

Background: Mimosine is a cell synchronization reagent used for arresting cells in late G1 and S phases.
Results: Replication fork assembly is reversibly blocked by ATM activation through mimosine-generated reactive oxygen species.
Conclusion: Mimosine induces cell cycle arrest strictly at the G1-S phase boundary, which prevents replication fork stalling-induced DNA damage.
Significance: These findings provide a novel mechanism of the mimosine-induced G1 checkpoint.
Keywords: Cell Cycle, Checkpoint Control, DNA Damage Response, DNA Replication, Oxidative Stress
Abstract
Mimosine is an effective cell synchronization reagent used for arresting cells in late G1 phase. However, the mechanism underlying mimosine-induced G1 cell cycle arrest remains unclear. Using highly synchronous cell populations, we show here that mimosine blocks S phase entry through ATM activation. HeLa S3 cells are exposed to thymidine for 15 h, released for 9 h by washing out the thymidine, and subsequently treated with 1 mm mimosine for a further 15 h (thymidine → mimosine). In contrast to thymidine-induced S phase arrest, mimosine treatment synchronizes >90% of cells at the G1-S phase boundary by inhibiting the transition of the prereplication complex to the preinitiation complex. Mimosine treatment activates ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related (ATR)-mediated checkpoint signaling without inducing DNA damage. Inhibition of ATM activity is found to induce mimosine-arrested cells to enter S phase. In addition, ATM activation by mimosine treatment is mediated by reactive oxygen species (ROS). These results suggest that, upon mimosine treatment, ATM blocks S phase entry in response to ROS, which prevents replication fork stalling-induced DNA damage.
Introduction
The transition from G1 to S phase of the cell cycle involves dynamic changes in protein complexes. The prereplication complex (pre-RC)3 is assembled on the replication origin during G1 phase. Pre-RC assembly is mediated by the replication licensing factor Cdt1. Cdt1 plays a role in loading the minichromosome maintenance (MCM) family of proteins onto the replication origin. In S phase, activation of the pre-RC triggers the unwinding of DNA, and a bidirectional replication fork is formed. Proliferating cell nuclear antigen (PCNA) binds to the replication fork, which leads to association with replicative polymerases (1, 2).
Excess thymidine is widely used for cell synchronization in G1/S phase (3). Mimosine, a rare plant amino acid derivative isolated from Leucaena seeds, is also used for cell synchronization in late G1 phase by preventing the formation of replication forks (4, 5). Mimosine has two modes of action in the cell cycle. Elongation of DNA replication is blocked at low concentrations (enrichment of cells in S phase), and entry into S phase is blocked at high concentrations (late G1 phase arrest) (5, 6). However, the mechanism underlying mimosine-induced late G1 phase arrest still remains unclear.
Mimosine is known to function as an iron chelator and inhibits the activity of ribonucleotide reductase (RNR) (7, 8). RNR inhibitors, such as hydroxyurea, block the elongation step of DNA replication and cause replication fork stalling, which results in S phase arrest (9). If mimosine inhibited DNA synthesis only through impairing the activity of RNR, the cell cycle would be arrested just in S phase. However, RNR inhibition cannot explain the effect of mimosine on late G1 phase arrest.
In this study, we examine the mechanism of mimosine-induced G1 phase arrest using highly effective cell synchronization methods. We show that ATM-mediated cell cycle checkpoint signaling blocks the activation of the pre-RC upon mimosine treatment. Moreover, we show that the activation of ATM upon mimosine treatment is induced in response to ROS-mediated hypoxic stress without DNA damage. These results suggest that mimosine treatment blocks S phase entry through ATM activation.
EXPERIMENTAL PROCEDURES
Chemicals
Mimosine (Sigma-Aldrich) was dissolved in 20 mm HEPES (pH 7.3). Thymidine, caffeine, and NAC (Wako Pure Chemical Industries, Osaka) were dissolved in MilliQ water. The pH of the NAC solution was adjusted to 7.0 before addition to the cells (10). Adriamycin (Sigma-Aldrich), microcystin-LR (Wako Pure Chemical Industries), and KU-55933 (Abcam) were dissolved in dimethyl sulfoxide.
Plasmids
The following plasmids were purchased from Addgene: pcDNA3.1(+)FLAG-His-ATM WT (Addgene plasmid 31985) and pcDNA3.1(+)FLAG-His-ATM kd (Addgene plasmid 31986).
Cells and Transfection
HeLa S3 (Japanese Collection of Research Bioresources, Osaka) and COS-1 cells were cultured in Iscove's modified Dulbecco's medium supplemented with 5% bovine serum. Cells were transiently transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen).
Cell Synchronization
To synchronize HeLa S3 cells in G1/S phase, cells were incubated with 0.5∼1 mm mimosine or 4 mm thymidine for 24 h. To release cells from synchronization, cells were washed with PBS and cultured in prewarmed, drug-free, fresh medium for the indicated times. For “thymidine → mimosine” synchronization, HeLa S3 cells were incubated with 4 mm thymidine for 15 h. After release for 9 h, cells were incubated with 1 mm mimosine for a further 15 h. “Thymidine → thymidine” synchronization (double thymidine block) was performed as described previously (11).
Antibodies
The following antibodies were used. PCNA (PC10), cyclin E (HE-12), Cdc45 (H-300), MCM3 (N-19), Cdt1 (H-300), lamin A/C (N-18), ATM (2C-1), and ATR (N-19) were purchased from Santa Cruz Biotechnology. Phospho-Ser-1981 ATM (10H11.E12), phospho-Thr-68 Chk2, Chk1 (DCS310), phospho-Ser-317 Chk1, phospho-Ser-345 Chk1 (133D3), and phospho-histone H2A.x (γH2AX, Ser-139, 20E3) were from Cell Signaling Technology. MCM2 and HIF-1α were from BD Biosciences. Phospho-Ser-41 MCM2, Chk2 (DCS273), replication protein A (NA19L), FLAG (polyclonal antibody), and actin (clone C4) were from Abcam, Medical and Biological Laboratories, Calbiochem, Sigma-Aldrich, and Chemicon International, respectively. HRP-conjugated F(ab′)2 fragments of anti-mouse IgG antibody, anti-rabbit IgG antibody, and anti-goat IgG antibody were from Amersham Biosciences. Alexa Fluor 488 anti-mouse IgG, Alexa Fluor 488 anti-rabbit IgG, Alexa Fluor 488 anti-goat IgG, and Alexa Fluor 647 anti-mouse IgG secondary antibodies were from BioSource International, Sigma-Aldrich, and Invitrogen, respectively.
Flow Cytometry
For cell cycle analysis, cells detached by trypsinization were fixed in 1.5% paraformaldehyde for 1 h and permeabilized with 70% ethanol for at least 1 h at −30 °C (11–13). For DNA staining, cells were treated with 200 μg/ml RNase A and 50 μg/ml propidium iodide at 37 °C for 30 min. A minimum of 10,000 cells/sample was analyzed by flow cytometry using a MoFlo cell sorter (Beckman Coulter) equipped with a 488-nm argon laser or a Guava easyCyte (Millipore) equipped with a 488-nm blue laser and a 640-nm red laser using liner amplification. Data acquired with Guava easyCyte were analyzed using Flowing Software version 2.5.0 (Perttu Terho, Centre for Biotechnology, Turku, Finland). Cell debris was excluded by gating on forward scatter and pulse width profiles.
Immunofluorescence
Confocal and Nomarski-differential interference contrast (DIC) images were obtained using a Fluoview FV500 confocal laser scanning microscope with a 60 × 1.00 water immersion objective (Olympus, Tokyo). One planar (xy) section slice was shown in all experiments. Immunofluorescence staining was performed as described (14–21). In brief, cells were washed with warmed PBS and fixed in 100% methanol for 5 min at −20 °C. For staining of γH2AX and phospho-ATM (Ser-1981), cells were fixed in PBS containing 4% paraformaldehyde and 20% methanol for 20 min and permeabilized in 100% methanol at −20 °C for 1 min (13). Fixed cells were permeabilized and blocked in PBS containing 0.1% saponin and 3% bovine serum albumin for 40 min and then incubated with a primary and secondary antibody for 1 h each. For DNA staining, cells were subsequently treated with 200 μg/ml RNase A and 20 μg/ml propidium iodide for 30 min. After washing with PBS containing 0.1% saponin, cells were mounted with ProLong antifade reagent (Molecular Probes). Composite figures were prepared using Photoshop 13.0 and Illustrator 16.0 (Adobe). For quantitation of PCNA, γH2AX, and RPA staining, fluorescence intensities of immunostaining were measured using ImageJ software. Results are presented as box plots, where the boxes represent the 25th to 75th percentiles. Numbers in parentheses and the lines within the boxes represent the median values, and lines outside of the boxes represent the minimum and maximum values. Because two or three independent experiments gave similar results, a representative experiment was shown in most experiments.
Western Blotting
Cell lysates were prepared in SDS-PAGE sample buffer, subjected to SDS-PAGE, and electrotransferred onto PVDF (Millipore). The Ser/Thr phosphatase inhibitor microcystin-LR (22) dissolved in SDS-PAGE sample buffer at 500 nm was used (Figs. 4C and 8, B and C). Immunodetection was performed by ECL (Amersham Biosciences) as described previously (18, 23–29). Sequential reprobing of membranes with a variety of antibodies was performed after inactivation of HRP by 0.1% NaN3, according to the instructions of the manufacturer. Results were analyzed using a LAS-1000plus image analyzer equipped with Science Lab software (Fujifilm, Tokyo) or a ChemiDoc XRSPlus analyzer (Bio-Rad) and quantitated with Quantity one software (Bio-Rad). Composite figures were prepared using Photoshop 13.0 and Illustrator 16.0 (Adobe).
FIGURE 4.

Decrease in the levels of γH2AX upon mimosine synchronization. HeLa S3 cells were treated with 20 ng/ml Adriamycin for 24 h (positive control) or synchronized by the indicated methods. A and B, cells were fixed and stained for γH2AX. Right panels, mean fluorescence intensities of γH2AX immunostaining in the nucleus were measured. DIC, Differential-interference-contrast; NS, not significant. Scale bars = 10 μm. C, whole cell lysates were analyzed by Western blotting. The results (percent) represent the mean ± S.D. from four independent experiments (right panel). **, p < 0.01; ***, p < 0.001; calculated by Student's t test.
FIGURE 8.

ATM activation through ROS-mediated hypoxic stress. A, HeLa S3 cells treated with 150 μm CoCl2 for 24 h (positive control) or cells synchronized by the indicated methods were analyzed by Western blotting. B, Thy → Mimo-synchronized cells released for the indicated times were analyzed by Western blotting. C, cells treated with 100 ng/ml Adriamycin for 1 h in the presence or absence of 50 mm NAC and cells synchronized by treatment with Thy → Mimo in the presence or absence of 50 mm NAC during mimosine treatment were analyzed by Western blotting. The results (percent) represent the mean ± S.D. from four independent experiments. *, p < 0.05; ***, p < 0.001; calculated by Student's t test. D, cells were analyzed for DNA content by flow cytometry. Shaded histograms represent DNA content of asynchronous cells. Right panel, overlay histogram of cells synchronized by Thy → Mimo in the presence (solid line) and absence (dotted line) of NAC during mimosine treatment. Asy, asynchronous cells.
Subcellular Fractionation
Cell pellets were washed with PBS and extracted in CSK buffer (10 mm PIPES (pH 6.8), 100 mm NaCl, 300 mm sucrose, 3 mm MgCl2, 50 mm NaF, 10 mm β-glycerophosphate, 10 mm Na3VO4, 50 μg/ml aprotinin, 100 μm leupeptin, 25 μm pepstatin A, and 2 mm PMSF) containing 0.5% Triton X-100 for 20 min on ice and then subjected to centrifugation at 2000 × g for 5 min. The insoluble material was washed once with CSK buffer. For Western blot analysis, the pellet was dissolved in SDS-PAGE sample buffer as the Triton-insoluble fraction. For flow cytometry analysis, the cell pellet was fixed in 1.5% paraformaldehyde on ice for 1 h and then permeabilized with 70% ethanol for at least 1 h at −30 °C. For microscopic analysis, cells were extracted in CSK buffer containing 0.1% Triton X-100 for 3 min at 4 °C and subsequently fixed in 4% paraformaldehyde (16, 17).
Measurement of DNA Replication by BrdU Incorporation
Measurement of incorporation of BrdU (Roche) into the genomic DNA was performed as described previously (30). In brief, HeLa S3 cells were incubated with 10 μm BrdU for 30 min before fixation, fixed with ethanol and 50 mm glycine (pH 2.0) for 45 min at room temperature, denatured in 4 n HCl for 15 min, and reacted with FITC-conjugated anti-BrdU antibody (1:5) according to the instructions of the manufacturer. Cells were treated with 200 μg/ml RNase A and 50 μg/ml propidium iodide at 37 °C for 30 min to stain DNA and analyzed by flow cytometry. For microscopic analysis, anti-BrdU antibody (B44, BD Biosciences) and Alexa Fluor 488 anti-mouse IgG were used.
RNA Interference
Knockdown of ATM was performed by shRNA for silencing ATM (GCACCAGTCCAGTATTGGCTT and GGATTTGCGTATTACTCAG) (11). The nucleotides for shRNA were annealed and subcloned into the SpeI site of the pEBmulti-Neo vector (Wako Pure Chemical Industries). To generate stable cell lines for shATM expression, HeLa S3 cells were transfected with pEBmulti-Neo/shATM and selected in 350 μg/ml G418.
RESULTS
Enrichment of Cells Having G1 Phase DNA Content upon Mimosine Synchronization
To examine the effect of mimosine on the cell cycle, asynchronous cells were treated with mimosine and analyzed for DNA content by flow cytometry. We used HeLa S3, a HeLa variant cell line that can be highly synchronized in various stages of the cell cycle (26, 31–33). Treatment with mimosine at a concentration of 0.5 mm for 24 h synchronized only 55% of the cells in G1 phase and the rest in S phase (33%) and G2/M phase (13%) (Fig. 1A). Then, we tested whether cells were highly synchronized in G1 phase upon treatment with a high concentration of mimosine. Treatment with mimosine at 1 mm for 24 h synchronized 78% of the cells in G1 phase (Fig. 1B, Mimosine). These results are consistent with previous studies showing that mimosine has two modes of action in the cell cycle: enrichment of cells in both S phase and G1 phase (5, 6). In this study, we used mimosine at 1 mm for synchronizing cells in G1 phase.
FIGURE 1.

Enrichment of cells in G1 phase using mimosine. A and B, HeLa S3 cells treated for 24 h with 0.5 mm mimosine (A) and with 1 mm mimosine or 4 mm thymidine (B) were released for the indicated times and analyzed for DNA content by flow cytometry. Percentages of cells with G1, S, and G2/M DNA content are shown. The results (percent) represent the mean ± S.D. from three independent experiments. The shaded histograms represent DNA content of asynchronous cells. B, right panel, overlay histogram of cells treated with 1 mm mimosine (solid line) and 4 mm thymidine (dotted line). C, schematic depiction of the two synchronization methods. Cells were treated with 4 mm thymidine for 15 h, washed with PBS, and incubated in drug-free medium for 9 h. Then, cells were incubated for a further 15 h with 1 mm mimosine (i) or 4 mm thymidine (ii). D and E, cells were synchronized by treatment with Thy → Mimo or Thy → Thy, released for the indicated times, and analyzed for DNA content by flow cytometry. Right panel, overlay histogram of cells treated with Thy → Mimo (solid line) and Thy → Thy (dotted line). Shaded histograms represent DNA contents of asynchronous cells (D) or Thy → Mimo-synchronized cells (release 0 h) (E).
Mimosine treatment at 1 mm for 24 h synchronized cells as effectively as thymidine treatment at 4 mm for 24 h (Fig. 1B, Thymidine). Although apoptotic cells (sub-G1 population) were not seen upon either mimosine or thymidine treatment, the majority of mimosine-synchronized cells had slightly less DNA content than thymidine-synchronized cells (Fig. 1B, right panel). These results suggest that treatment with 1 mm mimosine synchronizes cells in an earlier phase of the cell cycle than the phase in which cells are synchronized by treatment with 4 mm thymidine.
Thymidine → thymidine synchronization (double thymidine block, “Thy → Thy”) is an efficient synchronization method in which cells are treated twice with thymidine (3) (Fig. 1, C and D). We tested whether sequential treatment with mimosine after single thymidine treatment could improve the enrichment of cells in G1 phase. As described in Fig. 1C, HeLa S3 cells were cultured in the presence of 4 mm thymidine for 15 h. The cells were subsequently cultured in thymidine-free medium for 9 h, which allows cells to enter G2/M phase, and then cells were treated with 1 mm mimosine for a further 15 h. We found that thymidine → mimosine (“Thy → Mimo”) treatment increased populations in G1 phase in excess of 90% and drastically decreased populations in S phase (3%) and G2/M phase (2%). The majority of cells increased DNA content 2 h after release from mimosine treatment, subsequently progressed through S phase, and reached G2/M phase at 6 h (Fig. 1, D and E). These results suggest that Thy → Mimo treatment yields highly synchronized cell populations having G1 phase DNA content. After release from Thy → Mimo treatment, the cell populations can progress through the cell cycle in a highly synchronous manner.
Treatment with Thy → Thy efficiently synchronized cells in G1/S phase (Fig. 1D, Thymidine → Thymidine). However, the main peak in the DNA histogram of Thy → Thy-synchronized cells showed a slight shift toward S phase compared with the G1 peaks in the DNA histograms of asynchronous cells (Fig. 1D, Thymidine → Thymidine, 0 h) and Thy → Mimo-synchronized cells (Fig. 1D, right panel). In addition, cells released from Thy → Thy treatment increased DNA contents earlier than cells released from Thy → Mimo treatment (Fig. 1, D (1–3 h) and E). These results suggest that thymidine-synchronized cells are arrested at a point immediately after initiation of DNA synthesis. Given that cells released from mimosine treatment show a delay in initiating DNA synthesis, we can assume that the components required to initiate DNA synthesis are not completely loaded onto the replication origin in mimosine-synchronized cells.
Mimosine-induced Synchronization at the G1/S Phase Boundary
To precisely determine the phase of the cell cycle in which cells were synchronized by mimosine treatment, we asked whether replication origins were activated in mimosine-synchronized cells. First, we analyzed the levels of cyclin E, one of the regulatory subunits of S phase Cdk, because the protein level of cyclin E increases at the G1/S phase border (34, 35). The protein levels of cyclin E were low in cells treated with mimosine at 1 mm for 24 h, whereas they were high in cells treated with thymidine at 4 mm for 24 h (Fig. 2A). After release from mimosine treatment, cells showed increased levels of cyclin E. Western blot analysis showed that the protein levels of cyclin E in Thy → Mimo-synchronized cells were as low as those in asynchronous cells and G1 phase cells (14-h release from Thy → Thy treatment). In sharp contrast, Thy → Thy-synchronized cells showed high protein levels of cyclin E (Fig. 2B). Next, we examined the formation of PCNA foci on chromatin. PCNA is known to form chromatin-bound foci in early S phase, which reflects the formation of the replication fork (2, 36, 37). Quantitative analysis showed that cells treated with mimosine at 1 mm for 24 h did not form PCNA foci on chromatin, whereas cells treated with thymidine at 4 mm for 24 h formed PCNA foci. 1 h after release from mimosine treatment, the intensities of PCNA foci on chromatin were increased (Fig. 2C). To further ascertain the absence of PCNA foci in mimosine-synchronized cells, we examined the formation of PCNA foci in highly synchronized cell populations yielded by Thy → Mimo or Thy → Thy synchronization. Thy → Mimo-synchronized cells did not form PCNA foci on chromatin, whereas Thy → Thy-synchronized cells formed PCNA foci. 1 h after release from Thy → Mimo treatment, the intensities of PCNA foci on chromatin were increased (Fig. 2D). These results suggest that mimosine treatment synchronizes cells prior to replication fork assembly, whereas thymidine treatment arrests cells after the establishment of replication forks. Considering that the levels of PCNA foci were increased rapidly after release of cells from mimosine treatment (Fig. 2, C and D), these results also suggest that mimosine-synchronized cells are arrested immediately before replication fork assembly.
FIGURE 2.

Mimosine-induced synchronization at the G1/S phase boundary. HeLa S3 cells were synchronized by the indicated methods and released for the indicated times. A, cells were fixed in methanol and stained for cyclin E. DIC, differential interference contrast. Bottom panel, cell lysates were analyzed by Western blotting. Asy, asynchronous cells. Scale bars = 10 μm. B, cell lysates were analyzed by Western blotting. G1 phase cells were prepared by 14-h release from Thy → Thy treatment. The results (percent) represent the mean ± S.D. from three independent experiments (bottom panel). C and D, the Triton-resistant fraction was fixed and stained for PCNA and DNA. Right panels, mean fluorescence intensities of PCNA immunostaining in the nucleus were plotted. ***, p < 0.001, calculated by Student's t test.
It is known that pre-RC assembly, which occurs during G1 phase, is mediated by the replication licensing factor Cdt1. Cdt1 associates with the replication origin during G1 phase, which leads to the loading of the MCM complex onto chromatin. Cdt1 predominantly localizes to the nucleus in G1 phase because Cdt1 is degraded at the end of G1 and early S phase (38, 39). Thus, we compared pre-RC assembly between mimosine-synchronized cells and thymidine-synchronized cells. The levels of chromatin-bound MCM3, one of the components of the MCM complex, in Thy → Mimo-synchronized cells were as high as those in Thy → Thy-synchronized cells (Fig. 3A). Intriguingly, we found that Cdt1 was degraded in Thy → Mimo-synchronized cells (Fig. 3B). These results suggest that pre-RC assembly is completed in mimosine-synchronized cells.
FIGURE 3.

Inhibition of the activation of the pre-RC upon mimosine synchronization. HeLa S3 cells were synchronized by the indicated methods. A, the Triton-resistant fraction was fixed and stained for MCM3, PCNA, and DNA. Right panels, the Triton-resistant fraction was analyzed by flow cytometry. PCNA and MCM3 levels (y axis) are plotted against DNA contents (x axis). PCNA-positive or -negative cells (boxed areas) or MCM3-positive cells (boxed areas) are shown as percentages. DNA histograms are shown at the top. Asy, asynchronous cells; DIC, differential interference contrast. Scale bars = 10 μm. B, cells were fixed in methanol and stained for Cdt1, PCNA, and DNA. Right panel, whole cell lysates were analyzed by Western blotting. Scale bars = 10 μm. C and D, the Triton-resistant fraction was analyzed by Western blotting.
Cdc45, one of the pre-RC-activating factors, is loaded onto the pre-RC following phosphorylation of the MCM complex by the S phase kinases Cdk2 and Cdc7, and this event is required for the transition of the pre-RC to the preinitiation complex (2, 40). Because the behaviors of cyclin E, PCNA, MCM3, and Cdt1 (Figs. 2 and 3, A and B) suggest that mimosine treatment synchronizes cells at the G1/S phase boundary by preventing the activation of the pre-RC, we examined Cdc45 loading onto chromatin in mimosine-synchronized cells. Western blot analysis showed that Cdc45 was not loaded onto chromatin in Thy → Mimo-synchronized cells, whereas Thy → Thy-synchronized cells had high levels of Cdc45 on chromatin (Fig. 3C). In addition, Thy → Mimo-synchronized cells had low levels of phosphorylation of MCM2 at Ser-41, one of the phosphorylation sites by Cdk2 (41) (Fig. 3C). The levels of Cdc45 loading and MCM2 phosphorylation on chromatin increased 1 h after release from Thy → Mimo treatment (Fig. 3D). Taken together, these results suggest that mimosine treatment prevents the transition of the pre-RC to the preinitiation complex by blocking the activation of the pre-RC, which results in synchronization of cells at the G1/S phase boundary.
Prevention of DNA Damage upon Mimosine Synchronization
It has been reported that mimosine treatment induces DNA damage in several cell lines (42, 43). However, mimosine treatment of HeLa S3 cells did not generate apoptotic cells (sub-G1 populations) (Fig. 1), suggesting the possibility that mimosine treatment does not give rise to high levels of DNA damage. We examined whether mimosine treatment induced DNA damage in HeLa S3 cells. First, we treated asynchronous cells with mimosine at 1 mm for 24 h and analyzed them for phosphorylated histone H2AX (γH2AX), a sensitive marker of DNA damage (44). As a positive control, we treated cells with the DNA damaging agent Adriamycin at a subcytotoxic concentration (20 ng/ml) for 24 h, which does not induce cell death in HeLa cells (13). Microscopic analysis showed that treatment of asynchronous cells with mimosine induced γH2AX foci, consistent with previous observations (43). The levels of γH2AX in mimosine-synchronized cells were as high as those in thymidine-synchronized cells and Adriamycin-treated cells (Fig. 4A). Next, cells were synchronized by Thy → Mimo treatment and analyzed for γH2AX levels. We found that Thy → Mimo synchronization induced low levels of γH2AX foci. The majority of the γH2AX levels in Thy → Mimo-synchronized cells were as low as those in untreated cells. In contrast, thymidine synchronization induced high levels of γH2AX foci irrespective of single thymidine treatment or Thy → Thy synchronization (Fig. 4B). Western blot analysis confirmed that γH2AX levels in Thy → Mimo-synchronized cells were lower than those in Thy → Thy-synchronized cells (Fig. 4C). These results suggest that Thy → Mimo synchronization prevents the induction of DNA damage. It is known that inhibition of the elongation step of DNA replication causes replication fork stalling, which leads to DNA damage (45). Upon Thy → Mimo synchronization, the effect of mimosine treatment on DNA replication could be negligible because the majority of cells exited S phase and entered G2/M phase when cells were exposed to mimosine (Fig. 1). Given that thymidine treatment synchronizes cells after the initiation of DNA synthesis (Figs. 1 and 2), it is assumed that DNA damage induced by thymidine treatment is attributable to replication fork stalling.
Mimosine-induced Activation of ATM/ATR-mediated Cell Cycle Checkpoint Signaling without DNA Damage
It is known that cells are arrested at particular phases of the cell cycle by activation of cell cycle checkpoints in response to replication stress or DNA damage (46, 47). We asked whether Thy → Mimo synchronization activated cell cycle checkpoint signaling. The major regulators of the cell cycle checkpoints are ATM and ATR kinases. ATM and its substrate Chk2 are activated in response to DNA damage, and activation of ATM-mediated cell cycle checkpoint signaling inhibits the G1/S-phase transition (46, 48). ATR and its substrate Chk1 are activated in response to replication fork stalling, and the resulting ATR-mediated checkpoint signaling inhibits S phase progression (47, 49).
Using highly synchronized cell populations yielded by Thy → Thy or Thy → Mimo synchronization, we examined the phosphorylation status of these kinases for their activation: autophosphorylation of ATM at Ser-1981, phosphorylation of Chk2 at Thr-68 by ATM (50), and phosphorylation of Chk1 at Ser-317 and Ser-345 by ATR (49). Western blot analysis showed that phosphorylation of Chk1 was detected in Thy → Thy-synchronized cells (Fig. 5A, Thymidine → Thymidine, 0 h), suggesting that thymidine treatment activates S phase checkpoint signaling in response to replication fork stalling. In addition, Thy → Thy-synchronized cells showed slight phosphorylation of Chk2, despite the lack of phosphorylation of ATM at Ser-1981, in agreement with previous observations that thymidine treatment induces activation of ATM only in an early stage of checkpoint signaling (51). Thus, we assume that Chk2 activation may be attributed to DNA damage following replication fork stalling (Fig. 3B).
FIGURE 5.
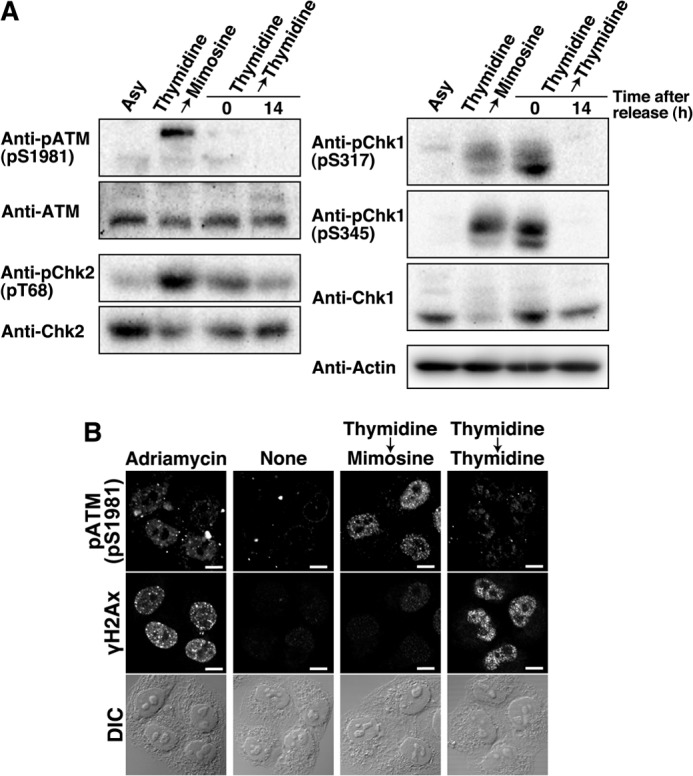
Phosphorylation status of the checkpoint kinases upon mimosine synchronization. A, HeLa S3 cells were synchronized by the indicated methods, and G1 phase cells were prepared by 14-h release from Thy → Thy. Cell lysates were analyzed by Western blotting. Asy, asynchronous cells. B, cells treated with 20 ng/ml Adriamycin for 24 h (positive control) or synchronized by the indicated methods were fixed and stained for phospho-ATM (Ser-1981) and γH2AX. DIC, differential interference contrast. Scale bars = 10 μm.
In marked contrast, Thy → Mimo-synchronized cells showed high levels of phosphorylation of ATM and Chk2 and moderate levels of phosphorylation of Chk1 (Fig. 5A, Thymidine → Mimosine), suggesting the activation of the ATM- and ATR-mediated checkpoint pathways despite the absence of the stalled replication fork (Figs. 1–3). Thy → Mimo synchronization induced ATM/Chk2 phosphorylation at much higher levels than Thy → Thy synchronization, although little DNA damage was induced by Thy → Mimo synchronization (Fig. 4). To exclude the possibility that the phosphorylation of ATM in Thy → Mimo-synchronized cells is caused by only a fraction of cells that underwent DNA damage, Thy → Mimo-synchronized cells were doubly stained for phosphorylation of ATM and γH2AX. Despite low levels of γH2AX, Thy → Mimo-synchronized cells had high levels of phosphorylation of ATM in the nucleus (Fig. 5B). Therefore, these results suggest that mimosine synchronization activates ATM/ATR-mediated cell cycle checkpoint signaling without replication fork stalling or DNA damage.
Role of ATM/ATR-mediated Cell Cycle Checkpoint Signaling upon Mimosine Synchronization
To examine the relationship between mimosine-induced checkpoint activation and cell cycle arrest, Thy → Mimo-synchronized cells were treated with the ATM/ATR inhibitor caffeine (52). The inhibition of ATM and ATR was confirmed by decreased levels of phosphorylations of Chk2 at Thr-68 and Chk1 at Ser-317 and Ser-345 (Fig. 6A, left panel). Treatment of Thy → Mimo-synchronized cells with caffeine increased the levels of Cdc45, PCNA, and MCM2 phosphorylation on chromatin (Fig. 6A, right panel), suggesting that caffeine treatment induces cells to enter S phase and initiate replication fork assembly in Thy → Mimo synchronization. In Thy → Thy-synchronized cells, caffeine treatment decreased the levels of Chk1 phosphorylation and increased the levels of Cdc45, PCNA, and MCM2 phosphorylation on chromatin, suggesting that ATR-mediated checkpoint signaling arrests cells in S phase upon thymidine synchronization. Although the level of Chk2 phosphorylation was increased even upon caffeine treatment, this may be attributed to the replication fork damage induced by thymidine treatment in the absence of Chk1 activity (53). Microscopic analysis ascertained that caffeine treatment induced the formation of PCNA foci on chromatin in Thy → Mimo and Thy → Thy-synchronized cells (Fig. 6B). The assembly of the replication fork was further ascertained by measuring the binding of RPA on chromatin, a marker of single-strand DNA on the replication fork (54, 55). Caffeine treatment induced the formation of RPA foci on chromatin in Thy → Mimo-synchronized cells (Fig. 6C).
FIGURE 6.

Inhibition of S phase entry by mimosine-activated cell cycle checkpoint signaling. A–D, HeLa S3 cells were synchronized by the indicated methods in the presence or absence of 10 mm caffeine during the last 3 h. A, cell lysates were analyzed by Western blotting. B and C, the Triton-resistant fraction was fixed and stained for DNA and PCNA (B) or RPA (C). Bottom panels, mean fluorescence intensities of PCNA (B) or RPA (C) immunostaining in the nucleus were measured. ***, p < 0.001, calculated by Student's t test. Scale bars = 10 μm. Asy, asynchronous cells; DIC, differential interference contrast. D, cells were pulse-labeled with BrdU during the last 30 min and analyzed by flow cytometry. BrdU levels (y axis) are plotted against DNA content (x axis), and BrdU-positive cells (boxed areas) are shown as percentages. DNA histograms are shown at the top. E, S phase cells prepared by 3-h release from Thy → Thy synchronization were further incubated in the presence or absence of 1 mm mimosine for the indicated times. Shaded histograms represent DNA content of cells released for 3 h from Thy → Thy synchronization. Bottom panels, overlay histograms of cells incubated in the presence (solid line) and absence (dotted line) of 1 mm mimosine.
To examine DNA synthesis in Thy → Mimo-synchronized cells, we used BrdU, a thymidine analog, which was incorporated into DNA during the elongation step of DNA replication (56). Treatment of Thy → Mimo-synchronized cells with caffeine did not increase DNA content and BrdU incorporation (Fig. 6D). Then, we analyzed the effect of mimosine on the elongation step of DNA replication. S phase cells were treated with 1 mm mimosine, and flow cytometry analysis showed that mimosine treatment strongly inhibited S phase progression (Fig. 6E). These results suggest that ATM/ATR-mediated cell cycle checkpoint signaling prevents cells from entering S phase upon mimosine synchronization. In addition, mimosine treatment in S phase inhibits the elongation step of DNA replication, irrespective of the generation of ATM/ATR-mediated cell cycle checkpoint signaling.
Inhibition of S Phase Entry by ATM-mediated Cell Cycle Checkpoint Signaling upon Mimosine Synchronization
Because mimosine treatment strongly activates ATM-mediated checkpoints (Fig. 5), we asked whether ATM inhibition could induce mimosine-arrested cells to enter S phase. We treated Thy → Mimo-synchronized cells with the ATM-specific inhibitor KU-55933 (57), and the inhibition of ATM was confirmed by a decreased level of Chk2 phosphorylation in Adriamycin-treated cells (Fig. 7A, Adriamycin). In Thy → Mimo-synchronized cells, KU-55933 treatment specifically decreased the level of Chk2 phosphorylation, whereas the levels of Chk1 phosphorylation by ATR were unchanged (Fig. 7A, Thymidine → Mimosine). Intriguingly, we found that the levels of PCNA and Cdc45 on chromatin were increased by KU-55933 treatment of Thy → Mimo-synchronized cells, suggesting that specific ATM inhibition by KU-55933 can induce cells to enter S phase and that the replication fork becomes assembled even in Thy → Mimo synchronization. The levels of MCM2 phosphorylation at Ser-41 were almost unchanged. Because ATR-mediated checkpoint signaling partly inhibits Cdk2 activity (49), it is assumed that MCM2 phosphorylation at Ser-41 by Cdk2 (41) is inhibited by ATR even upon KU-55933 treatment. In addition, KU-55933 treatment did not increase the DNA content of Thy → Mimo-synchronized cells (Fig. 7A, right panel). This is consistent with results showing that mimosine treatment inhibits the elongation step of DNA replication, irrespective of the generation of ATM/ATR-mediated cell cycle checkpoint signaling (Fig. 6).
FIGURE 7.

Inhibition of S phase entry by ATM-mediated checkpoint signaling. A, HeLa S3 cells treated with 100 ng/ml Adriamycin for 1 h in the presence or absence of 25 μm KU-55933 or cells synchronized by the indicated methods in the presence or absence of 25 μm KU-55933 during the last 3 h were analyzed by Western blotting. Right panel, top, cells were analyzed for DNA content by flow cytometry. Shaded histograms represent DNA content of asynchronous cells. Right panel, bottom, overlay histogram of cells synchronized by Thy → Mimo treatment in the presence (solid line) and absence (dotted line) of KU-55933 during the last 3 h. B, HeLa S3/Mock and HeLa S3/shATM cells were analyzed by Western blotting. Right panel, cells were analyzed for DNA content by flow cytometry. Shown is an overlay histogram of HeLaS3/Mock (dotted line) and HeLaS3/shATM (solid line). C, HeLaS3/Mock and HeLaS3/shATM cells were treated with 100 ng/ml Adriamycin for 1 h or synchronized by the indicated methods. Cell lysates were analyzed by Western blotting. Right panel, top, cells were analyzed for DNA content by flow cytometry. Right panel, bottom, overlay histogram of HeLaS3/Mock (dotted line) and HeLaS3/shATM (solid line). Asy, asynchronous cells. D, COS-1 cells transfected with vector, ATM-WT, or ATM-kd were cultured for 48 h in the presence of 1 mm mimosine during the last 24 h. The Triton-resistant fraction was fixed and stained for FLAG (expressed proteins) and PCNA. Right panel, mean fluorescence intensities of PCNA immunostaining in the nucleus were plotted. DIC, Differential-interference-contrast. Scale bars = 10 μm. ***, p < 0.001, calculated by Student's t test.
In contrast, KU-55933 treatment of Thy → Thy -synchronized cells did not affect the levels of PCNA, Cdc45, and MCM2 phosphorylation on chromatin (Fig. 7A, Thymidine → Thymidine), in agreement with results showing that ATR-mediated checkpoint signaling arrests cells in S phase upon thymidine synchronization (Fig. 6, A and B). In addition, KU-55933 treatment decreased not only the level of phosphorylation of Chk2 but also that of Chk1 in Thy → Thy-synchronized cells. Given that thymidine treatment induces DNA damage (Fig. 4), this may be attributed to the cross-talk between ATM and ATR signaling upon the DNA damage response (49).
To substantiate that ATM-mediated checkpoint signaling blocks S phase entry upon mimosine synchronization, we established ATM knockdown cell lines (HeLa S3/shATM) (Fig. 7B). Western blot analysis showed that ATM knockdown decreased Chk2 phosphorylation and increased the binding of PCNA and Cdc45 on chromatin upon Thy → Mimo synchronization (Fig. 7C). Consistent with the results of ATM inhibition by KU-55933 treatment (Fig. 7A), DNA content and the level of MCM2 were not affected by ATM knockdown upon Thy → Mimo synchronization. To verify that the blockade of S phase entry is mediated by ATM kinase activity, cells were transfected with ATM-WT or ATM-kinase-dead (ATM-kd). Because a very small number of cells showed the expression of ATM-WT or ATM-kd (<1%), we quantitatively analyzed the levels of PCNA on chromatin by microscopy (Fig. 7D). We found that the binding of PCNA to chromatin was increased in ATM-kd-expressing cells upon mimosine treatment, suggesting that ATM-kd functions as a dominant negative mutation. Taken together, these results suggest that the activation of ATM-mediated checkpoint signaling blocks S phase entry upon mimosine synchronization.
Activation of ATM through ROS-mediated Hypoxic Stress upon Mimosine Synchronization
It recently became known that ATM is activated in the absence of DNA damage in response to hypoxic stress (58, 59). Iron chelators, including mimosine and deferoxamine, are hypoxia-mimetic agents (60). Therefore, we examined whether the activation of ATM was induced by hypoxic stress upon mimosine treatment. In agreement with previous reports, Western blot analysis showed that the protein levels of Hif-1α, a marker of hypoxic stress (61, 62), were high in mimosine-synchronized cells (Fig. 8A). When Hif-1α was immediately degraded 10 min after release from Thy → Mimo synchronization, the levels of ATM phosphorylation were also decreased (Fig. 8B), suggesting the possibility that ATM phosphorylation is induced by hypoxic stress upon mimosine treatment.
It is known that hypoxic stress is mediated by reactive oxygen species (ROS) (63, 64). Because mimosine treatment has been reported to increase the production of ROS such as H2O2 (65, 66), we tested whether treatment with N-acetyl-l-cysteine (NAC), a ROS scavenger, could prevent ATM activation upon mimosine treatment. Consistent with the results that Adriamycin treatment activates ATM through ROS production (10), NAC treatment decreased the level of ATM phosphorylation in Adriamycin-treated cells (Fig. 8C, Adriamycin). In Thy → Mimo-synchronized cells, we found that NAC treatment decreased the level of ATM phosphorylation (Fig. 8C, Thy → Mimo). Moreover, flow cytometry analysis showed that NAC treatment upon Thy → Mimo synchronization increased the populations in S and G2/M phases, whereas it decreased the populations in G1 phase (Fig. 8D), suggesting that NAC treatment induces cells to progress through the cell cycle even upon Thy → Mimo synchronization. Taken together, these results suggest that mimosine treatment blocks S phase entry by activating ATM through ROS-mediated hypoxic stress.
Efficient BrdU Incorporation Following Release from Mimosine Treatment
It is known that treatment with excess thymidine induces the imbalance in deoxy-NTP (dNTP) pools and up-regulates the deoxy-TTP (dTTP) concentration and competitively interferes with BrdU incorporation (67). Thus, we compared the levels of BrdU incorporation in Thy → Mimo-synchronized cells with those in Thy → Thy-synchronized cells. Thy → Mimo-synchronized cells showed a rapid increase in the number of BrdU-positive cells (40%) 1 h after release and a further increase (64%) at 3 h (Fig. 9). In contrast, Thy → Thy-treated cells did not incorporate BrdU efficiently after release. BrdU-positive cells did not start to increase until 2 h after release from Thy → Thy treatment and increased (53%) at 3 h, although the DNA content of the cells was increased more than that of cells released from Thy → Mimo treatment. These results suggest that Thy → Mimo synchronization is suitable for measurement of an initial stage of DNA replication by BrdU incorporation.
FIGURE 9.

Efficient BrdU incorporation following release from mimosine treatment. HeLa S3 cells synchronized by the indicated methods were released for the indicated times and pulse-labeled with BrdU during the last 30 min of the release. BrdU levels (y axis) are plotted against DNA content (x axis), and BrdU-positive cells (boxed areas) are shown as percentages. DNA histograms are shown at the top. Bottom panels, cells were fixed and stained for BrdU and DNA. Asy, asynchronous cells; DIC, differential interference contrast. Scale bars = 10 μm.
DISCUSSION
In this study, we show that mimosine treatment synchronizes cells at the G1/S phase boundary by inhibiting the activation of the pre-RC. Mimosine treatment activates ATM-mediated checkpoint signaling, which blocks activation of the pre-RC and prevents cells from entering S phase. The activation of ATM is induced by ROS-mediated hypoxic stress without DNA damage.
We illustrate a model for cell cycle synchronization by mimosine treatment in Fig. 10. We show that mimosine treatment synchronizes cells at the G1/S phase boundary. Mimosine-synchronized cells do not show binding of PCNA and RPA to chromatin, suggesting that the replication fork and the resulting single-stranded DNA are not formed. Although pre-RC assembly is completed and Cdt1 is degraded, Cdc45, one of the pre-RC-activating factors, is not loaded onto chromatin in mimosine-synchronized cells. Therefore, mimosine treatment prevents the transition of the pre-RC to the preinitiation complex by blocking the activation of the pre-RC, which results in synchronization of cells at the G1/S phase boundary.
FIGURE 10.
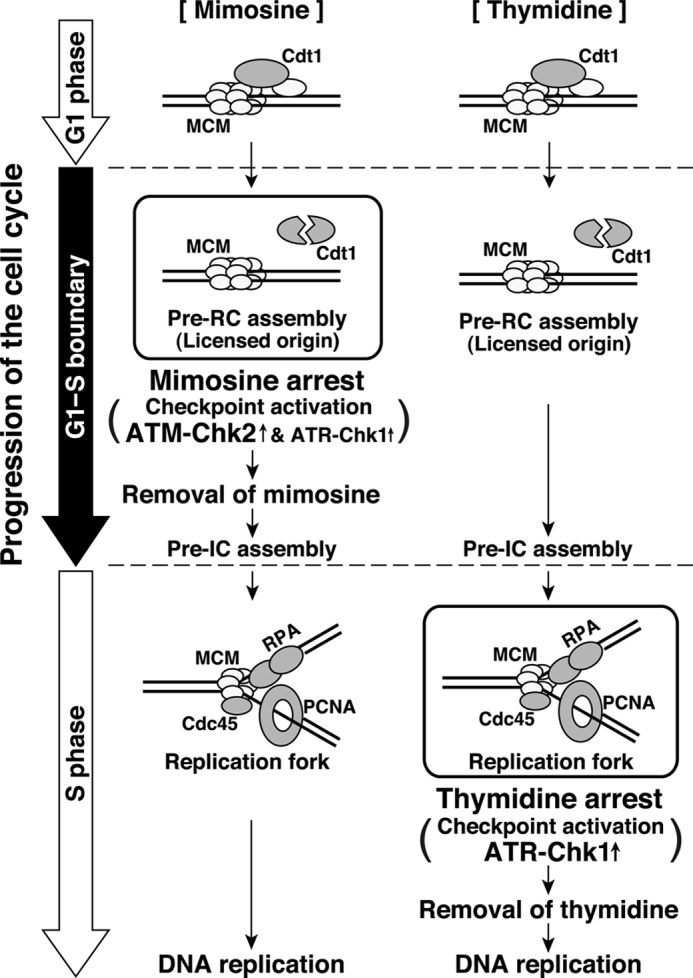
Model for the cell cycle synchronization by mimosine. Mimosine treatment synchronizes cells at the G1/S phase boundary by blocking the activation of the pre-RC. Mimosine treatment strongly activates ATM-mediated cell cycle checkpoint signaling, which prevents cells from entering S phase. In contrast, thymidine treatment synchronizes cells after the initiation of DNA replication and activates the S phase checkpoint. Pre-IC, preinitiation complex.
In contrast to mimosine treatment, thymidine treatment arrests cells after initiation of DNA synthesis (Figs. 1 and 2). Treatment with excess thymidine induces an imbalance in dNTP pools, which results in feedback inhibition of RNR (67, 68). RNR inhibition blocks the elongation step of DNA replication, which generates stalled replication forks. Then, stalled replication forks induce the activation of the S phase checkpoint (Fig. 4) (49). Therefore, these results suggest that thymidine treatment inhibits the elongation step of DNA replication and, subsequently, activates the S phase checkpoint, which results in synchronization of cells in S phase.
Mimosine treatment also inhibits the elongation step of DNA replication through impairing RNR (7, 8). However, this mechanism cannot explain the synchronization of cells at the G1/S phase boundary. We show that thymidine and mimosine differently activate the ATM/Chk2 and ATR/Chk1 kinases for cell cycle checkpoints (Fig. 5). The mimosine-activated cell cycle checkpoint prevents cells from entering S phase through inhibiting the activation of the pre-RC (Figs. 5 and 6). In addition, mimosine treatment inhibits the elongation step of DNA replication, irrespective of ATM/ATR-mediated cell cycle checkpoint signaling (Fig. 6, D and E). Therefore, we hypothesize that mimosine treatment synchronizes cells through two independent mechanisms: the activation of ATM/ATR-mediated cell cycle checkpoint signaling (enrichment of cells at the G1/S phase boundary) and the inhibition of the elongation step of DNA replication (enrichment of cells in S phase). In contrast to treatment of asynchronous cells with mimosine, Thy → Mimo treatment minimizes the inhibition of the elongation step of DNA replication because 9-h release from thymidine treatment depletes S phase cells, and the resulting G2/M-enriched cells are treated with mimosine (Fig. 1C, Thy → Mimo). Then, cells can progress through G1 phase in the presence of mimosine and are prevented from entering S phase by the mimosine-activated cell cycle checkpoint. Therefore, Thy → Mimo treatment highly enriches cells at the G1/S phase boundary.
Mimosine-synchronized cells show a strong activation of ATM/Chk2 compared with thymidine-synchronized cells (Fig. 5). We further show that specific ATM inhibition can induce cells to enter S phase and that the replication fork becomes assembled even upon mimosine synchronization (Fig. 7). The ATM/Chk2 pathway is known to inhibit the G1/S phase transition through inactivation of the cyclin E-Cdk2 complex, which has a role in activating replication origins (46, 48). We observed a decrease in the levels of cyclin E protein (Fig. 2, A and B), and previous reports showed that Cdk2 activity is down-regulated in mimosine-synchronized cells (69, 70). Therefore, mimosine-activated ATM/Chk2 may inactivate the cyclin E-Cdk2 complex and prevent cells from entering S phase. In addition, the ATR/Chk1 pathway may contribute to mimosine synchronization because ATR-mediated checkpoint signaling inhibits the activation of late origins (2, 46). However, given that ATR-mediated checkpoint signaling arrests cells after initiation of DNA synthesis upon thymidine treatment (Figs. 1, 2, and 4), we can assume that mimosine treatment blocks S phase entry by preventing the activation of early origins through ATM-mediated checkpoint signaling in addition to late origins through ATR-mediated checkpoint signaling.
Our results suggest that ATM/ATR-mediated cell cycle checkpoint signaling is activated without replication fork stalling or DNA damage upon mimosine synchronization (Figs. 2–5). It has recently become known that ATM is important not only for DNA damage responses but also for oxidative stress responses (59). For instance, ATM is activated in the absence of DNA damage in response to hypoxic stress (58, 59). Mimosine is a hypoxia-mimetic agent, and mimosine treatment has been reported to increase the production of ROS, one of the hypoxia mediators (60, 65, 66). We show that the activation of ATM upon mimosine treatment is induced in response to ROS-mediated hypoxic stress (Fig. 8, A–C). Moreover, treatment of cells with the ROS scavenger NAC is capable of inducing progression of the cell cycle even upon mimosine treatment (Fig. 8D). Therefore, we hypothesize that the activation of ATM in response to ROS-mediated hypoxic stress blocks S phase entry upon mimosine synchronization. In addition, the ATR/Chk1 pathway is activated in response to oxidative and hypoxic stress, and H2O2 treatment activates Chk1 in an ATM-dependent manner (58, 71). These results suggest the possibility that mimosine treatment activates the ATR/Chk1 pathway through ROS-mediated ATM activation.
In response to DNA damage, the activation of ATM-mediated checkpoint signaling is important for controlling cell cycle arrest, DNA repair, and apoptosis (46, 48). Similar to the roles of ATM in DNA damage responses, ATM has a protective role against oxidative stress upon mimosine treatment. Blockade of S phase entry by ATM prevents induction of DNA damage arising from replication fork stalling. The levels of DNA damage in Thy → Mimo-synchronized cells are much lower than those in Thy → Thy-synchronized cells (Fig. 4), although thymidine is less of a DNA-damaging agent compared with other G1/S synchronization reagents, such as hydroxyurea and aphidicolin (72, 73). In addition, apoptotic cells (sub-G1 population) are not seen upon mimosine treatment (Fig. 1). Our results provide new details on the protective role of ATM against oxidative stress in cell cycle arrest at the G1/S phase boundary. Further studies using mimosine synchronization will help us to gain new insights into the precise mechanisms that control the onset of DNA replication.
This work was supported by grants-in-aid for Scientific Research, Global Center of Excellence (COE) Program (Global Center for Education and Research in Immune Regulation and Treatment) and Special Funds for Education and Research (Development of single photon emission computed tomography (SPECT) probes for Pharmaceutical Innovation) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
- pre-RC
- prereplication complex
- MCM
- minichromosome maintenance
- PCNA
- proliferating cell nuclear antigen
- RNR
- ribonucleotide reductase
- ATM
- ataxia telangiectasia mutated
- ATR
- ataxia telangiectasia and Rad3-related
- RPA
- replication protein A
- Thy
- thymidine
- Mimo
- mimosine
- kd
- kinase-dead
- ROS
- reactive oxygen species
- NAC
- N-acetylcysteine.
REFERENCES
- 1. Moldovan G. L., Pfander B., Jentsch S. (2007) PCNA, the maestro of the replication fork. Cell 129, 665–679 [DOI] [PubMed] [Google Scholar]
- 2. Méchali M. (2010) Eukaryotic DNA replication origins. Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 11, 728–738 [DOI] [PubMed] [Google Scholar]
- 3. Harper J. V. (2005) Synchronization of cell populations in G1/S and G2/M phases of the cell cycle. Methods Mol. Biol. 296, 157–166 [DOI] [PubMed] [Google Scholar]
- 4. Lalande M. (1990) A reversible arrest point in the late-G1 phase of the mammalian cell cycle. Exp. Cell Res. 186, 332–339 [DOI] [PubMed] [Google Scholar]
- 5. Krude T. (1999) Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res. 247, 148–159 [DOI] [PubMed] [Google Scholar]
- 6. Kalejta R. F., Hamlin J. L. (1997) The dual effect of mimosine on DNA replication. Exp. Cell Res. 231, 173–183 [DOI] [PubMed] [Google Scholar]
- 7. Dai Y., Gold B., Vishwanatha J. K., Rhode S. L. (1994) Mimosine inhibits viral DNA synthesis through ribonucleotide reductase. Virology 205, 210–216 [DOI] [PubMed] [Google Scholar]
- 8. Gilbert D. M., Neilson A., Miyazawa H., DePamphilis M. L., Burhans W. C. (1995) Mimosine arrests DNA synthesis at replication forks by inhibiting deoxyribonucleotide metabolism. J. Biol. Chem. 270, 9597–9606 [DOI] [PubMed] [Google Scholar]
- 9. Levenson V., Hamlin J. L. (1993) A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Res. 21, 3997–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurz E. U., Douglas P., Lees-Miller S. P. (2004) Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 279, 53272–53281 [DOI] [PubMed] [Google Scholar]
- 11. Nakayama Y., Igarashi A., Kikuchi I., Obata Y., Fukumoto Y., Yamaguchi N. (2009) Bleomycin-induced over-replication involves sustained inhibition of mitotic entry through the ATM/ATR pathway. Exp. Cell Res. 315, 2515–2528 [DOI] [PubMed] [Google Scholar]
- 12. Nakayama Y., Yamaguchi N. (2005) Multi-lobulation of the nucleus in prolonged S phase by nuclear expression of Chk tyrosine kinase. Exp. Cell Res. 304, 570–581 [DOI] [PubMed] [Google Scholar]
- 13. Kikuchi I., Nakayama Y., Morinaga T., Fukumoto Y., Yamaguchi N. (2010) A decrease in cyclin B1 levels leads to polyploidization in DNA damage-induced senescence. Cell Biol. Int. 34, 645–653 [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi N., Fukuda M. N. (1995) Golgi retention mechanism of β-1,4-galactosyltransferase. Membrane-spanning domain-dependent homodimerization and association with α- and β-tubulins. J. Biol. Chem. 270, 12170–12176 [DOI] [PubMed] [Google Scholar]
- 15. Tada J., Omine M., Suda T., Yamaguchi N. (1999) A common signaling pathway via Syk and Lyn tyrosine kinases generated from capping of the sialomucins CD34 and CD43 in immature hematopoietic cells. Blood 93, 3723–3735 [PubMed] [Google Scholar]
- 16. Nakayama Y., Kawana A., Igarashi A., Yamaguchi N. (2006) Involvement of the N-terminal unique domain of Chk tyrosine kinase in Chk-induced tyrosine phosphorylation in the nucleus. Exp. Cell Res. 312, 2252–2263 [DOI] [PubMed] [Google Scholar]
- 17. Kasahara K., Nakayama Y., Sato I., Ikeda K., Hoshino M., Endo T., Yamaguchi N. (2007) Role of Src-family kinases in formation and trafficking of macropinosomes. J. Cell Physiol. 211, 220–232 [DOI] [PubMed] [Google Scholar]
- 18. Ikeda K., Nakayama Y., Togashi Y., Obata Y., Kuga T., Kasahara K., Fukumoto Y., Yamaguchi N. (2008) Nuclear localization of Lyn tyrosine kinase mediated by inhibition of its kinase activity. Exp. Cell Res. 314, 3392–3404 [DOI] [PubMed] [Google Scholar]
- 19. Takahashi A., Obata Y., Fukumoto Y., Nakayama Y., Kasahara K., Kuga T., Higashiyama Y., Saito T., Yokoyama K. K., Yamaguchi N. (2009) Nuclear localization of Src-family tyrosine kinases is required for growth factor-induced euchromatinization. Exp. Cell Res. 315, 1117–1141 [DOI] [PubMed] [Google Scholar]
- 20. Aoyama K., Fukumoto Y., Ishibashi K., Kubota S., Morinaga T., Horiike Y., Yuki R., Takahashi A., Nakayama Y., Yamaguchi N. (2011) Nuclear c-Abl-mediated tyrosine phosphorylation induces chromatin structural changes through histone modifications that include H4K16 hypoacetylation. Exp. Cell Res. 317, 2874–2903 [DOI] [PubMed] [Google Scholar]
- 21. Aoyama K., Yuki R., Horiike Y., Kubota S., Yamaguchi N., Morii M., Ishibashi K., Nakayama Y., Kuga T., Hashimoto Y., Tomonaga T., Yamaguchi N. (2013) Formation of long and winding nuclear F-actin bundles by nuclear c-Abl tyrosine kinase. Exp. Cell Res. 319, 3251–3268 [DOI] [PubMed] [Google Scholar]
- 22. Swingle M., Ni L., Honkanen R. E. (2007) Small-molecule inhibitors of Ser/Thr protein phosphatases. Specificity, use and common forms of abuse. Methods Mol. Biol. 365, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi N., Nakayama Y., Urakami T., Suzuki S., Nakamura T., Suda T., Oku N. (2001) Overexpression of the Csk homologous kinase (Chk tyrosine kinase) induces multinucleation. A possible role for chromosome-associated Chk in chromosome dynamics. J. Cell Sci. 114, 1631–1641 [DOI] [PubMed] [Google Scholar]
- 24. Kasahara K., Nakayama Y., Ikeda K., Fukushima Y., Matsuda D., Horimoto S., Yamaguchi N. (2004) Trafficking of Lyn through the Golgi caveolin involves the charged residues on αE and αI helices in the kinase domain. J. Cell Biol. 165, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsuda D., Nakayama Y., Horimoto S., Kuga T., Ikeda K., Kasahara K., Yamaguchi N. (2006) Involvement of Golgi-associated Lyn tyrosine kinase in the translocation of annexin II to the endoplasmic reticulum under oxidative stress. Exp. Cell Res. 312, 1205–1217 [DOI] [PubMed] [Google Scholar]
- 26. Kasahara K., Nakayama Y., Nakazato Y., Ikeda K., Kuga T., Yamaguchi N. (2007) Src signaling regulates completion of abscission in cytokinesis through ERK/MAPK activation at the midbody. J. Biol. Chem. 282, 5327–5339 [DOI] [PubMed] [Google Scholar]
- 27. Kuga T., Nakayama Y., Hoshino M., Higashiyama Y., Obata Y., Matsuda D., Kasahara K., Fukumoto Y., Yamaguchi N. (2007) Differential mitotic activation of endogenous c-Src, c-Yes, and Lyn in HeLa cells. Arch. Biochem. Biophys. 466, 116–124 [DOI] [PubMed] [Google Scholar]
- 28. Sato I., Obata Y., Kasahara K., Nakayama Y., Fukumoto Y., Yamasaki T., Yokoyama K. K., Saito T., Yamaguchi N. (2009) Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 122, 965–975 [DOI] [PubMed] [Google Scholar]
- 29. Kubota S., Fukumoto Y., Aoyama K., Ishibashi K., Yuki R., Morinaga T., Honda T., Yamaguchi N., Kuga T., Tomonaga T., Yamaguchi N. (2013) Phosphorylation of KRAB-associated protein 1 (KAP1) at Tyr-449, Tyr-458, and Tyr-517 by nuclear tyrosine kinases inhibits the association of KAP1 and heterochromatin protein 1α (HP1α) with heterochromatin. J. Biol. Chem. 288, 17871–17883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishibashi K., Fukumoto Y., Hasegawa H., Abe K., Kubota S., Aoyama K., Kubota S., Nakayama Y., Yamaguchi N. (2013) Nuclear ErbB4 signaling through H3K9me3 that is antagonized by EGFR-activated c-Src. J. Cell Sci. 126, 625–637 [DOI] [PubMed] [Google Scholar]
- 31. Knehr M., Poppe M., Enulescu M., Eickelbaum W., Stoehr M., Schroeter D., Paweletz N. (1995) A critical appraisal of synchronization methods applied to achieve maximal enrichment of HeLa cells in specific cell cycle phases. Exp. Cell Res. 217, 546–553 [DOI] [PubMed] [Google Scholar]
- 32. Matsui Y., Nakayama Y., Okamoto M., Fukumoto Y., Yamaguchi N. (2012) Enrichment of cell populations in metaphase, anaphase, and telophase by synchronization using nocodazole and blebbistatin. A novel method suitable for examining dynamic changes in proteins during mitotic progression. Eur. J. Cell Biol. 91, 413–419 [DOI] [PubMed] [Google Scholar]
- 33. Nakayama Y., Matsui Y., Takeda Y., Okamoto M., Abe K., Fukumoto Y., Yamaguchi N. (2012) c-Src but not Fyn promotes proper spindle orientation in early prometaphase. J. Biol. Chem. 287, 24905–24915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dulić V., Lees E., Reed S. I. (1992) Association of human cyclin E with a periodic G1-S phase protein kinase. Science 257, 1958–1961 [DOI] [PubMed] [Google Scholar]
- 35. Reed S. I. (2003) Ratchets and clocks. The cell cycle, ubiquitylation and protein turnover. Nat. Rev. Mol. Cell Biol. 4, 855–864 [DOI] [PubMed] [Google Scholar]
- 36. Leonhardt H., Rahn H. P., Weinzierl P., Sporbert A., Cremer T., Zink D., Cardoso M. C. (2000) Dynamics of DNA replication factories in living cells. J. Cell Biol. 149, 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang S. C., Nakajima Y., Yu Y. L., Xia W., Chen C. T., Yang C. C., McIntush E. W., Li L. Y., Hawke D. H., Kobayashi R., Hung M. C. (2006) Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 8, 1359–1368 [DOI] [PubMed] [Google Scholar]
- 38. Nishitani H., Taraviras S., Lygerou Z., Nishimoto T. (2001) The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem. 276, 44905–44911 [DOI] [PubMed] [Google Scholar]
- 39. Blow J. J., Dutta A. (2005) Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 6, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Labib K. (2010) How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 24, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montagnoli A., Valsasina B., Brotherton D., Troiani S., Rainoldi S., Tenca P., Molinari A., Santocanale C. (2006) Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 281, 10281–10290 [DOI] [PubMed] [Google Scholar]
- 42. Mikhailov I., Russev G., Anachkova B. (2000) Treatment of mammalian cells with mimosine generates DNA breaks. Mutat. Res. 459, 299–306 [DOI] [PubMed] [Google Scholar]
- 43. Szüts D., Krude T. (2004) Cell cycle arrest at the initiation step of human chromosomal DNA replication causes DNA damage. J. Cell Sci. 117, 4897–4908 [DOI] [PubMed] [Google Scholar]
- 44. Bonner W. M., Redon C. E., Dickey J. S., Nakamura A. J., Sedelnikova O. A., Solier S., Pommier Y. (2008) GammaH2AX and cancer. Nat. Rev. Cancer 8, 957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petermann E., Helleday T. (2010) Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 11, 683–687 [DOI] [PubMed] [Google Scholar]
- 46. Ben-Yehoyada M., Gautier J., Dupré A. (2007) The DNA damage response during an unperturbed S-phase. DNA Repair 6, 914–922 [DOI] [PubMed] [Google Scholar]
- 47. Jones R. M., Petermann E. (2012) Replication fork dynamics and the DNA damage response. Biochem. J. 443, 13–26 [DOI] [PubMed] [Google Scholar]
- 48. Ahn J., Urist M., Prives C. (2004) The Chk2 protein kinase. DNA Repair 3, 1039–1047 [DOI] [PubMed] [Google Scholar]
- 49. Cimprich K. A., Cortez D. (2008) ATR. An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lavin M. F., Kozlov S. (2007) ATM activation and DNA damage response. Cell Cycle 6, 931–942 [DOI] [PubMed] [Google Scholar]
- 51. Bolderson E., Scorah J., Helleday T., Smythe C., Meuth M. (2004) ATM is required for the cellular response to thymidine induced replication fork stress. Hum. Mol. Genet. 13, 2937–2945 [DOI] [PubMed] [Google Scholar]
- 52. Sarkaria J. N., Busby E. C., Tibbetts R. S., Roos P., Taya Y., Karnitz L. M., Abraham R. T. (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59, 4375–4382 [PubMed] [Google Scholar]
- 53. Gagou M. E., Zuazua-Villar P., Meuth M. (2010) Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell 21, 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fanning E., Klimovich V., Nager A. R. (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zou Y., Liu Y., Wu X., Shell S. M. (2006) Functions of human replication protein A (RPA). From DNA replication to DNA damage and stress responses. J. Cell Physiol. 208, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cardoso M. C., Leonhardt H. (1995) in Cell Cycle. Materials and Methods (Pagano M., ed.) pp 15–28, Springer Verlag, Berlin [Google Scholar]
- 57. Hickson I., Zhao Y., Richardson C. J., Green S. J., Martin N. M., Orr A. I., Reaper P. M., Jackson S. P., Curtin N. J., Smith G. C. (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64, 9152–9259 [DOI] [PubMed] [Google Scholar]
- 58. Bencokova Z., Kaufmann M. R., Pires I. M., Lecane P. S., Giaccia A. J., Hammond E. M. (2009) ATM activation and signaling under hypoxic conditions. Mol. Cell Biol. 29, 526–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ditch S., Paull T. T. (2012) The ATM protein kinase and cellular redox signaling. Beyond the DNA damage response. Trends Biochem. Sci. 37, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Correia S. C., Moreira P. I. (2010) Hypoxia-inducible factor 1. A new hope to counteract neurodegeneration? J. Neurochem. 112, 1–12 [DOI] [PubMed] [Google Scholar]
- 61. Warnecke C., Griethe W., Weidemann A., Jürgensen J. S., Willam C., Bachmann S., Ivashchenko Y., Wagner I., Frei U., Wiesener M., Eckardt K. U. (2003) Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J. 17, 1186–1188 [DOI] [PubMed] [Google Scholar]
- 62. Yang Y. T., Ju T. C., Yang D. I. (2005) Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. J. Neurochem. 93, 513–525 [DOI] [PubMed] [Google Scholar]
- 63. Semenza G. L. (1999) Perspectives on oxygen sensing. Cell 98, 281–284 [DOI] [PubMed] [Google Scholar]
- 64. Paddenberg R., Ishaq B., Goldenberg A., Faulhammer P., Rose F., Weissmann N., Braun-Dullaeus R. C., Kummer W. (2003) Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L710–719 [DOI] [PubMed] [Google Scholar]
- 65. Panopoulos A., Harraz M., Engelhardt J. F., Zandi E. (2005) Iron-mediated H2O2 production as a mechanism for cell type-specific inhibition of tumor necrosis factor α-induced but not interleukin-1β-induced IκB kinase complex/nuclear factor-κB activation. J. Biol. Chem. 280, 2912–2923 [DOI] [PubMed] [Google Scholar]
- 66. Hallak M., Vazana L., Shpilberg O., Levy I., Mazar J., Nathan I. (2008) A molecular mechanism for mimosine-induced apoptosis involving oxidative stress and mitochondrial activation. Apoptosis 13, 147–155 [DOI] [PubMed] [Google Scholar]
- 67. Julias J. G., Pathak V. K. (1998) Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 72, 7941–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eriksson S., Thelander L., Akerman M. (1979) Allosteric regulation of calf thymus ribonucleoside diphosphate reductase. Biochemistry 18, 2948–2952 [DOI] [PubMed] [Google Scholar]
- 69. Matsumoto Y., Hayashi K., Nishida E. (1999) Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 9, 429–432 [DOI] [PubMed] [Google Scholar]
- 70. Durcan T. M., Halpin E. S., Casaletti L., Vaughan K. T., Pierson M. R., Woods S., Hinchcliffe E. H. (2008) Centrosome duplication proceeds during mimosine-induced G1 cell cycle arrest. J. Cell Physiol. 215, 182–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Das K. C., Dashnamoorthy R. (2004) Hyperoxia activates the ATR-Chk1 pathway and phosphorylates p53 at multiple sites. Am. J. Physiol. Lung. Cell Mol. Physiol. 286, L87–97 [DOI] [PubMed] [Google Scholar]
- 72. Lundin C., Erixon K., Arnaudeau C., Schultz N., Jenssen D., Meuth M., Helleday T. (2002) Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell Biol. 22, 5869–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shields B. J., Hauser C., Bukczynska P. E., Court N. W., Tiganis T. (2008) DNA replication stalling attenuates tyrosine kinase signaling to suppress S phase progression. Cancer Cell 14, 166–179 [DOI] [PubMed] [Google Scholar]