

Background: Cannabinoid-1 (CB1) receptor allosteric modulator ORG27569 increases CP55,940 binding, yet antagonizes G protein signaling.
Results: ORG27569 binding sterically blocks movement in CB1 extracellular loops and transmembrane helix 6 (TMH6).
Conclusion: ORG27569 increases CP55,940 binding by promoting an intermediate receptor conformation where changes important for signaling are blocked.
Significance: This information may lead to the rational design of new allosteric modulators.
Keywords: Allosteric Regulation, Cannabinoid Receptors, Cannabinoids, G Protein-coupled Receptors (GPCR), Signaling
Abstract
The cannabinoid 1 (CB1) allosteric modulator, 5-chloro-3-ethyl-1H-indole-2-carboxylic acid [2-(4-piperidin-1-yl-phenyl)-ethyl]-amide) (ORG27569), has the paradoxical effect of increasing the equilibrium binding of [3H](−)-3-[2-hydroxyl-4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxylpropyl]cyclohexan-1-ol (CP55,940, an orthosteric agonist) while at the same time decreasing its efficacy (in G protein-mediated signaling). ORG27569 also decreases basal signaling, acting as an inverse agonist for the G protein-mediated signaling pathway. In ligand displacement assays, ORG27569 can displace the CB1 antagonist/inverse agonist, N-(piperidiny-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide(SR141716A). The goal of this work was to identify the binding site of ORG27569 at CB1. To this end, we used computation, synthesis, mutation, and functional studies to identify the ORG27569-binding site in the CB1 TMH3-6-7 region. This site is consistent with the results of K3.28192A, F3.36200A, W5.43279A, W6.48356A, and F3.25189A mutation studies, which revealed the ORG27569-binding site overlaps with our previously determined binding site of SR141716A but extends extracellularly. Additionally, we identified a key electrostatic interaction between the ORG27569 piperidine ring nitrogen and K3.28192 that is important for ORG27569 to act as an inverse agonist. At this allosteric site, ORG27569 promotes an intermediate conformation of the CB1 receptor, explaining ORG27569's ability to increase equilibrium binding of CP55,940. This site also explains ORG27569's ability to antagonize the efficacy of CP55,940 in three complementary ways. 1) ORG27569 sterically blocks movements of the second extracellular loop that have been linked to receptor activation. 2) ORG27569 sterically blocks a key electrostatic interaction between the third extracellular loop residue Lys-373 and D2.63176. 3) ORG27569 packs against TMH6, sterically hindering movements of this helix that have been shown to be important for receptor activation.
Introduction
The G protein-coupled receptor (GPCR)2 superfamily of integral membrane proteins is composed of ∼1000 members (1) and includes ∼3% of the human genome (2). Considering their ubiquity and fundamental importance to cellular function, it is not surprising that ∼60% of pharmaceuticals target GPCRs (3). Unfortunately, many of these drugs have numerous side effects, due to a lack of receptor subtype selectivity (4) and/or an interference with physiological signaling (5). This lack of receptor subtype selectivity is thought to be due (in part) to high sequence convergence at the orthosteric site, which is the binding site of most endogenous ligands and pharmaceutical drugs (6).
The discovery of ligands (i.e. allosteric modulators) that bind to an allosteric site has generated considerable interest. Allosteric sites are topographically distinct from the orthosteric site; classically, allosteric modulators are thought to influence the binding and/or efficacy of orthosteric ligands (7). Allosteric-based drugs could potentially have reduced side effects, due to the increased evolutionary divergence at allosteric sites (8). Additionally, allosteric modulators have been observed to direct signaling down specific second messenger pathways (i.e. biased agonism) (9). These results suggest that allosterically based therapies have the potential for unprecedented receptor subtype selectivity and functional control.
The discovery of an allosteric site at the cannabinoid 1 (CB1) receptor has generated significant interest, due to the anticipated therapeutic potential of allosteric-based drugs. The CB1 receptor is a member of the class A (rhodopsin-like) family of GPCRs; it is found primarily in the central nervous system (CNS) and is important in the regulation of neuronal activity. The CB1 receptor has been implicated in Alzheimer disease, cancer, obesity, and pain (10). Regrettably, drugs that target the orthosteric site of the CB1 receptor have failed due to unacceptable CNS-related side effects (11). CB1 allosteric modulators could potentially avoid these side effects, due to an anticipated increase in receptor subtype selectivity, as well as a predicted improvement in functional control (12). ORG27569 (see Fig. 1), the prototypical CB1 allosteric modulator, has the paradoxical effect of increasing the equilibrium binding of CP55,940 (a CB1 agonist, see Fig. 1), while concurrently antagonizing its G protein-mediated efficacy (13). Additionally, ORG27569 acts as an inverse agonist of G protein-mediated signaling (14).
FIGURE 1.

Compounds evaluated in this study. Ring letters are used in the description of aromatic interactions.
To understand these seemingly contradictory effects, we have used computational methods together with mutagenesis, synthesis, and pharmacological studies to identify an allosteric binding site for ORG27569 at the CB1 receptor and to probe its relationship to G protein signaling effects. We show here that the ORG27569-binding site is located in the TMH3-6-7 region of the CB1 receptor, partially overlapping the SR141716A-binding site but extending extracellularly. We identify receptor residues that are crucial not only for ORG27569 binding in the presence of CP55,940, but also residues important to the inverse agonism that ORG27569 exhibits when applied alone. Finally, we relate the location of this binding site to the conformational changes associated with G protein activation on the extracellular side of CB1 that are blocked by ORG27569.
EXPERIMENTAL PROCEDURES
Amino Acid Numbering
The numbering scheme suggested by Ballesteros and Weinstein was employed here (15). In this system, the most highly conserved residue in each TMH is assigned a locant of 0.50. This number is preceded by the TMH number and followed by the absolute sequence number in superscript. All other residues in a TMH are numbered relative to this residue. Sequence numbers used are human CB1 sequence numbers (16).
Chemistry
Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Anhydrous solvents were obtained from Aldrich and used directly. All reactions involving air- or moisture-sensitive reagents were performed under a nitrogen atmosphere. Numbers in boldface refer to compounds shown in Scheme 1.
SCHEME 1.

Analytical thin layer chromatography (TLC) was carried out on plates precoated with silica gel GHLF (250 μm thickness). TLC visualization was accomplished with a UV lamp. Silica gel chromatography was performed using RediSep prepacked silica gel cartridges. HPLC analyses were performed using a Waters Emperor chromatography system comprised of a 1525 Binary Pump, 2487 Dual 1 Absorbance Detector, and a 717 Plus Autosampler using a Waters C-18 reverse phase XBridge column (5 μm; 4.6 × 100 mm; 254 nm; 1 ml/min). 1H NMR spectra were run on a Bruker Advance 300 MHz NMR spectrometer. Low resolution mass spectra were run on a Sciex API 150 EX mass spectrometer (PerkinElmer Life Science) outfitted with atmospheric pressure chemical ionization or electrospray ionization (turbospray) sources in positive or negative modes. High resolution mass spectrometry (HRMS) was performed using a Waters Synapt HDMS quadrupole time of flight (Q-TOF) mass spectrometer interfaced to a Waters Acquity UPLC system. HRMS data were acquired in negative electrospray MS resolution mode.
4-Cyclohexylbenzaldehyde (2)
This compound was prepared according to the patent procedure described in Ref. 17. To a dry 250-ml round bottom flask was added 1-bromo-4-cyclohexylbenzene (2.6 ml, 14.0 mmol) and anhydrous THF (16 ml) under nitrogen. The solution was cooled in a dry ice/acetone bath, and n-butyllithium (1.6 m in hexane) (11 ml, 17.6 mmol, 1.26 eq) was added dropwise with stirring under nitrogen. The reaction mixture was stirred in the dry ice/acetone bath for 1 h. To the cold stirred reaction mixture was added dropwise anhydrous N,N-dimethylformamide (11 ml, 142 mmol, 10.1 eq). The reaction mixture was stirred in the dry ice/acetone bath for 1 h. The dry ice/acetone bath was replaced with an ice-water bath, and the reaction mixture was allowed to warm to 0 °C. To the stirred cold reaction mixture was added saturated ammonium chloride. The quenched reaction mixture was transferred to a separatory funnel and extracted with EtOAc. The organic phase was separated, dried (MgSO4), and filtered, and the filtrate was concentrated to give the crude product as a yellow oil. The crude product was purified by flash chromatography over silica (120 g) with a hexane/EtOAc gradient (100:0 to 50:50) to give 2.4 g (91%) of the desired product as a pale yellow oil. Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 9.97 (s, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 2.59 (m, 1H), 1.69–1.98 (m, 5H), 1.16–1.54 (m, 5H).
1-Cyclohexyl-4-[(E)-2-nitroethenyl]benzene (3)
This compound was prepared according to a procedure (18) described for another substituted benzaldehyde. 4-Cyclohexylbenzaldehyde (2.4 g, 0.0127 mol), ammonium acetate (1.74 g, 0.0226 mol, 1.78 eq), and nitromethane (25 ml) were combined in a round bottom flask, and the reaction mixture was heated at 100 °C with stirring under nitrogen for 2.5 h. The brown-orange reaction mixture was allowed to cool at room temperature and concentrated. The resulting oil was dissolved in EtOAc, and the solution was washed with 1 n HCl followed by saturated NaHCO3. The organic phase was separated, dried (MgSO4), and filtered, and the filtrate was concentrated to give an orange oil. The crude product was purified by flash chromatography over silica (80 g) with a hexane/EtOAc gradient (100:0 to 90:10) to give 1.27 g (43%) of the title compound as an orange oil. Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 8.00 (d, J = 13.7 Hz, 1H), 7.57 (d, J = 13.7 Hz, 1H), 7.48 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 2.56 (m, 1H), 1.84 (m, 5H), 1.50–1.16 (m, 5H).
2-(4-Cyclohexylphenyl)ethan-1-amine (4)
This compound was prepared according to a procedure described previously (19) for the reduction of a nitrovinyl-substituted indole. To a dry round bottom flask was added 1-cyclohexyl-4-[(E)-2-nitroethenyl]benzene (396 mg, 1.7 mmol) and anhydrous THF (24 ml). To the stirred solution was added dropwise lithium aluminum hydride (1 m in THF) (8 ml, 8 mmol, 4.7 eq) at room temperature under nitrogen. The reaction mixture was heated at reflux for 1 h. The reaction mixture was allowed to cool at room temperature and then cooled in an ice-water bath. To the stirred cold reaction mixture was slowly added water dropwise (0.3 ml) (H2 was evolved) followed by 15% NaOH (0.3 ml) and water (0.9 ml). The aqueous mixture was filtered through a pad of celite, and the pad was washed with EtOAc. The filtrate was partially concentrated in vacuo, washed with saturated NaHCO3 followed by brine, dried (MgSO4), and filtered, and the filtrate was concentrated to give 0.314 g (90%) of the crude product as a gold-yellow oil. The crude product was used directly without further purification. ES-MS was 204 (MH+).
Ethyl 3-Acetyl-5-chloro-1H-indole-2-carboxylate (6)
This compound was prepared according to a procedure described previously (20) for the corresponding 5-bromoindole analog. To a dry 500-ml round bottom flask was added ethyl-5-chloroindole-2-carboxylate (1.71 g, 0.0076 mol). The flask was capped with a rubber septum and purged with nitrogen. The indole ester was partially dissolved in anhydrous CH2Cl2 (40 ml). The stirred mixture was cooled in an ice-water bath, and diethylaluminum chloride (1 m in hexane) (15 ml, 0.015 mol, 2.0 eq) was added slowly dropwise. The reaction mixture was stirred with cooling for 30 min. To the cold stirred reaction mixture was added dropwise a solution of acetyl chloride (1.1 ml, 0.0154 mol, 2.0 eq) in CH2Cl2 (40 ml). The reaction mixture was stirred under nitrogen in the ice-water bath for 6 h and then allowed to slowly warm to room temperature overnight. The reaction mixture was cooled in an ice-water bath, and saturated NaHCO3 (100 ml) was slowly added. The quenched reaction mixture was extracted with CH2Cl2. The organic phase was separated, washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated to give a gold-yellow solid. The crude product was purified by flash chromatography over silica (80 g) with dichloromethane to give 1.12 g (56%) of the desired product as a pale yellow solid. Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 9.12 (br s, 1H), 8.12 (s, 1H), 7.34 (m, 2H), 4.48 (q, J = 7.1 Hz, 2H), 2.74 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H). ES-MS was 266 (M + H+), 288 (M + Na+), and 264 (M − H+).
Ethyl 5-Chloro-3-ethyl-1H-indole-2-carboxylate (7)
This compound was prepared according to a procedure described previously (20) for the corresponding 5-bromoindole analog. To a stirred solution of ethyl 3-acetyl-5-chloro-1H-indole-2-carboxylate (1.1 g, 0.0041 mol) in trifluoroacetic acid (10 ml) was added dropwise triethylsilane (2.6 ml, 0.0163 mol, 4 eq) at room temperature under nitrogen. After 4 h, the reaction mixture was poured into water, and the aqueous mixture was extracted with EtOAc. The organic extract was washed with water followed by brine, dried (MgSO4), and filtered, and the filtrate was partially concentrated to give a suspension. To the suspension was added EtOAc. Fumes were released upon addition of EtOAc, which suggested that trifluoroacetic acid remained. The organic solution was washed with saturated NaHCO3 followed by brine. The organic phase was separated, dried (MgSO4), and filtered, and the filtrate was concentrated to give a wet yellow solid. The solid was washed with hexane to give 0.893 g (87%) of the desired product as a pale yellow solid. Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 8.71 (br s, 1H), 7.66 (m, 1H), 7.30 (d, J = 8.7 Hz, 1H), 7.26 (dd, J = 1.9 Hz, 8.8 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 3.07 (q, J = 7.5 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H), 1.26 (t, J = 7.5 Hz, 3H). ES-MS was 250 (M − H+).
5-Chloro-3-ethyl-1H-indole-2-carboxylic Acid (8)
To a stirred solution of ethyl 5-chloro-3-ethyl-1H-indole-2-carboxylate (405 mg, 1.61 mmol) in 1,4-dioxane (12 ml) was added 1 n NaOH (8 ml, 8 mmol, 5 eq). The reaction mixture was heated under nitrogen at 120 °C for 30 min. The reaction mixture was allowed to cool at room temperature. To the stirred reaction mixture was added 1 n HCl to pH ∼3 (indicator strip). The acidic mixture was transferred to a separatory funnel and extracted with EtOAc. The organic phase was separated, washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated to give 0.39 g of a white solid. The solid was dried at 80 °C under vacuum to give 0.376 g. 1H NMR indicated the sample contains 1,4-dioxane (14% by weight). Final yield of the desired product (less 1,4-dioxane) was 0.323 g (90%). Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 8.73 (br s, 1H), 7.69 (m, 1H), 7.30 (m, 2H), 3.13 (q, J = 7.5 Hz, 2H), 1.30 (t, J = 7.5 Hz, 3H). ES-MS was 222 (M − H+).
5-Chloro-N-[2-(4-cyclohexylphenyl)ethyl]-3-ethyl-1H-indole-2-carboxamide (9)
To a stirred mixture of 5-chloro-3-ethyl-1H-indole-2-carboxylic acid (77 mg, 0.34 mmol), 1,3-dicyclohexylcarbodiimide (81 mg, 0.39 mmol, 1.15 eq), and 1-hydroxybenzotriazole hydrate (52 mg, 0.38 mmol, 1.13 eq) in anhydrous dichloromethane (12 ml) was added dropwise a solution of crude 2-(4-cyclohexylphenyl)ethan-1-amine (147 mg) in CH2Cl2 (4 ml) at room temperature under nitrogen. After 22 h, the reaction mixture was filtered through a pad of celite, and the pad was washed with dichloromethane. The filtrate was concentrated to give a cloudy yellow oil. To the oil was added dichloromethane, and the turbid mixture was filtered. The filtrate was applied to a silica column (24 g) and purified by flash chromatography with a hexane/EtOAc gradient (100:0 to 50:50) to give a white solid that was dried under vacuum at 70 °C.
HPLC Conditions
Waters XBridge C-18 reverse phase column (5 μm; 4.6 × 100 mm; 1 ml/min; CH3CN/water (90:10)) indicated the compound was 94% pure. The solid was triturated at room temperature with EtOAc followed by MeOH (two times) and hexane (two times) and then dried under vacuum at 70 °C to give 23.2 mg (17%) of the title compound as a white solid. HPLC analysis indicated the compound was 98.4% pure. Using 1H NMR (300 MHz; CDCl3), the following were observed: δ 7.57 (d, J = 1.9 Hz, 1H), 7.33 (d, J = 8.7 Hz, 1H), 7.17 (m, 5H), 3.63 (t, J = 7.3 Hz, 2H), 2.93 (m, 4H), 2.47 (m, 1H), 1.79 (m, 5H), 1.53–1.22 (m, 5H), 1.16 (t, J = 7.5 Hz, 3H). HRMS analysis was calculated for C25H28ClN2O as 407.1890 (M − H+) and observed as 407.1904.
HEK293 Cells
HEK293 cells were stably transfected with varying CB1 receptor mutations: K3.28192A, F3.36200A, W5.43279A, W6.48356A, F3.25189A, and wild-type (WT). These transfected cell lines express the receptor at ∼1 pmol/mg protein as described previously (21, 22). Cells were maintained at 37 °C and 5% CO2 in DMEM + 4.5g/liter glucose supplemented with 10% fetal bovine serum, 0.7 mg/ml G418, and 0.6% penicillin/streptomycin. These cells were passed approximately twice a week using nonenzymatic cell dissociation solution. When using these cells in the [35S]GTPγS binding assay, cells were starved of fetal bovine serum for 24 h before being scraped and frozen in a pellet at −20 °C.
[35S]GTPγS Binding Assay
0.5 mg/ml cell membranes were incubated with the agonist with vehicle or modulator for 60 min at 30 °C in assay buffer (50 mm Tris-HCl; 50 mm Tris base; 5 mm MgCl2; 1 mm EDTA; 100 mm NaCl; 1 mm DTT; 0.1% BSA) in the presence of 0.1 nm [35S]GTPγS and 30 μm GDP, in a final volume of 500 μl. Binding was initiated by the addition of [35S]GTPγS. Nonspecific binding was measured in the presence of 30 μm GTPγS. The reaction was terminated by rapid vacuum filtration (50 mm Tris-HCl; 50 mm Tris base; 0.1% BSA) using a 24-well sampling manifold (cell harvester; Brandel, Gaithersburg, MD) and GF/B filters (Whatman, Maidstone, UK) that had been soaked in buffer (50 mm Tris-HCl; 50 mm Tris base; 0.1% BSA) for at least 24 h. Each reaction tube was washed five times with a 1.2-ml aliquot of ice-cold wash buffer. The filters were oven-dried for at least 60 min and then placed in 4 ml of scintillation fluid (Ultima Gold XR, Packard). Radioactivity was quantified by liquid scintillation spectrometry.
Molecular Modeling: Conformational Analysis of Allosteric Modulators
A complete conformational analysis of ORG27569 and PHR015 was performed using ab initio Hartree-Fock calculations at the 6–31G* level as encoded in the Spartan molecular modeling program (Wavefunction, Inc., Irvine, CA), as described previously (23). Specifically, HF 6–31G* 6-fold conformer searches were performed for all rotatable bonds. In each conformer search, local energy minima were identified by rotation of a subject torsion angle through 360° in 60° increments (6-fold search), followed by HF 6–31G* energy minimization of each rotamer generated. To calculate the difference in energy between the global minimum energy conformer of each compound and its final docked conformation, rotatable bonds in the global minimum energy conformer were driven to their corresponding value in the final docked conformation, and the single-point energy of the resultant structure was calculated at the HF 6–31G* level. The global minimum energy conformers of ORG27569 and PHR015 were compared by superimposing the two structures on all heavy atoms (see Fig. 6C).
FIGURE 6.
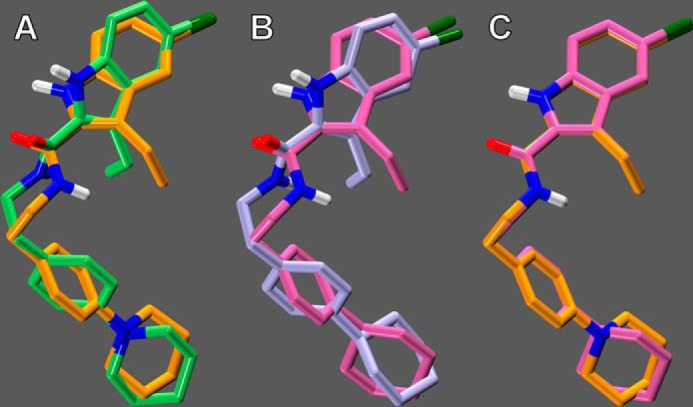
Comparison of the global minimum energy conformers (global min) and docked conformers of ORG27569 and PHR015. A, final docked conformer of ORG27569 (lime green) is shown overlaid on its global min (orange); the energy expense of ORG27569 to adopt its docked conformation was calculated to be 0.72 kcal/mol. B, final docked conformer of PHR015 (lavender) is shown overlaid on its global min (mauve); the energy expense of PHR015 to adopt its docked conformation was calculated to be 0.44 kcal/mol. C, global min of PHR015 (mauve) is shown overlaid with the global min of ORG27569 (orange).
Docking of ORG27569 in the Presence of CP55,940
The results of equilibrium binding assays from Kendall and co-workers (9), Ross and co-workers (13), and Fay and Farrens (24), as well as recent structural studies also from Fay and Farrens (24), strongly suggest that ORG27569 is inducing an R** conformation of the CB1 receptor (i.e. an intermediate receptor conformation that can bind agonists, yet does not signal in G protein-mediated pathways). However, before docking the allosteric modulators in our previously published model of the CB1R* (active state) receptor (with CP55,940 docked in its global minimum energy conformation, with respect to its ring systems) (25), the extracellular (EC) loops were temporarily removed from the model; this was done to allow a better exploration of potential allosteric binding sites. In addition, the N (residues 1–111) and C (residues 414–472) termini were truncated in our model, as mutation results from Fay and Farrens (24) have shown that ORG27569 is able to antagonize the efficacy of CP55,940 at WT levels (in G protein-mediated pathways) at mutant CB1 receptors in which both the N and C termini have been truncated.
ORG27569 (in its global minimum energy conformation) was manually docked in the TMH3-6-7 region of the receptor; this is consistent with the results of equilibrium binding assays that suggest that ORG27569 displaces SR141716A (but not other orthosteric ligands) (13). The automatic docking program, Glide (version 5.7, Schrödinger, LLC, New York), was then used to explore other possible receptor binding modes of ORG27569. Glide was used to generate a grid based on the centroid of select residues in the binding site (from the manual dock). Standard precision was selected for the docking setup. Recently, ORG27569 has been observed to act as an inverse agonist in G protein-mediated pathways (9, 14); thus, we hypothesized that ORG27569 may interact with K3.28192, a residue that we have previously reported to be critical to the inverse agonism of SR141716A (26, 27). The geometry of the global minimum energy conformation of ORG27569 (as well as receptor topography near K3.28192) suggested that its piperidine nitrogen would be the most likely hydrogen bond acceptor to interact with K3.28192. Therefore, the formation of a hydrogen bond between ORG27569's piperidine nitrogen and K3.28192 was the only constraint used for the automatic docking of ORG27569. The 26 lowest energy conformations (≤0.67 kcal/mol above global min) of ORG27569 were docked using Glide; the Glide dock with the best geometry between ORG27569's piperidine nitrogen and K3.28192 was selected for additional calculations.
Modeling of EC Loops
Using interactive computer graphics, the EC loops (EC-1, Phe-180–Ser-185; EC-2, Gly-254–Glu-273; and EC-3, Gly-369–Lys-376) were manually added to the chosen Glide dock. The program Modeler was then used to refine loop structures (29, 30). Because of their close spatial proximity, the conformations of all three EC loops were calculated together. Chosen loop conformations were those that produced a low value of the Modeler objective function. The model was then minimized using a three-stage minimization protocol (described below).
Docking of PHR015 in the Presence of CP55,940
The geometry of the global minimum energy conformers of ORG27569 and PHR015 is quite similar (see Fig. 6C). Therefore, we manually docked PHR015 using the ORG27569-selected Glide output as a guide. The primary difference between the ORG27569 and PHR015 docks is that PHR015 did not form an electrostatic interaction with K3.28192. The model was then minimized using a three-stage minimization protocol (described below).
Docking of ORG27569 Alone in the R (Inactive) Conformation of Wild-type and F3.25189A CB1
As mentioned previously, ORG27569 acts as an inverse agonist (i.e. reduces basal signaling) of the G protein-mediated pathway when applied alone. This suggests that ORG27569, when applied alone, preferentially binds to an inactive (R) conformation of CB1. However, before docking ORG27569 in our previously published model of CB1 in its inactive conformation (27), the helices and loops were pulled apart 2 Å in the x-y plane (a plane that would be parallel to the plane of a lipid bilayer). This was necessary because the binding region of the inactive model of CB1 was more compact than the active model, and thus pulling the helices apart allowed a better exploration of possible ORG27569 binding modes. The F3.25189A inactive model was constructed by mutating F3.25189 to an alanine after the helices had been pulled apart. Performing the mutation at this point (after pulling the model apart, but before subsequent calculations) provided the model the conformational freedom to respond to the structural consequences of the mutation.
In addition, in recent functional studies from Fay and Farrens (24), it was observed that ORG27569 did not act as an inverse agonist (when applied alone) when the N and C termini were truncated. Therefore, the N (Ser-88–Asn-112) and C (Ser-414–Leu-471) termini were modeled using Modeler (as described above). Structures with a low value of the Modeler objective function were chosen. In running Modeler on the C terminus, only the unstructured regions were explored; Ala-440–Met-461 were modeled as an α-helix that would be parallel to a lipid bilayer (i.e. the same plane as Helix 8); this is consistent with NMR results that suggest the existence of “Helix 9” in the C terminus (31).
Glide was then used to dock the 26 lowest energy conformations (≤0.67 kcal/mol above global min) of ORG27569, in both the WT R and F3.25189A R models, exactly as described above. The minimization protocol used to minimize these inactive models is described below, with one alteration as follows: only stages 2 and 3 were performed (stage 1 was omitted, as the TMHs needed to first pull together, before allowing the loops/termini to relax).
Receptor Model Energy Minimization Protocol
The energy of the ligand(s)-CB1 complex, including loop regions, was minimized using the OPLS 2005 force field in Macromodel 9.9 (Schrödinger LLC). An 8.0-Å extended nonbonded cutoff (updated every 10 steps), a 20.0-Å electrostatic cutoff, and a 4.0-Å hydrogen bond cutoff were used in each stage of the calculation. The minimization was performed in three stages. In the first stage of the calculation, the ligand(s) and TMH bundle were frozen, but the loops were allowed to relax. The generalized Born/surface area continuum solvation model for water as implemented in Macromodel was used. This stage of the calculation consisted of a Polak-Ribier conjugate gradient minimization in 1000-step increments until the bundle reached the 0.05 kJ/mol gradient. In the second stage, a harmonic constraint was placed on all the TMH backbone torsions (ϕ, ψ, and ω), with this constraint gradually reduced to zero in 500-step increments (using a total of 2500 steps to reach zero). In addition, a 500 kcal/mol harmonic constraint was placed on the backbone torsions of the loops. No constraints were placed on the ligand(s) during this stage. The minimization consisted of a conjugate gradient minimization using a distance-dependent dielectric, performed in 1000-step increments until the bundle reached the 0.05 kJ/mol gradient. The third stage was performed exactly like the first stage; this was done to allow the loops to adjust to changes that occurred in the TMH region (during the second stage of the minimization).
Assessment of Pairwise Interaction and Total Energies
Interaction energies between the allosteric modulator and the CB1R**-CP55,940, WT CB1R, and F3.25189A CB1R complexes were calculated using Macromodel, as described previously (25). Specifically, after defining the atoms of the allosteric modulator as one group (group 1) and the atoms corresponding to a residue that lines the binding site in the final ligand-CB1R** complex as another group (group 2), Macromodel was used to output the pairwise interaction energy (Coulombic and van der Waals) for a given pair.
Molecular Dynamics Simulation of ORG27569 Docked in the CB1R**-CP55,940 Complex and of CP55,940 Alone at CB1R*
The minimized model of ORG27569 docked in CB1 R** (in the presence of CP55,940), as well as the model of CP55,940 (alone) at CB1R*, was further studied using molecular dynamics. The OPLS2005 force field was utilized with a distance-dependent dielectric (coefficient of 2.0 to match the docking studies). The extended nonbonded treatment was employed, as in the minimization procedure discussed above. The dynamics module of Macromodel 9.1 was invoked, using stochastic dynamics at 300 K with the use of SHAKE constraints for bonds to hydrogen allowing a 1.5-fs time step. The models were first minimized for 500 steps with restraints on all the heavy atoms, using a large force constant of 4184 kJ/mol. The molecular dynamics was then initialized to 300 K, and an initial 100 ps of molecular dynamics was run. Subsequently, these restraints were slowly released for the side chain heavy atoms (4184–0.05 kJ/mol halving in each step for a total of 16 steps), and at each step 150 ps of dynamics was performed. Finally, a 22.5-ns molecular dynamics simulation was conducted. Because the goal of this simulation was to explore the dynamic behavior of both ORG27569 and CP55,940 in the binding pocket (as well as CP55,940 alone in its binding site), in these simulations only the amino acid side chains and ligands were free to move. Macromodel was used to calculate the docking energy of ORG27569 and CP55,940, as a function of simulation time, with a resolution of 1 ns.
RESULTS
Chemistry
A synthesis of a novel cyclohexyl analog of ORG27569, 5-chloro-N-[2-(4-cyclohexylphenyl)ethyl]-3-ethyl-1H-indole-2-carboxamide (PHR015, 9), wherein the piperidine ring is replaced with a cyclohexyl ring was developed for this study. The synthesis of the analog was not known and that of the parent ORG27569 had not yet been reported (32). Thus, commercially available 1-bromo-4-cyclohexylbenzene (1) was metallated via a metal-halogen exchange with n-butyllithium and the metallated intermediate captured with dimethylformamide to afford the corresponding aldehyde (2) following the patent procedure (17) in 91% yield after chromatographic purification. Condensation of 2 with nitromethane (18) provided the nitrostyrene 3 in 43% yield. The 1H NMR spectrum exhibited trans-coupled vinyl protons. Reduction (19) of the nitrostyrene 3 with lithium aluminum hydride gave the amine 4 in 90% unpurified yield. The electrospray mass spectrum (ES-MS) showed the expected M + 1 molecular ion.
The indole portion of the molecule was prepared from commercially available ethyl 5-chloroindole-2-carboxylate 5 in essentially the method recently reported (20, 32). Thus, 5 was acylated with acetyl chloride catalyzed by diethylaluminum chloride to provide the 3-acetylindole 6 in 56% yield after chromatographic purification (20). Selective reduction of the keto carbonyl of 6 with triethylsilane in the presence of trifluoroacetic acid gave the ethyl group of compound 7 an 87% yield after extractive work up (20). Saponification of the ester 7 afforded the acid 8 in a 90% yield. The 1H NMR and ES-MS supported the assigned structures of 5–7.
Coupling amine 4 with acid 8, mediated by dicyclohexylcarbodiimide and hydroxybenztriazole, provided the target amide 9 in 17% yield after purification by chromatography and trituration. The compound was >98% pure by HPLC analysis, and the structure was supported by 1H NMR and HRMS.
Generation of CB1 Receptor Mutants
Because previous studies have demonstrated that in ligand displacement assays ORG27569 can displace the CB1 antagonist/inverse agonist, SR141716A, mutation studies focused on residues shown to be important for SR141716A binding as follows: K3.28192A, F3.36200A, W6.48356A, and W5.43279A (21, 22, 26, 27), as well as an aromatic binding pocket residue shown not to be important for SR141716A binding, F3.25189A (21, 22).
[35S]GTPγS Binding Assays: Functional Analysis of ORG27569 at WT and Mutant CB1 Receptors
[35S]GTPγS binding assays were used to measure the stimulation of GTPγS binding at WT and mutant CB1 receptors upon addition of CP55,940, in the presence (or absence) of ORG27569; [35S]GTPγS binding assays were also used to measure the ability of ORG27569 to act as an inverse agonist at WT and mutant CB1 receptors. In WT cells, ORG27569 (1 μm) abolished the effect of CP55940, such that there was a reduction in basal [35S]GTPγS binding (see Fig. 2A). In line with this, ORG27569 behaved as an inverse agonist in WT cells, displaying a level of inverse efficacy in line with that of SR141716A (see Fig. 2B).
FIGURE 2.

Effect of ORG27569 on CP55,940-induced [35S]GTPγS binding, as well as the effect on basal levels of [35S]GTPγS binding, in wild-type and hCB1R cells expressing the K3.28192A mutant. A, CP55,940-induced [35S]GTPγS binding when applied alone (DMSO) or in the presence of ORG275659 (1 μm). The effect of CP55940 is expressed as a percentage of the Emax value of CP55940 in the presence of vehicle. B, effect of ORG27569 and SR141617A on basal levels of [35S]GTPγS binding in wild-type (WT) cells. C, CP55,940-induced [35S]GTPγS binding when applied alone (DMSO) or in the presence of ORG275659 (1 μm) in K3.28192A mutant hCB1R cells. The effect of CP55940 is expressed as a percentage of the Emax value of CP55940 in the presence of vehicle. D, effect of ORG27569 on basal levels of [35S]GTPγS binding in WT and K3.28192A mutant hCB1R cells is illustrated here. Data were analyzed via one sample t test compared with zero where **, p ≤0.01; ***, p ≤0.001.
K3.28192A
CP55,940 has an EC50 value of 225 nm (95% CL, 55–923) when applied alone at the K3.28192A mutant; this EC50 value is significantly larger than WT (1.3 nm; 95% CL, 0.3–5) and suggests that this mutation does influence the activity of CP55,940. This result is consistent with CB1 K3.28192A mutation studies that showed a significant loss of binding affinity and efficacy for the classical cannabinoid, HU-210, the nonclassical cannabinoid, CP55,940, and the endogenous cannabinoid, anandamide (33). In cells expressing the K3.28192A mutant, ORG27569 lost the ability to completely antagonize the efficacy of CP55,940 (see Fig. 2C). Thus, 1 μm ORG27569 produced a complete antagonism in WT cells (see Fig. 2A) but only an inhibition of 41% at the same concentration in cells expressing the K3.28192A mutant (see Table 1 and Fig. 2C).
TABLE 1.
CP55,940-induced [35S]GTPγS binding in wild-type and mutant hCB1R cells, when applied alone (DMSO) or in the presence of an allosteric modulator (1 μm)
| Cell line | DMSO/allosteric | Emaxa (%) (95% CL) |
|---|---|---|
| WT | DMSO | 100 (72–122) |
| ORG27569 | Inverse effectb | |
| PHR015 | 31 (9–53)c | |
| K3.28192A | DMSO | 100 (75–125) |
| ORG27569 | 41 (17–66)c | |
| PHR015 | 15 (−10–39)c | |
| F3.36200A | DMSO | 100 (56–144) |
| ORG27569 | Inverse effectb | |
| W5.43279A | DMSO | 100 (73–127) |
| ORG27569 | 103 (45–160) | |
| W6.48356A | DMSO | 100 (56–144) |
| ORG27569 | Inverse effectb | |
| F3.25189A | DMSO | 100 (78–123) |
| ORG27569 | 19 (−10–48)c |
a Maximal agonist effect is expressed as a % of the maximum effect of CP55940 in the presence of vehicle, as determined using nonlinear regression analysis. Values represent the mean with 95% confidence interval of four to six experiments.
b Data decrease in the basal binding indicative of inverse effect.
c Data are significantly different (nonoverlapping confidence limits) from the DMSO vehicle.
Fig. 2D shows that for WT CB1, ORG27569 at concentrations of 0.1, 1, and 10 μm produced a statistically significant decrease in basal [35S]GTPγS binding, rendering it an inverse agonist. Fig. 2D also suggests a trend toward inverse agonism for ORG27569 at the K3.28192A mutant; however, the decrease in basal [35S]GTPγS binding even at the highest concentration of 10 μm did not reach statistical significance. We were not able to test higher concentrations of ORG27569 due to solubility issues. Regardless, these results clearly illustrate that inverse agonism of ORG27569 is greatly attenuated at the K3.28192A mutant. Altogether, these results suggest that K3.28192 is part of the ORG27569-binding site; in addition, these results suggest that an interaction with K3.28192 may be important (although not unequivocally required) for ORG27569's inverse agonism.
F3.36200A and W6.48356A
ORG27569 completely antagonized the efficacy of CP55,940 at both the F3.36200A and W6.48356A mutant (see Table 1 and Fig. 3, A and C). If ORG27569 interacted significantly with either of these residues, one would expect that these mutations would reduce the ability of ORG27569 to antagonize the efficacy of CP55,940. Also like WT, ORG27569 acted as an inverse agonist when applied alone (at either of these mutants), as well as in the presence of CP55,940 (see Fig. 3, B and D and A and C, respectively). Therefore, these results suggest that neither F3.36200 nor W6.48356 is part of the ORG27569-binding site.
FIGURE 3.

A, CP55,940-induced [35S]GTPγS binding in F3.36200A mutant hCB1R cells, when applied alone (DMSO) or in the presence of ORG27569 (1 μm). B, effect of ORG27569 alone on [35S]GTPγS basal binding in WT and F3.36200A hCB1 cells. C, CP55,940-induced [35S]GTPγS binding in W6.48356A mutant hCB1R cells, when applied alone (DMSO) or in the presence of ORG27569 (1 μm). D, effect of ORG27569 alone on basal [35S]GTPγS binding in WT and W6.48356A hCB1 cells. Symbols represent mean values ±S.E. from three to six experiments carried out in duplicate. Effect of CP55940 is expressed as a percentage of the Emax value of CP55940 in the presence of vehicle.
W5.43279A
ORG27569 was unable to antagonize the efficacy of CP55,940 at the W5.43279A mutant at a concentration that abolished the effect of CP55940 in WT cells (see Table 1 and Fig. 4A). If ORG27569 interacted significantly with W5.43279, one would expect that the W5.43279A mutation would reduce the ability of ORG27569 to antagonize the efficacy of CP55,940, but not completely eliminate its ability to antagonize CP55,940. Also unlike WT, ORG27569 was unable to act as an inverse agonist when applied alone at the W5.43279A mutant (see Fig. 4B) nor in the presence of CP55,940 (see Fig. 4A). W5.43279 is a large central binding pocket residue in the TMH3-4-5 region of CB1. We have shown previously that although the W5.43279A mutation does not affect CP55,940 binding or signaling, it has a profound effect on SR141716A and WIN55,212-2 binding and function (21, 22). This is most likely because of a structural change in the TMH3-4-5 binding pocket region. The fact that ORG27569 was unable to affect CP55,940 signaling or act as an inverse agonist at this mutant suggests that it has also been impacted by the structural change produced by the w5.43279A mutation. This may be because the mutation has altered its binding pocket.
FIGURE 4.

A, CP55,940-induced [35S]GTPγS binding in W5.43279A mutant hCB1R cells, when applied alone (DMSO) or in the presence of ORG27569 (1 μm). B, effect of ORG27569 alone on [35S]GTPγS basal binding in WT and W5.43279A hCB1 cells. C, CP55,940-induced [35S]GTPγS binding in F3.25189A mutant hCB1R cells, when applied alone (DMSO) or in the presence of ORG27569 (1 μm). D, effect of ORG27569 alone on basal [35S]GTPγS binding in WT and F3.25189A cells. Symbols represent mean values ± S.E. from three to six experiments carried out in duplicate. Effect of CP55940 is expressed as a percentage of the Emax value of CP55940 in the presence of vehicle.
F3.25189A
CP55,940 has an EC50 value of 35 nm (95% CL, 9-131) (see Fig. 4C) when applied alone at the F3.25189A mutant; this EC50 value is significantly different from WT and suggests that this mutation does influence CP55,940's efficacy. In addition, ORG27569 antagonized the efficacy of CP55,940 at the F3.25189A mutant (see Table 1 and Fig. 4C). If ORG27569 interacted significantly with F3.25189, one would expect that the F3.25189A mutation would significantly reduce the ability of ORG27569 to antagonize the efficacy of CP55,940. Therefore, these results suggest that F3.25189 does not form a significant interaction with ORG27569 (while in the presence of CP55,940) that is important for the ability of ORG2759 to antagonize CP55,940's efficacy. However, unlike WT, ORG27569 was unable to act as an inverse agonist when applied alone at the F3.25189A mutant (see Fig. 4D). These results suggest that ORG27569, when applied alone, may form an interaction with F3.25189.
Functional Analysis of PHR015 at WT and K3.28192A CB1
The results of characterizing ORG27569 at the K3.28192A mutant suggested that ORG27569 may form an interaction with K3.28192 that is important for its ability to antagonize the efficacy of CP55,940, as well as act as an inverse agonist. Our computational results suggested that ORG27569's piperidine ring nitrogen was the most likely hydrogen bond acceptor to interact with K3.28192. Therefore, we synthesized and characterized (using [35S]GTPγS binding assays, at WT and K3.28192A CB1) an analog of ORG27569 (PHR015, see Fig. 1), in which ORG27569's piperidine ring has been replaced with a cyclohexyl ring, eliminating the hydrogen bond accepting capability in this ring.
PHR015 was unable to completely antagonize the efficacy of CP55,940 at WT CB1 (see Table 1 and Fig. 5A), and the results for PHR015 at the K3.28192A mutant were quite similar (see Table 1 and Fig. 5B). It should be noted that CP55940's signaling is affected by the K3.28192A mutation, so the curves in Fig. 5B are shifted to higher concentrations. Fig. 5C shows that PHR015 does not have a statistically significant effect on basal [35S]GTPγS binding either at WT CB1 or the K3.28192A mutant. Because the effects of PHR015 at WT CB1 and the K3.28192A mutant were quite similar, these results suggest that PHR015 may lack the ability to interact with K3.28192.
FIGURE 5.

Characterization of PHR015. A, CP55,940-induced [35S]GTPγS binding when applied alone (DMSO) or in the presence of PRH015 (1 μm). B, CP55,940-induced [35S]GTPγS binding when applied alone (DMSO) or in the presence of PHR015 (1 μm) in K3.28192A mutant cells. C, effect of PHR015 alone on basal [35S]GTPγS binding in WT and K3.28192A mutant hCB1R cells. Symbols represent mean values ± S.E. from three to six experiments carried out in duplicate. Effect of CP55940 is expressed as a percentage of the Emax of CP55940 in the presence of vehicle.
Mutant Cycle
Figs. 2, A and C, and 5, A and B, describe the equivalent of a mutant cycle set of experiments. Here, functional effects (rather than effects on binding affinity) are used to determine whether a direct interaction between ligand and receptor occurs. In a mutant cycle, a set of complementary chemical groups is deleted from both ligand (ORG27569 → PHR015) and receptor (WT CB1 → K3.28192A). If there is a direct interaction between the ORG27569 piperidine nitrogen and K3.28192, then one would expect similar effects if the ligand is “mutated,” i.e. ORG27569 → PHR015, and tested at WT CB1 or the amino acid is mutated, WT CB1 → K3.28192A, and tested with ORG27569.
As discussed previously, PHR015 was unable to completely antagonize the efficacy of CP55,940 at WT CB1 nor was ORG27569 able to completely antagonize the efficacy of CP55940 at the K3.28192A mutant. More importantly, these two Emax values are not statistically different from each other (Emax = 31% (9–53) for PHR015 and Emax = 41% (17–66) for ORG27569). The decrease in efficacy due to the deletion of ligand functionality may result from a loss in binding energy, whereas a decrease in efficacy due to receptor residue substitution may come from conformational contributions. These losses may have similar magnitudes, even if the deleted groups do not directly interact with each other (34). Therefore, the key to determination of whether deletions have occurred between two groups that interact indirectly or directly is the effect produced by simultaneous deletion of both groups (i.e. PHR015 tested at K3.28192A). If the modified groups do not interact directly with each other in the WT state then the effect of the two simultaneous changes will be additive. If the interaction is a direct one, deletion of ligand functionality plus mutation of the amino acid will not be additive but should be comparable with either of the single changes. PHR015 decreased the Emax value of CP55940 to an extent at the K3.28192A mutant (Emax = 15% (−10−39) (see Table 1 and Fig. 5B) that is not statistically significantly different from the single change cases listed above. Therefore, these results suggest that a direct interaction occurs between the ORG27569 piperidine nitrogen and residue K3.28192 in WT CB1.
The same conclusion can be reached by considering the effect on [35S]GTPγS binding produced by ORG27569 versus PHR015 at WT CB1 or the K3.28192A mutant when each compound is applied alone. Fig. 2D shows that at concentrations of 0.1, 1, and 10 μm, ORG27569 has a statistically significant inverse effect on basal [35S]GTPγS signaling. In contrast, although ORG27569 at the K3.28192A mutant showed a trend toward an inverse effect on basal [35S]GTPγS binding at higher concentrations, this effect did not achieve statistical significance (Fig. 2D). Fig. 5C shows that PHR015 has no dose-dependent effect upon basal [35S]GTPγS signaling at WT CB1 when applied alone (see Fig. 5C). Therefore, using the logic of the mutant cycle as described previously, the key to the determination of whether deletions have occurred between two groups that interact indirectly or directly is the effect produced by simultaneous deletion of both groups (i.e. PHR015 at CB1 K3.28192A). Fig. 5C shows that PHR015 has no dose-dependent effect on [35S]GTPγS binding at the K3.28192A mutant when applied alone. This result suggests a direct interaction between the piperidine nitrogen of ORG27569 and K3.28192 in WT CB1 is important for ORG27569 to act as an inverse agonist.
Modeling Studies: Conformational Analysis
Fig. 6A illustrates the global minimum energy conformer (hereafter named “global min”) of ORG27569 (orange), superimposed on its final docked conformer (lime green); it is clear from this figure that the docked conformation of ORG27569 is quite similar to its global min. The energetic cost of ORG27569 adopting its final docked conformation was calculated to be 0.72 kcal/mol.
Fig. 6B illustrates the global min of PHR015 (mauve), superimposed on its final docked conformer (lavender); it is clear from this figure that the docked conformation of PHR015 is quite similar to its global min. The energetic cost of PHR015 adopting its final docked conformation was calculated to be 0.44 kcal/mol.
Finally, Fig. 6C illustrates the global min of ORG27569 (orange), superimposed on the global min of PHR015 (mauve); it is clear from this figure that the global min of ORG27569 and PHR015 is quite similar. These results indicate that ORG27569 and PHR015 may assume similar conformations, suggesting that both ligands may bind at a similar region of CB1.
Creation of the CB1R** Complex: Docking ORG27569 (in the Presence of CP55,940) in the CB1R* Model
The CB1R** model represents a receptor state that is promoted by ORG27569 in the presence of CP55,940. As such, it exists only in complex with ORG27569 and CP55,940. This state is one that would promote the binding of agonist, yet not signal via G protein-mediated pathways. The CB1R** complex model was derived from our previously published activated state CB1R* model (25), and differs from the R* model primarily on the extracellular side of the receptor in the conformations of the EC-2 and EC-3 loops and the EC end of TMH6. The conformations of these components are restricted by the presence of ORG27569 and CP55,940.
Before docking ORG27569, the global min of CP55,940 (with respect to its ring systems) was docked at its previously identified binding site (35), i.e. the TMH1-2-3-7 region of CB1 (i.e. the “classical/endogenous cannabinoid” binding site). In general, CP55,940 is oriented so that its three hydroxyl groups are near the extracellular face of the receptor; in contrast, its hydrophobic dimethylheptyl tail is positioned lower, within the transmembrane core. Most interestingly, CP55,940 forms an important hydrogen bond with K3.28192; this is consistent with mutation studies that indicate CP55,940 has a reduced binding affinity at the K3.28192A mutant (33). For additional information regarding the CP55,940-binding site at the CB1 receptor, see Ref. 35.
Fig. 7 illustrates ORG27569 (lime green) docked at its binding site, in the presence of CP55,940 (cyan). Residues that contribute 5.5% (or more) of ORG27569's total interaction energy with the CB1R** receptor-CP55,940 complex are shown in orange; K3.28192 is shown in bright orange. ORG57569's net interaction energy with the receptor-CP55,940 complex is −56.22 kcal/mol. The binding site of ORG27569 was identified by Glide docking studies to be in the TMH3-6-7 region of CB1 (see Fig. 7); this is consistent with our recently published mutation results that suggest that ORG27569 does not bind at the W5.43279A mutant CB1 receptor (14). Ligands that bind at the TMH3-4-5-6 region of the CB1 receptor have little to no binding affinity at the W5.43279A mutant; this is likely due to a gross conformational change that occurs in this region of the receptor upon removal of this large central residue (21, 22).
FIGURE 7.
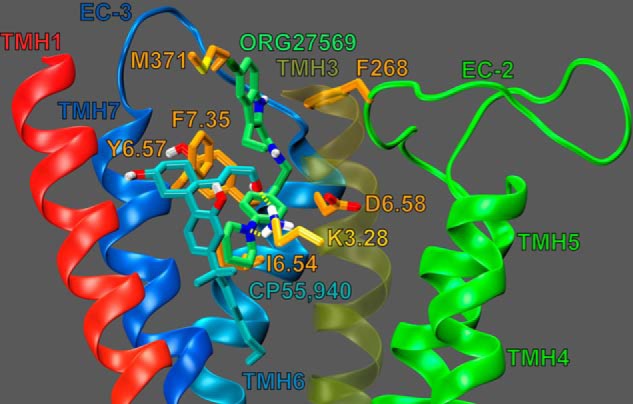
Dock of ORG27569 (lime green) at the CP55,940 (cyan)-CB1R** complex. The view is from the lipid bilayer looking toward TMH3. TMH2 and the EC-1 loop have been omitted for clarity. Residues that contribute ≥5.5% of ORG27569's total interaction energy are shown in orange. K3.28192 contributes 11.09% of ORG27569's total interaction energy and is shown in bright orange; K3.28192 is shown forming a hydrogen bond (yellow dashes) with both ORG27569 and CP55,940.
Fig. 8 illustrates that the ORG27569-binding site overlaps with our previously identified binding site for SR141716A (22, 26, 27); this is consistent with the results of equilibrium binding studies that suggest that ORG27569 displaces SR141716A (13). However, the ORG27569-binding site is more extracellular than the SR141716A-binding site. These results are consistent with the results of our mutation studies that illustrate that ORG27569's ability to influence CP55,940's binding and signaling is unaffected at the F3.36200A and W6.48356A mutants (see Table 1, Fig. 3, A and C), suggesting that ORG27569 does not bind low enough in the receptor to directly interact with F3.36200 or W6.48356. In contrast, we have previously reported that these residues are part of the SR141716A-binding site (22).
FIGURE 8.
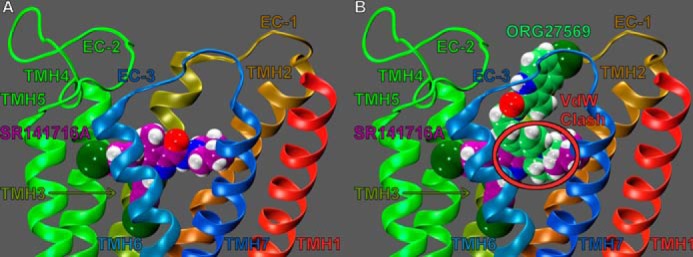
Overlay of ORG27569 (lime green) and SR141716A (purple) in the CB1R** model. A, SR141716A (contoured at its van der Waals radii) occupies the TMH3-4-5-6 aromatic microdomain of CB1, extending its piperidine ring into the TMH2/7 region. B, SR141716A and ORG27569 are illustrated here in their CB1 binding sites, with docks superimposed. As indicated by the red circle, there is severe steric overlap between these ligands. This steric overlap may explain why ORG27569 displaces SR141716A in equilibrium binding assays. Here, it is also clear that the binding site of ORG27569 extends higher in the TMH bundle toward EC loops than the binding site of SR141716A.
The results of our pairwise interaction energy calculations suggest that ORG27569's most important interaction is with residue F7.35379; this is likely because ORG27569 forms several aromatic stacking interactions with F7.35379 (see Fig. 7). F7.35379 forms an off-set parallel aromatic stack with ORG27569's indole ring (both rings A and B, see Fig. 1); the ring centroid to centroid distances are 6.02 and 4.75 Å, and the angles between the ring planes are 5.71 and 9.42° (for rings A and B, respectively). F7.35379 also forms an aromatic T-stack interaction with ORG27569's phenyl ring (ring C); the ring centroid to centroid distance is 5.60 Å, and the angle between the ring planes is 64.60° (see Figs. 1 and 7). Together, these aromatic interactions help position ORG27569 in the receptor, placing a significant amount of ORG27569's steric bulk against the EC end of TMH6. These results are consistent with recently published structural studies from Fay and Farrens (24) that suggest that ORG27569 antagonizes CP55,940's efficacy (in part) by preventing a necessary conformational change of TMH6 during receptor activation.
The ORG27569 indole ring (both rings A and B) also forms an aromatic T-stack interaction with Phe-268 (an EC-2 loop residue, see Fig. 7); the ring centroid to centroid distances are 5.63 and 4.95 Å, and the angles between the ring planes are 66.70 and 69.79° (for rings A and B, respectively). The importance of the interaction between ORG27569 and Phe-268 is discussed below (see under “ORG27569 Prevents Extracellular Loop Conformational Changes Critical for Signal Transduction”).
Additionally, the ORG27569 piperidine nitrogen forms an important hydrogen bond with K3.28192; the N–N distance is 3.00 Å and N-H··N angle is 124.5°. This interaction is almost as important to ORG27569's interaction energy as F7.35379 (−6.32 kcal/mol and −7.30 kcal/mol, respectively; see Fig. 7). The predicted importance of this residue's interaction with ORG27569 is consistent with our mutation results that suggest that ORG27569 has a significantly reduced ability to antagonize CP55,940's efficacy at the K3.28192A mutant (see Table 1 and Fig. 2, A and C). We also hypothesized that this interaction may be important for ORG27569's ability to act as an inverse agonist; this is because we have previously shown that K3.28192 was required for the inverse agonism of SR14716A (21, 26, 27). Our mutation results are consistent with this hypothesis; at concentrations up to 10 μm, ORG27569 was unable to significantly act as an inverse agonist at the K3.28192A mutant (see Fig. 2D).
Finally, ORG27569 has significant van der Waals interactions (in order of importance) with CP55,940, I6.54362, Met-371, and Y6.57365; ORG27569 also forms significant Columbic and van der Waals interactions with D6.58366 (see Fig. 7). Interestingly, with the exception of CP55,940, all of these residues are found on TMH6 or the EC-3 loop; again, this is consistent with results from Fay and Farrens (24) that suggest that ORG27569 antagonizes CP55,940's efficacy (in part) by preventing a necessary conformational change of TMH6 during receptor activation.
Docking of PHR015 (in the Presence of CP55,940) in the CB1R** Complex
The chemical structures of ORG27569 and PHR015 are extremely similar (the only difference between them is that ORG27569 has a piperidine ring and PHR015 has a cyclohexyl ring, see Fig. 1); therefore, it is not surprising that the global min of these two structures are nearly identical (see Fig. 6C). Thus, PHR015 was docked in the same general region (TMH3-6-7) of the receptor as ORG27569 (see Fig. 9).
FIGURE 9.
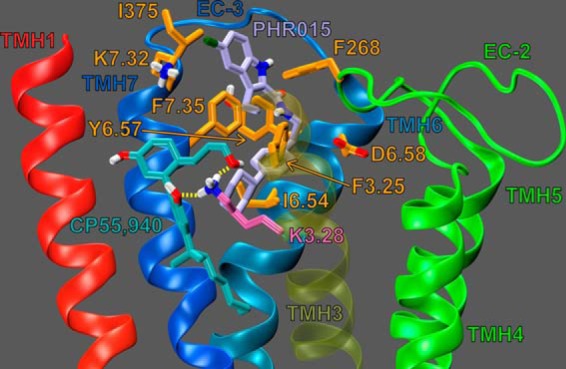
Dock of PHR015 (lavender) at the CP55,940 (cyan)-CB1R** complex. The view is from the lipid bilayer toward TMH3 with TMH2 and the EC-1 loop omitted for clarity. Residues that contribute ≥5.5% of the PHR015 total interaction energy are shown in orange. K3.28192 (mauve) forms two hydrogen bonds (yellow dashes) with CP55,940, but it does not interact with PHR015.
Fig. 9 illustrates PHR015 (lavender) docked at its binding site, in the presence of CP55,940 (cyan); residues that contribute 5.5% (or more) of PHR015's total interaction energy with the CB1R** receptor-CP55,940 complex are shown in orange; K3.28192 is shown in mauve (and does not significantly contribute to PHR015's total interaction energy). PHR015's net interaction energy with the CB1R** receptor-CP55,940 complex is −49.69 kcal/mol.
The results of our total interaction energy calculations suggest that PHR015 has a significantly reduced net interaction energy with the receptor-CP55,940 complex compared with ORG27569 (−49.69 and −56.22 kcal/mol, respectively). The results of our pairwise interaction energy calculations suggest that this difference in net interaction energy is due almost exclusively to each ligand's respective ability/inability to form a hydrogen bond with K3.28192 (i.e. PHR015 has a weaker net interaction energy than ORG27569, because its cyclohexyl ring cannot form a hydrogen bond with K3.28192; see Figs. 7 and 9). These observations are consistent with the results of the [35S]GTPγS binding assays; PHR015 was not able to completely antagonize the efficacy of CP55,940 (see Table 1 and Fig. 5A).
We have previously shown that K3.28192 is required for the inverse agonism of SR14716A (21, 26, 27), and we have shown here that ORG27569 also acts as an inverse agonist and interacts with K3.28192. Our docking studies indicate that although PHR015 occupies the same binding pocket location as ORG27569, removal of the piperidine nitrogen renders PHR015 incapable of interacting with K3.28192. This would suggest that PHR015 should not be an inverse agonist. Consistent with this hypothesis, in [35S]GTPγS binding assays (see Fig. 5C) PHR015 did not act as an inverse agonist. Together, these results for ORG27569 and PHR015 strongly suggest that an interaction between ORG27569's piperidine nitrogen and K3.28192 is important for the ability of ORG27569 to bind at the CB1 receptor and that this interaction is important for ORG27569 to act as an inverse agonist.
Analogous to ORG27569, the results of our pairwise interaction energy calculations suggest that PHR015's most important interaction is with residue F7.35379; this is likely because PHR015 forms several aromatic stacks with F7.35379 (see Fig. 9). F7.35379 forms an off-set parallel aromatic stack with PHR015's indole ring (both rings A and ring B, see Figs. 1 and 9); the ring centroid-ring centroid distances are 5.56 and 4.24 Å, and the angles between the ring planes are 7.50 and 8.26° (for rings A and B, respectively). F7.35379 also forms an aromatic T-stack interaction with PHR015's phenyl ring (ring C); the ring centroid-ring centroid distance is 6.00 Å, and the angle between the ring planes is 61.36° (see Fig. 1). Together, these aromatic interactions help position PHR015 in the receptor, placing a significant amount of PHR015's steric bulk against TMH6. These results suggest that (like ORG27569) PHR015 antagonizes CP55,940's efficacy (in part) by preventing a necessary conformational change of TMH6.
Like ORG27569, PHR015's indole ring (both rings A and B) also forms an aromatic T-stack interaction with Phe-268 (a EC-2 loop residue) (see Fig. 9); the ring centroid-ring centroid distances are 6.14 and 5.68 Å, and the angles between the ring planes are 86.46 and 87.54° (for rings A and B, respectively). The importance of the interaction between PHR015 and Phe-268 is discussed below (see “ORG27569 Prevents Extracellular Loop Conformational Changes Critical for Signal Transduction”). PHR015's phenyl ring also forms a T-stack aromatic interaction with F3.25189 (see Fig. 9); the ring centroid-ring centroid distance is 5.79 Å, and the angle between the ring planes is 84.95°.
Finally, PHR015 has significant van der Waals interactions (in order of importance) with I6.54362, CP55,940, Ile-375, and Y6.57365. PHR015 also forms significant Columbic and van der Waals interactions with D6.58366 and K7.32376 (see Fig. 9). Interestingly, with the exception of CP55,940 and K7.32376, all of these residues are found on TMH6 or the EC-3 loop; again, these results suggest that (like ORG27569) PHR015 antagonizes CP55,940's efficacy (in part) by preventing a necessary conformational change of TMH6.
ORG27569 Prevents Extracellular Loop Conformational Changes Critical for Signal Transduction
We have previously reported that two EC loop conformational changes must occur in order for the CB1 receptor to signal via G protein-mediated pathways (25). Specifically, our modeling results, based on mutation studies from Kendall and co-workers (36, 37), suggested that the EC-2 loop moves down toward the transmembrane core upon receptor activation (see Fig. 10A). Fig. 10A illustrates that this EC-2 loop conformational change places Phe-268 in close proximity to CP55,940 (25). In addition, we also reported that upon receptor activation an important ionic interaction forms between TMH2 and the EC-3 loop (specifically, an interaction forms between residues D2.63176 and Lys-373); this ionic interaction is necessary for signal transduction and promotes a conformation of the EC-3 loop that is pulled over the top (extracellular face) of the receptor (see Fig. 11A) (25).
FIGURE 10.
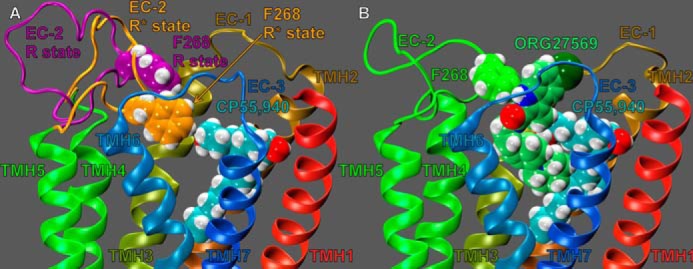
EC-2 conformations of various states of the CB1 receptor. A, EC-2 loop conformation of the CB1 receptor in its inactive (R; purple) and active (R*; orange) state conformations. CP55,940 is shown in cyan contoured at its van der Waals radii. The EC-2 loop residue, Phe-268 contoured at its van der Waals radii, is shown in purple for R and orange for R*. This residue is not in close proximity to CP55,940 in the CB1R state, but it is in close proximity to CP55,940 in the activated state. This image illustrates that the EC-2 loop moves down, toward the transmembrane core upon activation. B, EC-2 loop conformation in the presence of ORG27569 docked at the CP55,940-CB1 complex. ORG27569 is shown in lime green; CP55,940 is shown in cyan; the EC-2 loop and F268 are shown in green. This image illustrates that ORG27569 is sterically preventing the EC-2 loop from adopting its active conformation. Here, Phe-268 has formed an aromatic T-stack with the ORG27569 indole ring. This steric block of the EC-2 loop may explain how ORG27569 antagonizes the efficacy of CP55,940.
FIGURE 11.
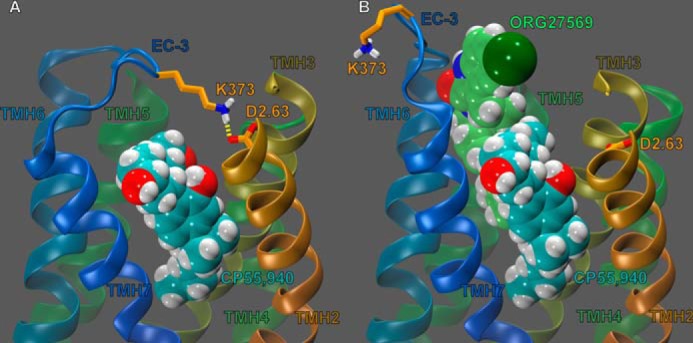
EC-3 loop conformations (and interactions) of various states of the CB1 receptor. The view is from lipid looking toward TMH3 with TMH1, the EC-1 loop, and the EC-2 loop omitted for clarity. A, EC-3 loop conformations of the CB1 receptor in its R* conformation. CP55,940 is shown in cyan, contoured at its van der Waals radii. D2.63176 and Lys-373 (orange tube) are shown forming a hydrogen bond (yellow dashes). We have recently reported (25) that, upon activation, this interaction promotes an EC-3 loop conformation that is pulled across the extracellular face of the CB1 receptor. B, EC-3 loop conformation of the CP55,940 (cyan, van der Waals)-CB1 complex, in the presence of ORG27569 (lime green, van der Waals). D2.63176 and Lys-373 shown here in orange. This image shows ORG27569 sterically blocking D2.63176 and Lys-373 from forming a hydrogen bond, preventing this interaction from promoting an active conformation of the EC-3 loop. This steric hindrance of the D2.63176 and Lys-373 (and indeed, the EC-3 loop in general) may provide an additional reason for ORG27569 antagonism of CP55,940 efficacy.
As described under “Experimental Procedures,” low energy EC loop conformations were added to our model of ORG27569 docked at the CB1R** receptor (in the presence of CP55,940). Importantly, none of the EC-2 loop Modeler output conformations placed the EC-2 loop near the transmembrane core nor Phe-268 in close proximity to CP55,940. Fig. 10B clearly illustrates why these results were observed; ORG27569's indole ring sterically blocks the EC-2 loop from moving toward the transmembrane core. Specifically, Phe-268 has formed an aromatic T-stack interaction with ORG27569's indole ring, preventing the EC-2 loop from adopting its active conformation.
Likewise, none of the EC-3 loop Modeler output conformations had the D2.63176 and Lys-373 interaction formed, nor did any of the output structures have the EC-3 loop pulled over the top (extracellular face) of the receptor. Fig. 11B clearly illustrates why these results were observed; ORG27569's indole ring sterically blocks the EC-3 loop from pulling across the top of the receptor. Consequently, the presence of ORG27569's steric bulk makes it essentially impossible for the interaction between D2.63176 and Lys-373 to form. Together, these results suggest that ORG27569 antagonizes the efficacy of CP55,940 by sterically blocking the necessary conformational changes of the EC-2 and EC-3 loops during activation (in addition to sterically blocking conformational changes of TMH6, as described previously).
CP55,940/Receptor Pairwise and Total Interaction Energies
The results of recently published binding assays illustrate that ORG27569 does not significantly affect the Kd value of CP55,940, but it does significantly increase the measured Bmax (in a concentration-dependent manner) (14). This suggests that ORG27569 may increase the equilibrium binding of CP55,940 by shifting the population of receptors from an inactive conformation (R) to an intermediate conformation (R**) and that ORG27569 is NOT increasing the binding affinity of CP55,940. To test if our models agreed with these results, CP55,940's total interaction energies were calculated for the following: CP55,940 alone at the CB1R* receptor; CP55,940 (in the presence of ORG27569) in the CB1R** complex, and CP55,940 (in the presence of PHR015) in the CB1R** complex. CP55,940's net interaction energy (includes conformational expense) was calculated to be −53.37, −53.38, and −51.87 kcal/mol for CP55,940 alone in the receptor and in the presence of ORG27569 and PHR015, respectively. These results suggest that neither the presence of ORG27569 nor PHR015 resulted in a significant change in the net interaction energy between CP55,940 and the receptor-allosteric modulator complex. Together, these results indicate that our computational models are consistent with the results of the equilibrium binding assays.
Molecular Dynamics Simulation of ORG27569 Docked in the CB1R**-CP55,940 Complex and of CP55,940 Alone at CB1R*
To elucidate the dynamic behavior of ORG27569 and CP55,940 with the receptor (as well as each other), a 22.5-ns molecular dynamics simulation was run on the final CB1R**-CP55,940-ORG27569 complex (shown in Fig. 7). Specifically, the simulations were run for three reasons as follows: 1) to observe if the interaction (as proposed in the static model) between ORG27569 and K3.28192 persists in a dynamic simulation; 2) to observe if the interaction (as proposed in the static model) between CP55,940 and K3.28192 persists in a dynamic simulation; and 3) to observe how the interaction energy of ORG27569 and CP55,940 evolves throughout the simulation.
As predicted by the static model (of CB1R**-CP55,940-ORG27569 complex), the interaction between ORG27569's piperidine nitrogen and K3.28192 persisted throughout the entire simulation. The average distance between ORG27569's piperidine nitrogen and K3.28192's terminal nitrogen was 3.19 Å; furthermore, this distance did not significantly deviate from the average over the course of the trajectory. Together, the static and dynamic models are consistent with our experimental results that suggest that ORG27569's piperidine nitrogen forms an important and persistent interaction with K3.28192.
Interestingly, the interaction between CP55,940 (in the presence of ORG27569) and K3.28192 did not persist throughout the simulation. This interaction breaks early in the simulation and does not reform. The average distance between CP55,940's alkyl-hydroxyl (southern aliphatic hydroxyl, SAH) oxygen and the terminal nitrogen of K3.28192 was 6.80 Å; the average distance between CP55,940's phenolic oxygen and the terminal nitrogen of K3.28192 was 5.83 Å. To determine whether the loss of this interaction was due to the presence of ORG27569, a molecular dynamics simulation was run of our static model of CP55,940 alone docked at CB1R* (employing the same methodology used for the CB1R**-CP55,940-ORG27569 complex). In this simulation, the interaction between CP55,940 and K3.28192 persisted throughout the entire simulation. The average distance between CP55,940's SAH oxygen and the terminal nitrogen of K3.28192 was 3.41 Å (note, the hydrogen bond between CP55,940's SAH and K3.28192 breaks briefly between 15 and 18 ns (observed as a sharp increase in distance); however, this interaction quickly reforms and persists for the rest of the simulation). The average distance between CP55,940's phenolic oxygen and the terminal nitrogen of K3.28192 was 3.12 Å. Furthermore, these distances did not significantly deviate from their averages over the course of the trajectory. This result is consistent with our experimental results that suggest that when CP55,940 is applied alone, it forms a significant interaction with K3.28192. Together, these results suggest that ORG27569 may weaken the interaction between CP55,940 and K3.28192, resulting in a more transient interaction between CP55,940 and K3.28192. Intriguingly, the loss of the interaction between CP55,940 and K3.28192 did not decrease CP55,40's total interaction energy with the model, as described below.
To determine whether the presence of ORG27569 affects the interaction energy of CP55,940 in a dynamic system, we calculated the interaction energy of CP55,940 in the CB1R**-CP55,940-ORG27569 complex as a function of time. As predicted by the static model, CP55,940's interaction energy did not significantly change over the course of the trajectory; CP55,940's average interaction energy (over the course of the trajectory) was −54.62 kcal/mol. Furthermore, the energy did not significantly deviate from the average over the course of the trajectory. This was surprising considering CP55,940's loss of an interaction with K3.28192. However, after inspecting the simulation, the reason became clear; the tremendous flexibility of CP55,940's SAH substituent allows it to easily and consistently find other hydrogen bond partners to compensate.
In addition, the interaction energy of CP55,940 alone in CB1R* was calculated over the course of its trajectory; CP55,940's average interaction energy (over the course of the trajectory) was −53.19 kcal/mol. Consistently, CP55,940's interaction energy did not significantly change over the course of the trajectory nor was it significantly different from CP55,940's interaction energy in the CB1R**-CP55,940-ORG27569 complex. Finally, the interaction energy of ORG27569 (in the CB1R**-CP55,940-ORG27569 complex) was calculated as a function of time; ORG27569's average interaction energy (over the course of the trajectory) was −53.19 kcal/mol. ORG27569's interaction energy did not significantly change over the course of the trajectory. Together, the results of our static and dynamic models are consistent with our experimental results that suggest that ORG27569 does not influence CP55,940's binding affinity for the CB1 receptor.
Docking of ORG27569 (Alone) in the WT and F3.25189A CB1R (Inactive) Models
As discussed previously, our experimental results suggest that when applied alone at wild-type CB1, ORG27569 acts as an inverse agonist (in G protein-mediated pathways). In addition, the results of our mutant cycle suggest that this inverse agonism is related to the formation of an interaction between ORG27569's piperidine nitrogen and K3.28192. However, we observed that ORG27569 was also unable to act as an inverse agonist at the F3.25189A mutant, whereas it was still able to antagonize CP55,940's efficacy at this mutant. These results may suggest that ORG27569 may form somewhat different interactions when applied alone as opposed to in the presence of CP55,940. To explore this possibility, we used Glide to dock ORG27569 at our WT and F3.25189A CB1R (inactive) models.
Fig. 12 illustrates ORG27569 (lime green) docked alone at its binding site in the WT CB1R (inactive) model. Residues that contribute 5.5% (or more) of ORG27569's total interaction energy with the CB1R model are shown in orange in Fig. 12; K3.28192 is shown in bright orange in Fig. 12. ORG57569's net interaction energy with the receptor is −52.06 kcal/mol. The ORG27569-binding site in CB1R is quite similar to its binding site in the CB1R**-CP55,940-ORG27569 complex. The binding site of ORG27569 was identified to be in the TMH3-6-7 region of CB1 (see Fig. 12); this is consistent with our recently published mutation results that suggest that ORG27569 does not bind at the W5.43279A mutant CB1 receptor (14). Ligands that bind at the TMH3-4-5-6 region of the CB1 receptor have little to no binding affinity at the W5.43279A mutant: this is likely due to a gross conformational change that occurs in this region of the receptor upon removal of this large central residue (21, 22).
FIGURE 12.
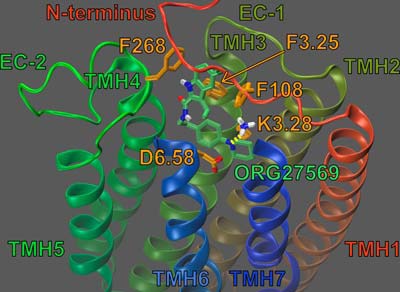
Dock of ORG27569 (lime green) at the WT CB1R (inactive) model. The view is from the lipid bilayer looking toward TMH6-7. The EC ends of TMH6-7 and the EC-3 loop have been omitted for clarity. Residues that contribute ≥5.5% of ORG27569's total interaction energy are shown in orange. This includes F3.25189. K3.28192 is shown in bright orange; K3.28192 is shown forming a hydrogen bond (yellow dashes) with ORG27569.
As in the CB1R**-CP55,940 model, the ORG27569-binding site (when docked alone) is more extracellular than the SR141716A-binding site. These results are consistent with the results of our mutation studies that illustrate that ORG27569's ability to act as an inverse agonist is unaffected at the F3.36200A and W6.48356A mutants (see Table 1 and Fig. 3, B and D), suggesting that ORG27569 does not bind low enough in the receptor to directly interact with F3.36200 or W6.48356. In contrast, we have previously reported that these residues are part of the SR141716A-binding site (22).
The results of our pairwise interaction energy calculations suggest that, when docked alone, ORG27569's most important interaction is with residue D6.58366. In addition, as in the CB1R** complex, ORG27569's piperidine nitrogen forms an important hydrogen bond with K3.28192 (see Figs. 7 and 12, respectively). The N-N distance is 2.90 Å and N-H··N angle is 152.8°. This interaction is almost as important to ORG27569's interaction energy as D6.58366 (−7.43 and −9.39 kcal/mol, respectively; see Fig. 12). We also hypothesized that this interaction may be important for ORG27569's ability to act as an inverse agonist; this is because we have previously shown that K3.28192 was required for the inverse agonism of SR14716A (21, 26, 27). Our mutation results are consistent with this hypothesis; at concentrations up to 10 μm, ORG27569 was unable to significantly act as an inverse agonist at the K3.28192A mutant (see Fig. 2D).
In addition, ORG27569 forms aromatic interactions with three residues. First, ORG27569's indole ring (both rings A and B) also forms an aromatic T-stack interaction with Phe-268 (an EC-2 loop residue; see Fig. 12); the ring centroid to centroid distances are 5.25 and 4.86 Å, and the angles between the ring planes are 84.28° and 95.60° (for rings A and B, respectively). Second, ORG27569's indole ring (ring A) forms an aromatic T-stack interaction with Phe-108 (an N terminus residue); the ring centroid to centroid distance is 4.97 Å, and the angle between the ring planes is 128.48°.
Finally, unlike in the CB1R** model, Fig. 12 illustrates that in the CB1R model, the indole ring of ORG27569 (ring A, see Fig. 1); forms a significant aromatic stack with F3.25189: the ring centroid to centroid in this interaction is 5.51 Å, and the angle between the ring planes is 43.71°. F3.25189 also forms an aromatic T-stack interaction with ORG27569's phenyl ring (ring C); the ring centroid to centroid distance is 5.36 Å, and the angle between the ring planes is 119.72° (see Fig. 12). Although this aromatic stack does significantly contribute to ORG27569's interaction energy with the CB1R model, it may serve a more important structural role. Specifically, this aromatic interaction between ORG27569 and F3.25189 may help sterically orient ORG27569 in the receptor so that it can form a hydrogen bond with K3.28189.
To test this hypothesis, we also used Glide to dock ORG27569 at the F3.25189A mutant CB1R model, using the exact same protocol as used for the WT model. Fig. 13 illustrates ORG27569 (lime green) docked alone at its binding site in the F3.25189A mutant CB1R model. Residues that contribute 5.5% (or more) of ORG27569's total interaction energy with the CB1R receptor are shown in orange; K3.28192 is shown in mauve (and does form a modest hydrophobic interaction with ORG27569, although it does not form a hydrogen bond with ORG27569's piperidine nitrogen). ORG57569's net interaction energy with the receptor is −37.17 kcal/mol. Fig. 13 illustrates that ORG27569- binding site at the F3.25189A mutant is similar to WT. Nonetheless, there is a profound difference; without the steric bulk provided by F3.25189, ORG27569 positions its indole ring much closer the backbone of TMH3. This orientation greatly damages ORG27569's ability to form several aromatic interactions that form in the WT model, resulting in a significantly reduced interaction energy compared with the WT model. However, ORG27569 does form an aromatic T-stack interaction with Phe-268; the ring centroid-centroid distance is 5.74 Å, and the angle between the ring planes is 89.73°. In addition, this orientation also makes it sterically impossible for ORG27569's piperidine nitrogen to form a significant interaction with K3.28192. This lack of an interaction with K3.28192 is consistent with our mutation results that suggest ORG27569 cannot act as an inverse agonist at the F3.25189A mutant receptor. Altogether, our computational and experimental results suggest that ORG27569 forms a significant interaction with F3.25189 when applied alone (that it does not necessarily form in the presence of CP55,940) and that this interaction is important in sterically orienting ORG27569 so that its piperidine nitrogen can form a hydrogen bond with K3.28192.
FIGURE 13.
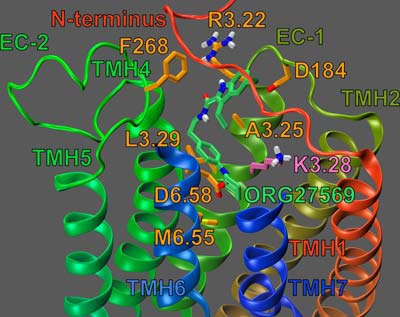
Dock of ORG27569 (lime green) at the F3.25189A mutant CB1R (inactive) model. The view is from the lipid bilayer looking toward TMH6-7. The EC ends of TMH6-7 and the EC-3 loop have been omitted for clarity. The net result of the F3.25189A mutation is a change in the WT CB1 binding position of ORG27569 (compare with Fig. 12). Residues that contribute ≥5.5% of ORG27569's total interaction energy are shown in orange. K3.28192 is shown in mauve (and does not form a hydrogen bond with ORG27569 due to the relocation of ORG27569 in this mutant.
DISCUSSION
Identification of the ORG27569-binding Site at the CB1 Receptor
In this study, we have used computational methods together with mutagenesis, synthesis, and pharmacological studies to identify an allosteric binding site for ORG27569 at the CB1 receptor and to probe its relationship to G protein signaling effects. Our results suggest that ORG27569 binds in the TMH3-6-7 region of the CB1 receptor. In this region, the ORG27569-binding site overlaps with our previously reported binding site for SR141716A (but not CP55,940) (21, 26, 27), consistent with the results of equilibrium binding assays that illustrate that SR141716A is displaced by ORG27569 (13). This allosteric site is also consistent with our results that suggest that, at the W5.43279A mutant, ORG27569 cannot antagonize the efficacy of CP55,940 nor act as an inverse agonist. To our knowledge, all cannabinoids that bind in the TMH3-4-5-6 region have little or no affinity (nor efficacy) at the W5.43279A mutant, suggesting that this mutation may cause a gross conformational change in this region of the receptor (21, 22). Finally, our results suggest that ORG27569 forms a distinct interaction with F3.25189 when applied alone (that it does not necessarily form when applied with CP55,940). More importantly, we observed that this interaction is important for sterically orienting ORG27569 so that its piperidine nitrogen can form a hydrogen bond with K3.28192.
Interaction between ORG27569 Piperidine Nitrogen and K3.28192 Is Important for ORG27569 Potent Antagonism of CP55,940 and Important for Its Inverse Agonism
We report here that ORG27569 acts as an inverse agonist at CB1 when applied alone by reducing basal activity. Our previous combined mutation cycle/modeling studies have identified the binding site of the CB1 antagonist, SR141716A (21, 22, 26). These studies identified an interaction between the carboxamide oxygen of SR141716A and K3.28192 that is possible only in the inactive state of CB1. It is this interaction that gives SR141716A a higher affinity for the inactive state of CB1, thereby rendering it an inverse agonist. At a K3.28192A mutant, SR141716A acts as a neutral antagonist. Analogs that lack the carboxamide oxygen such as 5-(4-chlorophenyl)-3-[(E)-2-cyclohexylethenyl]-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole (VCHSR) and 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-{lsqb](E)-piperidinoiminomethyl]-1H-pyrazole (PIMSR) are neutral antagonists at WT CB1 (26, 27).
Similarly, we report here the results of a mutant cycle performed to study the model-proposed interaction between ORG27569's piperidine nitrogen and K3.28192. We observed that ORG27569's ability to antagonize CP55,940 was reduced at the K3.28192A mutant (see Fig. 2C). This result suggests that an interaction with K3.28192 is important for ORG27569's ability to antagonize CP55,940 efficacy. PHR015 was designed to test if the piperidine nitrogen was the interaction site for K3.28192, and the results reported here are consistent with this hypothesis. Specifically, PHR015 did not antagonize CP55,940 as well as ORG27569 at WT CB1 (see Fig. 5A). Importantly, PHR015's ability to antagonize CP55,940 was unaffected by the K3.28192A mutation (see Fig. 5). These results strongly suggests that an interaction between ORG27569's piperidine nitrogen and K3.28192 is important for ORG27569's ability to antagonize CP55,940; this is because removal of either ORG27569's piperidine nitrogen or removal of K3.28192 reduced ORG27569's ability to antagonize CP55,940, but the effects of removing both the piperidine nitrogen and K3.28192 were not additive.
Likewise the same mutant cycle was performed to explore whether an interaction with K3.28192 is important for ORG27569's ability to reduce basal signaling. Although ORG27569 is an inverse agonist at WT CB1, at the K3.28192A mutant, ORG27569 shows a trend toward inverse agonism at high concentrations, but the effect did not reach statistical significance (see Fig. 2D). Furthermore, PHR015 did not act as an inverse agonist at WT CB1 (see Fig. 5C) nor at the K3.28192A mutant. Using the same logic of the mutant cycle (as just described), these results suggest that an interaction between ORG27569's piperidine nitrogen and K3.28192 may be important (although not unequivocally required) for ORG27569 to act as an inverse agonist. Thus, there is a parallel between SR141716A and ORG27569 action, suggesting that ORG27569 is both a negative allosteric modulator in the presence of CP55,940, but also an inverse agonist when applied alone.
Binding Site of ORG27569 Is More Extracellular than the Binding Site of SR141716A
Our results suggest that the ORG27569-binding site overlaps with the SR141716A-binding site, but it extends more extracellularly than that of SR141716A; specifically, ORG27569 does not bind low enough in the receptor to significantly interact with F3.36200 or W6.48356. In contrast, we have reported that both of these residues are part of the binding site of SR141716A (21, 22). Therefore, these results support our hypothesis that the ORG27569-binding site is more extracellular than the binding site of SR141716A.
ORG27569 Does Not Sterically Block CP55,940 from Exiting the CB1 Receptor
It is not surprising that our results suggest that ORG27569 binds at a partially extracellular region of the CB1 receptor, considering that many allosteric binding sites of Class A GPCRs have also been found in EC regions (4). This is perhaps best exemplified by the muscarinic family of receptors, in which a common allosteric binding site has been reported in all five muscarinic receptor subtypes; this site is located in an EC region (38). It has been hypothesized that by binding more extracellularly than orthosteric agonists, allosteric modulators may sterically hinder an orthosteric agonist from exiting the receptor (39–41).
However, unlike many GPCRs that bind hydrophilic ligands that enter the receptor via the receptor's extracellular surface, the cannabinoid receptors bind hydrophobic lipid-derived ligands; this fundamental difference in hydrophobicity may suggest that these hydrophobic ligands enter by an alternative route. For example, we have previously reported that sn-2-arachidonoylglycerol enters the CB2 receptor (via TMH6-7) from the lipid bilayer (42). Likewise, Hanson et al. (43) have reported that ligands may enter the sphingosine 1-phosphate receptor (another lipid-binding GPCR) between TMH1 and TMH7 (from the lipid bilayer). These observations may suggest that an allosteric ligand that binds in an extracellular region of a GPCR may not block the entry or exit of orthosteric ligands that enter/exit from the lipid bilayer.
Consistent with this hypothesis, we have recently reported that ORG27569 does not significantly decrease CP55,940's dissociation rate (koff) nor does ORG27569 significantly affect CP55,940's equilibrium binding constant (14). The results of our docking studies are consistent with the results of the dissociation experiments. Specifically, our models suggest that ORG27569 docks in an extracellular region of CB1. By docking in an extracellular region, ORG27569 would not be able to hinder the entry/exit of orthosteric ligands that enter/exit via the lipid bilayer.
In addition, because ORG27569 does not significantly impact the Kd value of CP55,940, this can be taken as evidence that ORG27569 does not sterically block CP55,940 from exiting the CB1 receptor. This is because if ORG27569 sterically blocked CP55,940 from exiting, this would be observed as a significant change in the Kd value of CP55,940 (i.e. if ORG27569 blocked CP55,940 from exiting, the Kd value of CP55,940 would increase). The results of our energy calculations performed on both the static models and dynamic simulations are consistent with these observations. Specifically, CP55,940 was found to have a similar total interaction energy, regardless of whether it was docked alone in the receptor or in the presence of ORG27569 (or PHR015). Altogether, these results suggest that ORG27569 does not sterically block CP55,940's exit from the CB1 receptor.
ORG27569 Promotes an Intermediate (R**) Conformation of the CB1 Receptor
In contrast, ORG27569 was observed to significantly increase the Bmax of CP55,940; this may suggest that ORG27569 increases the number of available binding sites for CP55,940 (14). These observations suggest that ORG27569 may be promoting a shift in the receptor population, i.e. from receptors in an inactive (R) conformation to receptors in an intermediate (R**) conformation (i.e. a conformation that preferentially binds agonist but cannot signal in G protein-mediated pathways). This hypothesis is consistent with structural results from the work of Fay and Farrens (24) that suggest that ORG27569 promotes an intermediate receptor conformation (i.e. a receptor conformation that is in between inactive and active). Interestingly, Kendall and co-workers (9) have reported that ORG27569 causes a modest (but significant) increase in CP55,940's binding affinity for the CB1 receptor; in addition, they did not observe a significant change in the Bmax values of CP55,940 when in the presence of ORG27569. The origins of these conflicting results are unclear at the present time. This difference will likely be resolved by the characterization of more potent CB1 allosteric modulators (which may have a more obvious pharmacological profile).
ORG27569 Sterically Blocks EC Loop Movements and Interactions Critical to Signal Transduction
Recently, the relationship between where an allosteric modulator binds and its mechanism of action has enjoyed intense interest (4, 44, 45). For example, the EC-2 and EC-3 loops have been reported to be important for the binding and efficacy of allosteric modulators at the adenosine A1 receptor (46). In the muscarinic field, not only have the EC loops been implicated in the binding and efficacy of allosteric modulators, but they have also been reported to play a role in receptor subtype specificity (i.e. sequence differences in the EC loop regions help determine allosteric modulator-receptor subtype preference) (47). This correlation between allosteric modulator binding/efficacy and the EC region of GPCRs is not surprising, given the fundamental importance of the EC loops to signal transduction (48).
Our results suggest that upon receptor activation, the EC-2 loop moves down toward the transmembrane core; this movement places Phe-268 in close proximity to CP55,940. In addition, we have presented evidence that suggests that ORG27569 may sterically block this movement, specifically by the formation of an aromatic interaction between ORG27569's indole ring and Phe-268. This correlation between a conformational change in the EC-2 loop and receptor activation has been observed in numerous GPCRs. For example, the results of NMR studies have suggested that upon activation, rhodopsin's EC-2 loop undergoes a necessary conformational change that is coupled to the breaking of the intracellular “ionic lock” (i.e. an ionic interaction between R3.50 and D/E6.30 that promotes an inactive GPCR conformation) (49). Additionally, the results of recent mutation studies of the angiotensin II type 1 receptor suggest that the EC-2 loop undergoes ligand-specific conformational changes that are required for receptor activation (50, 51). Finally, circular dichroism and steady-state fluorescence studies have been used to illustrate that the EC-2 loop of the serotonin 5-HT4(a) receptor adopts specific loop conformations that are determined by the receptor's activation state (52).
We have recently reported that the EC-3 loop and TMH2 form an ionic interaction (specifically, Lys-373 and D2.63176) that is necessary for G protein-mediated signaling of CB1 (25). Our computational results suggest that ORG27569 may sterically block the EC-3 loop from being able to reach across the top (extracellular face) of the receptor, preventing this interaction from forming. In analogy to the EC-2 loop, there appears to be a strong correlation between conformational changes of the EC-3 loop and GPCR activation. For example, mutation studies of the CXC chemokine receptor type 4 (i.e. CXCR4) suggest that an interaction between the EC-3 loop and the N terminus may form an “activation microswitch”; additionally, these results suggest that the conformation of the EC-3 loop may influence both basal and agonist-induced G protein-mediated signaling (53). In addition, mutation studies of the δ-opioid receptor suggest that its EC-3 loop may form a tight hairpin structure and that the disruption of this conformation may trigger receptor activation (54).
ORG27569 May Sterically Prevent TMH6 from Undergoing an Important Conformational Change
The hallmark of Class A GPCR activation by an agonist is the “tripping” of the toggle switch within the binding pocket that allows TMH6 to flex in the CWXP hinge region and straighten. This straightening breaks the “ionic lock” between R3.50 and D/E6.30 at the intracellular end of the receptor. The result is the formation of an intracellular opening of the receptor, exposing residues that can interact with the C terminus of the G protein's Gα subunit (55).
Our docking studies suggest that ORG27569 interacts with several residues on TMH6-7 and the EC-3 loop. These interactions position ORG27569 so that it packs tightly against TMH6; these results may suggest that ORG27569 antagonizes the efficacy of CP55,940 by preventing TMH6 from undergoing a necessary conformational change. These results are consistent with structural studies by Fay and Farrens (24) that suggest ORG27569 prevents an agonist-induced conformational change at the intracellular end of TMH6. Indeed, there is a significant amount of evidence in the literature that indicates that the extracellular end of TMH6 moves toward the transmembrane core, as the intracellular end of TMH6 moves away from the receptor (56). Therefore, ORG27569 may antagonize the efficacy of CP55,940 by preventing TMH6 from undergoing necessary conformational changes.
Non-G Protein-mediated Signaling by ORG27569
In addition to its effects on G protein-mediated signaling, ORG27569 has recently been reported to also signal via the ERK pathway when applied alone or in the presence of an orthosteric agonist (9, 14). This signaling has been reported to be mediated specifically by β-arrestin 1 (28). In this work, we have confined our study to how ORG27569 exerts its effects on G protein-mediated signaling only. Future studies will explore conformational changes induced by ORG27569 in other receptor regions that may correlate with β-arrestin-mediated ERK signaling.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 DA003934 and KO5 DA021358 (to P. H. R.).
- GPCR
- G protein-coupled receptor
- CB1
- cannabinoid-1
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- TMH
- transmembrane helix
- EC
- extracellular
- HRMS
- high resolution mass spectrometry
- SAH
- southern aliphatic hydroxyl
- CL
- confidence limit.
REFERENCES
- 1. Lagerström M. C., Schiöth H. B. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357 [DOI] [PubMed] [Google Scholar]
- 2. Insel P. A., Tang C. M., Hahntow I., Michel M. C. (2007) Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim. Biophys. Acta 1768, 994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCusker E. C., Bane S. E., O'Malley M. A., Robinson A. S. (2007) Heterologous GPCR expression: A bottleneck to obtaining crystal structures. Biotechnol. Prog. 23, 540–547 [DOI] [PubMed] [Google Scholar]
- 4. Wang C. I., Lewis R. J. (2013) Emerging opportunities for allosteric modulation of G-protein coupled receptors. Biochem. Pharmacol. 85, 153–162 [DOI] [PubMed] [Google Scholar]
- 5. Kenakin T. (2005) New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat. Rev. Drug Discov. 4, 919–927 [DOI] [PubMed] [Google Scholar]
- 6. Langmead C. J., Christopoulos A. (2006) Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol. Sci. 27, 475–481 [DOI] [PubMed] [Google Scholar]
- 7. May L. T., Leach K., Sexton P. M., Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51 [DOI] [PubMed] [Google Scholar]
- 8. Conn P. J., Christopoulos A., Lindsley C. W. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 8, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahn K. H., Mahmoud M. M., Kendall D. A. (2012) Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J. Biol. Chem. 287, 12070–12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pertwee R. G. (2009) Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br. J. Pharmacol. 156, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christopoulou F. D., Kiortsis D. N. (2011) An overview of the metabolic effects of rimonabant in randomized controlled trials: potential for other cannabinoid 1 receptor blockers in obesity. J. Clin. Pharm. Ther. 36, 10–18 [DOI] [PubMed] [Google Scholar]
- 12. Ross R. A. (2007) Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol. Sci. 28, 567–572 [DOI] [PubMed] [Google Scholar]
- 13. Price M. R., Baillie G. L., Thomas A., Stevenson L. A., Easson M., Goodwin R., McLean A., McIntosh L., Goodwin G., Walker G., Westwood P., Marrs J., Thomson F., Cowley P., Christopoulos A., Pertwee R. G., Ross R. A. (2005) Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 68, 1484–1495 [DOI] [PubMed] [Google Scholar]
- 14. Baillie G. L., Horswill J. G., Anavi-Goffer S., Reggio P. H., Bolognini D., Abood M. E., McAllister S., Strange P. G., Stephens G. J., Pertwee R. G., Ross R. A. (2013) CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol. Pharmacol. 83, 322–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ballesteros J. A., Weinstein H. (1995) in Methods in Neuroscience (Sealfon S. C. ed) pp. 366–428, Academic Press, San Diego [Google Scholar]
- 16. Bramblett R. D., Panu A. M., Ballesteros J. A., Reggio P. H. (1995) Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 56, 1971–1982 [DOI] [PubMed] [Google Scholar]
- 17. Ripka A., Shapiro G., Chesworth R. (January 14, 2010) U. S. Patent WO2010/006130
- 18. Okazaki K., Uchida M., Mukaiyama H., Kobayashi H., Kai Y., Takeuchi H., Yokoyama K., Terao Y., Hoyano Y., Shiohara H., Kikuchi N. (July 30, 2003) Patent EP1331221A1
- 19. Al-Awar R. S., Hecker K. A., Ray J. E., Huang J., Joseph S., Li T., Paal M., Rathnachalam R., Shih C., Wald P. P., Zhou X., Zhu G. (December 11, 2003) U. S. Patent 2003/0229026 A1
- 20. Pelcman B., Olofsson K., Katkevics M., Ozola V., Suna E., Kalvins I., Trapencieris P., Katkevica D., Schaal W. (February 2, 2006) U. S. Patent Application 20120029016 A1
- 21. McAllister S. D., Hurst D. P., Barnett-Norris J., Lynch D., Reggio P. H., Abood M. E. (2004) Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J. Biol. Chem. 279, 48024–48037 [DOI] [PubMed] [Google Scholar]
- 22. McAllister S. D., Rizvi G., Anavi-Goffer S., Hurst D. P., Barnett-Norris J., Lynch D. L., Reggio P. H., Abood M. E. (2003) An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J. Med. Chem. 46, 5139–5152 [DOI] [PubMed] [Google Scholar]
- 23. Kotsikorou E., Madrigal K. E., Hurst D. P., Sharir H., Lynch D. L., Heynen-Genel S., Milan L. B., Chung T. D., Seltzman H. H., Bai Y., Caron M. G., Barak L., Abood M. E., Reggio P. H. (2011) Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 50, 5633–5647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fay J. F., Farrens D. L. (2012) A key agonist-induced conformational change in the cannabinoid receptor CB1 is blocked by the allosteric ligand org 27569. J. Biol. Chem. 287, 33873–33882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marcu J., Shore D. M., Kapur A., Trznadel M., Makriyannis A., Reggio P. H., Abood M. E. (2013) Novel insights into CB1 cannabinoid receptor signaling: a key interaction identified between EC3-loop and TMH2. J. Pharmacol. Exp. Ther. 345, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hurst D. P., Lynch D. L., Barnett-Norris J., Hyatt S. M., Seltzman H. H., Zhong M., Song Z. H., Nie J., Lewis D., Reggio P. H. (2002) N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor. Mol. Pharmacol. 62, 1274–1287 [DOI] [PubMed] [Google Scholar]
- 27. Hurst D., Umejiego U., Lynch D., Seltzman H., Hyatt S., Roche M., McAllister S., Fleischer D., Kapur A., Abood M., Shi S., Jones J., Lewis D., Reggio P. (2006) Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J. Med. Chem. 49, 5969–5987 [DOI] [PubMed] [Google Scholar]
- 28. Ahn K. H., Mahmoud M. M., Shim J. Y., Kendall D. A. (2013) Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor one (CB1). J. Biol. Chem. 288, 9790–9800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 31. Ahn K. H., Pellegrini M., Tsomaia N., Yatawara A. K., Kendall D. A., Mierke D. F. (2009) Structural analysis of the human cannabinoid receptor one carboxyl terminus identifies two amphipathic helices. Biopolymers 91, 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piscitelli F., Ligresti A., La Regina G., Coluccia A., Morera L., Allarà M., Novellino E., Di Marzo V., Silvestri R. (2012) Indole-2-carboxamides as allosteric modulators of the cannabinoid CB(1) receptor. J. Med. Chem. 55, 5627–5631 [DOI] [PubMed] [Google Scholar]
- 33. Song Z. H., Bonner T. I. (1996) A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol. Pharmacol. 49, 891–896 [PubMed] [Google Scholar]
- 34. Ambrosio C., Molinari P., Cotecchia S., Costa T. (2000) Catechol-binding serines of β(2)-adrenergic receptors control the equilibrium between active and inactive receptor states. Mol. Pharmacol. 57, 198–210 [PubMed] [Google Scholar]
- 35. Kapur A., Hurst D. P., Fleischer D., Whitnell R., Thakur G. A., Samaniego P., Makriyannis A., Reggio P. H., Abood M. E. (2007) Mutation studies of Ser7.39 and Ser2.60 in the human CB1 receptor: evidence for a serine-induced bend in cb1 transmembrane helix 7. Mol. Pharmacol. 71, 1512–1524 [DOI] [PubMed] [Google Scholar]
- 36. Ahn K. H., Bertalovitz A. C., Mierke D. F., Kendall D. A. (2009) Dual role of the second extracellular loop of the cannabinoid receptor 1: ligand binding and receptor localization. Mol. Pharmacol. 76, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bertalovitz A. C., Ahn K. H., Kendall D. A. (2010) Ligand binding sensitivity of the extracellular loop two of the cannabinoid receptor 1. Drug Dev. Res. 71, 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gregory K. J., Sexton P. M., Christopoulos A. (2007) Allosteric modulation of muscarinic acetylcholine receptors. Curr. Neuropharmacol. 5, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wess J. (1993) Mutational analysis of muscarinic acetylcholine receptors: structural basis of ligand/receptor/G protein interactions. Life Sci. 53, 1447–1463 [DOI] [PubMed] [Google Scholar]
- 40. Birdsall N. J., Lazareno S., Matsui H. (1996) Allosteric regulation of muscarinic receptors. Prog. Brain Res. 109, 147–151 [DOI] [PubMed] [Google Scholar]
- 41. Wess J. (2005) Allosteric binding sites on muscarinic acetylcholine receptors. Mol. Pharmacol. 68, 1506–1509 [DOI] [PubMed] [Google Scholar]
- 42. Hurst D. P., Grossfield A., Lynch D. L., Feller S., Romo T. D., Gawrisch K., Pitman M. C., Reggio P. H. (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 285, 17954–17964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., Sanna M. G., Han G. W., Kuhn P., Rosen H., Stevens R. C. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Keov P., Sexton P. M., Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60, 24–35 [DOI] [PubMed] [Google Scholar]
- 45. Schwartz T. W., Holst B. (2007) Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends Pharmacol. Sci. 8, 366–373 [DOI] [PubMed] [Google Scholar]
- 46. Peeters M. C., Wisse L. E., Dinaj A., Vroling B., Vriend G., Ijzerman A. P. (2012) The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem. Pharmacol. 84, 76–87 [DOI] [PubMed] [Google Scholar]
- 47. Huang X. P., Ellis J. (2007) Mutational disruption of a conserved disulfide bond in muscarinic acetylcholine receptors attenuates positive homotropic cooperativity between multiple allosteric sites and has subtype-dependent effects on the affinities of muscarinic allosteric ligands. Mol. Pharmacol. 71, 759–768 [DOI] [PubMed] [Google Scholar]
- 48. Peeters M. C., van Westen G. J., Li Q., IJzerman A. P. (2011) Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 32, 35–42 [DOI] [PubMed] [Google Scholar]
- 49. Ahuja S., Hornak V., Yan E. C., Syrett N., Goncalves J. A., Hirshfeld A., Ziliox M., Sakmar T. P., Sheves M., Reeves P. J., Smith S. O., Eilers M. (2009) Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat. Struct. Mol. Biol. 16, 168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Unal H., Jagannathan R., Bhat M. B., Karnik S. S. (2010) Ligand-specific conformation of extracellular loop-2 in the angiotensin II type 1 receptor. J. Biol. Chem. 285, 16341–16350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Unal H., Jagannathan R., Bhatnagar A., Tirupula K., Desnoyer R., Karnik S. S. (2013) Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J. Biol. Chem. 288, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Banères J. L., Mesnier D., Martin A., Joubert L., Dumuis A., Bockaert J. (2005) Molecular characterization of a purified 5-HT4 receptor: a structural basis for drug efficacy. J. Biol. Chem. 280, 20253–20260 [DOI] [PubMed] [Google Scholar]
- 53. Rana S., Baranski T. J. (2010) Third extracellular loop (EC3)-N terminus interaction is important for seven-transmembrane domain receptor function: implications for an activation microswitch region. J. Biol. Chem. 285, 31472–31483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Décaillot F. M., Befort K., Filliol D., Yue S., Walker P., Kieffer B. L. (2003) Opioid receptor random mutagenesis reveals a mechanism for G protein-coupled receptor activation. Nat. Struct. Biol. 10, 629–636 [DOI] [PubMed] [Google Scholar]
- 55. Hamm H. E., Deretic D., Arendt A., Hargrave P. A., Koenig B., Hofmann K. P. (1988) Site of G protein binding to rhodopsin mapped with synthetic peptides from the α subunit. Science 241, 832–835 [DOI] [PubMed] [Google Scholar]
- 56. Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., Elling C. E. (2006) Molecular mechanism of 7TM receptor activation–a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]