L. (2012) Redox modulation of the DNA damage response. Biochem. Pharmacol. 84, 1292–1306 [DOI] [PubMed] [Google Scholar]
- 33. Wang Q., Chen W., Xu X., Li B., He W., Padilla M. T., Jang J. H., Nyunoya T., Amin S., Wang X., Lin Y. (2013) RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis 34, 2119–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Epperly M. W., Melendez J. A., Zhang X., Nie S., Pearce L., Peterson J., Franicola D., Dixon T., Greenberger B. A., Komanduri P., Wang H., Greenberger J. S. (2009) Mitochondrial targeting of a catalase transgene product by plasmid liposomes increases radioresistance in vitro and in vivo. Radiat. Res. 171, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bauer G. (2012) Tumor cell-protective catalase as a novel target for rational therapeutic approaches based on specific intercellular ROS signaling. Anticancer Res. 32, 2599–2624 [PubMed] [Google Scholar]
- 36. LaCasse E. C., Mahoney D. J., Cheung H. H., Plenchette S., Baird S., Korneluk R. G. (2008) IAP-targeted therapies for cancer. Oncogene 27, 6252–6275 [DOI] [PubMed] [Google Scholar]
- 37. Krepler C., Chunduru S. K., Halloran M. B., He X., Xiao M., Vultur A., Villanueva J., Mitsuuchi Y., Neiman E. M., Benetatos C., Nathanson K. L., Amaravadi R. K., Pehamberger H., McKinlay M., Herlyn M. (2013) The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin. Cancer Res. 19, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dynek J. N., Goncharov T., Dueber E. C., Fedorova A. V., Izrael-Tomasevic A., Phu L., Helgason E., Fairbrother W. J., Deshayes K., Kirkpatrick D. S., Vucic D. (2010) c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Varfolomeev E., Goncharov T., Fedorova A. V., Dynek J. N., Zobel K., Deshayes K., Fairbrother W. J., Vucic D. (2008) c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vanlangenakker N., Vanden Berghe T., Vandenabeele P. (2012) Many stimuli pull the necrotic trigger. An overview. Cell Death Differ. 19, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]