

Background: The Y4R is involved in regulation of food intake and gastrointestinal transport.
Results: Mutagenesis studies revealed several residues displaying a significant loss of potency for hPP.
Conclusion: Tops of TM2, TM6, and TM7 interact with the hY4R native agonist hPP.
Significance: Characterizing the structure of the Y4R binding pocket is crucial for the development of new anti-obesity drugs.
Keywords: 7-Helix Receptor, Computer Modeling, G Protein-coupled Receptors (GPCR), Molecular Modeling, Neuropeptide, Binding Pocket, Comparative Model, Pancreatic Polypeptide
Abstract
Structural characterization of the human Y4 receptor (hY4R) interaction with human pancreatic polypeptide (hPP) is crucial, not only for understanding its biological function but also for testing treatment strategies for obesity that target this interaction. Here, the interaction of receptor mutants with pancreatic polypeptide analogs was studied through double-cycle mutagenesis. To guide mutagenesis and interpret results, a three-dimensional comparative model of the hY4R-hPP complex was constructed based on all available class A G protein-coupled receptor crystal structures and refined using experimental data. Our study reveals that residues of the hPP and the hY4R form a complex network consisting of ionic interactions, hydrophobic interactions, and hydrogen binding. Residues Tyr2.64, Asp2.68, Asn6.55, Asn7.32, and Phe7.35 of Y4R are found to be important in receptor activation by hPP. Specifically, Tyr2.64 interacts with Tyr27 of hPP through hydrophobic contacts. Asn7.32 is affected by modifications on position Arg33 of hPP, suggesting a hydrogen bond between these two residues. Likewise, we find that Phe7.35 is affected by modifications of hPP at positions 33 and 36, indicating interactions between these three amino acids. Taken together, we demonstrate that the top of transmembrane helix 2 (TM2) and the top of transmembrane helices 6 and 7 (TM6–TM7) form the core of the peptide binding pocket. These findings will contribute to the rational design of ligands that bind the receptor more effectively to produce an enhanced agonistic or antagonistic effect.
Introduction
G protein-coupled receptors (GPCRs)5 are the most prominent group of cell surface proteins. They are formed by seven transmembrane helices (TM) that are connected by intracellular and extracellular loops (ECL). GPCRs can be activated by several stimuli such as hormones, light, or odorant molecules (1). It is estimated that ∼30% of all prescribed pharmaceuticals modify the activity of GPCRs (2) indicating that these receptors are fundamental drug targets in modern pharmacology.
The Y4 receptor (Y4R) is a member of the NPYR, a class A GPCR family composed of Y1R, Y2R, Y4R, and Y5R receptors in humans. The NPYR is closely related to other class A GPCR families such as the neuropeptide FF receptor family and the orexin receptor family (3). NPY receptors are physiologically coupled to the Gi or Go proteins; however, other reports show that rabbit Y2R and rabbit Y4R are also coupled to the Gq protein, triggering an increase in inositol phosphate (4). These receptors are activated by the NPY family of peptide hormones, consisting of NPY, peptide YY (PYY), and pancreatic polypeptide (PP). NPY peptides and receptors form a multiligand/multireceptor system that plays a role in several physiological and pathological processes such as obesity and cancer (5). NPY peptides consist of 36 amino acids, are C-terminally amidated, and share high sequence identity. Shared structural features include a C-terminal helix. Despite high sequence homology and common structural features among NPY receptors and peptide hormones, however, there are significant differences in the affinity of these peptide hormones to the different receptor subtypes as well as differences in how the peptides bind their receptor (6).
The Y4R, cloned in 1995 (7), has 375 amino acids and was found to be expressed in the colon, small intestine, pancreas, prostate (8), brain, and coronary arteries (7). Physiologically, the Y4R is involved in the regulation of food intake (9), colonic anion transport (10), and adipose tissue and bone formation synergistically with Y2R (11). The Y4R sequence is one of the least conserved members of the NPYR family among different species, making it the fastest evolving functional member of the family (12). This makes it difficult to transfer conclusions from other Y receptor members to this subtype. Its main agonist, hPP, is produced by endocrine cells of the Langerhans islets of the duodenal part of the pancreas. These cells are also found in the gastrointestinal tract (13), albeit in much lower numbers. hPP was the first member of the NPY family of peptides to be identified. It is secreted after food ingestion in proportion to its caloric content (14), and it promotes appetite suppression and inhibition of gastric emptying (15). This ligand was already found in ancient tetrapod evolution and appears to be one of the fastest developing peptides of the family (12). Because of its role in appetite suppression, this system is a very attractive target for the design of new therapeutic compounds for fighting obesity.
Detailed knowledge of the receptor-peptide interaction is essential for rational structure-based drug design. Although several studies characterizing the binding pocket of NPY receptors have been published in the past years (6, 16–19), little is known about the Y4R. To our knowledge, only one study describes a subtype-selective interaction between Y4R and its ligand hPP (20) where the conserved residue Asp6.59 of human Y1R/Y4R binds to Arg35 of the peptides pNPY or hPP. In contrast, Asp6.59 of hY2R and hY5R interacts via Arg33 of pNPY and pPYY. The importance of this conserved residue was later identified in other systems closely related to the NPYR, such as the NPFF receptor 1 and 2 systems (3) or the prolactin-releasing peptide receptor (21).
In this study, we characterize in detail for the first time structural determinants of the hPP-hY4R interaction. Residues located at extracellular regions of the hY4R, chosen according to their location in the receptor sequence and in comparative models of hY4R, were mutated to determine their role in hPP binding. Simultaneously, a set of hPP analogs was also developed to pinpoint specific interactions between hPP and hY4R. We identified Tyr2.64, Asp2.68, Asn6.55, Asn7.32, and Phe7.35 as members of the hY4R binding pocket. Furthermore, hPP analogs with modifications in residues 27, 33, or 36 revealed these positions to be interaction partners with the receptor. These results clearly demonstrate the importance of the top of transmembrane helix 2 (TM2) and the top of transmembrane helices 6 and 7 (TM6 and -7) for hPP binding and illustrate the complexity of the intermolecular interactions within the hY4R subtype.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
Peptides were synthesized by automated solid-phase peptide synthesis on a Syro II peptide synthesizer (MultiSynTech, Bochum, Germany) and manual coupling steps following a 9-fluorenylmethoxycarbonyl-tert-butyl (Fmoc/tBu) strategy as described previously with minor modifications (3). All peptides were synthesized on Rink amide resin (15 μmol scale) to obtain C-terminally amidated peptides. Amino acid coupling steps were performed with equimolar amounts of N,N′-diisopropylcarbodiimide/hydroxybenzotriazole or N,N′-diisopropylcarbodiimide/ethylcyanoglyoxylat-2-oxime. The cleavage from the resin was carried out using a mixture of trifluoroacetic acid (TFA)/thioanisole/ethanedithiol (90:7:3 v/v/v). After full cleavage, a reducing mixture containing TFA/ethanedithiol/trimethylsilyl bromide was applied to reduce oxidized methionine. Crude peptides were purified using preparative reversed-phase HPLC on a Phenomenex C18 Jupiter 10u column (Proteo, 250 × 21.20 mm, 90 Å).
Peptide characterization was achieved by matrix-assisted laser desorption/ionization (Ultraflex III MALDI-ToF/ToF, Bruker Daltonics, Billerica, MA) and by electrospray ionization mass spectrometry. Peptide purities were determined on two analytical reversed-phase HPLC systems using a linear gradient of 0.1% (v/v) TFA in H2O (eluent A) and 0.08% (v/v) TFA in acetonitrile (eluent B). The gradient used was 20–70% of eluent B in eluent A in 40 min. The purity of the synthetic peptides was higher than 92% (data not shown).
Preparation of hY4R Mutants
The single mutations were inserted by site-directed mutagenesis into the protein sequence. Pfu Turbo DNA polymerase (Agilent) (2.5 units/μl) was used as a reaction enzyme in combination with 10× reaction buffer. The plasmid hY4_EYFP_N1 was used as a template (50–100 ng), and sense/antisense oligonucleotides were used in 2.5–10 pmol/μl concentrations depending on the reaction conditions. The deoxyribonucleotide (dNTP) mix was added in 10 mm concentration, and dimethyl sulfoxide (DMSO) was used occasionally to reduce secondary structures. DpnI (Thermo Fischer Scientific) was used to eliminate the original dsDNA template. Constructs were transformed in semi-competent Escherichia coli DH5α or E. coli JM109 cells, and the plasmid DNA was isolated using a Wizard plus Mini or Midi DNA purification system kit (Promega). The desired mutations were confirmed by sequencing of the complete coding sequence. The mutated positions are named after the system of Ballesteros and Weinstein (22).
Cell Culture
HEK293 cells (human embryonic kidney) and COS-7 cells (African green monkey) were cultured as described previously (21).
Fluorescence Microscopy Studies
HEK293 cells were seeded and transfected with cDNA encoding hY4R constructs as earlier described (23). The nuclei were stained with Hoechst 33342 (0.5 mg/ml) for 10 min after starving the cells for 20 min in Opti®-MEM medium. Fluorescence microscopy pictures were captured using an ApoTome Imaging system with an Axio Observer microscope (Carl Zeiss, Jena, Germany).
Signal Transduction Assays
Signal transduction assays were performed on 24- or 48-well plates as described previously with minor changes (3, 23). As transfection reagents, Metafectene and Metafectene Pro (Biontex) were used. The analysis of the data obtained was performed using the GraphPad Prism 5.03 software (GraphPad Software, San Diego). For each hypothesis, the data were processed using a nonlinear regression analysis, obtaining concentration-response curves displaying EC50 and Emax values. Furthermore, EC50 ratios were calculated using the global curve fitting function from GraphPad Prism 5.03. All the experiments were performed in duplicates of at least two independent experiments.
Fourteen Experimental GPCR Structures Were Considered as Templates for hY4R Comparative Modeling
A comparative model of hY4R was constructed using the protein structure prediction software package Rosetta, version 3.4 (24). Fourteen experimental GPCR structures from the Protein Data Bank (PDB) were considered as possible templates. These structures include the following: rhodopsin (PDB code 1U19) (25); β2-adrenergic receptor (PDB code 2RH1) (26); β1-adrenergic receptor (PDB code 2VT4) (27); A2A-adenosine receptor (PDB code 3EML) (28); CXC chemokine receptor type 4 (PDB code 3ODU) (29); D3 dopamine receptor (PDB code 3PBL) (30); H1 histamine receptor (PDB code 3RZE) (31); M2 muscarinic receptor (PDB code 3UON) (32); sphingosine 1-phosphate receptor (PDB code 3V2W) (33); M3 muscarinic receptor (PDB code 4DAJ) (34); κ-opioid receptor (PDB code 4DJH) (35); μ-opioid receptor (PDB code 4DKL) (36); nociceptin/orphanin FQ opioid receptor (PDB code 4EA3) (37), and δ-opioid receptor (PDB code 4EJ4) (38).
These structures were aligned with MUSTANG (39), and the resulting multiple sequence alignment was aligned with a multiple sequence alignment of hY1R, hY2R, hY4R, and hY5R using ClustalW (40). Sequence alignments were adjusted to remove gaps within transmembrane α-helices and ensure that highly conserved residues remain aligned (supplemental Fig. S1). hY4R residues were threaded onto the three-dimensional coordinates of aligned residues in each of the 14 GPCRs.
Missing Atom Coordinates Were Constructed Using Rosetta Loop Construction Protocols
Missing density and loop regions were reconstructed using Monte Carlo Metropolis fragment replacement and cyclic coordinate descent loop closure algorithms in Rosetta (41). All models underwent repacking and gradient minimization with RosettaMembrane (42). An additional constraint was included to account for the expected disulfide bond between hY4R residues Cys3.25 and Cys5.25.
The final set of models was clustered based on r.m.s.d. using bcl::Cluster (43). The top scoring models from the five largest clusters were used for docking studies.
Docking of Pancreatic Polypeptide (PP) into the Comparative Model of hY4R
A set of NMR structure conformations of bovine pancreatic polypeptide (PDB code 1LJV) (44) was docked into the hY4R comparative models. Bovine pancreatic polypeptide differs only on positions 6 and 23 with respect to hPP and has similar affinity for the hY4R as earlier reported (45, 46). The use of ILJV provided a guide for the structural distinction between the peptide's helical region and dynamic tail region. The helical region (residues 14PEQMAQYAAELRRYINML31) was first docked into the hY4R models. Four distinct helix conformations were docked into 37 hY4R comparative models without ECLs, guided by a predicted interaction between hY4R Tyr2.64 and hPP Tyr27.
C-terminal Residues of hPP Were Added Using de Novo Folding with Experimental Restraints
The five C-terminal residues of hPP (TRPRY) were constructed using Rosetta's low resolution de novo folding algorithm where residues are represented as “centroids” (47). Three experimentally derived restraints between hY4R and PP residues were used to guide this step using an 8-Å distance cutoff between residues Asp6.59 and Arg35, Phe7.35 and Arg33, and Asn7.32 and Arg33 (20, 48). All restraints are detailed in Table 1.
TABLE 1.
Experimental restraints used to guide docking of PP with hY4R
| hY4R residue | PP residue | Low resolution restraint | High resolution restraint | Proposed interaction | Steps imposed | Experimental evidence |
|---|---|---|---|---|---|---|
| Tyr2.64 | Tyr27 | C-β atoms within 8 Å | None | Unknown | hPP helix placement | Table 2 (Tyr2.64 and Tyr27 single mutants) |
| Asp6.59 | Arg35 | C-β atoms within 8 Å | Asp6.59 O-δ and Arg35 NH within 4 Å | Salt bridge | hPP C-terminal folding (low resolution), hY4R loop building (low resolution), final relaxation (high resolution) | Ref. 20 (Asp6.59 and Arg35 single mutants) |
| Asn7.32 | Arg33 | C-β atoms within 8 Å | Asn7.32 O-δ and Arg33 NH within 4 Å | Hydrogen bond | hPP C-terminal folding (low resolution), hY4R loop building (low resolution), final relaxation (high resolution) | Table 5, Fig. 5B (Asp7.32 and Arg33 single mutants) |
| Phe7.35 | Arg33 | C-β atoms within 8 Å | None | π-cation stacking | hPP C-terminal folding (low resolution), hY4R loop building (low resolution), final relaxation (high resolution) | Ref. 50, Table 4 (Phe7.35 and Arg33 single mutants) |
| Phe7.35 | Tyr36 | None | Phe7.35 CZ and Tyr36 CZ within 4 Å | Unknown | Final relaxation (high resolution) | Table 6, Fig. 6 (Phe7.35 and Tyr36 single mutants) |
The ECLs were rebuilt as described for the comparative modeling of hY4R, with the addition of these experimental constraints. Additionally, these models were refined to atomic detail, replacing centroids with side chain rotamers based on a backbone-dependent rotamer library and energy minimization with RosettaMembrane (49–51).
Models Were Relaxed Using Atomic Resolution Experimental Restraints
Models were again clustered based on r.m.s.d. Top scoring models from the largest clusters were visually inspected for binding poses that preserved the experimental restraints. Selected models underwent an additional relaxation step with constraints adjusted to reflect atomic level interactions between residues Asp6.59 and Arg35 (3 Å distance between the two δ-oxygen atoms on Asp6.59 and the side chain nitrogen atoms on Arg35), and residues Asn7.32 and Arg33 (4 Å distance between the δ-oxygen atom on Asn7.32 and the two side chain nitrogen atoms on Arg33). These constraint distances allow for possible hydrogen bonding and salt bridge interactions. An additional restraint between hY4R Phe7.35 and PP Tyr36 was introduced. Final models were clustered and visually inspected, and nine representative models were selected. The overall workflow for receptor modeling and peptide docking is summarized in Fig. 1.
FIGURE 1.
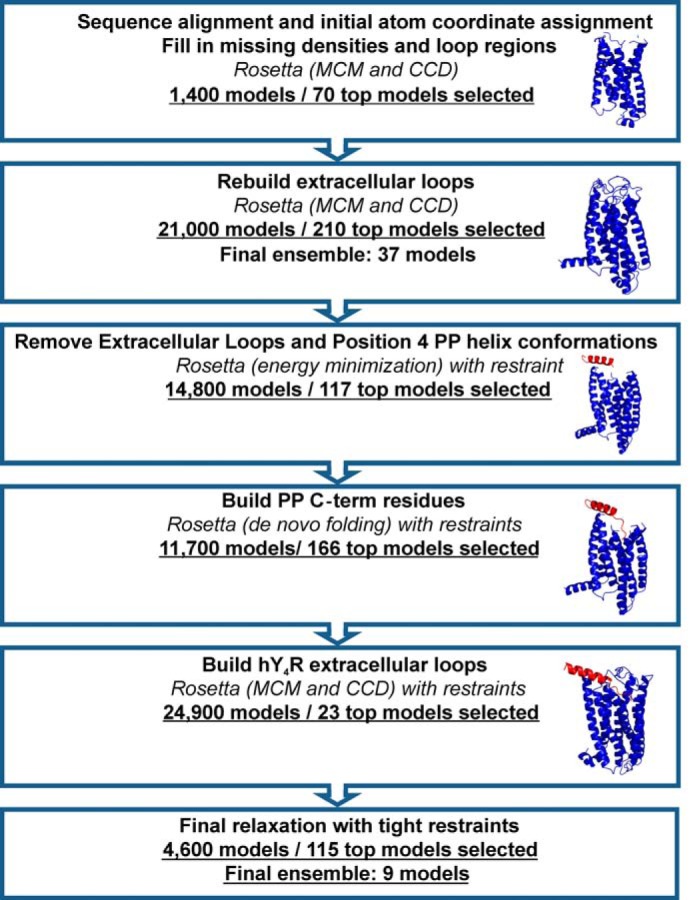
hY4R comparative model and PP docking work flow. An ensemble of hY4R comparative models was constructed through several rounds of loop building and energy minimization followed by selection of the best models. Alongside the flowchart are representative models to illustrate the evolution of the comparative model. hPP was docked through the placement of the PP helix, de novo addition of the C-terminal residues, and finally the addition of the ECLs of hY4R. Those steps were guided by experimentally derived restraints and followed by selection of the top models.
RESULTS
The comparative models presented here reflect an iterative process where multiple rounds of modeling were performed in parallel with in vitro experiments. Early models were generated based on comparative modeling with only seven GPCR templates and limited experimental restraints. The number of templates eventually increased to 14 as more GPCR structures became available. Additionally, predicted interactions seen in earlier models were used to guide some of the mutational assays. These assays provided additional restraints that were included in later models. The final models represent one plausible way that binding between hPP and hY4R can occur based on all experimental evidence available.
Tyr2.64 of TM2 Interacts with Tyr27
Tyr2.64 is located on top of TM2 and was found to be important in hY1R (16). This amino acid is conserved in all receptor subtypes except for hY5R (Fig. 2A); therefore, we hypothesized that this amino acid would be relevant in hY4R. To investigate Tyr2.64, this position was mutated to Ala and showed a significant shift of the EC50 value with 65-fold loss of potency but with 130% of efficacy (Fig. 3A). This high efficacy value matches with excellent membrane localization of the receptors revealed by fluorescence microscopy studies. Furthermore, the modification of Tyr2.64 to the larger aliphatic amino acid, Leu, was much better tolerated and displayed wild type-like activity with slightly reduced efficacy (83%). Furthermore, to test the importance of the hydroxyl moiety at this position, the mutant Y2.64F was constructed. The native ligand hPP displayed a 4-fold loss of potency and wild type-like efficacy (94%).
FIGURE 2.

A, alignment of the NPY receptor sequences. Highlighted residues are conserved or partially conserved amino acids within the receptor subfamily. B, snake plot showing the sequence of hY4R. Residues in light gray show residues investigated with no influence in activity. Residues in dark gray show an influence in receptor activity. Residues in black have an interaction partner on the peptide side. C, alignment of the NPY ligand sequences.
FIGURE 3.

Concentration-response curves of hY4R receptor mutants and hPP analogs determined with an immunoprecipitation accumulation assay. A, functional characterization of Y4R mutant Y2.64A with the modified ligands [Ala27]hPP and [Cha27]hPP to study the influence between Tyr2.64 of hY4R and Tyr27 of hPP. B, functional investigation of hY4R mutant D2.68A.
Because alanine-scan studies of pNPY (52) revealed Tyr27 as an important position for pNPY binding on hY4R, this led to the hypothesis this residue might be the interaction partner of Tyr2.64. To investigate the role of Tyr27, the analogs [Ala27]hPP, [Leu27]hPP, [Cha27]hPP, and [Phe27]hPP were synthesized. The substitution of Tyr27 to Leu pursued the aim of introducing a longer aliphatic amino acid, whereas the introduction of Cha was constructed to investigate the effects of a more bulky hydrophobic amino acid. Additionally, the substitution of Tyr27 to Phe was made to investigate the relevance of the hydroxyl group and to discard a possible hydrogen bond. [Ala27]hPP displayed an 8-fold loss of activity (EC50 11.78 nm), whereas the Leu and Cha variants showed wild type like potency on hY4R (Table 2). To investigate the effect of modifications on position Tyr27 with a hypothetical interaction to Tyr2.64, the mutants Y2.64A and Y2.64L were also tested with the peptide analogs in a double-cycle mutagenesis approach. [Ala27]hPP revealed a dramatic activity shift when tested on Y2.64A (424-fold), whereas [Leu27]hPP displayed no further loss on Y2.64A (Fig. 3A). In contrast, [Cha27]hPP activated Y2.64A with higher potency (EC50 22.34 nm) compared with hPP. However, testing [Ala27]hPP on Y2.64L displayed a great activity loss of 138-fold (EC50 205.20 nm), whereas[Leu27]hPP and [Cha27]hPP activated Y2.64L with a moderate loss of potency of 21–23-fold compared with hY4R with hPP (Table 2). Besides these findings, [Phe27]hPP was tested with Y2.64F to further investigate the interaction type. [Phe27]hPP displayed only a 9-fold loss of potency on Y2.64F. Taken together, this confirms that the presence of a bulky hydrophobic amino acid is favorable for this interaction site of the binding pocket and suggests that Tyr2.64 might interact with a second amino acid. This is supported by the comparative models, because seven models have Tyr2.64 within 8 Å of Tyr27 and eight models have Tyr2.64 within 8 Å of Leu31, which might be the second interaction point. Nevertheless, experimental data are needed to confirm this second interaction point.
TABLE 2.
Signal transduction of hY4 receptor mutants, mutated residues located on TM2
The immunoprecipitation accumulation assays were performed using increasing concentrations of hPP, [Ala27]hPP, [Leu27]hPP, [Cha27]hPP, and [Phe27]hPP. The incubation time lasted for 1 h. EC50 values from dose-response curves were determined. n represents the number of independent experiments, each performed in duplicate.
| Peptides | Wild type |
Y2.64A |
Y2.64L |
Y2.64F |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nm),a (pEC50 ± S.E.) | EC50 ratio,b (mutant/wild type) | n | EC50 (nm),a (pEC50 ± S.E.) | EC50 ratio,b (mutant/wild type) | n | EC50 (nm),a (pEC50 ± S.E.) | EC50 ratio,b (mutant/wild type) | n | EC50 (nm),a (pEC50 ± S.E.) | EC50 ratio,b (mutant/wild type) | n | |
| hPP | 1.44 (8.84 ± 0.04) | 1 | 39 | 93.20 (7.03 ± 0.09) | 65 | 7 | 1.30 (8.89 ± 0.11) | 0.9 | 4 | 6.13 (8.21 ± 0.09) | 4 | 5 |
| [Ala27]hPP | 11.78 (7.93 ± 0.08) | 8 | 4 | 579.90 (6.24 ± 0.15) | 424 | 4 | 205.20 (6.69 ± 0.09) | 138 | 3 | NTc | ||
| [Leu27]hPP | 1.19 (8.92 ± 0.17) | 0.85 | 4 | 90.63 (7.04 ± 0.14) | 63 | 4 | 32.20 (7.49 ± 0.09) | 21 | 3 | NT | ||
| [Cha27]hPP | 2.81 (8.55 ± 0.09) | 2 | 7 | 22.34 (7.65 ± 0.17) | 14 | 4 | 33.45 (7.48 ± 0.14) | 23 | 2 | NT | ||
| [Phe27]hPP | 0.852 (9.07 ± 0.12) | 0.6 | 4 | NT | NT | 11.26 (7.95 ± 0.11) | 9 | 4 | ||||
a EC50 and pEC50 values were determined from the mean ± S.E. of n independent experiments, each performed in duplicate.
b The ratio was calculated using the global fitting function for EC50 shift determination of Prism 5.03.
c NT represents not tested.
Other Positions Highlight the Importance of ECL1
Another amino acid investigated was Asp2.68. This residue is conserved in all receptor subtypes except in hY2R (Fig. 2A), which contains Gly at this position. Asp2.68 was found to be important in mutagenesis studies on the hY1R (17), and it is one of the interaction points between receptor and NPY on the hY5R system (6). In hY4R, the exchange of Asp2.68 to Ala led to a 94-fold loss in activity and decreased the efficacy dramatically to 39% of the wild type receptor response (Fig. 3B and Table 3). The loss in efficacy fits with the high intracellular accumulation of receptors demonstrated by fluorescence microscopy (Fig. 4). In contrast, the exchange to Glu or Asn regained the efficacy (93 to 87%, respectively) and displayed a loss of potency for hPP of only 9- and 16-fold, respectively.
TABLE 3.
Signal transduction of hY4 receptor mutants, mutated residues located on the ECL1 and TM3
The immunoprecipitation accumulation assays were performed using increasing concentrations of hPP for 1 h. EC50 values from dose-response curves were determined. n represents the number of independent experiments, each performed in duplicate.
| Y4R mutants | hPP |
|||
|---|---|---|---|---|
| EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | |
| Wild type | 1.44 (8.84 ± 0.04) | 1 | 100 | 39 |
| D2.68A | 135 (6.87 ± 0.24) | 94 | 39 ± 2 | 3 |
| D2.68E | 13.66 (7.87 ± 0.18) | 9 | 93 ± 4 | 2 |
| D2.68N | 22.88 (7.64 ± 0.13) | 16 | 87 ± 3 | 2 |
| W2.70A | 157.7 (6.80 ± 0.36) | 107 | 95 ± 9.5 | 6 |
| W2.70Y | 3.06 (8.52 ± 0.45) | 2 | 108 ± 7 | 2 |
| Q3.32A | 1.25 (8.90 ± 0.22) | 0.87 | 95 ± 4 | 3 |
a EC50 and pEC50 values were determined from the mean ± S.E. of n independent experiments, each performed in duplicate.
b The ratio was calculated using the global fitting function for EC50 shift determination of Prism 5.03.
c The Emax was determined at the highest peptide concentration.
FIGURE 4.
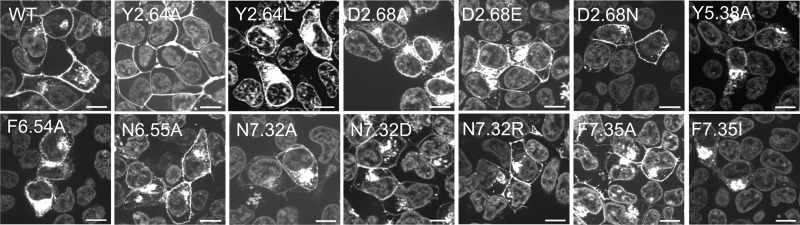
Cell surface expression of Y4R and Y4R mutants, Y2.64A, Y2.64L, D2.68A, D2.68E, D2.68N, Y5.38A, N6.55A, N7.32A, N7.32D, N7.32R, F7.35A, and F7.35I. HEK293 cells were transiently transfected with hY4R constructs C-terminally fused to enhanced YFP. The nucleus was developed using Hoechst 33342. Scale bar, 10 μm.
Additionally, position Trp2.70, which is conserved in all receptor subtypes, was described in some GPCR to belong to the motif WXFG and to be important for receptor activation (53). To prove its relevance, Trp was mutated to Ala, leading to a 107-fold loss in potency. Mutation to Tyr displayed only a 2-fold loss of potency (Table 3). These exchanges reveal that an aromatic or bulky side chain is necessary at this position to keep wild type-like activity.
Tested Residues in TM3, ECL2, and TM5 Do Not Play a Relevant Role in the Binding Pocket
The single residue tested in TM3 was Gln3.32. This position has been shown to participate in the binding pocket of nearly all crystallized class A GPCRs (2). In the hY4R, the exchange of Gln3.32 to Ala displayed a wild type-like potency when tested with hPP.
The ECL2 is the least conserved region between receptor subtypes. To elucidate the role of this ECL, several amino acids were mutated to Ala (Fig. 2B). To investigate polar interactions with the positively charged residues of the peptide Arg33 and Arg35, amino acids with negatively charged side chains Glu4.67, Glu4.79, and Asp4.83 were mutated to Ala. These three Ala mutants revealed wild type-like activity with EC50 values from 1- to 2-fold over hY4R. Furthermore, preliminary comparative models suggested that the residues Lys4.72, Phe4.80, Trp5.29, His5.34, Tyr5.38, and Phe5.41 located on ECL2 and the top of TM5 might be involved in the receptor binding pocket. The exchange of Lys4.72, Trp5.29, and Phe5.41 to Ala displayed only a 3–4-fold loss of potency when tested with hPP. Phe4.80 and His5.34 revealed wild type-like potency for hPP when exchanged to Ala. Only Tyr5.38 showed a reduced efficacy for hPP (43%, Table 4) but wild type-like potency. A further mutation to Ser at this position led to a partial regain of efficacy and displayed a 3-fold loss of potency for hPP, whereas an additional exchange to Phe at this position, to prove the relevance of the hydroxyl moiety, led to wild type-like potency for hPP.
TABLE 4.
Signal transduction results of ECL2 and TM5 mutants of the hY4 receptor
The immunoprecipitation accumulation assays were performed using increasing concentrations of hPP for 1 h. EC50 values from dose-response curves were determined. n represents the number of independent experiments, each performed in duplicate.
| Y4R mutants | hPP |
|||
|---|---|---|---|---|
| EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | |
| Wild type | 1.44 (8.84 ± 0.04) | 1 | 100 | 39 |
| E4.67A | 2.74 (8.56 ± 0.30) | 2 | 88 ± 6 | 2 |
| K4.72A | 5.07 (8.30 ± 0.17) | 4 | 105 ± 5 | 2 |
| E4.79A | 1.77 (8.75 ± 0.20) | 1 | 114 ± 6 | 2 |
| F4.80A | 1.58 (8.8 ± 0.24) | 1 | 100 ± 6 | 2 |
| D4.83A | 2.58 (8.59 ± 0.15) | 2 | 88 ± 3 | 3 |
| W5.29A | 4.03 (8.40 ± 0.17) | 3 | 96 ± 4 | 3 |
| H5.34A | 1.26 (8.89 ± 0.33) | 0.9 | 86 ± 7 | 3 |
| Y5.38A | 1.36 (8.87 ± 0.11) | 0.9 | 44 ± 3 | 5 |
| Y5.38S | 4.19 (8.38 ± 0.18) | 3 | 73 ± 3 | 5 |
| Y5.38F | 1.637 (8.79 ± 0.29) | 1 | 109 ± 6 | 5 |
| F5.41A | 5.02 (8.30 ± 0.24) | 4 | 92 ± 6 | 3 |
a EC50 and pEC50 values were determined from the mean ± S.E. of n independent experiments, each performed in duplicate.
b The ratio was calculated using the global fitting function for EC50 shift determination of Prism 5.03.
c The Emax was determined at the highest peptide concentration.
TM6 Plays a Crucial Role in Building the Binding Pocket
Asp6.59 on top of TM6 has already been shown to form a direct contact to the ligand (20). To further characterize the role of this part of the receptor, residues in close proximity to Asp6.59 were chosen for further mutagenesis studies. Phe6.54 was mutated to Ala because of its importance in other GPCRs closely related to the hY4R, its aromatic character, and its close proximity to Asp6.59. F6.54A displayed a 4-fold loss of hPP potency with reduced efficacy (73%, Table 4) that corresponds to poor membrane localization observed by fluorescence microscopy (Fig. 4). Asn6.55 was found to be involved in the hY1R binding pocket (18), and because initial comparative models oriented the side chain of this amino acid toward the interior of the hY4R binding pocket, this indicated its possible involvement in ligand-receptor interactions. Stimulation of N6.55A with hPP resulted in an 8-fold loss in activity and 79% of efficacy (Fig. 5A and Table 5) suggesting that this position is important in the receptor pocket.
FIGURE 5.

Concentration-response curves obtained with an immunoprecipitation accumulation assay using increasing concentrations of the ligands. A, functional characterization of hY4R mutants N6.55A. B, investigation of the relationship between position 33 of hPP and the amino acids Asn7.32 of hY4R using the ligands hPP, [ADMA33]hPP, and [Lys33]hPP.
TABLE 5.
Signal transduction of hY4 receptor mutants, mutated residues located on the TM6, ECL3, and TM7
The immunoprecipitation accumulation assays were performed using increasing concentrations of hPP, [Lys33]hPP, [ADMA33]hPP, or [SDMA33]hPP. The incubation time lasted for 1 h. EC50 values from dose-response curves were determined, each performed in duplicate. n represents the number of independent experiments.
| Y4R mutants | hPP |
[Lys33]hPP |
[ADMA33]hPP |
[SDMA]hPP |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | n | EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | n | EC50 (nM)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | n | |
| Wild type | 1.44 (8.84 ± 0.04) | 1 | 100 | 39 | 1.68 (8.78 ± 0.07) | 1 | 9 | 9.67 (8.02 ± 0.07) | 7 | 9 | 9.13 (8.04 ± 0.08) | 6 | 4 |
| F6.54A | 4.46 (8.35 ± 0.34) | 4 | 73 ± 6 | 3 | NTd | NT | NT | ||||||
| H6.62A | 0.42 (9.37 ± 0.24) | 0.33 | 98 ± 5 | 2 | NT | NT | NT | ||||||
| N6.55A | 11.39 (7.94 ± 0.10) | 8 | 79 ± 2 | 2 | NT | NT | NT | ||||||
| N7.32A | 6.92 (8.16 ± 0.26) | 5 | 84 ± 5 | 7 | 87.78 (7.06 ± 0.17) | 60 | 3 | 1416 (5.85 ± 0.14) | 979 | 4 | 2963 (5.53 ± 0.17) | 1892 | 3 |
| N7.32R | 24.54 (7.61 ± 0.12) | 18 | 107 ± 3 | 4 | NT | NT | NT | ||||||
| N7.32D | 2.02 (8.69 ± 0.16) | 1 | 97 ± 4 | 3 | 3.34 (8.48 ± 0.21) | 2 | 3 | 4.24 (8.37 ± 0.23) | 3 | 3 | NT | ||
a EC50 and pEC50 values were determined from the mean ± S.E. of n independent experiments, each performed in duplicate.
b The ratio was calculated using the global fitting function for EC50 shift determination of Prism 5.03.
c The Emax value was determined at the highest peptide concentration.
d NT represents not tested.
Additionally, His6.62 was investigated because of its proximity to Asp6.59 at the beginning of ECL3. H6.62A revealed an EC50 of 0.4 nm, which is moderately better than wild type.
TM7 Is a Contact Point of hPP in the hY4R and Asn7.32 Interacts with Arg33 of hPP
The first position investigated in TM7 was Asn7.32. This residue was found to be relevant for PYY binding on the Y1R (54), and initial comparative models indicated the possible importance of this position as well. Asn7.32 was mutated to Ala, Arg, and Asp. The effect of these substitutions increased in the following manner: Arg > Ala > Asp (Table 5 and Fig. 5B). This suggested that the introduction of a positive charge might cause a repulsion that was eliminated with the introduction of the negative charge Asp. Hence, we supposed that one of the C-terminal positively charged arginines might be the interaction partner. Because Arg35 of hPP was already identified to interact with hY4R Asp6.59, Arg33 was suggested as a possible interaction partner of Asn7.32. To clarify the binding hypothesis, the hPP analogs [ADMA33]hPP, [SDMA33]hPP, and [Lys33]hPP were synthesized. Position 33 of hPP was modified to SDMA and ADMA to maintain the positive charge and simultaneously reduce the possibility of hydrogen bond formation. Whereas [Lys33]hPP revealed wild type-like activity, the exchange of Arg to ADMA or SDMA displayed a 7- to 6-fold loss of activity (Table 5). Next, the hPP variants were tested on the receptor mutants N7.32A and N7.32D. [Lys33]hPP displayed a great loss of activity on N7.32A (60-fold, EC50 87.78 nm), and [ADMA33]hPP and [SDMA33]hPP displayed a dramatic activity loss compared with the wild type peptide with EC50 values of 1416 and >2000 nm, respectively (Table 5 and Fig. 5B). These experiments demonstrated that the introduction of a shorter amino acid such as Lys was better tolerated than the double methylation of Arg, which was not tolerated at all. Unlike N7.32A, N7.32D showed only a 2–3-fold loss of potency when tested with [Lys33]hPP and [ADMA33]hPP, revealing that the introduction of the double methylation or the reduction of the side chain length did not affect the binding pocket.
Phe7.35 Interacts with Arg33 as Well as Tyr36
Phe7.35 on the top of TM7 was also investigated, because this conserved position might be a suitable interaction point because of its aromatic characteristics and location. Furthermore, in the hY5R, Tyr7.35 was found to be relevant for the receptor and was suggested to belong to the receptor binding pocket (6). The initial comparative models also suggested its orientation to the interior of the proposed binding pocket. The amino acid was mutated to Ala and Ile, displaying a moderate loss in potency (7-fold) for the Ala mutant, whereas the Ile mutant revealed 41-fold loss of potency compared with wild type (Table 6). The higher potency loss caused by the Ile variant might indicate that the distance or the space available between Phe7.35 and the ligand or other positions in the receptor is important. F7.35I also showed reduced efficacy that corresponds to high intracellular receptor localization as demonstrated by fluorescence microscopy (Fig. 4). Preliminary comparative hY4R models suggested that this residue was in close proximity to Arg33 and Tyr36. Furthermore, Arg33 and Tyr36 revealed to be critical residues for pNPY binding on the hY4R (52). Taken all these facts together, we hypothesized that Phe7.35 might interact with one or both amino acids, Arg33 or Tyr36, of the peptide. As described above, the exchange of Phe7.35 to Ile displayed higher impact on receptor activity than the Ala substitution (Table 6). [Lys33]hPP and [ADMA33]hPP were used to investigate the relationship between Phe7.35 and Arg33. [Lys33]hPP displayed a dramatic loss of 451-fold in activation (EC50 640.4 nm) on F7.35A. Following tests on F7.35A with [ADMA33]hPP revealed a 107-fold loss of activity compared with hPP on hY4R. This corresponds to the potency loss produced by the mutant plus the loss produced by the analog. Finally, F7.35I leads to a dramatic loss in activity when tested with both analogs.
TABLE 6.
Signal transduction of wild type, F7.35A, and F7.35I receptors
The immunoprecipitation accumulation assays were performed using increasing concentrations of hPP as well as with hPP ligands with modifications on position Arg33 or Tyr36 as follows: [ADMA33]hPP or [Lys33]hPP, [Ala36]hPP, [Ile36]hPP, [Phe36]hPP, [Cha36]hPP, or [Nle36]hPP. The incubation time lasted for 1 h. EC50 values from concentration-response curves were determined. n represents the number of independent experiments, each performed in duplicate.
| Peptides | Wild type |
F7.35A |
F7.35I |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | EC50 (nm)a (pEC50 ± S.E.) | EC50 ratiob (mutant/wild type) | Emax ± S.E.c | n | |
| hPP | 1.44 (8.84 ± 0.04) | 1 | 100 | 39 | 11.63 (7.93 ± 0.11) | 8 | 99 ± 3 | 11 | 57.76 (7.24 ± 0.11) | 41 | 58 ± 2 | 7 |
| [ADMA33]hPP | 9.67 (8.02 ± 0.07) | 7 | 97 ± 2 | 9 | 160.5 (6.79 ± 0.15) | 107 | 78 ± 4 | 2 | >5000 | ND | 37 ± 3 | 3 |
| [Lys33]hPP | 1.68 (8.78 ± 0.07) | 1 | 98 ± 2 | 9 | 640.4 (6.19 ± 0.14) | 451 | 93 ± 4 | 3 | 1153 (5.95 ± 0.10) | 762 | 48 ± 1 | 3 |
| [Ala36]hPP | >2000 | NDd | 104 ± 11 | 3 | NTe | NT | ||||||
| [Ile36]hPP | 191 (6.72 ± 0.15) | 123 | 99 ± 4 | 3 | ND | ND | 3 | NT | ||||
| [Phe36]hPP | 2.07 (8.68 ± 0.23) | 1 | 102 ± 4 | 4 | 25.29 (7.60 ± 0.43) | 17 | 138 ± 15 | 3 | NT | |||
| [Cha36]hPP | 0.76 (9.14 ± 0.18) | 0.6 | 107 ± 4 | 3 | 211.2 (6.67 ± 0.09) | 138 | 93 ± 3 | 3 | N | |||
| [Nle36]hPP | 14.40 (7.84 ± 0.16) | 10 | 100 ± 4 | 5 | 1030 (5.99 ± 0.14) | 679 | 81 ± 4 | 4 | NTd | |||
a EC50 and pEC50 values were determined from the mean ± S.E. of n independent experiments, each performed in duplicate.
b The ratio was calculated using the global fitting function for EC50 shift determination of Prism 5.03.
c The Emax was determined at the highest peptide concentration.
d ND represents nondetermined because the curve could not reach the plateau.
e NT represents not tested.
Preliminary models suggested Tyr36 as a second interaction partner to Phe7.35. To characterize this hypothetical interaction, several analogs with modifications on Tyr36 were synthesized (Table 6). The introduction of Phe36 was well tolerated on the hY4R, indicating that the hydroxyl group of the side chain of Tyr was not playing a relevant role. The introduction of a nonaromatic amino acid such Ile brought a dramatic loss in potency on the wild type receptor (123-fold). Shortening the length of the side chain to Ala led to an even higher loss in potency (>2000-fold). Surprisingly, the introduction of unnatural amino acids such as Cha and Nle was better tolerated (EC50 values 0.6 and 14.40 nm, respectively, see Table 6 and Fig. 6). These hPP analogs were tested on F7.35A to investigate the type of interaction between these two positions. F7.35A showed a 17-fold loss of activity when tested with [Phe36]hPP (EC50 25.29 nm). [Ile36]hPP was not tolerated at all with F7.35A. This peptide displayed a dramatic loss of activity (EC50 not determinable), whereas Nle was slightly better tolerated revealing an EC50 value 679-fold over wild type. Finally, the second unnatural amino acid, [Cha36]hPP, displayed a 138-fold loss of activity over wild type (EC50 of 211.2 nm) on F7.35A and was thereby substantially better tolerated than Ile and Nle. Taken together, this indicates the need for a bulky hydrophobic amino acid at this position (Table 6 and Fig. 6).
FIGURE 6.

Concentration-response curves were determined with an immunoprecipitation accumulation assay with increasing concentrations of the analogs. Functional investigations are shown for position 36 of hPP and Phe7.35 of hY4R. COS-7 cells were transiently co-transfected with hY4R constructs and the chimeric G protein GαΔ6qi4myr.
The final comparative models support these results because within the nine best models obtained, 8 out of 9 showed Asn7.32 within 8 Å of distance to Arg33, and 9 out of 9 models showed Phe7.35 within 8 Å of proximity to Arg33 (Fig. 8).
FIGURE 8.

hY4R and PP residues within an 8 Å distance (based on C-β atoms) represent possible binding interactions. Neighboring residue pairs were collected across the nine final PP-hY4R docked models and presented as a heatmap indicating the most represented neighbors. hY4R residues are listed on the x axis with their secondary structure indicated (orange = TM and blue = ECL). PP residues are listed on the y axis with similar secondary structure indications. Numbers represent the number of models (out of nine) from which these residue pairs were within 8 Å. ICL, intracellular loop; ECL, extracellular loop.
In addition to the residues mentioned above, the conserved residue His7.39, which was one helix turn deeper in TM7, was also investigated. The Ala mutant did not reveal any detectable activity, and fluorescence microscopy pictures confirmed intracellular localization of the receptor. No further studies have been performed as this receptor variant is stuck in trafficking.
Docking of PP to the hY4R Comparative Model
Pancreatic polypeptide was docked into the comparative model of hY4R to assist interpretation of experimental results. Because inactive GPCR structures were used for our templates, it was important to consider the effects this may have on docking an agonist to this model.
Rosetta's comparative modeling protocol is insensitive to the state of GPCR templates. Templates are used only in the initial transmembrane helix positioning. Several relaxation steps allow for energy-based adjustments to these placements. Additionally, all extracellular loops are rebuilt in accordance with Rosetta's de novo folding algorithm. It is conceivable, however, that the helical conformations of an active template may be altered enough to fall outside of the conformational area explored with inactive templates. We compared the r.m.s.d. of our templates with the latest agonist-bound GPCR crystal structures. Pairwise alignments using the structure-based alignment tool MAMMOTH revealed that the average r.m.s.d. value of our inactive structures (2.9 ± 0.6) is not significantly different from that of the active structures (2.8 ± 0.6). Importantly, the average r.m.s.d. value is unchanged when combining the two groups (3.1 ± 0.5).
In addition to our analysis, Tautermann and Pautsch (55) examined the binding sites of active and inactive β2-adrenergic receptors. They show that the binding site is very similar between the inactive and active states. Previous modeling studies with the inactive structure predicted the binding mode of an agonist that overlapped well with that seen in the agonist-bound crystal structure (55).
The initial placement of the PP helix was guided specifically by the altered activity of hY4R Tyr2.64 and hPP Tyr27 mutants (Table 2). This placement provided a starting position from which the dynamic ECLs and C-terminal tail of hPP might be folded to simulate additional interactions suggested by the mutational data. These interactions specifically include a predicted salt bridge between hY4R Asp6.59 and PP Arg35, a predicted hydrogen bond between hY4R Asn7.32 and PP Arg33, a predicted cation-π interaction between hY4R Phe7.35 and PP Arg33, and an interaction between hY4R Phe7.35 and PP Tyr36.
The restraints imposed by these experimental results were included initially as low resolution restraints based on residue proximity. To complete the model, several restraints were adjusted to higher resolution atom level restraints in an attempt to capture the proposed interactions on an atomic level. The specific restraints imposed and their corresponding steps are described in Table 1. When PP was docked using the low resolution restraints, 81% of the generated models did not significantly violate any of the restraints. In the final step, when the high resolution restraints were imposed, 29.8% of the models generated were able to fit these restraints with no significant violations. This was encouraging in that a significant portion of our models were capable of fitting proposed atom-level interactions. A subset of nine top-scoring models that showed no significant violation of high resolution restraints was selected as the final ensemble for discussion. These models fit well with the majority of the experimental results, accurately portraying residues found to affect activity as well as those residues that failed to show any effect on activity. Specifically, the predicted salt bridge between Asp6.59 and Arg35 is well represented in eight of the nine models. All models show less than a 4.0 Å distance between both inter-residue oxygen-nitrogen pairs, providing possible salt bridge interactions or hydrogen bonding. Six of the nine models demonstrate a distance of less than 3.2 Å between the oxygen in hY4R Asn7.32 and amine group in PP Arg33, providing for the possibility of a hydrogen bond between these residues. hY4R Phe7.35 and PP Arg33 point toward each other in all nine models, which is conducive to the proposed cation-π interaction. Additionally, hY4R Phe7.35 and hPP Tyr36 were oriented toward each other in four models. Finally, hY4R Asp2.68 is within 8 Å and points toward the PP helix in five models, suggesting an interaction between the hPP helix and hY4R Asp2.68. One of the nine models is shown in Fig. 7, A and B, highlighting the binding site and residues important for PP-hY4R binding.
FIGURE 7.

Characterization of the binding pocket of PP docked in the hY4R comparative model. A, side view of PP (purple) docked to hY4R (cyan). Residues found to be important in the activation of hY4R by hPP are labeled. Predicted interactions are indicated by dotted red lines (salt bridge between Asp6.59 and Arg35 and hydrogen bond between Arg33 and Asn7.32). B, top-down view of the same docked model. C, two docked models show the variability in ECL1. The model shown in gray has a significantly longer ECL1 than that shown in cyan. Trp2.70, which was experimentally shown to be important in hY4R activation by PP, is shown to be in different proximity to PP depending on the size of ECL1. D, side view of the same docked model shown in A and B. Residues experimentally shown to be inactive in the binding of hPP to hY4R are indicated in black. The disulfide bond in ECL2 is also shown in yellow. a = His7.39; b = Gln3.32; c = Phe6.54; d = His6.62; e = Tyr5.38; f = His5.34; g = Trp5.29; h = Phe4.80; i = Glu4.67; j = Glu4.79; k = Lys4.72; and l = Asp4.83.
The importance of hY4R Trp2.70 for hPP binding is the only experimental finding not well reflected in the models. In all but one of the nine models, it is pointing away and/or not in close proximity to hPP. Possible explanations include inaccuracy of the model in this region, increased dynamics of this region as displayed in our models, or an indirect effect that involves a second site on the receptor that interacts with both hPP and Trp2.70. It is interesting that the length of TM2 varies in the models, thereby changing the length of the first intracellular loop dramatically from three residues in two of the models, 9–11 residues in five models, and 12–13 residues in two models. Because the models did not converge on a consistent length of ECL1, and precision is a prerequisite for accuracy, we expect that accuracy in this region might be low. This discrepancy in loop length is shown in Fig. 7C.
The residues that failed to show a significant effect on activity in the mutational assays are generally not contacting PP in the model. The majority of these residues are located in ECL2, which is consistently localized on the edge of the receptor away from hPP. Specifically, Lys4.72, Glu4.79, Phe4.80, Asp4.83, His5.34, and Phe6.54 are further than 8 Å away from any PP residue. Gln3.32, Glu4.67, Trp5.29, His6.62, and His7.39 are within 8 Å of an hPP residue in only three of the nine models, and Tyr5.38 is within 8 Å of an hPP residue in only two of the nine models. ECL2 and the residues not involved in PP binding are shown in Fig. 7D.
The ensemble of nine models was analyzed for ligand-receptor interactions. These predictions can serve as hypotheses to direct future mutational assays. Residue pairs between PP and hY4R with a distance of less than 8 Å were collected across all nine models. The total counts are shown in Fig. 8. This map can serve as a foundation from which to identify the residues that line the binding pocket. For example, five of nine models show that hY4R Ser5.28 and PP Thr32 are within 8 Å of each other, suggesting a possible interaction between these two residues.
DISCUSSION
In the NPY receptor family, the ECL1 and TM6 were regions described to form the binding pocket and interact with the peptide (6, 20). Additionally, hY1R, which shares high sequence homology with hY4R, has been extensively characterized in the past. Many amino acids located on ECL1, TM6, and TM7 are crucial for the interaction (16, 19, 54). Taking all these data into consideration, we expected the hY4R binding pocket to be composed of amino acids located in these areas of the receptor. Furthermore, it was also expected that hY4R has a second interaction site with the peptide on top of TM2 or beginning of ECL1, as suggested for hY5R (6).
We have now identified a binding pocket for the hY4R system that is composed of several residues located on TM2, TM6, and TM7. The first identified position in the pocket, Tyr2.64, is conserved in hY1R, hY2R, and hY4R. It is also present in the prolactin-releasing peptide receptor from several species, including human, rat, and mouse. Tyr2.64 was found to be involved in ligand binding on the hY1R and was suggested to belong to a hydrophobic pocket (19). In the hY4R, Tyr2.64 demonstrated that bulkiness and not aromaticity is critical for the interaction with hPP.
Y2.64L displayed a small decrease in efficacy for hPP. This might be caused by a small portion of receptors being trapped intracellularly (Fig. 4). The substantial amount of intracellular accumulation could be due to high expression levels of the mutant receptor, although it is not very likely because all constructs share the same promoter. It is more probable that this intracellular increase is due to an impaired folding of the mutant. However, the signal intensities suggest that enough active receptors are present in the cell membrane.
To elucidate a candidate position on the peptide side to interact with position Tyr2.64 of hY4R, earlier Ala-scanning mutagenesis studies on the NPY peptide family were considered (56). Among others, Tyr27 of NPY and PYY is relevant for binding in all NPY receptor subtypes (52, 56). This conserved residue in the three peptide ligands of the NPY family was thought to be a likely candidate to interact with Tyr2.64 of hY4R. Because Leu and Cha at position 27 of hPP have a nonplanar configuration compared with the wild type Tyr, in the presence of Leu or Cha on position 2.64 of hY4R the interaction might be slightly impeded, and a lack of space between Tyr2.64 and Tyr27 seems to be a limiting factor for the interaction to take place. This fact would support the close distance between these two positions suggested by the comparative models (Fig. 7A). Additionally, a hydrogen bonding interaction could be discarded between hY4R Tyr2.64 and hPP Tyr27. Besides this, we could not explain the relevance of the hydroxyl moiety of Tyr2.64 because an aromatic amino acid lacking the hydroxyl moiety like Phe is not as well tolerated as a hydrophobic amino acid like Leu. One hypothesis could be that Phe would adopt a slightly different orientation than Tyr or Leu, and therefore the interaction with the ligand could be slightly impeded. Overall, our data are most consistent with a hydrophobic interaction between hY4R Tyr2.64 and hPP Tyr27. Furthermore, the fact that the activity shift obtained for Y2.64A with hPP was larger than the shift obtained for [Ala27]hPP on hY4R would indicate that Tyr2.64 might interact with another position in the peptide or within the receptor.
Confirming the importance of ECL1, the nearby residues Asp2.68 and Trp2.70 proved critical for the hY4R-hPP interaction. D2.68A displayed high loss of potency and efficacy for hPP. Further mutations on Asp2.68 suggest that perhaps a polar or negatively charged amino acid is needed for correct export to or stability in the membrane. The relevance of this position is supported by the fact that on the hY5R Asp2.68 has been proven to interact with Arg25 of pNPY (6). Additionally this residue was hypothesized to form electrostatic interactions with NPY in the hY1R (17, 19). On hY4R, Asp2.68 may form hydrogen bonds with the peptide. Also, a polar effect on the structure that stabilizes the receptor binding pocket might be a feasible function for this position. The nearby residue Trp2.70 needs a bulky hydrophobic amino acid. Although the models show this residue not directly pointing to the peptide, further data are needed to elucidate the role of this residue on the hY4R binding pocket. This position could participate on direct ligand binding or have a more structural role affecting the nearby important positions when mutated.
On TM6, Asn6.55 participates in the binding pocket of the hY4R. Our results are supported by the loss in NPY binding displayed by N6.55A on studies with the hY1R (18). The comparative models demonstrate this amino acid pointing to the inner side of the proposed binding pocket close to Phe7.35 (Fig. 7, A and B). Asn6.55 is a candidate for interaction with Arg35 of hPP as in six models these residues are within a distance of 8 Å (Fig. 8). Moreover, as reported recently, this position is involved in ligand receptor interactions of many crystallized class A GPCRs (2). This fact strongly supports our data and confirms the role that Asn6.55 of hY4R has in the hPP binding pocket.
The results obtained on position Asn7.32 suggest that this residue is a key player in the binding pocket of hY4R. The mutation to Ala displays a small loss in potency for hPP. Prior studies on the hY1R, PYY and 1229U91 (GR231118), a Y4R agonist and a Y1R antagonist, displayed a loss in binding for N7.32A (54). It could be shown that Asn7.32 might be in close proximity with a positively charged residue, probably one of the two Arg of the C-terminal segment of hPP. To characterize the relationship between hY4R Asn7.32 and Arg33 of hPP, position 33 was modified to Lys to investigate the influence of the side chain length. Also the asymmetric and symmetric side chain dimethylations were tested at this position. Side chain methylations block hydrogen bond donor positions and increase hydrophobicity and bulkiness of the residue (57). Furthermore, the ability to form polar interactions such as dipole-dipole interactions might be impeded by double side chain methylation. The asymmetric and symmetric double methylation on position Arg33 of hPP produced a potency loss (6–7-fold) on hY4R, probably by blocking potential hydrogen bonding positions, potential dipoles, or due to steric hindrance. On N7.32A, the double side chain methylation at position Arg33 of hPP had a more dramatic effect. This might cause conformational changes in doubly methylated Arg33 of hPP impeding interactions with close by residues such as Phe7.35. These data are in agreement with the shortening of the side chain in [Lys33]hPP that resulted in a smaller potency loss on N7.32A, suggesting that Asn7.32 and Arg33 of hPP are in very close proximity. The fact that these three peptide analogs displayed potencies similar to wild type on N7.32D also supports an interaction with Arg33, because Asp maintains the hydrogen bonding capability and incorporates a negative charge able to form an ionic bond with position Arg33. Accordingly, we were able to demonstrate that Asn7.32 interacts with Arg33 possibly by hydrogen bonding or polar interactions. This hypothesis is supported by the great relevance of Arg33 as already demonstrated in the Ala scan (52). Comparative models where Arg33 of hPP is located between Asn7.32 and Phe7.35 of hY4R nicely reflect this hypothesis (Fig. 7B).
The last residue of the proposed binding pocket is Phe7.35. The exchange of Phe7.35 to Ile led to a higher potency and efficacy loss than the exchange to Ala, possibly due to steric hindrance. This position has been found to belong to the binding pocket of several class A GPCRs, among them the peptide receptors human CXC chemokine receptor type 4 and the rat neurotensin receptor 1 (2). Furthermore, this position might highlight the singularity of the Y4R binding pocket with respect to the Y1R. To investigate the role of hydrophobicity and size of Arg33 of hPP toward Phe7.35 of hY4R, [ADMA33]hPP was tested on F7.35A. The obtained results fit with the higher potency of [ADMA33]hPP compared with [Lys33]hPP on F7.35A, because the methyl groups can reduce the distance between both positions. A second interaction point of Phe7.35 was suggested by preliminary models to be Tyr36 as hypothesized in previous studies on the Y1R (58). The fact that an aliphatic amino acid such as Ile with a branched β-carbon is not tolerated in contrast to Cha or Phe could suggest a need for space close to the peptide backbone. Moreover, the effect of these ligands on F7.35A indicates that in the absence of Phe at position 7.35 an aromatic amino acid must be present at position 36 of hPP. This may arise for conformational reasons as only an aromatic amino acid with a planar structure might be able to contact position 7.35 in the absence of Phe. So, in the presence of both aromatic groups a π-π interaction might be established between Phe7.35 and Tyr36. In the absence of the aromatic group at position 36 of hPP, this residue might form hydrophobic interactions instead.
Our data provide the first insights into the complex binding pocket of the hY4R system derived from a combination of modeling and mutagenesis. As it may not be possible to solve the structure of all GPCRs, we demonstrate that this iterative method of study is very promising for understanding structurally uncharacterized receptors. As the model is in agreement with experimental data, it can be used to generate further testable hypotheses regarding the receptor-peptide interaction contributing to the development of ligands with enhanced hY4R activity.
Supplementary Material
Acknowledgments
We thank Regina Reppich-Sacher, Kristin Löbner, and Janet Schwesinger for technical support. We also thank Daniel Rathmann and René Meier for research discussions, Stephanie DeLuca for assistance in developing the peptide docking protocols, and Eric Dawson for developing preliminary comparative models.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM080403, R01 MH090192, R01 DK020593, and R01 GM099842 and Grant R01 DK097376 (to J. M. and A. B.-S.). This work was also supported by German Research Foundation Grants SFB 610/3-A1 and BE 1264-11 (to A. B.-S), National Science Foundation Grant Career 0742762 (to J. M.), a pilot and feasibility study from the Vanderbilt Diabetes Research and Training Center, and National Science Foundation Award OISE 1157751 to explore international collaboration.

This article contains supplemental Fig. S1.
- GPCR
- G protein-coupled receptor
- hY4R
- human Y4R
- hPP
- human pancreatic polypeptide
- TM
- transmembrane helix
- ECL
- extracellular loop
- NPY
- neuropeptide Y
- NPYR
- neuropeptide Y receptor
- PYY
- peptide YY
- PDB
- Protein Data Bank
- SDMA
- symmetrically dimethylated Arg
- ADMA
- asymmetrically dimethylated Arg
- r.m.s.d.
- root mean square deviation
- Cha
- β-cyclohexylalanine.
REFERENCES
- 1. Zhang R., Xie X. (2012) Tools for GPCR drug discovery. Acta Pharmacol. Sin. 33, 372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 3. Findeisen M., Rathmann D., Beck-Sickinger A. G. (2011) Structure-activity studies of RFamide peptides reveal subtype-selective activation of neuropeptide FF1 and FF2 receptors. Chem. Med. Chem. 6, 1081–1093 [DOI] [PubMed] [Google Scholar]
- 4. Misra S., Murthy K. S., Zhou H., Grider J. R. (2004) Coexpression of Y1, Y2, and Y4 receptors in smooth muscle coupled to distinct signaling pathways. J. Pharmacol. Exp. Ther. 311, 1154–1162 [DOI] [PubMed] [Google Scholar]
- 5. Pedragosa Badia X., Stichel J., Beck-Sickinger A. G. (2013) Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. 10.3389/fendo.2013.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindner D., van Dieck J., Merten N., Mörl K., Günther R., Hofmann H. J., Beck-Sickinger A. G. (2008) GPC receptors and not ligands decide the binding mode in neuropeptide Y multireceptor/multiligand system. Biochemistry 47, 5905–5914 [DOI] [PubMed] [Google Scholar]
- 7. Bard J. A., Walker M. W., Branchek T. A., Weinshank R. L. (1995) Cloning and functional expression of a human Y4 subtype receptor for pancreatic polypeptide, neuropeptide Y, and peptide YY. J. Biol. Chem. 270, 26762–26765 [DOI] [PubMed] [Google Scholar]
- 8. Lundell I., Blomqvist A. G., Berglund M. M., Schober D. A., Johnson D., Statnick M. A., Gadski R. A., Gehlert D. R., Larhammar D. (1995) Cloning of a human receptor of the NPY receptor family with high affinity for pancreatic polypeptide and peptide YY. J. Biol. Chem. 270, 29123–29128 [DOI] [PubMed] [Google Scholar]
- 9. Li J. B., Asakawa A., Terashi M., Cheng K., Chaolu H., Zoshiki T., Ushikai M., Sheriff S., Balasubramaniam A., Inui A. (2010) Regulatory effects of Y4 receptor agonist (BVD-74D) on food intake. Peptides 31, 1706–1710 [DOI] [PubMed] [Google Scholar]
- 10. Tough I. R., Holliday N. D., Cox H. M. (2006) Y(4) receptors mediate the inhibitory responses of pancreatic polypeptide in human and mouse colon mucosa. J. Pharmacol. Exp. Ther. 319, 20–30 [DOI] [PubMed] [Google Scholar]
- 11. Sainsbury A., Baldock P. A., Schwarzer C., Ueno N., Enriquez R. F., Couzens M., Inui A., Herzog H., Gardiner E. M. (2003) Synergistic effects of Y2 and Y4 receptors on adiposity and bone mass revealed in double knockout mice. Mol. Cell. Biol. 23, 5225–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wraith A., Törnsten A., Chardon P., Harbitz I., Chowdhary B. P., Andersson L., Lundin L. G., Larhammar D. (2000) Evolution of the neuropeptide Y receptor family: gene and chromosome duplications deduced from the cloning and mapping of the five receptor subtype genes in pig. Genome Res. 10, 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ekblad E., Sundler F. (2002) Distribution of pancreatic polypeptide and peptide YY. Peptides 23, 251–261 [DOI] [PubMed] [Google Scholar]
- 14. Perry B., Wang Y. (2012) Appetite regulation and weight control: the role of gut hormones. Nutr. Diabetes 2, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L., Bijker M. S., Herzog H. (2011) The neuropeptide Y system: pathophysiological and therapeutic implications in obesity and cancer. Pharmacol. Ther. 131, 91–113 [DOI] [PubMed] [Google Scholar]
- 16. Sautel M., Martinez R., Munoz M., Peitsch M. C., Beck-Sickinger A. G., Walker P. (1995) Role of a hydrophobic pocket of the human Y1 neuropeptide Y receptor in ligand binding. Mol. Cell. Endocrinol. 112, 215–222 [DOI] [PubMed] [Google Scholar]
- 17. Walker P., Munoz M., Martinez R., Peitsch M. C. (1994) Acidic residues in extracellular loops of the human Y1 neuropeptide Y receptor are essential for ligand binding. J. Biol. Chem. 269, 2863–2869 [PubMed] [Google Scholar]
- 18. Sautel M., Rudolf K., Wittneben H., Herzog H., Martinez R., Munoz M., Eberlein W., Engel W., Walker P., Beck-Sickinger A. G. (1996) Neuropeptide Y and the nonpeptide antagonist BIBP 3226 share an overlapping binding site at the human Y1 receptor. Mol. Pharmacol. 50, 285–292 [PubMed] [Google Scholar]
- 19. Sjödin P., Holmberg S. K., Akerberg H., Berglund M. M., Mohell N., Larhammar D. (2006) Re-evaluation of receptor-ligand interactions of the human neuropeptide Y receptor Y1: a site-directed mutagenesis study. Biochem. J. 393, 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Merten N., Lindner D., Rabe N., Römpler H., Mörl K., Schöneberg T., Beck-Sickinger A. G. (2007) Receptor subtype-specific docking of Asp6.59 with C-terminal arginine residues in Y receptor ligands. J. Biol. Chem. 282, 7543–7551 [DOI] [PubMed] [Google Scholar]
- 21. Rathmann D., Lindner D., DeLuca S. H., Kaufmann K. W., Meiler J., Beck-Sickinger A. G. (2012) Ligand-mimicking receptor variant discloses binding and activation mode of prolactin-releasing peptide. J. Biol. Chem. 287, 32181–32194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 23. Rathmann D., Pedragosa-Badia X., Beck-Sickinger A. G. (2013) In vitro modification of substituted cysteines as tool to study receptor functionality and structure-activity relationships. Anal. Biochem. 439, 173–183 [DOI] [PubMed] [Google Scholar]
- 24. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V. (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J. Mol. Biol. 342, 571–583 [DOI] [PubMed] [Google Scholar]
- 26. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. (2008) Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaakola V. P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., Stevens R. C. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chien E. Y., Liu W., Zhao Q., Katritch V., Han G. W., Hanson M. A., Shi L., Newman A. H., Javitch J. A., Cherezov V., Stevens R. C. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimamura T., Shiroishi M., Weyand S., Tsujimoto H., Winter G., Katritch V., Abagyan R., Cherezov V., Liu W., Han G. W., Kobayashi T., Stevens R. C., Iwata S. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haga K., Kruse A. C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W. I., Okada T., Kobilka B. K., Haga T., Kobayashi T. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., Sanna M. G., Han G. W., Kuhn P., Rosen H., Stevens R. C. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kruse A. C., Hu J., Pan A. C., Arlow D. H., Rosenbaum D. M., Rosemond E., Green H. F., Liu T., Chae P. S., Dror R. O., Shaw D. E., Weis W. I., Wess J., Kobilka B. K. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson A. A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X. P., Trapella C., Guerrini R., Calo G., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Granier S., Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Weis W. I., Kobilka B. K. (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konagurthu A. S., Whisstock J. C., Stuckey P. J., Lesk A. M. (2006) MUSTANG: a multiple structural alignment algorithm. Proteins 64, 559–574 [DOI] [PubMed] [Google Scholar]
- 40. Thompson J. D., Gibson T. J., Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics 00:2.3.1–2.3.22 [DOI] [PubMed] [Google Scholar]
- 41. Canutescu A. A., Dunbrack R. L., Jr. (2003) Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 12, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yarov-Yarovoy V., Schonbrun J., Baker D. (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alexander N., Woetzel N., Meiler J. (2011) 1st IEEE International Conference on Computational Advances in Bio and Medical Sciences, Orlando, FL, February 3–5, 2011 ((Mandoiu I. I., Miyano S., Przytycka T. A., Rajasekaran S., eds) IEEE Computer Society [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lerch M., Gafner V., Bader R., Christen B., Folkers G., Zerbe O. (2002) Bovine pancreatic polypeptide (bPP) undergoes significant changes in conformation and dynamics upon binding to DPC micelles. J. Mol. Biol. 322, 1117–1133 [DOI] [PubMed] [Google Scholar]
- 45. Gehlert D. R., Schober D. A., Gackenheimer S. L., Beavers L., Gadski R., Lundell I., Larhammar D. (1997) [125I]Leu31, Pro34-PYY is a high affinity radioligand for rat PP1/Y4 and Y1 receptors: evidence for heterogeneity in pancreatic polypeptide receptors. Peptides 18, 397–401 [DOI] [PubMed] [Google Scholar]
- 46. Walker M. W., Smith K. E., Bard J., Vaysse P. J., Gerald C., Daouti S., Weinshank R. L., Branchek T. A. (1997) A structure-activity analysis of the cloned rat and human Y4 receptors for pancreatic polypeptide. Peptides 18, 609–612 [DOI] [PubMed] [Google Scholar]
- 47. Simons K. T., Kooperberg C., Huang E., Baker D. (1997) Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 [DOI] [PubMed] [Google Scholar]
- 48. Lindner D., Stichel J., Beck-Sickinger A. G. (2008) Molecular recognition of the NPY hormone family by their receptors. Nutrition 24, 907–917 [DOI] [PubMed] [Google Scholar]
- 49. Dunbrack R. L., Jr., Karplus M. (1993) Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230, 543–574 [DOI] [PubMed] [Google Scholar]
- 50. Bradley P., Misura K. M., Baker D. (2005) Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871 [DOI] [PubMed] [Google Scholar]
- 51. Misura K. M., Baker D. (2005) Progress and challenges in high-resolution refinement of protein structure models. Proteins 59, 15–29 [DOI] [PubMed] [Google Scholar]
- 52. Eckard C. P., Cabrele C., Wieland H. A., Beck-Sickinger A. G. (2001) Characterisation of neuropeptide Y receptor subtypes by synthetic NPY analogues and by anti-receptor antibodies. Molecules 6, 448–467 [Google Scholar]
- 53. Klco J. M., Nikiforovich G. V., Baranski T. J. (2006) Genetic analysis of the first and third extracellular loops of the C5a receptor reveals an essential WXFG motif in the first loop. J. Biol. Chem. 281, 12010–12019 [DOI] [PubMed] [Google Scholar]
- 54. Kannoa T., Kanatani A., Keen S. L., Arai-Otsuki S., Haga Y., Iwama T., Ishihara A., Sakuraba A., Iwaasa H., Hirose M., Morishima H., Fukami T., Ihara M. (2001) Different binding sites for the neuropeptide Y Y1 antagonists 1229U91 and J-104870 on human Y1 receptors. Peptides 22, 405–413 [DOI] [PubMed] [Google Scholar]
- 55. Tautermann C. S., Pautsch A. (2011) The implication of the first agonist bound activated GPCR X-ray structure on GPCR in silico modeling. ACS Med. Chem. Lett. 2, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beck-Sickinger A. G., Wieland H. A., Wittneben H., Willim K. D., Rudolf K., Jung G. (1994) Complete Ll-alanine scan of neuropeptide Y reveals ligands binding to Y1 and Y2 receptors with distinguished conformations. Eur. J. Biochem. 225, 947–958 [DOI] [PubMed] [Google Scholar]
- 57. Raman B., Guarnaccia C., Nadassy K., Zakhariev S., Pintar A., Zanuttin F., Frigyes D., Acatrinei C., Vindigni A., Pongor G., Pongor S. (2001) N(ω)-arginine dimethylation modulates the interaction between a Gly/Arg-rich peptide from human nucleolin and nucleic acids. Nucleic Acids Res. 29, 3377–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sylte I., Andrianjara C. R., Calvet A., Pascal Y., Dahl S. G. (1999) Molecular dynamics of NPY Y1 receptor activation. Bioorg. Med. Chem. 7, 2737–2748 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.