

Background: The Ptp4a gene family encodes cancer-associated phosphatases with poorly understood in vivo functions.
Results: Mice deficient for PTP4A3 exhibit reduced tumor angiogenesis and decreased VEGF-mediated endothelial cell motility and vascular permeability.
Conclusion: PTP4A3 has an important role in endothelial cell response to proangiogenic VEGF signaling.
Significance: PTP4A3 appears to be an attractive molecular target for impeding angiogenesis in addition to tumor progression.
Keywords: Angiogenesis, Cancer, Endothelial Cell, Endothelium, Phosphatase, Src, Vascular Endothelial Growth Factor (VEGF), PTP4A3 (PRL-3), Vascular Permeability
Abstract
Protein-tyrosine phosphatase 4A3 (PTP4A3) is highly expressed in multiple human cancers and is hypothesized to have a critical, albeit poorly defined, role in the formation of experimental tumors in mice. PTP4A3 is broadly expressed in many tissues so the cellular basis of its etiological contributions to carcinogenesis may involve both tumor and stromal cells. In particular, PTP4A3 is expressed in the tumor vasculature and has been proposed to be a direct target of vascular endothelial growth factor (VEGF) signaling in endothelial cells. We now provide the first in vivo experimental evidence that PTP4A3 participates in VEGF signaling and contributes to the process of pathological angiogenesis. Colon tumor tissue isolated from Ptp4a3-null mice revealed reduced tumor microvessel density compared with wild type controls. Additionally, vascular cells derived from Ptp4a3-null tissues exhibited decreased invasiveness in an ex vivo wound healing assay. When primary endothelial cells were isolated and cultured in vitro, Ptp4a3-null cells displayed greatly reduced migration compared with wild type cells. Exposure to VEGF led to an increase in Src phosphorylation in wild type endothelial cells, a response that was completely ablated in Ptp4a3-null cells. In loss-of-function studies, reduced VEGF-mediated migration was also observed when human endothelial cells were treated with a small molecule inhibitor of PTP4A3. VEGF-mediated in vivo vascular permeability was significantly attenuated in PTP4A3-deficient mice. These findings strongly support a role for PTP4A3 as an important contributor to endothelial cell function and as a multimodal target for cancer therapy and mitigating VEGF-regulated angiogenesis.
Introduction
Protein-tyrosine phosphatase 4A3 (PTP4A3,3 also known as PRL-3) is a prenylated dual-specificity phosphatase with poorly understood enzymology and functionality (1). Mice in which Ptp4a3 is genetically ablated are healthy, fertile, and phenotypically similar to wild type littermates, although adult male homozygous knock-out mice exhibit a slightly decreased body mass (2). Loss of functional PTP4A3 partially suppresses colon carcinogenesis in a mouse model of colitis-associated colon cancer with no obvious differences in gross tumor histology (2). High gene expression levels of Ptp4a3, as well as its closely related family members Ptp4a1 and Ptp4a2, are often associated with tumor growth and metastasis of many human cancer types (3). Furthermore, poor patient prognosis and increased tumor invasiveness are commonly observed in many different malignancies expressing high levels of PTP4A3 (4, 5). Although the specific PTP4A substrates have remained elusive, several downstream signaling pathways have been proposed including: PI3K/AKT (6), Src (7), ERK1/2 (8), and Rho GTPases (9). Considering the multitude of proposed signaling effectors, it is likely that the function of PTP4A3 is tightly regulated by cell type and specific cues from the extracellular environment.
Preclinical and clinical findings have suggested that in addition to a role in cancer cells, PTP4A3 may be fundamentally involved in tumor angiogenesis. High Ptp4a3 gene expression levels are observed in tumor endothelium, pointing to its potential involvement in the pathological angiogenesis required for tumor progression as well as metastatic colonization (10, 11). Ptp4a3 is increased 10-fold in the vasculature of invasive breast tumors relative to normal vasculature (12). High PTP4A3 protein levels are also observed in developing heart tissue and blood vessels but not in their mature forms, suggesting a general role for PTP4A3 in cardiovascular system development (13).
Angiogenesis is a multifaceted process dependent on a complex network of growth factors and signaling pathways. Ectopic overexpression of PTP4A3 increases tube formation by endothelial cells, a phenotype associated with angiogenesis (14), whereas genetic or pharmacological reduction of PTP4A3 expression or activity leads to a decrease in tube formation (15). PTP4A3-expressing tumor cells can recruit endothelial cells both in vitro and in vivo (13). Vascular endothelial growth factor (VEGF) is a well characterized proangiogenic factor capable of inducing proliferation, migration, and sprouting of endothelial cells and is necessary for the creation of new vasculature. When VEGF binds to its cognate VEGFR2 receptor on endothelial cells, several key pathways that promote angiogenic signaling are activated. Known downstream effectors of VEGF signaling include Src (16) and MAP kinase-associated proteins (17). Interestingly, Ptp4a3 gene expression in cultured endothelial cells appears to be regulated by VEGF through its VEGFR2 receptor and the transcription factor myocyte enhancer factor 2C (15).
Because of the potential role of PTP4A3 in vascular function during angiogenesis and metastases, we tested the hypothesis that PTP4A3 is a mediator of the angiogenic phenotype of vascular cells in tumor and nontumor microenvironments. Accordingly, blood vessel development was contrasted in experimental colon tumors from wild type and Ptp4a3-null mice using CD31 immunocytochemistry. Aspects of wound repair were quantified in primary murine pulmonary endothelial cells from wild type and Ptp4a3-null mice. Furthermore, the contribution of PTP4A3 to VEGF signaling and its downstream signaling components, notably phosphorylation of Src, was examined. To complement previous siRNA studies (18–20), we confirmed disruption of human microvascular endothelium wound repair with a small molecule PTP4A3 inhibitor. Finally, we documented diminution of in vivo VEGF-mediated vascular permeability in mice lacking PTP4A3.
EXPERIMENTAL PROCEDURES
Ptp4a3 Mutant Mice
Creation of Ptp4a3 mutant mice, global gene deletion, and colon tumorigenesis experiments were performed as described previously (2). These mice have been donated to the Jackson Laboratory mutant strain repository (stock 21159). Experimental mice were produced by mating heterozygous breeding pairs, and genotyping was performed by Southern blotting of genomic DNA with a radiolabeled probe corresponding to exon 6 of the Ptp4a3 genomic locus. All animal experiments were performed in accordance with the guidelines of the University of Pittsburgh and University of Virginia Animal Care and Use Committees.
Measurement of Blood Pressure and Cardiovascular Output
Mouse blood pressure and cardiovascular output measurements were assayed with the CODA noninvasive tail cuff system (Kent Scientific). An occlusion cuff was placed at the base of the tail, and a volume pressure recording sensor was placed around the tail vein. Blood volume was measured by the differential pressure transducer, which utilizes a volumetric assay to determine blood flow and blood volume. Adult male mice (n = 5/genotype, 8–10 weeks) under normal diet and environmental conditions were used for the experiment. All tail cuff experiments were performed on restrained, conscious mice at approximately the same time of day, and body temperature was controlled with a heat pad at 37 °C. Mice were trained on the equipment daily for 4 consecutive days before experimental data were collected. Cardiovascular statistics for each mouse were determined as the average of at least three measurements taken on day 5.
Immunohistochemistry and Microvessel Density Quantification
Tissues from four mice per genotype as well as tumors from mice 16 weeks after azoxymethane treatment (2) (14 wild type and 11 Ptp4a3-null) were isolated and fixed in 10% neutral buffered formalin overnight at room temperature. Samples were then embedded in paraffin and sectioned onto glass slides. Slides were deparaffinized and antigenicity retrieved by steaming in EDTA, pH 8.0, for 30 min. For the microvessel density assay, an antibody against CD31 (clone M-20; Santa Cruz Biotechnology) was used at a concentration of 1:300 at room temperature overnight. For each colon cancer sample blinded and randomly chosen CD31-stained fields (n = 5/sample) from multiple tumors were imaged, positively stained vessels were counted, and vessel density was determined as a function of tissue area (mm2).
Western Blotting and Protein Quantification
Cell and tissue samples were lysed using radioimmuneprecipitation assay buffer, protein concentration was quantified by Bradford assay (Bio-Rad), and experiments were repeated in triplicate. Lysates (30 μg each) were separated using the Novex SDS-PAGE system and transferred to nitrocellulose membranes (Invitrogen). Membranes were blocked in Odyssey buffer (LI-COR Biosciences) and incubated with primary antibodies overnight followed by secondary fluorescent antibodies according to the manufacturers' instructions. Detection and signal quantification were performed with an Odyssey infrared imager (LI-COR Biosciences). We used the following commercially available primary antibodies: PTP4A3 clone 318 (Santa Cruz Biotechnology); VEGFR2, p-Src (Tyr-416), Src, p-ERK1/2 (Thr-202/Tyr-204), ERK1/2, FAK, p-FAK (Tyr-576/Tyr-577), paxillin, p-paxillin (Tyr-118) (Cell Signaling Technology); β-tubulin (Cedarlane Laboratories).
The phosphorylation status of VEGFR2 was measured using an ELISA-based antibody array according to the manufacturer's protocol (Full Moon Biosciences). Whole protein lysates (60 μg) from wild type and Ptp4a3-null endothelial cells were biotinylated and bound to individual antibody array slides. Secondary fluorescence was performed with Cy3-labeled streptavidin (Amersham Biosciences), and slides were shipped to the manufacturer for imaging and data analysis. Relative signal intensities were determined by comparing Ptp4a3-null median-centered values with wild type.
Tissue Explant Assay
Skeletal muscle tissue biopsies (pectoralis major muscle) were isolated from mice (n = 3/genotype, male, 6–8 weeks), placed into prewarmed microvascular endothelial growth medium (EGM-MV; Lonza) and cut into small (<2 mm) pieces. Collagen matrix was prepared with type I collagen containing Medium 199, l-glutamine, penicillin-streptomycin, sodium bicarbonate, and NaOH (Sigma-Aldrich) as reported previously (21). Tissue pieces were embedded into the matrix in individual wells (n = 14–33/mouse) of a 96-well tissue culture plate and overlaid with EGM-MV. The samples were incubated at 37 °C in 5% CO2 for 72 h. For each well, the distance from the tissue edge to the furthest migrating vascular cell was measured under a brightfield microscope with a total of 69 and 84 determinations performed from wild type and null tissues, respectively.
Primary Endothelial Cell Culture
Mouse primary endothelial cells were isolated from pulmonary tissue (n = 12 mice/genotype) as described previously (22). Cells were cultured on a collagen matrix in 2% O2, 5% CO2, 93% N2 in a Coy hypoxic glove box/chamber (Coy Laboratories, Inc.) in Opti-MEM (Invitrogen), 10% FBS, 2 mm glutamine, 0.2% retinal derived growth factor (ENDOGRO; Vec Technologies), 10 units/ml heparin, 0.1 mm nonessential amino acid supplement (Invitrogen), and 55 μm β-mercaptoethanol. Commercially available human microvascular endothelial cells (HMVECs; ScienCell or Lonza) were cultured in EGM-2 under normoxic conditions (5% CO2) in collagen-coated tissue culture flasks. The primary growth factor components of EGM-2, namely IGF, EGF, FGF, and VEGF, were purchased from Lonza and used at the manufacturer's recommended concentrations.
The PTP4A3 inhibitor BR-1 (Santa Cruz Biotechnology) was solubilized in tissue culture grade dimethyl sulfoxide (Sigma) and added to HMVECs at the indicated concentrations in 6- or 12-well plates. Lysates from BR-1-treated HMVECs were collected following 24 h of treatment.
In Vitro Wound Healing Assay
Primary mouse endothelial cells and HMVECs were grown to confluence in collagen-coated 12-well tissue culture plates (BD Biosciences). Each well (n = 6/genotype or treatment) was scratched longitudinally with a pipette tip and incubated for 16 h to allow gap closure. Cell migration distance was determined by measuring the gap distance between cell fronts following inward migration.
VEGF-mediated Vascular Permeability Assay
Adult male mice (n = 6–8/genotype, 8–12 weeks) under normal diet and environmental conditions were injected intravenously with Evans blue dye (100 μl of a 1% solution in 0.9% NaCl). After 20 min, 400 ng of recombinant murine VEGF (Peprotech) dissolved in 20 μl of PBS and 0.1% BSA or 20 μl of vehicle alone was injected intradermally into the shaved right or left flank of the mouse, respectively. After 20 min, the animals were euthanized and perfused with 10 ml of saline via the left ventricle. The area of skin containing the injection site was removed using a 5-mm biopsy punch (Miltex). Evans blue dye was extracted from the skin by incubation with 20 μl of formamide for 24 h at 57 °C, and the absorbance of the extracted dye was measured at 620 nm in triplicate in a 96-well plate.
Statistics
Statistical analysis of cell and tissue based assays, as well as Western blot quantification, was analyzed using the two-tailed Student's t test. For all experiments significance was defined as p < 0.05.
RESULTS
Expression and Knockout of Ptp4a3 in Cardiovascular Tissue
High levels of PTP4A3 expression have previously been observed in fetal heart and the immature blood vessels of tumor tissue (13). Western blot assay on mouse tissue samples revealed high PTP4A3 protein in lysates from fetal heart and adult spleen, whereas detectable levels were also found in adult heart, skeletal muscle, pancreas, lung, brain, and fetal intestine (Fig. 1A). As expected, fetal heart tissue (from whole heart lysates) exhibited the highest level of PTP4A3 protein in the tissues analyzed. Compared with adult heart and skeletal muscle tissues (pectoralis major), fetal heart had 5-fold greater protein levels (Fig. 1B). When fetal and adult heart tissues from wild type and Ptp4a3-null mice were compared, however, these tissues appeared overtly normal and healthy (Fig. 1C). Furthermore, immunohistochemical staining for the endothelial cell marker CD31 revealed similar blood vessel density in skeletal muscle tissue in both genotypes (Fig. 1C) under basal conditions consistent with our previous observation of no significant gross abnormalities in the mice (2).
FIGURE 1.

Loss of Ptp4a3 did not alter basal cardiovascular functionality. A, several wild type tissue lysates were assayed for PTP4A3 by Western blotting. B, fetal heart tissue exhibited 5-fold higher PTP4A3 protein than either adult heart or adult skeletal muscle (n = 3/tissue). C, wild type and Ptp4a3-null heart tissues did not display abnormalities when examined histologically. Skeletal muscle tissue was stained with antibodies against CD31 and demonstrated the presence of similar vasculature (arrows). Bars = 10 μm D, blood pressure was measured in wild type and Ptp4a3-null mice by tail cuff assay and did not produce a significant difference between genotypes (n = 5/genotype). E and F, the same animals tested normally for both heart rate (E) and blood flow through the tail vein (F). Error bars, S.E. for all panels.
We next examined cardiovascular function in adult wild type and knock-out mice. Blood pressure, heart rate, and tissue blood flow were measured using a noninvasive tail cuff system. Mice were acclimated to the assay system for four preliminary trials throughout the course of 1 week. Systolic and diastolic blood pressure and heart rate were not significantly different between genotypes (Fig. 1, D and E). Tail blood flow rate also was unaffected by the loss of PTP4A3 (Fig. 1F). Collectively, these results suggest that loss of PTP4A3 activity does not overtly affect heart and vessel development and is not required for basal function of the cardiovascular system in adult mice under nonstressed laboratory conditions.
Loss of PTP4A3 Decreased Tumor-driven Angiogenesis
Expression of human PTP4A3 has been reported in tumor endothelium (10, 12) and potentially could contribute to the formation of the extensive vascular network necessary for tumor growth. Furthermore, PTP4A3-expressing tumor cells have the ability to functionally recruit endothelial cells in vitro and in vivo (13). Mice deficient for PTP4A3 develop fewer tumors in a carcinogenesis model of colitis-associated colon cancer driven by azoxymethane and dextran sodium sulfate (2). The adenocarcinomas that form in the wild type and Ptp4a3-null mice do not exhibit gross histological differences (2). We found that both wild type and knock-out tumors were vascularized as evidenced by CD31-positive staining of endothelium in both genotypes (Fig. 2, A and B). Ptp4a3-null tumors, however, had a reduced microvessel presence compared with tumors from the wild type mice, which was evident when the tissues were examined under higher magnification (Fig. 2, C and D). On average, microvessel density was decreased 30% in Ptp4a3-null tissue compared with wild type (Fig. 2E). Because of inherent skewness of vessel density in autochthonous-growing tumors (23), we also performed log transformation of the data set and continued to observe a statistically significant (p = 0.001) difference between the tumors from wild type and PTP4A3-null mice. These findings support a critical role for PTP4A3 in the development of the tumor vasculature in this mouse model of colon cancer.
FIGURE 2.
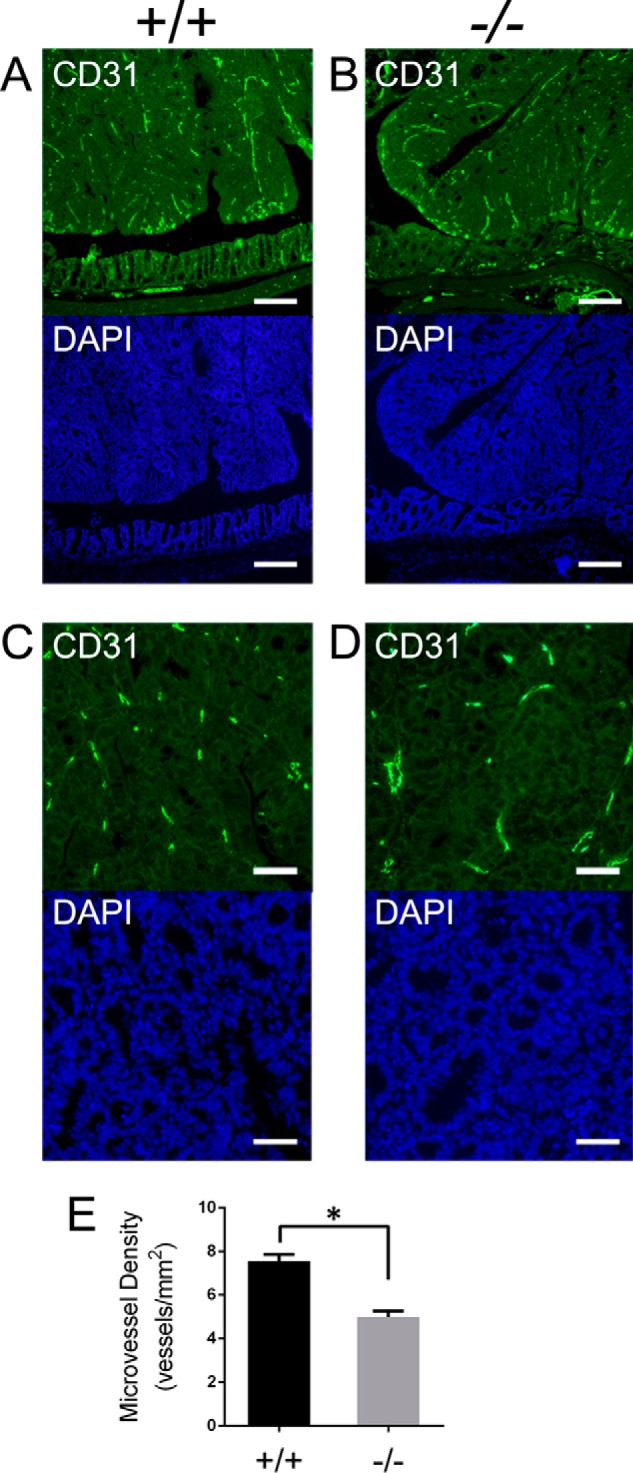
Microvessel density was decreased in azoxymethane-derived Ptp4a3-null tumors. A and B, colon tumor and adjacent normal tissue from wild type and Ptp4a3-null mice were stained for CD31 (FITC) to visualize microvessel presence. Colon tumor and adjacent normal tissue were also stained with DAPI (×4 magnification; scale bars, 50 μm). C and D, wild type and Ptp4a3-null tumor tissue examined under higher magnification revealed less vessel formation in knock-out tissue compared with wild type by CD31 staining. Tumor tissue was also stained with DAPI (×20 magnification; scale bars, 10 μm). E, for each colon sample (n = 4/genotype) the average of 5 fields taken from multiple tumors (n = 11 wild type and n = 14 null) demonstrated a 30% reduction in CD31+ microvessel density (*, p < 0.001). The mean vessel density of the tumors from wild type mice ranged from 37.4 ± 4.7 (S.E., error bars) mm2 to 45.2 ± 2.7 mm2. The mean vessel density of the tumors from PTP4A3-null mice ranged from 25.6 ± 2.7 mm2 to 32.8 ± 3.9 mm2.
Decreased Vascular Cell Invasion from PTP4A3 Knock-out Tissues
Examination of tumor angiogenesis in vivo is complex because of inherent variability in vascular density but also due to differences in intratumor and extratumor vasculature and vessel size (23, 24). Therefore we more precisely examined the angiogenic phenotype of vascular cells from wild type and Ptp4a3-null mice. An ex vivo invasion assay was implemented using skeletal muscle tissue biopsies that were capable of producing outgrowth of vascular cells in response to external stimuli. Tissue samples were embedded in a collagen matrix containing microvascular endothelial growth medium, and vascular cell invasion was determined after 72 h (Fig. 3, A and B). The resulting outgrowth of cells into the matrix has been commonly used to measure capacity for wound healing or angiogenesis (21), although the migrating cells may not be exclusively endothelial in origin. When examined under higher magnification, the morphology of invasive cells in the three-dimensional matrix became apparent (Fig. 3C). Compared with wild type tissue, Ptp4a3-null samples produced a reduction in outgrowth as measured by the distance from the solid tissue to the farthest invading cell. The relative invasion distance after incubation for 72 h was 20% less in Ptp4a3 knock-out cells compared with wild type cells (Fig. 3D). This result suggested that PTP4A3 was a contributing factor to vascular cell invasiveness that is important during angiogenesis.
FIGURE 3.
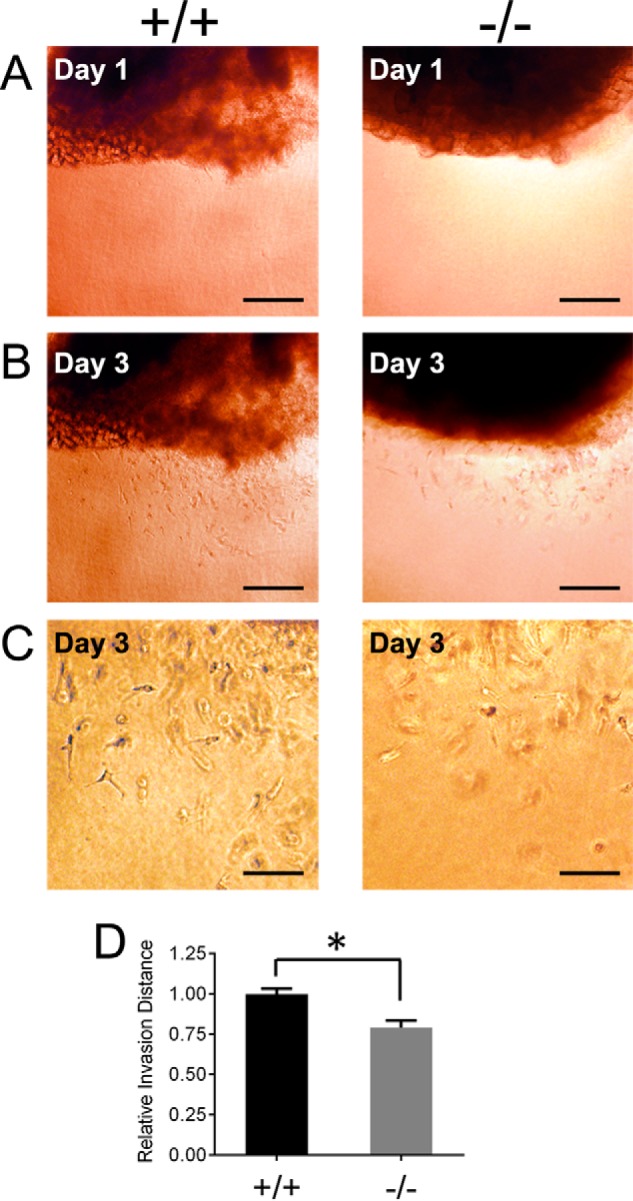
Ptp4a3 knock-out tissue exhibited reduced vascular cell invasion ex vivo. A and B, skeletal muscle tissue biopsies from wild type and Ptp4a3-null mice were embedded in a three-dimensional collagen matrix allowing for outgrowth of vascular cells (scale bars, 50 μm). C, invasive vascular cells examined under higher magnification displayed an apical and invasive appearance when migrating in the matrix. D, cells from Ptp4a3-null tissue exhibited a 20% decrease in invasion distance compared with cells expressing PTP4A3 (*, p < 0.02). Error bars, S.E.
Ptp4a3 Knock-out Endothelial Cells Exhibit Reduced Cell Migration
The cellular function of PTP4A3 was further examined with cultured pulmonary endothelium from adult wild type and Ptp4a3-null mice. Cells were cultured, and purity was assessed as described under “Experimental Procedures.” Cell migration was measured using an in vitro wound healing assay in which endothelial cells were grown to confluence and a longitudinal gap was created. These cells were incubated for 16 h to permit gap closure via endothelial migration (Fig. 4, A–D). Following incubation, migration was determined as the relative gap closure exhibited by the cells. On average, Ptp4a3-null cells migrated 50% less than the corresponding wild type cells (Fig. 4E). This was strong evidence for a functional role for PTP4A3 in endothelial migration, which is necessary for angiogenesis.
FIGURE 4.
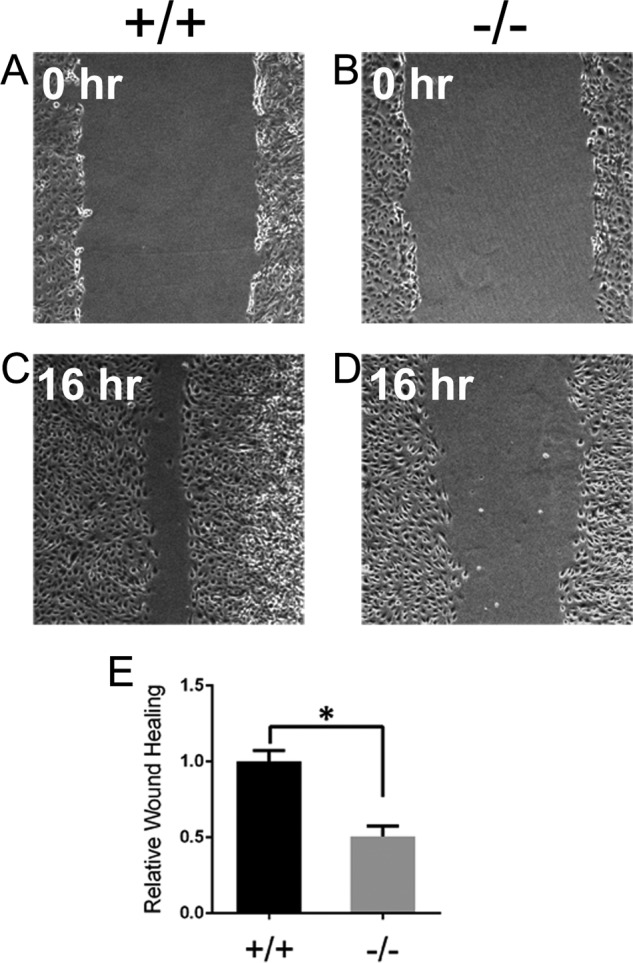
Knock-out of Ptp4a3 decreased in vitro migration of endothelial cells. A–D, pulmonary endothelial cells were purified from wild type and Ptp4a3-null mice, seeded evenly, and grown to confluence in collagenized cell culture plates. A wound was created in the cell layer by scratching longitudinally with a pipette tip, and migration, in the presence of complete medium and growth factors, was measured after 16 h. E, relative wound healing was determined by measuring the gap closure between cell fronts following the 16-h migration period. Ptp4a3-null cells migrated 50% less compared with wild type endothelial cells (*, p < 0.001). Error bars, S.E.
Loss of Ptp4a3 Alters Angiogenic Signaling Pathways
Although it has been reported that Ptp4a3 gene expression is induced in human umbilical vein endothelial cells following VEGF treatment (15), the distal effects of PTP4A3 in the endothelium have not yet been explored. To investigate the molecular effects of eliminating PTP4A3 expression in endothelial cells, we assayed the activation state of several known angiogenesis-associated pathways following VEGF treatment. Endothelial cells from wild type and Ptp4a3-null pulmonary tissue were cultured and incubated in serum-free medium for 4 h. VEGF was added to the culture medium (50 ng/ml), and protein samples were collected at indicated time points for up to 8 h after treatment. Two known VEGF-dependent mediators of the angiogenic phenotype, Src (16) and ERK1/2 (17), have also been associated with PTP4A3 signaling (7, 8). Activation of both proteins was examined in VEGF-treated wild type and Ptp4a3-null endothelial cells by Western blotting (Fig. 5A). As expected, mature VEGFR2 protein levels decreased in both genotypes following VEGF treatment, presumably due to internalization of the receptor upon activation. Reproducible differences in basal Src protein phosphorylation (Tyr-416) prior to VEGF treatment were not observed (Fig. 5A). A significant increase in Src protein phosphorylation (Tyr-416) was observed in wild type cells as early as 15 min following VEGF treatment and persisted up to 8 h (Fig. 5B). Comparatively, Ptp4a3-null endothelial cells did not exhibit an increase in Src phosphorylation at any time point following VEGF exposure. An increase in phosphorylation of ERK1/2 protein was observed in both genotypes following treatment with VEGF (Fig. 5A). Levels of total Src and ERK1/2 protein were unchanged throughout this time course. We also observed modest increased phosphorylation of FAK (Tyr-576/Tyr-577) and paxillin (Tyr-118), when wild type endothelial cells were treated with VEGF (Fig. 5D). This was not observed with the Ptp4a3-null endothelial cells (Fig. 5D). Several tyrosine sites have been identified as important in the transduction of extracellular signals by VEGFR2 (25). We, therefore, examined the tyrosine phosphorylation status of VEGFR2 with a commercially available antibody array and observed a 50% decrease in Tyr-1054 phosphorylation in Ptp4a3-null endothelial cells 15 min after VEGF treatment compared with the wild type cells and small decrease in Tyr-951 phosphorylation (Fig. 5C). Thus, PTP4A3 appeared to be important for VEGF-induced activation of SRC, but not necessary for ERK1/2 activation in endothelial cells.
FIGURE 5.

PTP4A3 mediated the endothelial cell response to VEGF exposure. A, wild type and Ptp4a3-null endothelial cells were grown in culture and treated with VEGF (50 ng/ml) for up to 8 h. Protein lysates were collected at indicated time points, and Western blot analysis was performed to determine the activation levels of two known signaling effectors of the VEGF pathway (Src and ERK1/2). B, in wild type cells, relative p-Src (activated) was increased ∼2-fold following 15 min of VEGF treatment, and phosphorylation was significantly increased at all time points up to 8 h later. Increased activation of Src was not observed at any time point when Ptp4a3-null cells were treated with VEGF. C, VEGFR2 tyrosine phosphorylation was assayed from protein lysates of wild type and Ptp4a3-null endothelial cells by an ELISA-based antibody array. Protein values are the result of six determinations from a single sample. D, wild type and Ptp4a3-null endothelial cells were grown in culture and treated with VEGF (50 ng/ml) for 8 h. Protein lysates were collected as in A, and Western blot analysis was performed to assay activation of the downstream Src targets FAK and paxillin.
Pharmacological Inhibition of PTP4A3 Impairs Human Endothelial Cell Migration
Recent studies indicate that suppression of Ptp4a3 expression results in altered lamellipodia formation and cellular morphology that are necessary for cellular migration and invasion (25). Therefore, we next examined the role of PTP4A3 in HMVEC migration using the pharmacological tool BR-1, which is a previously reported PTP4A3 inhibitor (26) with limited inhibitory activity against other related phosphatases (27). The essential role of VEGF in HMVEC migration was demonstrated by addition/removal studies of the complete growth medium, which contained IGF-1, EGF, FGF, and VEGF (Fig. 6, A and B). Using the in vitro wound healing assay, we observed almost complete closure of the HMVEC wound within 16 h in the presence of complete growth factors (Fig. 6, B and C). Removal of VEGF from the complete growth factor mixture resulted in an almost complete loss of migration whereas removal of FGF had a minimal effect (Fig. 6A). Exclusion of either EGF or IGF-1 produced an intermediate diminution in migration. Unlike IGF-1, VEGF alone was competent in reproducing most of the migration seen with the complete growth factor mixture, indicating its central role in the process of endothelial migration. The PTP4A3 inhibitor BR-1 produced a concentration-dependent decrease in endothelial cell motility when examined in a wound healing assay but had no effect on the basal migration in the absence of growth factors or with complete growth factors lacking only VEGF (Fig. 6, B and D). Moreover, 10 μm BR-1 significantly reduced migration stimulated by VEGF alone (Fig. 6B). These results demonstrate further the importance of the VEGF and PTP4A3 interactions.
FIGURE 6.

Pharmacological inhibition of PTP4A3 altered human microvascular endothelial cell properties. A, growth factors stimulated migration of HMVECs. Confluent endothelial cells were scratched longitudinally as in Fig. 4, and wound healing was measured 16 h after exposure to various growth factors. B, effect of PTP4A3 inhibitor BR-1 on wound closure was mediated by complete growth factors, growth factors lacking VEGF, or VEGF alone. C, BR-1 concentration-dependent inhibition of complete growth medium-mediated HMVEC wound closure is shown. D, BR-1 inhibition of complete growth factor-stimulated migration is quantified. Native HMVECs exhibit a significant decrease in migration when treated with BR-1 relative to dimethyl sulfoxide (DMSO) vehicle control (*, p < 0.02). Error bars, S.E.
Loss of PTP4A3 Inhibits VEGF-mediated Vascular Permeability
It is well established that tumors exhibit vascular hyperpermeability, which is induced by elevated levels of VEGF (28). The enhanced permeability accompanies pathogenic angiogenesis and facilities the extravasation of tumor cells and movement of growth factors and other cell types. Although VEGF-induced vascular permeability was initially believed to be independent of Src (29), more recent studies (30) reveal a fundamental role for Src in regulating VEGF-mediated vascular permeability. We therefore examined VEGF-induced vascular permeability in Ptp4a3 wild type, heterozygous, and homozygous null mice using a modified Miles assay (31, 32). Wild type mice exhibited a robust response to VEGF intradermal injection compared with vehicle alone (Fig. 7). In contrast, Ptp4a3-null mice were almost nonresponsive to VEGF treatment with an intermediate response with the heterozygous mice (Fig. 7). These in vivo results strongly support our conclusion that PTP4A3 has a previously unrecognized role in controlling VEGF signaling and could have significance in tumor metastasis.
FIGURE 7.

Loss of PTP4A3 reduced VEGF-mediated vascular permeability. Adult male mice (n = 6–8 per genotype) were injected intravenously with Evans blue dye and 20 min later injected intradermally with 400 ng of recombinant murine VEGF (gray bars) or PBS (black bars) in the flank. Total dye in a 5-mm biopsy punch was extracted, and dye absorbance was determined with a spectrophotometer. Ptp4a3-null mice exhibited significantly reduced vascular permeability in response to EGF relative to wild type mice (*, p < 0.01). Error bars, S.E.
DISCUSSION
Although the precise function of PTP4A3 (a phosphatase of regenerating liver family enzyme) remains unclear, it is closely associated with tumor growth and metastasis (11). Elevated levels of PTP4A3, especially in gastrointestinal tumors, are useful biomarkers and may be a novel therapeutic target (5). It is becoming clear that PTP4A3 can functionally contribute to multiple aspects of malignant disease including tumor formation, metastasis, and angiogenesis. Accordingly, the current study further examines a previously reported whole animal genetic ablation model of Ptp4a3 with respect to the vasculature component of carcinogenesis. Primary cultures of murine and human endothelium were used to determine the role of endothelial PTP4A3 in cell migration and VEGF response.
Ptp4a3-null mice are phenotypically similar to their wild type littermates under standard conditions in terms of cardiac and skeletal muscle morphometry, vascularity, blood pressure, heart rate, and tail vein blood flow (Fig. 1). Nonetheless, Ptp4a3 gene deletion produced colon tumors with similar gross histopathology but with reduced microvessel density compared with wild type tumors (Fig. 2). The absence of differences in gross histopathology between the tumors of the wild type and Ptp4a3-null mice can be rationalized by the complexities and redundancies of the angiogenic process. In addition to the potential involvement of extratumoral vasculature and different size vessels in autochthonous tumor development, vessel density measurements have a debatable value in predicting patient outcomes (24). Nevertheless, tumor cells expressing high levels of PTP4A3 can recruit endothelial cells both in vitro and in vivo (13). In our study endothelial cells lacking PTP4A3 were less invasive and migratory when assayed ex vivo. The tissue explant assay used models an important early step in the angiogenic process: the period when cells invade the basal lamina as a precursor to vessel sprouting. Although not all of the invading cells are likely to be of endothelial origin, tissue samples from Ptp4a3-null mice displayed significantly less invasive capacity measured by this assay (Fig. 3). Likewise, the in vitro wound healing assay demonstrated that when PTP4A3 has been genetically ablated or pharmacologically inhibited, endothelial cells are significantly deficient in gap closure (Fig. 4). Because of the length of this assay (16 h) and the required doubling time of these primary endothelial cells (>30 h), this effect is likely due to an impairment of cellular migration, although at this time we cannot rule out an effect on cell proliferation as well.
The exact mechanism by which PTP4A3 alters endothelial function remains uncertain in the absence of validated substrate(s). Although a reduction in protein phosphorylation with the depletion of a phosphatase might initially appear paradoxical, there are several reports showing that an increase in PTP4A3 activates phosphorylation-dependent responses, and a decrement in its activity leads to suppression. PTP4A3 activates the NK-κB signaling pathway by interacting with repressor/activator protein 1 (RAP1) (33). Reduction in PTP4A3 reduces Rho signaling pathways (9). PTP4A3 has been found to induce EGF receptor activation and distal phosphorylation events possibility due to transcriptional down-regulation of PTP1B (34). Additionally, PTP4A3 activates Src kinase by down-regulating C-terminal Src kinase (Csk) (7).
The results presented here suggested that several signaling proteins, notably Src, FAK, and paxillin, were affected by the absence of PTP4A3 in this cell system. PTP4A3 overexpression has been previously associated with increased migration and invasion in cancer cell lines (35) and conceivably could have a role in these processes in nonmalignant stromal cell types such as endothelial cells. The current results support this hypothesis and suggest that Src is a downstream effector of PTP4A3 in endothelial cells, as Ptp4a3-null cells completely lost the ability to increase Src activation following VEGF exposure (Fig. 5). VEGFR2 is known to be regulated by intracellular tyrosine phosphorylation on multiple sites including Tyr-951, Tyr-1054,Tyr-1059, Tyr-1175, and Tyr-1214 (30). VEGFR2 phosphorylation on Tyr-951 facilitates binding of VEGFR2 to the Src homology-2 domain of T cell-specific adaptor, TSAd, which regulated VEGF-induced activation of Src kinase (30). Tyr-1059 serves as a positive regulator and is located on the kinase activation loop of VEGFR2 (30). It is therefore interesting that loss of PTP4A3 resulted in a reduction of phosphotyrosine 951 and 1059. This presumably indirect effect seems worthy of further investigation. To further validate PTP4A3 as a potential pharmacological target, we examined a human endothelial cell model. Following treatment of HMVECs with increasing concentrations of the PTP4A3 inhibitor BR-1, we observed a concentration-dependent decrease in cell migration (Fig. 6). These results corroborate siRNA studies in other cell types (18–20). Recent data (19) indicate that Src may regulate PTP4A3 phosphorylation on Tyr-53. This is consistent with the growing appreciation for reciprocal regulation of signaling pathways.
Because PTP4A3 is expressed in the heart and developing cardiovascular system, there has been concern over the safety and potential cardiotoxic side effects of PTP4A3 inhibitors. Mice without a functional Ptp4a3 allele are able to develop normally and do not have an overt cardiovascular phenotype under standard conditions. Moreover, Ptp4a3-null mice do not exhibit altered blood pressure or heart rate, suggesting that the function of the cardiovascular system, at least under basal conditions, was not adversely affected by the embryonic loss of PTP4A3. Nevertheless, examination of the long term cardiovascular effects of PTP4A3 inhibitors are desirable because of the established importance of VEGF on endothelial cell biology.
PTP4A3 is an attractive potential target for the treatment of multiple forms of malignancy due to high expression levels observed in human tumors and frequent associations with cancer cell invasiveness (5). Importantly, PTP4A3 is also expressed in the tumor vasculature, and our data suggest that it may be an important mediator of pathological angiogenesis and vascular permeability, which are essential processes for tumor progression and metastasis. It is possible that efforts to therapeutically target PTP4A3 in cancer cells may also have the added benefit of inhibiting tumor angiogenesis by decreasing PTP4A3 activity in the vasculature. Our results support this hypothesis by demonstrating that Ptp4a3-deficient cells and tissues have a reduced angiogenic phenotype. Targeting the VEGF signaling pathway is a clinically established effective means of treating multiple human cancers including primary and metastatic tumors of the colon (36), lung (37), and kidney (38). Unfortunately, even malignancies that benefit from antiangiogenic therapy typically relapse and become resistant to treatments targeting VEGF signaling (39). For this reason, downstream VEGF targets are actively being pursued as multiple opportunities for therapeutic intervention likely exist.
The results presented in this paper add novel in vivo evidence that PTP4A3 has a vital role in controlling the migratory and invasive properties of nonmalignant cells, specifically endothelial cells. PTP4A3 appears to be an attractive therapeutic target downstream of VEGF and could theoretically be involved in many conditions involving pathological angiogenesis including cancer, diabetes, macular degeneration, and cardiovascular dysfunction.
Acknowledgments
We thank Carolyn Ferguson (University of Pittsburgh) for technical expertise and Rob Powers for providing tail-cuff assay equipment (University of Pittsburgh).
This work was supported by National Institute of Alcohol Abuse and Alcoholism Grant R37AA10422 and Fellowship F31AA019597 and National Institutes of Health Pharmacological Sciences Training Grant Predoctoral Fellowship T32GM007055.
- PTP4A3
- protein-tyrosine phosphatase 4A3
- EGM-MV
- microvascular endothelial growth medium
- HMVEC
- human microvascular endothelial cell.
REFERENCES
- 1. Zeng Q., Si X., Horstmann H., Xu Y., Hong W., Pallen C. J. (2000) Prenylation-dependent association of protein-tyrosine phosphatases PRL-1, -2, and -3 with the plasma membrane and the early endosome. J. Biol. Chem. 275, 21444–21452 [DOI] [PubMed] [Google Scholar]
- 2. Zimmerman M. W., Homanics G. E., Lazo J. S. (2013) Targeted deletion of the metastasis-associated phophatase Ptp4a3 (PRL-3) suppresses murine colon cancer. PLoS One 8, e58300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stephens B. J., Han H., Gokhale V., Von Hoff D. D. (2005) PRL phosphatases as potential molecular targets in cancer. Mol. Cancer Ther. 4, 1653–1661 [DOI] [PubMed] [Google Scholar]
- 4. Radke I., Götte M., Kersting C., Mattsson B., Kiesel L., Wülfing P. (2006) Expression and prognostic impact of the protein-tyrosine phosphatases PRL-1, PRL-2, and PRL-3 in breast cancer. Br. J. Cancer 95, 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Molleví D. G., Aytes A., Padullés L., Martínez-Iniesta M., Baixeras N., Salazar R., Ramos E., Figueras J., Capella G., Villanueva A. (2008) PRL-3 is essentially overexpressed in primary colorectal tumours and associates with tumour aggressiveness. Br. J. Cancer 99, 1718–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang H., Quah S. Y., Dong J. M., Manser E., Tang J. P., Zeng Q. (2007) PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 67, 2922–2926 [DOI] [PubMed] [Google Scholar]
- 7. Liang F., Liang J., Wang W. Q., Sun J. P., Udho E., Zhang Z. Y. (2007) PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J. Biol. Chem. 282, 5413–5419 [DOI] [PubMed] [Google Scholar]
- 8. Ming J., Liu N., Gu Y., Qiu X., Wang E. H. (2009) PRL-3 facilitates angiogenesis and metastasis by increasing ERK phosphorylation and up-regulating the levels and activities of Rho-A/C in lung cancer. Pathology 41, 118–126 [DOI] [PubMed] [Google Scholar]
- 9. Fiordalisi J. J., Keller P. J., Cox A. D. (2006) PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res. 66, 3153–3161 [DOI] [PubMed] [Google Scholar]
- 10. St Croix B., Rago C., Velculescu V., Traverso G., Romans K. E., Montgomery E., Lal A., Riggins G. J., Lengauer C., Vogelstein B., Kinzler K. W. (2000) Genes expressed in human tumor endothelium. Science 289, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 11. Bardelli A., Saha S., Sager J. A., Romans K. E., Xin B., Markowitz S. D., Lengauer C., Velculescu V. E., Kinzler K. W., Vogelstein B. (2003) PRL-3 expression in metastatic cancers. Clin. Cancer Res. 9, 5607–5615 [PubMed] [Google Scholar]
- 12. Parker B. S., Argani P., Cook B. P., Liangfeng H., Chartrand S. D., Zhang M., Saha S., Bardelli A., Jiang Y., St Martin T. B., Nacht M., Teicher B. A., Klinger K. W., Sukumar S., Madden S. L. (2004) Alterations in vascular gene expression in invasive breast carcinoma. Cancer Res. 64, 7857–7866 [DOI] [PubMed] [Google Scholar]
- 13. Guo K., Li J., Wang H., Osato M., Tang J. P., Quah S. Y., Gan B. Q., Zeng Q. (2006) PRL-3 initiates tumor angiogenesis by recruiting endothelial cells in vitro and in vivo. Cancer Res. 66, 9625–9635 [DOI] [PubMed] [Google Scholar]
- 14. Rouleau C., Roy A., St Martin T., Dufault M. R., Boutin P., Liu D., Zhang M., Puorro-Radzwill K., Rulli L., Reczek D., Bagley R., Byrne A., Weber W., Roberts B., Klinger K., Brondyk W., Nacht M., Madden S., Burrier R., Shankara S., Teicher B. A. (2006) Protein-tyrosine phosphatase PRL-3 in malignant cells and endothelial cells: expression and function. Mol. Cancer Ther. 5, 219–229 [DOI] [PubMed] [Google Scholar]
- 15. Xu J., Cao S., Wang L., Xu R., Chen G., Xu Q. (2011) VEGF promotes the transcription of the human PRL-3 gene in HUVEC through transcription factor MEF2C. PLoS One 6, e27165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He H., Venema V. J., Gu X., Venema R. C., Marrero M. B., Caldwell R. B. (1999) Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J. Biol. Chem. 274, 25130–25135 [DOI] [PubMed] [Google Scholar]
- 17. Rousseau S., Houle F., Landry J., Huot J. (1997) p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15, 2169–2177 [DOI] [PubMed] [Google Scholar]
- 18. Fagerli U. M., Holt R. U., Holien T., Vaatsveen T. K., Zhan F., Egeberg K. W., Barlogie B., Waage A., Aarset H., Dai H. Y., Shaughnessy J. D., Jr., Sundan A., Børset M. (2008) Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood 111, 806–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fiordalisi J. J., Dewar B. J., Graves L. M., Madigan J. P., Cox A. D. (2013) Src-mediated phosphorylation of the tyrosine phosphatase PRL-3 is required for PRL-3 promotion of Rho activation, motility and invasion. PLoS One 8, e64309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ooki A., Yamashita K., Kikuchi S., Sakuramoto S., Katada N., Waraya M., Kawamata H., Nishimiya H., Nakamura K., Watanabe M. (2011) Therapeutic potential of PRL-3 targeting and clinical significance of PRL-3 genomic amplification in gastric cancer. BMC Cancer 11, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isenberg J. S., Ridnour L. A., Perruccio E. M., Espey M. G., Wink D. A., Roberts D. D. (2005) Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 102, 13141–13146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tyurina Y. Y., Tyurin V. A., Kapralova V. I., Wasserloos K., Mosher M., Epperly M. W., Greenberger J. S., Pitt B. R., Kagan V. E. (2011) Oxidative lipidomics of γ-radiation-induced lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Radiat. Res. 175, 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson H. J., McGinley J. N., Knott K. K., Spoelstra N. S., Wolfe P. (2002) Vascular density profile of rat mammary carcinomas induced by 1-methyl-1-nitrosourea: implications for the investigation of angiogenesis. Carcinogenesis 23, 847–854 [DOI] [PubMed] [Google Scholar]
- 24. Preusser M., Heinzl H., Gelpi E., Schonegger K., Haberler C., Birner P., Marosi C., Hegi M., Gorlia T., Hainfellner J. A., and European Organization for Research and Treatment of Cancer Brain Tumor Group (2006) Histopathologic assessment of hot-spot microvessel density and vascular patterns in glioblastoma: Poor observer agreement limits clinical utility as prognostic factors: a translational research project of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Cancer 107, 162–170 [DOI] [PubMed] [Google Scholar]
- 25. Liu H., Omer Al-aidaroos A. Q., Wang H., Guo K., Li J., Zhang H. F., Zeng Q. (2013) PRL-3 suppresses c-FOS and integrin a2 expression in ovarian cancer cells. BMC Cancer 13, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ahn J. H., Kim S. J., Park W. S., Cho S. Y., Ha J. D., Kim S. S., Kang S. K., Jeong D. G., Jung S. K., Lee S. H., Kim H. M., Park S. K., Lee K. H., Lee C. W., Ryu S. E., Choi J. K. (2006) Synthesis and biological evaluation of rhodanine derivatives as PRL-3 inhibitors. Bioorg. Med. Chem. Lett. 16, 2996–2999 [DOI] [PubMed] [Google Scholar]
- 27. Min G., Lee S. K., Kim H. N., Han Y. M., Lee R. H., Jeong D. G., Han D. C., Kwon B. M. (2013) Rhodanine-based PRL-3 inhibitors blocked the migration and invasion of metastatic cancer cells. Bioorg. Med. Chem. Lett. 23, 3769–3774 [DOI] [PubMed] [Google Scholar]
- 28. Nagy J. A., Dvorak A. M., Dvorak H. F. (2012) Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harb. Perspect. Med. 2, a006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eliceiri B. P., Paul R., Schwartzberg P. L., Hood J. D., Leng J., Cheresh D. A. (1999) Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4, 915–924 [DOI] [PubMed] [Google Scholar]
- 30. Sun Z., Li X., Massena S., Kutschera S., Padhan N., Gualandi L., Sundvold-Gjerstad V., Gustafsson K., Choy W. W., Zang G., Quach M., Jansson L., Phillipson M., Abid M. R., Spurkland A., Claesson-Welsh L. (2012) VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J. Exp. Med. 209, 1363–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miles A. A., Miles E. M. (1952) Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea pigs. J. Physiol. 118, 228–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Auerbach R., Akhtar N., Lewis R. L., Shinners B. L. (2000) Angiogenesis assays: problems and pitfalls. Cancer Metastasis Rev. 19, 167–172 [DOI] [PubMed] [Google Scholar]
- 33. Lian S., Meng L., Liu C., Xing X., Song Q., Dong B., Han Y., Yang Y., Peng L., Qu L., Shou C. (2013) PRL-3 activates NF-κB signaling pathway by interacting with RAP1. Biochem. Biophys. Res. Commun. 430, 196–201 [DOI] [PubMed] [Google Scholar]
- 34. Al-Aidaroos A. Q., Yuen H. F., Guo K., Zhang S. D., Chung T. H., Chng W. J., Zeng Q. (2013) Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. J. Clin. Invest. 123, 3459–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zeng Q., Dong J. M., Guo K., Li J., Tan H. X., Koh V., Pallen C. J., Manser E., Hong W. (2003) PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 63, 2716–2722 [PubMed] [Google Scholar]
- 36. Hurwitz H., Fehrenbacher L., Novotny W., Cartwright T., Hainsworth J., Heim W., Berlin J., Baron A., Griffing S., Holmgren E., Ferrara N., Fyfe G., Rogers B., Ross R., Kabbinavar F. (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342 [DOI] [PubMed] [Google Scholar]
- 37. Sandler A., Gray R., Perry M. C., Brahmer J., Schiller J. H., Dowlati A., Lilenbaum R., Johnson D. H. (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 355, 2542–2550 [DOI] [PubMed] [Google Scholar]
- 38. Escudier B., Eisen T., Stadler W. M., Szczylik C., Oudard S., Siebels M., Negrier S., Chevreau C., Solska E., Desai A. A., Rolland F., Demkow T., Hutson T. E., Gore M., Freeman S., Schwartz B., Shan M., Simantov R., Bukowski R. M. (2007) Sorafenib in advanced clear-cell renal cell carcinoma. N. Engl. J. Med. 356, 125–134 [DOI] [PubMed] [Google Scholar]
- 39. Casanovas O., Hicklin D. J., Bergers G., Hanahan D. (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309 [DOI] [PubMed] [Google Scholar]