

Background: BACE1 (β-secretase) is an important enzyme in Alzheimer disease pathology, but how it is regulated remains unclear.
Results: Rheb GTPase binds and regulates BACE1 levels and its activity, in an mTOR independent manner.
Conclusion: Rheb GTPase is a novel physiological regulator of BACE1 pathway.
Significance: This study establishes a novel molecular link between Rheb and BACE1 that may have a role in age-related biology and disease.
Keywords: Aging, Amyloid, GTPase, Protein Stability, Secretases
Abstract
The β-site amyloid precursor protein (APP)-cleaving enzyme 1 (β-secretase, BACE1) initiates amyloidogenic processing of APP to generate amyloid β (Aβ), which is a hallmark of Alzheimer disease (AD) pathology. Cerebral levels of BACE1 are elevated in individuals with AD, but the molecular mechanisms are not completely understood. We demonstrate that Rheb GTPase (Ras homolog enriched in brain), which induces mammalian target of rapamycin (mTOR) activity, is a physiological regulator of BACE1 stability and activity. Rheb overexpression depletes BACE1 protein levels and reduces Aβ generation, whereas the RNAi knockdown of endogenous Rheb promotes BACE1 accumulation, and this effect by Rheb is independent of its mTOR signaling. Moreover, GTP-bound Rheb interacts with BACE1 and degrades it through proteasomal and lysosomal pathways. Finally, we demonstrate that Rheb levels are down-regulated in the AD brain, which is consistent with an increased BACE1 expression. Altogether, our study defines Rheb as a novel physiological regulator of BACE1 levels and Aβ generation, and the Rheb-BACE1 circuitry may have a role in brain biology and disease.
Introduction
Ras homolog enriched in brain (Rheb) belongs to the small GTPase family of proteins (1) and can be induced upon synaptic activity (2). Rheb has been implicated in numerous functions, including cell growth, apoptosis, and autophagy (3). In addition, Rheb has a role in development as its deletion causes embryonic lethality in mice (4). Recent studies implicate Rheb in myelination (5) and in the protection of dopaminergic neurons in Parkinson disease (6). Rheb is a master regulator of mTOR3 signaling, which is implicated in several human diseases, including cancer, obesity, immune, and neurodegenerative disorders (7). Although an endogenous interaction between Rheb and mTOR has yet to be demonstrated (8), Rheb readily binds with mTOR when overexpressed, in a GTP-dependent manner (9). Like many other GTPases, Rheb consists of a C-terminal farnesylation domain that supports binding to the membranes (10), but is not essential for mTOR activation (11). Interestingly, mTOR-independent roles for Rheb have been demonstrated in the endocytic trafficking pathway and in the activation of B-Raf signaling (12, 13). These studies indicate that Rheb plays a variety of important roles, through both mTOR-dependent and mTOR-independent pathways.
β-Site APP-cleaving enzyme 1 (BACE1, β-secretase) is the rate-limiting and principal enzyme responsible for Aβ generation in neurons (14). Although its depletion in wild-type mice promotes hypomyelination (15), its depletion in mouse models of Alzheimer disease (AD) abolishes Aβ production and rescues AD-like phenotypes (16–18). Moreover, BACE1 activities toward Aβ are controlled by its intracellular localization (19). This suggests that BACE1 protein stability and localization must be tightly regulated in a healthy brain. In the AD brain, the BACE1 protein, not its mRNA levels, have been shown to be up-regulated (20, 21). Several proteins, such as translation initiation factor eIF2α (22, 23), Golgi-localized γ-ear-containing ADP-ribosylation factor binding (GGA), the reticulon/Nogo family of proteins, Arf6, and sortilins, are implicated in the stability and intracellular localization of BACE1 (24–27), but the mechanism, as well as their putative coordinated actions, remains unclear. Efforts are underway to develop drugs that would block BACE1 to prevent the generation of Aβ and alleviate the associated cognitive symptoms in AD (28, 29). As BACE1 has normal physiological functions, a detailed understanding of the molecular mechanisms of BACE1 stability is crucial to identify novel drug targets for BACE1-related disorders, such as AD (30).
In this study, we report that Rheb GTPase physiologically modulates the stability and the activity of BACE1. Rheb overexpression reduces BACE1 levels and Aβ generation, whereas Rheb depletion causes accumulation of BACE1. Mechanistically, we found that Rheb requires the GTP-bound state, but not mTOR activity, to destabilize BACE1 through proteasomal and lysosomal pathways. Moreover, we found for the first time that the Rheb levels are reduced in postmortem AD brain samples consistent with increased BACE1 levels. Altogether, our study reveals a hitherto unknown role for Rheb as a physiological regulator of BACE1 protein stability and activity, independent of mTOR activity. Thus, Rheb-BACE1 circuitry may have a role in age-related biology and disease.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise noted, the chemicals, including, rapamycin, MG132, cycloheximide, and chloroquine, were purchased from Sigma. BACE1 and other primary antibodies (PDGFR, PERK, mTOR, TSC2, phospho-S6K (Thr-389), S6K, HES1) were from Cell Signaling. 3D5 monoclonal BACE1 antibody gift was from Robert Vassar (University of Chicago). Rheb and β-actin antibody were from Santa Cruz Biotechnology. APP antibody (C-terminal specific) was from Sigma. Soluble APPβ (sAPPβ) antibody was from IBL International. The secondary antibodies conjugated with HRP were from Jackson Immunochemicals. Cell culture reagents were from Invitrogen. The Myc-Rheb WT and Myc-Rheb D60K, Rheb Q64L were obtained from Kun-Liang Guan. The Myc-Rheb C181S was cloned in using an appropriate protocol with site-directed mutagenesis, as described previously (31, 32). The Rheb WT, D60K, and Rheb C181S were further subcloned in a pCMV-GST vector, as before (31, 32). The human BACE1 clone was obtained from Open Biosystems and was subcloned in a pCMV-Myc vector. The human/rat β amyloid ELISA kit was from Wako, and was used for the estimation of Aβ (x-40, catalog number 294-64701) and Aβ (x-42, catalog number 292-64501) levels. Recombinant Rheb proteins were purchased from Prospecbio. The protease inhibitor tablets were from Roche Applied Science, and phosphatase inhibitor cocktails were from Sigma. Premade adenoviral-Rheb or adeno-control (empty) particles were purchased from Applied Biological Materials Inc. BACE1 knock-out mice were purchased from The Jackson Laboratory (stock no: 004714).
Cell Culture, Transfections, and Infections
Primary cortical neuronal cultures were prepared from embryonic day 17–18 C57BL/6 mice (Charles River Laboratories), as described previously (33). Animals were maintained and treated in accordance with the Institutional Animal Care and Use Committees (IACUC) at The Scripps Research Institute, Jupiter, FL. All experiments were performed on neurons cultured for 14 days in vitro. Primary cortical neurons were infected with 5 μl of Ad-Rheb or Ad-control virus (1 × 106pfu/ml). HEK293 cells were grown in DMEM with 10% FBS (fetal bovine serum) and 5 mm glutamine. Transfections of cDNA into the cells, using a PolyFect (Qiagen) reagent, were carried out according to the manufacturer's instructions. Ectopic expression of Rheb begins as early as 8 h in our cultured cells that are ∼95% confluent by 48 h. We routinely used either 36 h or 48 h after transfection of cDNA. The 36 h was chosen, in some cases, for example, as in the cycloheximide chase experiments, which involve time course 0–9 h. We avoided going beyond 48 h as the HEK 293 cells might become overconfluent, which might interfere with Rheb-mediated BACE1 regulation.
Immunoblotting, Immunoprecipitation (IP), and in Vitro Binding
Western blotting, IP, and in vitro binding experiments were carried out essentially as described previously (31, 32, 34). Briefly, at the indicated time points after transfection, cells were pelleted and lysed in IP buffer (50 mm Tris, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, and 10% glycerol with protease and phosphatase inhibitor). Protein concentration was measured with a BCA protein assay reagent (Pierce), or the cells were directly lysed in 2× SDS loading buffer (NuPAGE LDS loading buffer). Equal amounts of protein or equal volume of cell lysates were loaded and separated by 4–12% Bis-Tris gel (Invitrogen). The blots were probed for β-actin to estimate the total protein loaded. All the primary antibodies were used in the range of 1:3000 dilutions, whereas the secondary antibodies were used at 1:10,000. GST-tagged Rheb was pulled down with glutathione beads, as described before (31, 32), and the binding of endogenous BACE1 was detected by Western blotting. BACE1 was immunoprecipitated, after a preclearance step, from P25 mouse brain homogenate using a BACE1 antibody followed by Protein G Plus/Protein A-Agarose beads (Calbiochem), washed three times with IP buffer, and then incubated with 1 μg of recombinant Rheb (∼250 nm) in 200 μl of IP buffer for 4 h. The beads were washed in IP buffer, and the bound Rheb was detected using Western blotting. Primary antibodies were diluted in 2% fish gelatin in TBS-T (Sigma-G7765). We found that fish gelatin, which is less expensive than BSA, works as effectively as BSA for primary antibody dilutions.
Measurement of APP Processing by BACE1
Primary cortical neurons were infected with Ad-control and Ad-Rheb. The medium was collected and centrifuged, and the cell pellet was resuspended in lysis buffer and loaded onto the gel to measure APP-FL and APP-C-terminal fragment (CTF). The sAPPβ levels in the medium were determined using an antibody against sAPPβ and were quantified after normalizing to APP-FL. Similarly, Aβ (x-40 and x-42) levels in the medium were estimated using a commercially available ELISA kit (Wako) according to the manufacturer's protocol.
Immunostaining
Staining for Rheb and BACE1 was performed essentially as described before (31). Briefly, ∼75,000 HEK293 cells were seeded on 35-mm glass-bottom dishes. After 24 h, the cells were transfected with the indicated vectors. After 48 h, the cells were fixed with 4% paraformaldehyde (20 min) and membrane-permeabilized with 0.2% Triton X-100 (5 min). For Rheb/BACE1 co-staining, the transfected HA-Rheb and Myc-BACE1 were stained with antibodies against HA (1:200, rabbit polyclonal) and Myc (1:150, mouse monoclonal), and each was incubated for 12 h at 4 °C. Appropriate secondary antibodies conjugated to Alexa Fluor 488 and 568 (Molecular Probes) were incubated together with the nuclear DAPI stain for 1 h at room temperature. Glass dishes were covered with antifade Fluoromount G (Southern Biotech). The images were obtained by a Leica TCS SP8 confocal microscope.
RT-PCR for BACE1 mRNA
The RNA transcripts for BACE1 mRNA were estimated using the forward primer, GCCTTCCCAGTTGGAGCCGTTGAT, and the reverse primer, CGCAGCGGCCTGGGGGGCGCCCC, and the RNA transcripts for GAPDH mRNA as internal control were estimated using the forward primer, GAGTCAACGGATTTGGTCGT, and the reverse primer, TTGATTTTGGAGGGATCTCG, as indicated previously (35, 36).
Rheb Knockdown Experiments
Cultured cortical neurons on days in vitro 14 were infected with lentiviral particle, produced using Addgene protocol, expressing control shRNA (scrambled) or Rheb shRNA1 specific to human and mouse (TRCN0000010424, Sigma) at multiplicity of infection 1–3. After 48 h, Rheb deletion was confirmed by Western blotting.
Postmortem AD Samples
The prefrontal cortex of the postmortem AD and control brain tissue (n = 10) was obtained from The Harvard Brain Tissue Resource Center (McLean Hospital, Belmont, MA). Table 1 indicates the subject's code (AN No.), diagnosis (Dx), and AD severity, according to Braak staging, age, sex, postmortem interval (PMI), and the brain region, Brodmann area 9 or 10 (BA 9 OR BA 10), a part of the prefrontal cortex. Tissue was lysed in radioimmunoprecipitation assay buffer using a hand sonicator, protein estimation was performed using the BCA method, and equal proteins were loaded in the Western gel.
TABLE 1.
The demographics and the pathology of the cases
PMI, postmortem interval; Dx, diagnosis.
| AN No. | Distributive Dx | Age | Sex | PMI | Frozen tissue received |
|---|---|---|---|---|---|
| AN17557 | AD/Braak 5 | 67 | M | 25.75 | BA 9 |
| AN01605 | AD/Braak 6 | 79 | F | 10.07 | BA 9 |
| AN05806 | AD/Braak 5 | 71 | F | 27.00 | BA 9 |
| AN08641 | AD/Braak 6 | 69 | M | 20.91 | BA 9 |
| AN12697 | AD/Braak 5 | 94 | M | 25.00 | BA 9 |
| AN11154 | AD/Braak 4 | 91 | F | 9.62 | BA 9 |
| AN09050 | AD/Braak 5 | 73 | F | 15.50 | BA 9 |
| AN09155 | AD/Braak 6 | 64 | M | 20.32 | BA 9 |
| AN18776 | AD/Braak 6 | 72 | F | 17.15 | BA 9 |
| AN16547 | AD/Braak 5 | 80 | M | 24.75 | BA 9 |
| AN06429 | Control (Braak 2) | 77 | F | 23.25 | BA 9 |
| AN08385 | Control (Braak 1) | 69 | F | 23.33 | BA 9 |
| AN11253 | Control (Braak 1) | 91 | F | 30.10 | BA 10 |
| AN07243 | Control | 73 | M | 21.67 | BA 9 |
| AN06669 | Control (Braak 2) | 81 | M | 23.33 | BA 10 |
| AN03324 | Control | 97 | M | 17.00 | BA 9 |
| AN18028 | Control | 67 | M | 23.25 | BA 9 |
| AN04917 | Control | 65 | M | 21.00 | BA 10 |
| AN13687 | Control (Braak 1) | 73 | F | 24.88 | BA 10 |
| AN08704 | Control | 70 | M | 23.50 | BA 9 |
Generation of AAV-Rheb
AAV1-Rheb viral particles were produced essentially as described before (6). Adeno-associated virus (AAVs) were produced by the University of North Carolina Vector Core. The genomic titer of AAV wild-type hRheb (WT) was 4 × 1012 viral genomes/ml. Enhanced GFP, used as a control, was subcloned into the same viral backbone, and viral stocks were produced at titers of 1.0 × 1012 viral genomes/ml.
Intrahippocampal AAV Injection
Adult (P90) male C57BL/6 mice (Charles River Laboratories) were anesthetized with ketamine/xylazine solution and placed in a stereotaxic frame (Kopf Instruments) with a mouse adapter. Two microinjections of 1 and 0.5 μl of AAVs were made per hemisphere, targeting the dorsal and ventral hippocampus, respectively. The coordinates targeting the dorsal and ventral hippocampus were based on the mouse brain stereotaxic atlas (55). The coordinates (anterior-posterior (AP), measured from the bregma; medio-lateral (ML), measured from the midline of the central sinus; and dorso-ventral (DV), measured from the dura surface) were as follows: −2.0 mm AP, ±1.7 mm ML, and −2.0 mm DV for the dorsal hippocampus; −3.3 mm AP, ±3.0 mm ML, and −3.2 mm DV for the ventral hippocampus. Stepwise delivery (0.25 μl) of the virus was performed by retracting the needle to −1.4 mm (DV: dorsal) and −2.6 mm (DV: ventral) to achieve optimal vector spread in the hippocampus. The virus was delivered at 0.2 μl/min. At the end of the infusion, the needle was left in place for 5 min before being slowly withdrawn from the brain.
Statistical Analysis
Data were expressed as means ± S.E. All of the experiments were performed at least in triplicate and repeated twice, at a minimum. Statistical analysis was performed using the Student's t test or analysis of variance, and the data were plotted using Microsoft Excel software. The correlation analyses for AD versus control postmortem brain samples were assessed using Pearson's linear regression analysis by GraphPad Prism software.
RESULTS
Rheb Physiologically Regulates BACE1 Levels
Rheb plays an important role in protein turnover through the mTOR pathway that controls protein translation as well as autophagy, an intracellular bulk protein degradation process (37, 38). Here, we found that Rheb selectively modulates BACE1 protein. We overexpressed Myc (vector control) or Myc-Rheb wild-type (WT) in HEK293 cells and estimated the levels of BACE1 and other proteins by Western blotting. As shown in Fig. 1A, endogenous BACE1 protein levels were significantly reduced (∼60% loss versus Myc, n = 6, ***, p < 0.001, Student's t test) by Myc-Rheb expression, but the levels of other proteins, mTOR, TSC2, or membrane-associated proteins such as PDGFR, PERK, or full-length APP (APP-FL), were not affected (Fig. 1A). We also found that Rheb attenuates BACE1 levels in a dose-dependent manner (Fig. 1B). To confirm the specificity of the BACE1 antibody D10E5 (Cell Signaling Technology, catalog number 5606), we employed BACE1 knock-out (BACE1−/−) mice (The Jackson Laboratory) lysate and corresponding age-matched C57BL/6 wild-type (BACE1+/+) controls. D10E5, as well as 3D5, another well known BACE1 antibody (from Dr. Robert Vassar), detected BACE1 band (∼62 kDa) only in the BACE1+/+, but not in the BACE1−/− mice, confirming the specificity of the BACE1 antibody used in this study (Fig. 1C). We also found that 3D5 antibody, like D10E5 (Fig. 1, A and B), detected BACE1 suppression by Rheb (Fig. 1D). Next, we investigated whether Rheb could modulate BACE1 levels in the brain. We stereotaxically injected adeno-associated virus expressing Rheb (AAV-Rheb) into the right hippocampus and injected AAV-GFP into the left hippocampus of P90 mice and found that BACE1 levels and activity, as measured by a significant loss of sAPPβ or Aβ (x-40 and x-42) generation, are markedly reduced in the AAV-Rheb-injected side of the hippocampus when compared with the AAV-GFP-injected side (Fig. 2A). We next tested whether this effect could be observed in primary cortical neurons by infecting the neurons with adenovirus null (Ad-control) or adenovirus Rheb (Ad-Rheb). As expected, Rheb overexpression effectively attenuated the endogenous BACE1 protein levels and also reduced BACE1 activity, as measured by a significant loss of sAPPβ, APP-CTFβ, or Aβ (x-40 and x-42) generation (Fig. 2B). Note that the levels of other proteins, such as mTOR or membrane-associated proteins such as PDGFR, or full-length APP (APP-FL), were not affected in these experiments.
FIGURE 1.

Rheb GTPase regulates BACE1 levels in HEK293 cells. A, Myc (control) or Myc-Rheb cDNAs (2 μg each) transfected in HEK293 cells. After 48 h, the cells were lysed, and the equal proteins were loaded on the Western blot gel and probed for the proteins of interest, as indicated under “Experimental Procedures.” Note that BACE1 levels were significantly reduced in Myc-Rheb transfected cells (n = 6, ***, p < 0.001 versus control, Student's t test). B, Rheb reduces BACE1 protein levels in a dose-dependent manner in HEK293 cells. Myc (1 μg) or Myc-Rheb cDNA (0.1, 0.5, 1 μg) was transfected in HEK293 cells, and, after 48 h, the cells were lysed and probed for BACE1 and PERK (protein kinase RNA-like endoplasmic reticulum kinase). ***, p < 0.001, **, p < 0.01. C, BACE1 antibody, D10E5, is as specific as 3D5 to detect BACE1. BACE1+/+ and BACE1−/− mice brain lysates were probed for BACE1 and EIF2α. D10E5 or 3D5 antibody (1:1000) detected ∼62-kDa bands only in BACE1+/+, but not in BACE1−/− mice brain lysates. D, 3D5 antibody detects Rheb-mediated BACE1 suppression. Myc or Myc-Rheb were transfected in HEK293 cells, and BACE1 was detected using 3D5 antibody, as in panel A.
FIGURE 2.

Rheb GTPase regulates BACE1 levels in brain cells. A, Rheb overexpression reduces BACE1 levels and its activity in brain. Adeno-associated virus (AAV)-GFP or AAV-Rheb-Flag was injected into the left and right hippocampus of P90 mice, respectively. After 3 weeks, the hippocampus was dissected and processed to detect BACE1 and other proteins, as indicated under “Experimental Procedures.” BACE1 levels, sAPPβ, and Aβ (x-40 and x-42) were selectively down-regulated by AAV-Rheb (n = 6, ***, p < 0.001, *, p < 0.05 versus AAV-GFP, Student's t test). B, Rheb overexpression reduces BACE1 levels and its activity in cultured neurons. Adenovirus null (Ad-Control) or adenovirus Rheb (Ad-Rheb) was infected in the primary cortical neurons at 14 days in vitro, and BACE1 and other proteins were detected in lysates, as in Fig. 1A. sAPPβ was measured in the medium, and Aβ was detected by ELISA, as indicated under “Experimental Procedures.” Note that Ad-Rheb markedly inhibited BACE1 levels and reduced BACE1 activity, as demonstrated by decrease in sAPPβ, APP-CTFβ, and Aβ (x-40 and x-42) levels (n = 8, ***, p < 0.001, **, p < 0.01 versus Ad-control). C, depletion of Rheb enhanced BACE1 in the cultured neurons. Primary cortical neurons (14 days in vitro) were infected with lentiviral particles expressing scrambled shRNA or Rheb shRNA1, and after 3 days, the neurons were lysed to detect BACE1, sAPPβ, APP-CTFβ, APP-CTFα, and Aβ (x-40 and x-42) (n = 6, ***, p < 0.001, *, p < 0.05 versus control shRNA (Con shRNA)).
Next, we hypothesized, based on the overexpression data above, that Rheb depletion should enhance BACE1 protein levels. To test this, we depleted endogenous Rheb in primary cortical neurons using lentiviral shRNA. We found that shRNA for Rheb that effectively reduced Rheb levels also increased BACE1 levels and its activity, but had no effect on PDGFR, APP-FL, or mTOR levels in cultured cortical neurons (Fig. 2C). This indicates that Rheb physiologically regulates BACE1 protein stability.
At present, we do not know whether Rheb also affects α- or γ-secretases, which also cleave APP (39, 40). We found that the production of APP-CTFα of APP, which is a target of α-secretase (41), or the Notch-HES1 pathway, which is a target for γ-secretase (42), is not altered in cells that overexpress Rheb (Figs. 1A and 2B). This suggests that Rheb selectively modulates BACE1 levels and activity.
Rheb Reduces BACE1 Protein Half-life
Next, we investigated whether Rheb affects BACE1 gene expression, using RT-PCR, as described previously (35). We found no significant changes in the levels of BACE1 mRNA between Myc and Myc-Rheb-expressing cells (Fig. 3A), indicating that Rheb down-regulates BACE1 protein, but not its mRNA. To further confirm this, we tested whether Rheb destabilizes BACE1 by enhancing its protein degradation. We blocked protein synthesis with cycloheximide in cells that were transfected with Myc (control) or Myc-Rheb, for 3, 6, or 9 h. Treatment of cycloheximide showed an accelerated degradation of BACE1 in Myc-Rheb-overexpressing cells, as the rate of BACE1 degradation is reduced to ∼6 h when compared with ∼8 h in Myc alone transfected cells (Fig. 3B). Overall, this indicates that Rheb reduces BACE1 stability through the protein degradation pathway. Because the effect is relatively small (∼2 h decrease in the half-life of BACE1), we predict that Rheb might suppress BACE1 through additional means, for example, through translational control, but that remains to be determined.
FIGURE 3.

Rheb GTPase degrades BACE1 protein in an mTOR-independent manner. A, Myc or Myc-Rheb cDNA (2 μg each) was transfected in HEK293 cells, and after 48 h, the cells were lysed and RNA was isolated to detect BACE1 levels and GAPDH by RT-PCR, as indicated under “Experimental Procedures.” B, Myc or Myc-Rheb was transfected in HEK293 cells. At 36 h (0-h chase), the cycloheximide (100 μm) was added into the culture medium, and the cells were chased for 3, 6, and 9 h. The rate of half-life of BACE1 was significantly reduced in the Myc-Rheb cells when compared with vector alone, indicating that Rheb degrades BACE1 (*, p < 0.05 versus Myc alone, analysis of variance). PERK was not altered by Myc-Rheb. n.s., not significant. C, Myc or Myc Rheb cDNAs (WT, D60K, C181S, or Q64L, 2 μg each) were transfected into HEK293 cells, and after 48 h, they were lysed to detect BACE1 and other proteins, as well as mTOR signaling (by phosphorylation of S6K (ps6K) at Thr-389), as indicated under “Experimental Procedures.” Rheb D60K was unable to induce BACE1 degradation, whereas Rheb C181S was as effective as Rheb WT (***, p < 0.001 versus Myc alone, Student's t test). D, Myc or Myc-Rheb cDNA (2 μg each) was transfected in HEK293 cells after 36 h, and DMSO (0.1%) or rapamycin (Rapa, 100 nm) was added for 12 h. Rapamycin blocked mTOR activation (as measured by phosphorylation of S6K at Thr389), but did not appreciably affect the Rheb-mediated degradation of BACE1 (***, p < 0.001 versus DMSO, Student's t test).
Rheb Promotes BACE1 Degradation Independent of mTOR, but Requires GTP
All these data establish that Rheb is a novel regulator of BACE1 protein stability and mediates BACE1 degradation in cultured cells and intact mice hippocampus. We next asked how Rheb mediates BACE1 degradation. Rheb consists of a GTP binding domain that is essential for its mTOR activity, and has a farnesylation domain that is required for binding to the membranous structures such as vesicles (43). We found that the Rheb D60K, a mutant that cannot bind to GTP, failed to promote BACE1 degradation, whereas the Rheb C181S mutant, which cannot be farnesylated, induced BACE1 degradation similar to WT (Fig. 3C). Another mutant Rheb Q64L, in which GTP is constitutively bound, suppressed BACE1 similar to WT. This indicates that although GTP binding of Rheb is essential for its BACE1 regulation, a constitutively bound GTP itself cannot further potentiate BACE1 regulation, thus hinting for unknown signals that in addition to GTP binding orchestrate Rheb-mediated BACE1 regulation. Overall, these data indicate that GTP binding to Rheb, but not farnesylation contributes to BACE1 suppression. Note that Rheb D60K failed to activate mTOR (assessed by the extent of S6K phosphorylation), whereas Rheb C181S activated mTOR similar to Rheb WT (Fig. 3C), as shown before (11). Because the GTP binding mutant (D60K) was unable to promote BACE1 degradation and this mutant is essential for its mTORC1 activity (Fig. 3C), we wondered whether Rheb promotes BACE1 degradation via mTOR activity. To test this, we transfected HEK293 cells with Myc or Myc-Rheb as in Fig. 1A and treated them with DMSO or rapamycin, a potent inhibitor of TORC1 activity. We found that rapamycin, which robustly inhibited mTOR activation (as measured by the phosphorylation of S6K), had no effect on Rheb-mediated BACE1 loss (Fig. 3D). This is consistent with a previous study by Vassar and co-workers (23) that showed rapamycin had no appreciable effect on the levels of BACE1 protein. Together, these data suggest that GTP binding to Rheb, but not its mTORC1 activity is necessary for BACE1 degradation.
Rheb Interacts with BACE1
We next hypothesized that Rheb promotes BACE1 degradation by physically interacting with BACE1. To test the binding between these two proteins, first, we carried out co-immunoprecipitation (Co-IP) studies in the brain with a BACE1 antibody that was suitable for IP, using a previously published protocol (34). However, we failed to detect endogenous Rheb Co-IP with BACE1 (data not shown). We reasoned that the interaction could be weaker or transient, and thus, could not be readily detected. To investigate whether BACE1 and Rheb interact in vitro, we incubated the BACE1 IgG or control IgG immunoprecipitates from BACE1+/+ or BACE1−/− mouse brains with recombinant Rheb, and we could readily detect an interaction between BACE1 and Rheb (Fig. 4A). This interaction was observed only in BACE1+/+ brain lysates, supporting the notion that BACE1 and Rheb interaction is specific. Immunocytochemistry revealed that overexpressed Rheb and BACE1 co-localize readily within cells displaying perinuclear morphology (Fig. 4B), consistent with previous studies (44, 45). Next, we overexpressed GST-Rheb in HEK293 cells and performed GST pulldown assays. We found that GST-Rheb interacted readily with endogenous BACE1 (Fig. 4C). Note that this interaction is specific to Rheb WT, as the Rheb D60K mutant fails to bind with BACE1, indicating that GTP binding to Rheb is required for association with BACE1, and presumably, for the degradation of BACE1. We found that Rheb C181S, which promoted BACE1 degradation like Rheb WT (Fig. 3C), also interacted with BACE1 in a manner similar to that of Rheb WT (Fig. 4C). Taken together, these data suggest that Rheb binds to BACE1 and that this interaction is necessary for BACE1 degradation.
FIGURE 4.
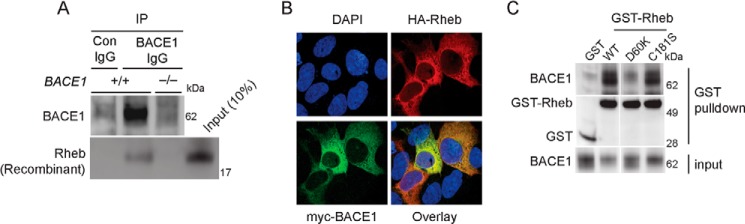
Rheb binds with BACE1. A, BACE1+/+ and BACE1−/− mice brain lysates were treated with BACE1 IgG or control IgG (Con IgG), precipitated with Sepharose beads, and incubated with recombinant Rheb. Rheb co-precipitated with the BACE1 IgG sample, which effectively pulled down BACE1, indicating Rheb and BACE1 interactions. B, HEK293 cells were transfected with HA-Rheb or Myc-BACE1 for 36 h and fixed for the microscopy work, as indicated under “Experimental Procedures.” Note that Rheb and BACE1 readily co-localized in cells with perinuclear localization. C, GST or GST-Rheb (WT, D60K, or C181S) was transfected in HEK293 cells, and after 48 h, lysed and incubated with glutathione beads to detect the interaction of Rheb with endogenous BACE1. BACE1 interacted with Rheb WT or Rheb C181S, but not with Rheb D60K.
Rheb Promotes BACE1 Degradation through Lysosomal and Proteasomal Pathways
Next, we investigated how Rheb mediates BACE1 degradation. Previous studies have established that BACE1 can be degraded through two major degradation pathways: the ubiquitin-proteasome and the lysosomal degradation pathways (46–48). We tested whether these pathways are involved in Rheb-mediated BACE1 stability (46–48), using a pharmacological approach in HEK293 cells. We found that the lysosomal inhibitor, chloroquine (10–100 μm), as well as the proteasomal inhibitor, MG132 (1–25 μm), both inhibited Rheb-mediated reduction of BACE1 in a dose-dependent manner (Fig. 5, A–C). This effect of the inhibitors is specific to BACE1, as they did not appreciably affect the levels of PDGFR in these cells. These data suggest that Rheb employs two major degradation pathways to degrade BACE1.
FIGURE 5.
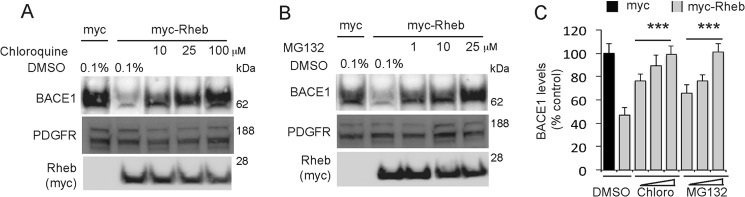
Rheb degrades BACE1 through the lysosomal and proteasomal degradation pathway, and Rheb levels are reduced in AD postmortem tissue. A and B, HEK293 cells were transfected with Myc or Myc-Rheb cDNA (2 μg each) for 36 h and treated with DMSO or with different doses of lysosomal inhibitor (10–100 μm), chloroquine (A), or the proteasomal inhibitor (1–25 μm), MG132 (B), for 9 h, and then lysed to detect BACE1 or PDGFR, as described under “Experimental Procedures.” C, chloroquine (Chloro) and MG132 both blocked Rheb-mediated degradation of BACE1 (***, p < 0.001 versus DMSO-treated Myc-Rheb, analysis of variance).
Rheb Is Dysregulated in AD Brain
Because BACE1 is up-regulated in AD, we wondered whether the dysregulation of Rheb might contribute to the elevated cerebral BACE1 levels in AD. We evaluated the expression of Rheb protein by Western blotting in the prefrontal cortex of AD postmortem brain tissue, and corresponding non-AD (control) tissue. Although variable, there was a significant loss of Rheb levels, which is correlated with enhanced BACE1 levels, in the AD samples when compared with non-AD controls (Fig. 6), whereas actin levels were not markedly affected in these samples. This suggests that diminution of Rheb may contribute to the enhanced BACE1 levels in AD.
FIGURE 6.
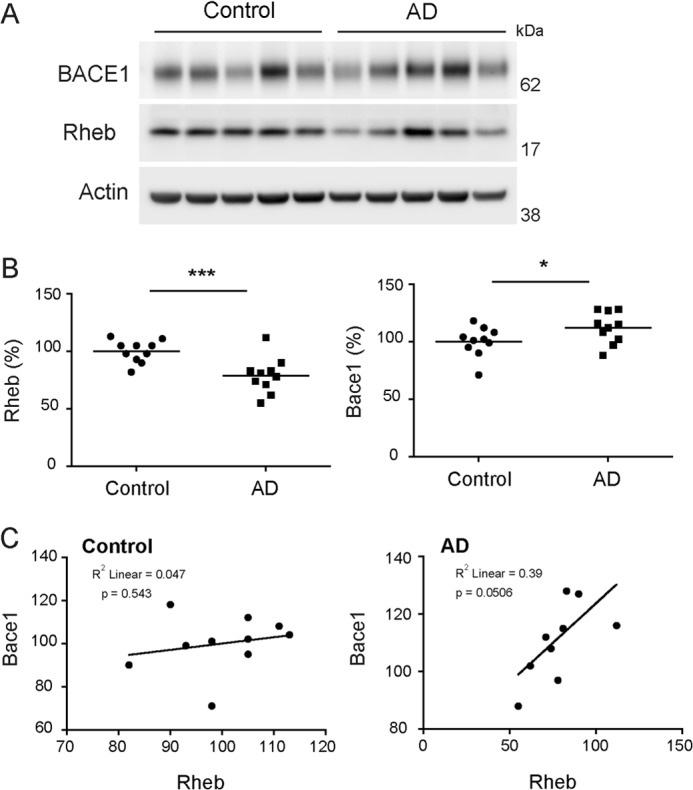
Rheb levels are decreased in AD prefrontal cortex. A, representative Western blot of BACE1 and Rheb in AD versus control samples. B, quantitative analysis of the Western blots showed that Rheb levels are significantly reduced, whereas BACE1 levels significantly increased in AD groups relative to control group (n = 10/group, ***, p < 0.001, *, p < 0.05 versus control, Student's t test). C, Rheb reduction is correlated to BACE1 increase in AD and not in control group.
DISCUSSION
The most significant and novel findings of this study are as follows. (a) We have identified Rheb GTPase as a novel physiological regulator of BACE1 stability and activity. (b) Rheb, according to the classical view, promotes protein translation through mTOR, but we have found Rheb reduces the half-life of BACE1, an effect that is independent of mTOR activity, revealing hitherto unknown dual but opposite roles of Rheb. (c) To degrade BACE1, we found that Rheb must interact with BACE1 in a GTP-dependent manner, as the Rheb mutant (defective in binding to GTP) fails to interact with, or degrade, BACE1. This is an intriguing finding because the importance of GTP-bound Rheb to the functions other than mTOR activity is largely unknown. Hence, this is the first demonstration of the role of GTP binding in Rheb to destabilizing protein, other than activating mTOR. (d) We demonstrate, for the first time, that Rheb levels are down-regulated in the AD brain, which is consistent with an increased BACE1 expression, indicating a potential relevance of Rheb-BACE1 circuitry in AD pathology.
Our findings that Rheb induces BACE1 degradation are somewhat paradoxical because, as indicated before, Rheb is a potent activator of mTOR signaling, which, in general, enhances protein turnover either by increasing protein translation or by decreasing autophagy. Our data establish that the effect of Rheb on BACE1 is independent of mTOR activity, suggesting unknown mTOR-independent regulations. However, what distinguishes the mTOR-dependent and -independent effects of Rheb is currently unclear. Our preliminary data indicate that Rheb promotes BACE1 degradation in a serum-dependent manner.4 This leads us to speculate that some unknown phosphorylation switch on Rheb might be controlling BACE1 degradation. Nevertheless, this is the first study, to the best of our knowledge, that has implicated Rheb in protein degradation. As this effect by Rheb is specific to BACE1, but not to APP or other membrane proteins that we tested, it raises two important questions. What determines the selectivity of Rheb toward BACE1 degradation? Also, are there other targets that can be degraded by Rheb? In the future, we would want to address these questions.
There is an emerging view that the physiological regulation of BACE1 degradation involves both lysosome and proteasome pathways (46, 48, 49). Based on our findings, we predict that BACE1 degradation through these two pathways is not mutually exclusive. Evidence is accumulating to suggest a cross-talk between these two degradative pathways (reviewed in Ref. 50); for example, ubiquitin-tagged proteins can be degraded through autophagy pathways (51). Recent work reveals that BACE1 is also ubiquitinated (49). Although the mechanisms by which Rheb recruits intracellular machinery to degrade BACE1 is currently unclear, we predict that Rheb may be involved in endocytic-lysosomal trafficking of BACE1. This is consistent with the already known role of Rheb in endocytic trafficking, which, like BACE1 degradation, also depends on GTP binding, but not on mTOR signaling (12). Because the lysosomal inhibitor chloroquine completely blocked Rheb-mediated BACE1 degradation, we posit that an endocytic-lysosomal fusion is a principal pathway by which Rheb degrades BACE1. In support of this view, other small GTPases are known to mediate the degradation of proteins through the endocytic degradation pathway. For example, Rab7 overexpression induces low density lipoprotein degradation (15); Rab12 GTPase induces constitutive degradation of the transferrin receptor (17); and Rab7 degrades epidermal growth factor receptor by enhancing the late endosome-lysosomal fusion (1). Although it is clear that Rheb reduces the half-life of BACE1 in dividing human cells in culture (Fig. 3B), we acknowledge, however, that the mechanisms could be different in brain neurons and dividing cells. In addition to reducing the half-life of BACE1, a possibility that there are other modes of BACE1 protein regulation, such as translational control, by Rheb, cannot be ruled out. Previous studies have demonstrated a molecular coupling of protein translation and protein degradation (52, 53). Whether Rheb promotes such coupling to regulate BACE1 stability remains to be determined.
A broader significance of our findings is the potential role of Rheb-BACE1 circuitry in aging and age-related dysfunctions, such as AD. Genetic deletion of BACE1 confirmed its dual role in the age-related decline of cognitive functions in normal aging and in AD; targeted deletion of Bace1 alleles in mice promotes age-related cognitive decline in normal aging, but also alleviates AD-related pathology as well as cognitive symptoms, such as learning and memory, in a transgenic mouse model of AD (17, 18). Although the role of Rheb in mediating the age-related cognitive functions in mammals is currently unknown, a dual role for Rheb in aging has been demonstrated in Caenorhabditis elegans (21); although Rheb is required for the intermittent fasting-induced longevity, the inhibition of Rheb mimics the caloric restriction-induced longevity. The biological basis for these dual, but opposite, roles of BACE1 and Rheb remains enigmatic. Because our study demonstrates a novel molecular link between Rheb and BACE1, the two aging-related proteins, we speculate that Rheb-BACE1 circuitry may play an important role in age-related brain functions. Because there was a reduction of Rheb levels in the AD postmortem tissue, in correlation with increased BACE1 (Fig. 6), which is linked to AD pathogenesis (18, 21, 54), we posit that Rheb overexpression may alleviate age-related abnormalities in AD through the inhibition of the BACE1-Aβ pathway.
Acknowledgments
We are grateful to Dr. Solomon H. Snyder of Johns Hopkins University for sharing reagents during the preliminary investigation of the study and to Dr. Louis Fernandes of The Harvard Brain Tissue Resource Center, which is supported in part by Public Health Service Grant R24 MH068855, for the generous sharing of human postmortem samples and Dr. Robert Vassar for the 3D5 BACE1 antibody. We are thankful to Natalie Defraene, Nancy Norton, and Trina Miles for the administrative support. We are grateful to the people in The Scripps Research Institute, Florida, especially in the Department of Neuroscience, for their continuous support for setting up the laboratory and providing technical support whenever needed.
This work was supported by an O'Keeffe Neuroscience Scholar Award (to W. P.) and by Scripps startup funds (to S. Subramaniam).
N. Shahani, W. Pryor, S. Swarnkar, and S. Subramaniam, unpublished results.
- mTOR
- mammalian target of rapamycin
- BACE1
- β-site APP-cleaving enzyme 1
- APP
- amyloid precursor protein
- sAPPβ
- soluble APPβ
- Aβ
- amyloid β
- AD
- Alzheimer disease
- IP
- immunoprecipitation
- Co-IP
- co-immunoprecipitation
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- PDGFR
- platelet-derived growth factor receptor
- AAV
- adeno-associated virus
- FL
- full-length
- CTF
- C-terminal fragment
- Ad
- adenovirus
- AP
- anterior-posterior
- ML
- medio-lateral
- DV
- dorso-ventral
- BA
- Brodmann area
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Aspuria P. J., Tamanoi F. (2004) The Rheb family of GTP-binding proteins. Cell. Signal. 16, 1105–1112 [DOI] [PubMed] [Google Scholar]
- 2. Yamagata K., Sanders L. K., Kaufmann W. E., Yee W., Barnes C. A., Nathans D., Worley P. F. (1994) rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 269, 16333–16339 [PubMed] [Google Scholar]
- 3. Neuman N. A., Henske E. P. (2011) Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol. Med. 3, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goorden S. M., Hoogeveen-Westerveld M., Cheng C., van Woerden G. M., Mozaffari M., Post L., Duckers H. J., Nellist M., Elgersma Y. (2011) Rheb is essential for murine development. Mol. Cell. Biol. 31, 1672–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zou J., Zhou L., Du X. X., Ji Y., Xu J., Tian J., Jiang W., Zou Y., Yu S., Gan L., Luo M., Yang Q., Cui Y., Yang W., Xia X., Chen M., Zhao X., Shen Y., Chen P. Y., Worley P. F., Xiao B. (2011) Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 20, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S. R., Kareva T., Yarygina O., Kholodilov N., Burke R. E. (2012) AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 20, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avruch J., Long X., Ortiz-Vega S., Rapley J., Papageorgiou A., Dai N. (2009) Amino acid regulation of TOR complex 1. Am. J. Physiol. Endocrinol. Metab. 296, E592–E602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Long X., Ortiz-Vega S., Lin Y., Avruch J. (2005) Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 280, 23433–23436 [DOI] [PubMed] [Google Scholar]
- 10. Clark G. J., Kinch M. S., Rogers-Graham K., Sebti S. M., Hamilton A. D., Der C. J. (1997) The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J. Biol. Chem. 272, 10608–10615 [DOI] [PubMed] [Google Scholar]
- 11. Li Y., Inoki K., Guan K. L. (2004) Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol. Cell. Biol. 24, 7965–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saito K., Araki Y., Kontani K., Nishina H., Katada T. (2005) Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 137, 423–430 [DOI] [PubMed] [Google Scholar]
- 13. Karbowniczek M., Cash T., Cheung M., Robertson G. P., Astrinidis A., Henske E. P. (2004) Regulation of B-Raf kinase activity by tuberin and Rheb is mammalian target of rapamycin (mTOR)-independent. J. Biol. Chem. 279, 29930–29937 [DOI] [PubMed] [Google Scholar]
- 14. Vassar R., Kandalepas P. C. (2011) The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer's disease. Alzheimers Res. Ther. 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willem M., Garratt A. N., Novak B., Citron M., Kaufmann S., Rittger A., DeStrooper B., Saftig P., Birchmeier C., Haass C. (2006) Control of peripheral nerve myelination by the β-secretase BACE1. Science 314, 664–666 [DOI] [PubMed] [Google Scholar]
- 16. Luo Y., Bolon B., Kahn S., Bennett B. D., Babu-Khan S., Denis P., Fan W., Kha H., Zhang J., Gong Y., Martin L., Louis J. C., Yan Q., Richards W. G., Citron M., Vassar R. (2001) Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 4, 231–232 [DOI] [PubMed] [Google Scholar]
- 17. Ohno M., Sametsky E. A., Younkin L. H., Oakley H., Younkin S. G., Citron M., Vassar R., Disterhoft J. F. (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41, 27–33 [DOI] [PubMed] [Google Scholar]
- 18. Laird F. M., Cai H., Savonenko A. V., Farah M. H., He K., Melnikova T., Wen H., Chiang H. C., Xu G., Koliatsos V. E., Borchelt D. R., Price D. L., Lee H. K., Wong P. C. (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 25, 11693–11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vetrivel K. S., Meckler X., Chen Y., Nguyen P. D., Seidah N. G., Vassar R., Wong P. C., Fukata M., Kounnas M. Z., Thinakaran G. (2009) Alzheimer disease Aβ production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 284, 3793–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. (2002) β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 21. Yang L. B., Lindholm K., Yan R., Citron M., Xia W., Yang X. L., Beach T., Sue L., Wong P., Price D., Li R., Shen Y. (2003) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4 [DOI] [PubMed] [Google Scholar]
- 22. Dislich B., Lichtenthaler S. F. (2012) The membrane-bound aspartyl protease BACE1: Molecular and functional properties in Alzheimer's disease and beyond. Front. Physiol. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Connor T., Sadleir K. R., Maus E., Velliquette R. A., Zhao J., Cole S. L., Eimer W. A., Hitt B., Bembinster L. A., Lammich S., Lichtenthaler S. F., Hébert S. S., De Strooper B., Haass C., Bennett D. A., Vassar R. (2008) Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He X., Li F., Chang W. P., Tang J. (2005) GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J. Biol. Chem. 280, 11696–11703 [DOI] [PubMed] [Google Scholar]
- 25. Sannerud R., Declerck I., Peric A., Raemaekers T., Menendez G., Zhou L., Veerle B., Coen K., Munck S., De Strooper B., Schiavo G., Annaert W. (2011) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. U.S.A. 108, E559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okada H., Zhang W., Peterhoff C., Hwang J. C., Nixon R. A., Ryu S. H., Kim T. W. (2010) Proteomic identification of sorting nexin 6 as a negative regulator of BACE1-mediated APP processing. FASEB J. 24, 2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He W., Lu Y., Qahwash I., Hu X. Y., Chang A., Yan R. (2004) Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat. Med. 10, 959–965 [DOI] [PubMed] [Google Scholar]
- 28. Mandrekar-Colucci S., Karlo J. C., Landreth G. E. (2012) Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J. Neurosci. 32, 10117–10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeppsson F., Eketjäll S., Janson J., Karlström S., Gustavsson S., Olsson L. L., Radesäter A. C., Ploeger B., Cebers G., Kolmodin K., Swahn B. M., von Berg S., Bueters T., Fälting J. (2012) Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of Alzheimer disease. J. Biol. Chem. 287, 41245–41257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kandalepas P. C., Vassar R. (2012) Identification and biology of β-secretase. J. Neurochem. 120, Suppl. 1, 55–61 [DOI] [PubMed] [Google Scholar]
- 31. Subramaniam S., Sixt K. M., Barrow R., Snyder S. H. (2009) Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramaniam S., Napolitano F., Mealer R. G., Kim S., Errico F., Barrow R., Shahani N., Tyagi R., Snyder S. H., Usiello A. (2012) Rhes, a striatal-enriched small G protein, mediates mTOR signaling and l-DOPA-induced dyskinesia. Nat. Neurosci. 15, 191–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hilgenberg L. G., Smith M. A. (2007) Preparation of dissociated mouse cortical neuron cultures. J. Vis. Exp. 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuzuya A., Uemura K., Kitagawa N., Aoyagi N., Kihara T., Ninomiya H., Ishiura S., Takahashi R., Shimohama S. (2007) Presenilin 1 is involved in the maturation of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1). J. Neurosci. Res. 85, 153–165 [DOI] [PubMed] [Google Scholar]
- 35. Nawrot B., Antoszczyk S., Maszewska M., Kuwabara T., Warashina M., Taira K., Stec W. J. (2003) Efficient inhibition of β-secretase gene expression in HEK293 cells by tRNAVal-driven and CTE-helicase associated hammerhead ribozymes. Eur. J. Biochem. 270, 3962–3970 [DOI] [PubMed] [Google Scholar]
- 36. Miyashita M., Oshiumi H., Matsumoto M., Seya T. (2011) DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 31, 3802–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inoki K., Li Y., Xu T., Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rubinsztein D. C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]
- 39. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., Kopan R. (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 [DOI] [PubMed] [Google Scholar]
- 40. Colombo A., Wang H., Kuhn P. H., Page R., Kremmer E., Dempsey P. J., Crawford H. C., Lichtenthaler S. F. (2012) Constitutive α- and β-secretase cleavages of the amyloid precursor protein are partially coupled in neurons, but not in frequently used cell lines. Neurobiol. Dis. 49C, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Efferson C. L., Winkelmann C. T., Ware C., Sullivan T., Giampaoli S., Tammam J., Patel S., Mesiti G., Reilly J. F., Gibson R. E., Buser C., Yeatman T., Coppola D., Winter C., Clark E. A., Draetta G. F., Strack P. R., Majumder P. K. (2010) Downregulation of Notch pathway by a γ-secretase inhibitor attenuates AKT/mammalian target of rapamycin signaling and glucose uptake in an ERBB2 transgenic breast cancer model. Cancer Res. 70, 2476–2484 [DOI] [PubMed] [Google Scholar]
- 43. Hanker A. B., Mitin N., Wilder R. S., Henske E. P., Tamanoi F., Cox A. D., Der C. J. (2010) Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene 29, 380–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi K., Nakagawa M., Young S. G., Yamanaka S. (2005) Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J. Biol. Chem. 280, 32768–32774 [DOI] [PubMed] [Google Scholar]
- 45. Yan R., Han P., Miao H., Greengard P., Xu H. (2001) The transmembrane domain of the Alzheimer's β-secretase (BACE1) determines its late Golgi localization and access to β-amyloid precursor protein (APP) substrate. J. Biol. Chem. 276, 36788–36796 [DOI] [PubMed] [Google Scholar]
- 46. Qing H., Zhou W., Christensen M. A., Sun X., Tong Y., Song W. (2004) Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 18, 1571–1573 [DOI] [PubMed] [Google Scholar]
- 47. Koh Y. H., von Arnim C. A., Hyman B. T., Tanzi R. E., Tesco G. (2005) BACE is degraded via the lysosomal pathway. J. Biol. Chem. 280, 32499–32504 [DOI] [PubMed] [Google Scholar]
- 48. Gong B., Chen F., Pan Y., Arrieta-Cruz I., Yoshida Y., Haroutunian V., Pasinetti G. M. (2010) SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer's disease amyloidosis and improves synaptic function. Aging Cell 9, 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang E. L., Cameron A. N., Piazza F., Walker K. R., Tesco G. (2010) Ubiquitin regulates GGA3-mediated degradation of BACE1. J. Biol. Chem. 285, 24108–24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Korolchuk V. I., Menzies F. M., Rubinsztein D. C. (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398 [DOI] [PubMed] [Google Scholar]
- 51. Kirkin V., McEwan D. G., Novak I., Dikic I. (2009) A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269 [DOI] [PubMed] [Google Scholar]
- 52. Chuang S. M., Chen L., Lambertson D., Anand M., Kinzy T. G., Madura K. (2005) Proteasome-mediated degradation of cotranslationally damaged proteins involves translation elongation factor 1A. Mol. Cell. Biol. 25, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dimitrova L. N., Kuroha K., Tatematsu T., Inada T. (2009) Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 284, 10343–10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Holsinger R. M., McLean C. A., Beyreuther K., Masters C. L., Evin G. (2002) Increased expression of the amyloid precursor β-secretase in Alzheimer's disease. Ann. Neurol. 51, 783–786 [DOI] [PubMed] [Google Scholar]
- 55. Franklin K. B. J., Paxinos G. (2008) The Mouse Brain in Stereotaxic Coordinates, Compact 3rd Ed., Elsevier Academic Press, San Diego [Google Scholar]