

Background: Metabolic homeostasis is central to normal cardiac function. The molecular mechanisms underlying metabolic plasticity in the heart remain poorly understood.
Results: Kruppel-like factor 15 (KLF15) is a direct and independent regulator of myocardial lipid flux.
Conclusion: KLF15 is a core component of the transcriptional circuitry that governs cardiac metabolism.
Significance: This work is the first to implicate the KLF transcription factor family in cardiac metabolism.
Keywords: Cardiac Metabolism, Cardiovascular, Kruppel-like Factor (KLF), Lipid Metabolism, Transcription Factors
Abstract
The mammalian heart, the body's largest energy consumer, has evolved robust mechanisms to tightly couple fuel supply with energy demand across a wide range of physiologic and pathophysiologic states, yet, when compared with other organs, relatively little is known about the molecular machinery that directly governs metabolic plasticity in the heart. Although previous studies have defined Kruppel-like factor 15 (KLF15) as a transcriptional repressor of pathologic cardiac hypertrophy, a direct role for the KLF family in cardiac metabolism has not been previously established. We show in human heart samples that KLF15 is induced after birth and reduced in heart failure, a myocardial expression pattern that parallels reliance on lipid oxidation. Isolated working heart studies and unbiased transcriptomic profiling in Klf15-deficient hearts demonstrate that KLF15 is an essential regulator of lipid flux and metabolic homeostasis in the adult myocardium. An important mechanism by which KLF15 regulates its direct transcriptional targets is via interaction with p300 and recruitment of this critical co-activator to promoters. This study establishes KLF15 as a key regulator of myocardial lipid utilization and is the first to implicate the KLF transcription factor family in cardiac metabolism.
Introduction
The working mammalian heart requires enormous amounts of energy to maintain contractile function and blood flow. Under physiologic conditions, the majority this energy production is derived from oxidative catabolism of fatty acids (FAs)3 (∼70%), with the remainder being derived from glucose (∼20%) and other substrates, such as lactate and ketones (∼10%) (1). In order to maintain contractile performance under a wide range of physiologic and pathophysiologic settings, the myocardium has evolved critical molecular mechanisms that match fuel supply with energy demand and nutrient availability (2–4).
A major form of the heart's metabolic plasticity is its ability to augment lipid utilization as a means to guard against energy depletion and circulatory failure during periods of transient carbohydrate deprivation (e.g. fasting) or heightened energy needs (e.g. exercise, postnatal growth). Defects in specific genes critical for efficient myocardial lipid utilization are known to cause metabolic cardiomyopathies in both animal models and humans (5). In addition, loss of metabolic plasticity has been observed in common forms of human heart failure (5–8), a leading cause of hospitalization and death worldwide (5). Importantly, recent studies strongly support the contention that dysregulated lipid utilization and energy homeostasis play a causal role in heart failure progression (9, 10). As recently highlighted by Goldberg et al. (2), although the heart is far and away the most energy-requiring organ in the body, studies of cardiac lipid metabolism, especially in vivo are relatively scarce compared with investigations in adipose tissue, liver, or skeletal muscle. A better understanding of the fundamental mechanisms governing metabolic plasticity in the myocardium may therefore provide key insights into heart failure pathogenesis and pave the way for novel therapeutic approaches for this common and lethal condition.
Efficient myocardial lipid flux involves coordination of several key enzymatic steps, including sarcolemmal FA uptake and activation, maintenance of the intracellular triglyceride (TG) pool, acyl-CoA import into the mitochondrial matrix, β-oxidation, TCA cycle, and oxidative phosphorylation (1, 2, 5, 11). The enzymatic machinery regulating each of these steps is under robust allosteric and transcriptional control in a manner that tightly couples lipid flux with energy demand and nutrient availability (12, 13). Despite the fundamental importance of metabolic plasticity in the heart, there is surprisingly little known about the transcriptional pathways that directly regulate myocardial substrate flux. When compared with the vast body of work that has defined gene-regulatory networks governing myocyte hypertrophy (14), the identification of transcription factors that are bona fide and direct regulators of myocardial nutrient flux is limited to a few members of the nuclear receptor superfamily (PPARs and ERRs) and the forkhead box factor FoxO1 (12, 15–17).
Kruppel-like factors (KLFs) are DNA-binding transcriptional regulators that contain three conserved zinc fingers within the carboxyl terminus that bind a putative consensus 5′-C(A/T)CCC-3′ motif in the promoters and enhancers of various genes (18, 19). The amino terminus is involved in transcriptional activation and repression as well as protein-protein interaction. Previous studies from our group have defined KLF15 as a transcriptional repressor of pathologic cardiac hypertrophy (20–22). In contrast, the broad transcriptional programs directly regulated by KLF15 in the heart remain unknown.
Initial insights linking the KLF transcription factor family to metabolic homeostasis were gleaned from our studies defining KLF15 as a regulator of adipogenesis (23). Subsequent studies in liver and skeletal muscle in the context of circadian cycles, fasting, and exercise now identify KLF15 as a central component of the transcriptional circuitry that regulates physiologic flux of all three major nutrient classes (glucose, lipids, and amino acids) (24–26). Despite these important observations, no role for any member of the KLF family in cardiac metabolism has been elucidated.
Here, we demonstrate that Kruppel-like factor 15 (KLF15) is a direct and independent regulator of myocardial lipid flux in vivo through a molecular mechanism involving recruitment of the chromatin remodeling enzyme p300 to target promoters. This study introduces an entirely new class of transcriptional regulators (KLFs) to the core molecular circuitry that governs myocardial metabolism.
EXPERIMENTAL PROCEDURES
Animal Models
All protocols concerning animal use were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Klf15−/− mice have been described previously (23). Studies were performed with age- and sex-matched littermate controls (12–14 weeks old, male, pure C57Bl/6 background). Mice were housed in a temperature- and humidity-controlled barrier facility with a 12-h light/dark cycle and ad libitum access to water and standard laboratory rodent chow (Laboratory diet P3000; 4.5% fat by weight, 14% kcal).
Cell Culture
Neonatal rat ventricular myocytes (NRVM) were isolated from 2-day-old rat pups and isolated and maintained under standard conditions as described previously (20, 21, 27). Isolated NRVM were cultured for 24–48 h under quiescent conditions by the inclusion of serum-free DMEM supplemented with insulin, transferrin, and selenium prior to experiments. C2C12 myoblasts were purchased from ATCC (Manassas, VA) and grown in DMEM supplemented with 10% FBS. C2C12 myoblasts were passaged prior to reaching 60% confluence to maintain cells in the undifferentiated state. All studies used low passage C2C12 (passages 3–6). P300−/− mouse embryonic fibroblasts (28) were cultured in DMEM supplemented with 10% FBS as described previously (29).
Plasmids and Adenoviruses
Expression plasmids containing the mouse KLF15 cDNA have been described previously (20). The −1.2 kb promoter region of mouse Slc27a1 (Fatp1) was PCR-amplified from purified genomic DNA (Promega) and subcloned into pGL3-basic (Promega). The Pdk4 promoter-luciferase construct was a gift from D. Kelly (30). For shRNA against mouse/rat Klf15, the hairpin sequence 5′-GCGGTAAGATGTACATCAAACGTGTGCTGTCCGTTTGGTGTACATCTTGCTGC-3′ (loop sequence is underlined) was subcloned into the pEQ adenoviral shRNA vector (Welgen, Inc.). Recombinant adenoviruses for sh-Klf15 and sh-scrambled were amplified and purified by Welgen, Inc.
Isolated Working Heart
The isolated mouse working heart perfusion was performed at the Washington University Metabolic Core as described previously (31, 32). In brief, hearts were excised and placed in ice-cold Krebs-Henseleit solution. Retrograde perfusion of the aorta was first performed via the Langendorff method followed by left atrial cannulation. Working mode was maintained in oxygenated buffer containing 0.4 mm palmitate, 5.0 mm glucose, and 100 microunits insulin/ml with a preload pressure of 11.5 mm Hg and afterload pressure of 50 mm Hg. Palmitate and glucose oxidation rates were determined using radiolabeled [3H]palmitate and [U-14C]glucose, respectively. Samples were collected every 10 min, and 3H2O and 14CO2 radioactivity was counted to determine palmitate and glucose oxidation rates, respectively. In addition, functional measurements such as cardiac output, peak systolic pressure, and heart rate were measured as described previously (31).
Human Left Ventricular Samples
Human left ventricular RNA samples were obtained as described previously (6). In brief, myocardial specimens were collected before and after left ventricular assisted device (LVAD) implantation and explantation as a bridge to transplantation for end-stage HF patients. Control heart samples were obtained from non-failing hearts as described previously (6). Fetal left ventricular samples were obtained at the time of elective termination immediately following tissue extraction. The use of all human samples was approved by the Institutional Review Board of Columbia University and Case Western Reserve University (IRB-AAAE7393).
Measurement of Plasma Parameters and Tissue Lipids
Hearts and plasma were rapidly harvested and flash frozen from WT and KO mice. Lipids were extracted from ventricles by the Folch method. Cardiac FA and TG content was quantified by thin layer chromatography by the National Institutes of Health-Mouse Metabolic Phenotyping Core/Lipid Analytic Core at Vanderbilt University. Plasma free fatty acids and TGs were quantified by the Vanderbilt National Institutes of Health Mouse Metabolic Phenotyping Core.
Mitochondrial Physiology and Electron Microscopy
Myocardia of four individual WT or KO mice were pooled for each experiment. The data presented are from four independent experiments. Fresh myocardial subsarcolemmal mitochondria were isolated as described previously (33). Mitochondrial protein concentration, citrate synthase activity, and oxygen consumption (using a Clark-type oxygen electrode at 30 °C) were assayed as described previously (33, 34). Mouse heart tissue was freshly dissected and rapidly processed for transmission electron microscopy as described previously (35).
Mitochondrial Genome Quantification
Total tissue DNA was prepared from hearts of WT versus KO mice using the QIAamp DNA minikit (Qiagen) according to the manufacturer's instructions. SYBR Green qPCR of purified DNA was performed using primers specific for the mouse mitochondrial Mt-Cox2 gene. Values were normalized to the nucleus-encoded mouse Rplp0 gene using the ΔΔCt method. Primer sequences are available upon request.
Chromatin Immunoprecipitation
Chromatin isolated from 15 million NRVM were used for each immunoprecipitation. NRVM were infected with empty virus or Ad-KLF15-FLAG, fixed with 1.1% formaldehyde for 10 min, and quenched with 0.125 m glycine for 5 min. Chromatin was extracted and sonicated using a BioRuptor (Diagnode, Sparta, NJ). The sonicated chromatin was immunoprecipitated with 5 μg of anti-FLAG(M2) antibodies (Sigma) bound to Dynabeads (Invitrogen) followed by extensive washing and elution. For heart tissue, chromatin isolated from two hearts was pooled for a single immunoprecipitation. Tissue was finely minced in PBS supplemented with protease inhibitors, fixed with 1.1% formaldehyde (10 min), and quenched with 0.125 m glycine (5 min). Tissues were disrupted using a Dounce homogenizer followed by chromatin extraction and sonication with a Bioruptor. Approximately 500 μg of chromatin was used in each immunoprecipitation with 5 μg of anti-p300 antibody (C-20, Santa Cruz Biotechnology, Inc.) bound to Dynabeads followed by extensive washing and elution. Chromatin was then reverse cross-linked followed by purification of genomic DNA. Target and non-target regions of genomic DNA were amplified by qPCR in both the immunoprecipitates and input samples. For NRVM samples, the rat Alb1 locus was used as a non-target control. For mouse heart tissue samples, the 28 S genomic locus was used as a non-target control. Relative abundance in immunoprecipitates was expressed as the percentage of abundance in input samples as described previously (26, 36). ChIP primer sequences are available upon request.
RNA Extraction and qPCR
Tissue samples were disrupted/homogenized in PureZOLTM (Bio-Rad) in a Tissue-lyzer (Qiagen), using stainless steel beads (30 Hz for a total of 4 min). Total RNA was isolated using the AurumTM (Bio-Rad) RNA isolation kit according to the manufacturer's directions. For cellular samples, total RNA from NRVM was isolated using the high pure RNA isolation kit (Roche Applied Science) according to the manufacturer's directions. For qPCR, total RNA was deoxyribonuclease-treated on-column and transcribed to complementary DNA using iScriptTM (Bio-Rad) following the manufacturer's protocol. qPCR was performed with the TaqMan method (using the Roche Universal Probe Library System) on an ABI Step One Plus real-time PCR system. Relative expression was calculated using the ΔΔCt method with normalization to constitutive genes as indicated in each figure. Specific primer/probe sequences are available on request.
Co-Immunoprecipitation
Co-IP was performed from nuclear protein extracts. Nuclear protein from 293 cells or mouse tissues was prepared using the Ne-PER kit (Pierce) according to the manufacturer's instructions. For each IP, 100–200 μg of nuclear protein was loaded in IP dilution buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.2% Nonidet P-40, supplemented with protease inhibitors) and immunoprecipitated with 2–5 μg of antibody bound to Dynabeads. Antibodies used for IP were anti-Myc (9E1, Santa Cruz Biotechnology) and goat polyclonal anti-KLF15 (Abcam). Immune complexes were washed extensively and eluted in SDS sample buffer. Immunoprecipitated and input proteins were run on SDS-PAGE and immunoblotted with the following antibodies as indicated: anti-p300 (N-15, Santa Cruz Biotechnology), anti-HA (HA-7, Sigma), anti-Myc (9E10, Santa Cruz Biotechnology), and goat polyclonal anti-KLF15 (Abcam). Secondary HRP-conjugated antibodies and ECL-plus chemiluminescent detection reagent were from Amersham Biosciences.
Microarray Analysis and Bioinformatics
Total ventricular tissue RNA was purified from wild-type and Klf15−/− mice (n = 4), which were subject to an 18-h overnight fast (4 p.m. to 10 a.m. the following morning). Labeling, hybridization, and scanning using an Affymetrix Genechip (Mouse Genome 430A 2.0 Array) were performed at the Harvard Partners Center for Genetics and Genomics. Raw microarray data were subject to background subtraction and multiarray normalization. Microarray data were log-transformed before differential expression analysis using the limma package (37). Probes with a p value of less than 0.05 after adjusting for false discovery rate using Benjamini and Hochberg method (38) were considered significant in differential expression. Enrichment in biological processes was analyzed using the DAVID bioinformatics suite (39, 40). The data set was submitted to the NCBI Gene Expression Omnibus with the accession number GSE53231.
Promoter-Luciferase Studies
Transient transfections of the indicated promoter plasmids using cultured NRVM or C2C12 cells at 70% confluence were carried out using Fugene6 (Roche Applied Science and Promega) following the manufacturer's instructions, and bioluminescence was recorded on a Veritas luminometer (Turner Biosystems) as described previously (20, 26).
Data Analysis
All results are expressed as means, and error bars depict S.E. Statistical analysis of microarray data is described separately above. For experiments comparing the means of two normally distributed groups, two-tailed Student's t test for unpaired data was used. Statistical significance was defined as p < 0.05.
RESULTS
KLF15 Regulates Myocardial Lipid Utilization
Because KLF15 has been demonstrated to be important for fasting-induced gluconeogenesis in the liver (24), we first assessed its expression pattern in hearts of adult mice in the fed, fasted, and refed states. As with previously identified metabolic regulators, such as Pdk4 and Fatp1, Klf15 was robustly induced with fasting with a return to baseline after refeeding (Fig. 1A), suggesting a role in cardiac metabolic function. Klf15 expression was very low in neonatal mouse hearts and significantly induced with postnatal maturation in a pattern that paralleled other important metabolic genes (Pdk4 and Fatp1) during this transition from neonatal cardiac metabolism (glucose utilization) to that of the adult (predominance of lipid oxidation) (7) (Fig. 1B). In a similar manner, KLF15 expression was induced during the fetal to adult transition in human heart tissue (Fig. 1C). Conversely, KLF15 has been shown to be substantially down-regulated in rodent models of heart failure (20, 21), a pathologic state where myocardial lipid utilization is reduced (5, 6). Consistent with this rodent data, we found KLF15 to be significantly reduced in failing human hearts with a significant recovery of expression after mechanical unloading with a left ventricular assist device (Fig. 1D) (6). Although we have shown that KLF15 functions as a transcriptional repressor of prohypertrophic signaling in cardiomyocytes (20, 21), the gene networks directly regulated by KLF15 in the myocardium remain largely unknown. Based on the data above, we hypothesized that KLF15 might play an important role in metabolic function of the adult heart.
FIGURE 1.

KLF15 is regulated in physiological and pathological conditions. A, Klf15, Ep300, Pdk4, and Fatp1 expression in mouse heart during fed, fasted (24 h), and refed (24 h of fasting followed by 24 h of refeeding). n = 5. *, p < 0.05 versus Fed. **, p < 0.05 versus fasted. B, Klf15, Ep300, Pdk4, Fatp1, Glut4, and Glut1 expression in mouse heart during postnatal maturation (n = 4). *, p < 0.05 versus day 1. C, KLF15 expression in fetal versus adult human heart tissue (n = 3–4). p < 0.05. D, KLF15 expression in human hearts that are non-failing, with advanced heart failure (prior to LVAD placement), and post-LVAD placement (n = 3–4). *, p < 0.05 versus non-HF. **, p < 0.05 versus Pre-LVAD. Rodent values are normalized to cyclophilin-B (Ppib) and human values to 18 S rRNA. Error bars, S.E.
To establish a tissue-intrinsic role for KLF15 in cardiac metabolism, we performed ex vivo isolated working heart (IWH) studies in wild-type (WT) and Klf15-deficient (KO) hearts. We focused these IWH studies on fatty acid and glucose utilization because these substrates are the major fuel sources in the working mammalian heart (1). KO hearts had a striking reduction in fatty acid oxidation with a parallel increase in glucose oxidation (Fig. 2A). Importantly, there was no significant difference in hemodynamic indices between KO and WT hearts during the IWH experiments (Fig. 2B). Furthermore, KO hearts are structurally normal and have preserved systolic function under basal conditions (20). These results suggest that the observed defect in lipid utilization is not occurring during a state of severe contractile dysfunction. In addition, KO mice are characterized by reduced intramyocardial FA and TG abundance (Fig. 3A), despite elevated plasma concentration of FAs and TGs (Fig. 3B). This observation was corroborated by electron microscopic examination, which revealed a striking paucity of intramyocellular lipid droplets in the hearts of KO mice (Fig. 3C). Hence, KLF15 deficient hearts are unable to adequately partition plasma lipids despite adequate delivery to the myocardium.
FIGURE 2.
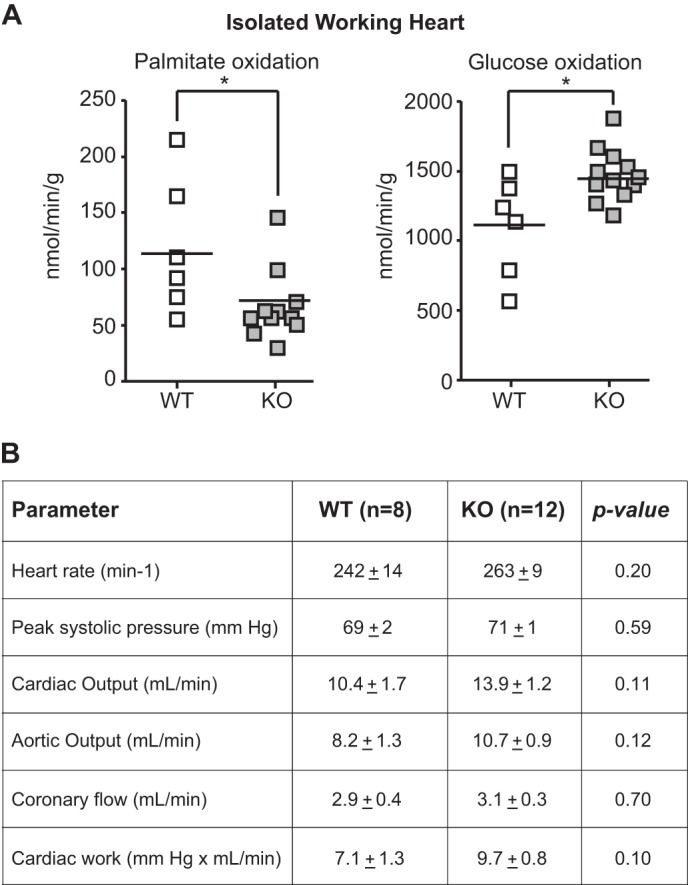
KLF15 is essential for myocardial lipid utilization. A, fatty acid (palmitate) and glucose oxidation rates in WT versus KO hearts in isolated working heart preparation (n = 8–12). *, p < 0.05 versus WT. B, hemodynamic parameters in isolated working heart studies. WT and KO hearts (n = 8–12) were perfused in isolated working mode, and hemodynamic parameters were measured as listed. There was no statistically significant difference in hemodynamic performance between genotypes.
FIGURE 3.
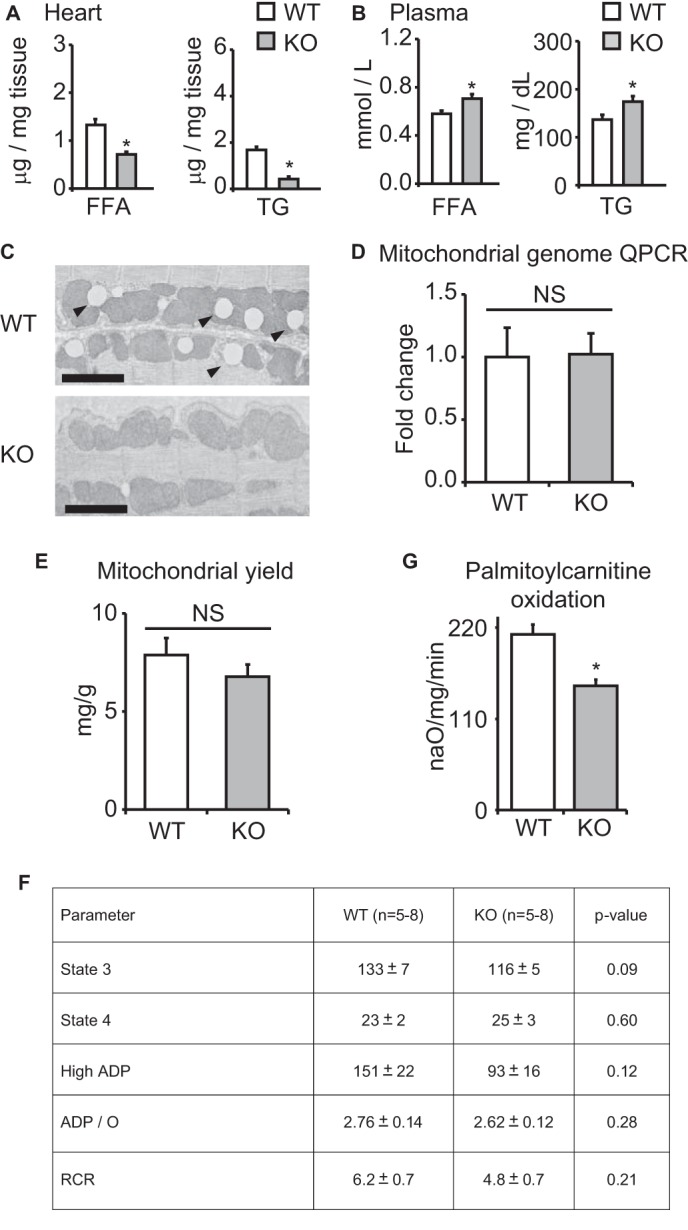
Mitochondrial parameters in KLF15 KO. Shown are FA and TG content in WT versus KO heart tissue (A) and plasma (B) (n = 5). *, p < 0.05 versus WT. C, representative transmission electron micrographs of WT versus KO heart tissue demonstrating marked paucity of intramyocellular lipid droplets (arrowheads) in KO tissue. Bar, 2 μm. D, qPCR for relative mitochondrial genome content in WT versus KO heart tissue (n = 4). Values are normalized to Rplp0 (36B4) genomic DNA. E, mitochondrial yield from whole heart tissue of WT versus KO mice, expressed as mg of mitochondrial protein/g of heart tissue (n = 4). F, rates of glutamate oxidation in mitochondria freshly isolated from WT versus KO hearts (n = 5–8). G, palmitoylcarnitine oxidation rates in freshly isolated mitochondria from WT versus KO hearts (n = 4). *, p < 0.05. Error bars, S.E.
To demonstrate an additional downstream defect in mitochondrial lipid catabolism independent of the above defect in cytosolic lipid bioavailability, we directly assessed substrate utilization in mitochondria freshly isolated from the hearts of WT and KO mice. We did not detect differences in mitochondrial genome copy number in KO hearts (Fig. 3D). Concordant with this finding, mitochondrial yield was equivalent between WT and KO hearts, thus providing additional confirmation that mitochondrial quantity was unchanged (Fig. 3E). Furthermore, WT and KO cardiac mitochondria exhibited no significant differences in glutamate oxidation rates, reflecting unaltered function of the electron transport chain (Fig. 3F). Last, transmission electron microscopy at the mitochondrial level revealed no gross differences in mitochondrial ultrastructure (Fig. 3C). However, when we directly assessed mitochondrial lipid oxidation rates, we found that freshly isolated mitochondria from KO hearts had a significant defect in fatty acid oxidation (Fig. 3G). Thus, in addition to proximal abnormalities in sarcolemmal lipid partitioning (Fig. 3, A and B), KLF15 is also critical for the distal step of mitochondrial lipid oxidation.
To gain further insight into the broad defects in myocardial lipid utilization seen above, we performed transcriptome-wide gene expression profiling of KO heart tissue. These microarray analyses showed that KO hearts have a transcriptomic signature characterized by a broad and significant reduction in genes critical for myocardial substrate metabolism, including genes involving lipid utilization (Fig. 4A and supplemental Table 1). Because lipids are the dominant fuel source for the heart under physiologic conditions, the above transcriptomic signature and our IWH data (Fig. 2A) prompted us to directly assess a broad panel of lipid-related transcripts by qRT-PCR in WT versus KO heart tissue. These analyses confirmed significant reductions in genes encompassing nearly all major components of the myocardial lipid flux pathway, including sarcolemmal FA partitioning/activation, intramyocellular storage, mitochondrial transport and β-oxidation, and peroxisomal function (Fig. 4B). Genes directly involved in the mitochondrial electron transport chain and oxidative phosphorylation were not reduced in KO hearts (Fig. 4C). Importantly, the observed reductions in lipid flux targets seen in KO hearts occurred without significant alterations in the expression of known transcriptional regulators of myocellular substrate flux and energetics (Fig. 4D). Moreover, we did not observe significant differences in key genes involved in myocardial glucose uptake and glycolysis (Fig. 4E) (12, 16, 41). Acute silencing as well as overexpression of KLF15 in cultured neonatal rat cardiomyocytes significantly reduced and augmented, respectively, expression of a parallel panel of lipid flux targets, confirming a cell-autonomous role for KLF15 in cardiomyocyte gene expression (Fig. 4, G and H).
FIGURE 4.

KLF15 regulates the myocardial lipid flux gene program. A, microarray heat map of transcript expression profiles from WT/KO hearts (n = 3–4). B, expression (qPCR) in WT versus KO hearts of critical transcripts involved in myocardial lipid flux; C, mitochondrial oxidative phosphorylation; D, transcriptional regulation; E, glycolysis (n = 4–5). *, p < 0.05. Expression (qPCR) of a parallel panel of lipid flux transcripts in cultured neonatal rat ventricular cardiomyocytes after shRNA-mediated Klf15 silencing (F) and adenovirus-mediated Klf15 overexpression (G) (n = 5–6). *, p < 0.05. Values normalized to Ppib. Error bars, S.E.
KLF15 Transactivates Direct Targets via Recruitment of p300
We next sought to establish the principle that KLF15 could regulate key targets in this pathway via a direct mechanism of action. Pdk4 and Fatp1 (Slc27a1) were used as exemplary targets, given the essential role of these genes in myocellular lipid flux and extensive functional knowledge of their proximal promoters (30, 42–45). The proximal promoters of both genes were induced by KLF15 in cultured cardiomyocytes (Fig. 5A). ChIP analysis in cardiomyocytes confirmed enrichment of KLF15 at conserved KLF response elements on these promoters (Fig. 5B). Thus, although Fatp1 and Pdk4 are only representative of a larger array of KLF15-regulated metabolic targets, these focused analyses serve to establish the principle that KLF15 can regulate important genes via direct occupancy of target promoters.
FIGURE 5.

KLF15 directly regulates metabolic targets. A, induction of Pdk4(−1.6kb)-luc and Fatp1(−1.2kb)-luc in transfected NRVM (n = 6). *, p < 0.05. B, ChIP against exogenously expressed KLF15 in NRVM on the proximal promoter regions of the endogenous rat Pdk4 and Fatp1 loci in the vicinity of highly conserved KLF consensus sites (n = 3). *, p < 0.05. C, domain map of KLF15 protein depicting putative p300-interacting transactivation domain (residues 140–160). ZF, zinc finger. D, alignment of KLF15 protein sequence across species in the vicinity of highly conserved p300-interacting transactivation domain (residues 140–160). E, cooperative promoter induction between KLF15 and p300 on Pdk4(−1.6kb)-luc and Fatp1(−1.2kb)-luc in C2C12 cells (n = 5). *, p < 0.05 versus mock transfection. **, p < 0.05 versus K15 full-length. F, expression of the indicated transcripts in p300+/+ versus p300−/− MEFs after KLF15 overexpression (n = 5). Values normalized to Ppib. *, p < 0.05 versus EV p300+/+. #, p < 0.05 versus K15 p300+/+. Error bars, S.E.
In order to gain deeper insight into a generalizable mechanism underlying KLF15-mediated target gene transactivation, we turned to structure-function analyses. We were intrigued by a recent biophysical study that demonstrated significant similarity between the p300-interacting domain of p53 and a homologous domain within the erythroid-specific KLF family member, EKLF/KLF1 (46). Indeed, inspection of the KLF15 gene revealed a putative p300-interacting transactivation domain (Fig. 5C) that was highly conserved from humans to zebrafish (Fig. 5D). The possibility of cooperativity between these two regulators on metabolic targets was further supported by our observation that myocardial Ep300 and Klf15 expression were induced in parallel during fasting and postnatal maturation (Fig. 1, A and B). Indeed, we observed significant cooperativity between KLF15 and p300 upon induction of target promoters (Fig. 5E). Studies in p300−/− cells revealed that optimal target gene induction by KLF15 required endogenous p300 (Fig. 5F). We next demonstrated interaction between epitope-tagged KLF15 and p300 when co-expressed in 293 cells and also confirmed interaction between endogenous KLF15 and p300 in mouse heart and liver tissues (Fig. 6, A–C). Ablation of the KLF15 putative p300 binding domain (KLF15Δp300) abrogated the interaction with endogenous p300 (Fig. 6C) and the cooperative induction of target promoters (Fig. 6D). Importantly, KLF15Δp300 remains nucleus-localized and is expressed as a protein of expected mobility with abundance identical to that of identical full-length KLF15 (Fig. 6C). Finally, ChIP against endogenous p300 in KLF15-deficient mouse heart tissue revealed reduced p300 enrichment in the proximal promoters of target genes (Fig. 6E). Taken together, the above data establish KLF15 as a direct regulator of the myocardial lipid flux pathway in vivo. In addition, we demonstrate that a major mechanism of KLF15-mediated transactivation involves interaction with p300 and site-specific recruitment of this critical co-activator to target promoters.
FIGURE 6.
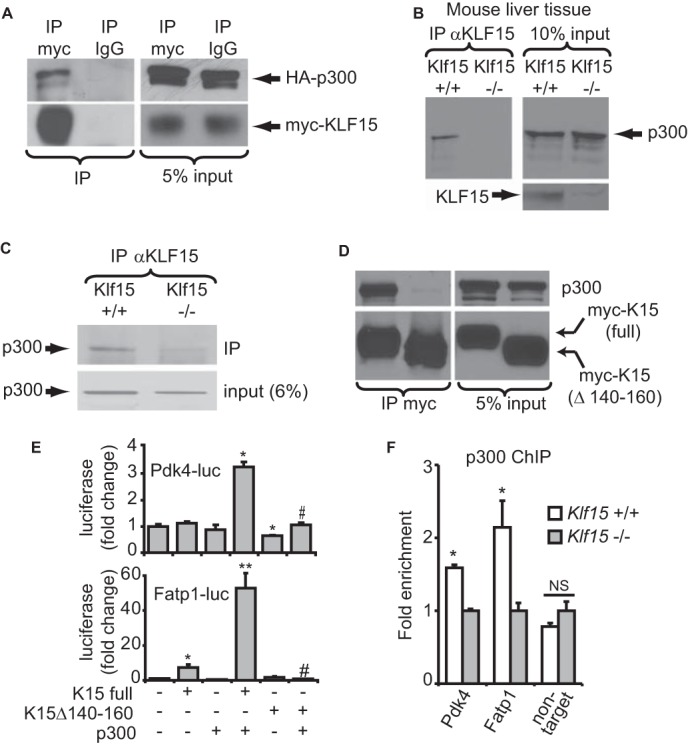
KLF15 and p300 interact to regulate metabolic targets. A, co-IP of heterologously expressed, epitope-tagged KLF15 and p300 in 293 cells. B, co-IP of endogenous KLF15 and p300 in mouse liver tissue nuclear extracts. C, co-IP of endogenous KLF15 and p300 in mouse heart tissue nuclear extracts. Klf15−/− tissue was used as a negative control for co-IPs. D, co-IP between overexpressed KLF15 and endogenous p300 in nuclear extracts of 293 cells. E, transfections demonstrating loss of cooperativity with K15Δ140–160 in C2C12 cells. n = 5. *, p < 0.05 versus mock transfection. **, p < 0.05 versus K15 full-length. #, p < 0.05 versus K15 full-length + p300. F, ChIP against p300 in WT versus KO mouse heart tissue in the proximal promoters of Pdk4 and Fatp1 in the vicinity of the KLF15 binding site (n = 3). *, p < 0.05. Error bars, S.E.
DISCUSSION
Our current findings establish KLF15 as a direct and independent regulator of myocardial nutrient flux and thereby introduce an entirely new class of factors (KLFs) as core components of the transcriptional circuitry that governs cardiac metabolism (schematized in Fig. 7). Using unbiased gene expression profiling, we elucidate a large number of KLF15-dependent targets in the heart in vivo and confirm its essential role in regulating a transcriptional program essential for efficient myocardial lipid flux. Furthermore, we establish a fundamental molecular mechanism of KLF15-mediated target gene transactivation, which involves its ability to interact with and recruit the chromatin remodeling enzyme p300. Because the mammalian heart's ability to maintain blood flow across a wide range of physiologic and pathophysiologic circumstances depends on tightly coupled control of fuel supply with energy demand (5), this work suggests that KLF15 is essential for the heart's ability to metabolically adapt to stress. Consistent with this critical role in myocardial adaptation, we have found that Klf15-deficient mice develop dramatically accelerated heart failure after hemodynamically (pressure overload) and neurohormonally (chronic angiotensin II infusion) induced stress (20, 21). When considered alongside our prior observations that implicate KLF15 as a regulator of fasting hepatic gluconeogenesis and skeletal muscle substrate catabolism (24–26), the current work implicates KLF15 as a nodal integrator of nutrient flux across diverse organ systems.
FIGURE 7.
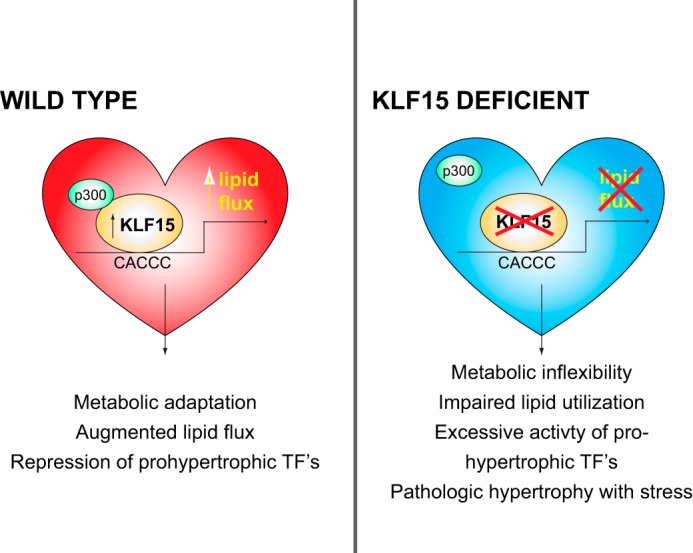
Schematic representing KLF15 as a regulator of cardiac lipid flux.
We show that cardiac KLF15, which is robustly induced during physiologic states, such as fasting and postnatal development, cooperates with p300 to regulate key metabolic targets. Previous studies have implicated p300 acetyltransferase as a positive regulator of pathologic cardiomyocyte hypertrophy, possibly via its ability to acetylate prohypertrophic transcription factors, such as GATA4, MEF2, and p53 (21, 47–50). Prior work from our group has shown that Klf15 expression is dramatically reduced during pathologic cardiac hypertrophy, an event that releases repression of GATA4, MEF2, and the p300-p53 transcriptional module (20, 21). Therefore, our current findings, when taken in the context of previously published work, support a model in which increased levels of KLF15 (e.g. during fasting) favor recruitment of p300 to specific metabolic targets, whereas reduced levels of KLF15 (e.g. during pressure overload) decrease expression of these metabolic genes and simultaneously liberate molecules of p300 to co-activate prohypertrophic transcription factors, such as GATA4 and MEF2. In this manner, increased levels of KLF15 may recruit rate-limiting quantities of p300 to transactivate its direct targets while simultaneously repressing the function of p300-regulated prohypertrophic transcriptional effectors.
Our IWH experiments show that Klf15-deficient hearts have not only a 47% reduction in palmitate oxidation but also a 32% increase in glucose oxidation rate. These IWH data parallel those observed in Ppara null hearts (15). Although our studies demonstrate that KLF15, like PPARα, directly regulates genes critical for efficient myocardial lipid utilization, there are several potential explanations for the concomitant increase in glucose oxidation rates. We did not observe increased expression of key genes that directly participate in myocardial glucose uptake or glycolysis (Fig. 4E). However, KO hearts did have reduced expression of Pdk4, an important inhibitor of the pyruvate dehydrogenase complex. Decreased PDK4 would increase pyruvate dehydrogenase activity, leading to increased rates of glucose oxidation. In addition, it is possible that a primary reduction in myocardial long chain fatty acid oxidation rates can secondarily increase glucose oxidation rates via the well described allosteric mechanisms of the Randle cycle (51). Finally, it remains possible that KLF15 might regulate the metabolism of other myocardial fuels (e.g. amino acids) in a manner that could influence glucose homeostasis.
Because cardiac KLF15 is induced during fasting in vivo (Fig. 1A), delineation of the precise molecular events that govern its expression will provide additional insights into its physiologic function. Recent studies by our group and others have identified several neurohormonal pathways as important regulators of KLF15 expression in non-cardiac tissue. Signals known to drive nutrient catabolism, such as glucocorticoids (52, 53) and cAMP-response element-binding protein activation (54), have been shown to induce KLF15 expression, whereas anabolic signals, such as insulin, can suppress its expression (55). In addition, we have recently demonstrated that cardiac KLF15 expression is directly regulated by BMAL1 (27), a central component of the mammalian circadian clock. Therefore, it is likely that balance between such catabolic and anabolic upstream signals, with additional synchronization via the circadian clock, regulates cardiac KLF15 function. In addition to its newly defined role in cardiac lipid metabolism, KLF15 also regulates the catabolism of skeletal muscle branched chain amino acids, the body's major store of glucogenic precursors (24, 25). Because cardiac and skeletal muscle share similar substrate flux machinery, it is possible that KLF15 might also regulate myocardial amino acid homeostasis in a manner that influences metabolism of other substrates and cardiac function. Given the intense interest in the interplay between lipid and branched chain amino acid metabolism during physiological and pathophysiological states (56), it will be interesting to explore whether these aspects of KLF15 function are perturbed in heart failure and other cardiometabolic diseases.
Our current work in cardiac metabolism is also interesting to consider alongside our recent observation that KLF15 regulates cardiac repolarization in vivo (27). Studies in large animal models of heart failure demonstrate that regional electromechanical dyssynchrony is associated with profound changes in genes critical for myocardial lipid and amino acid metabolism (57). Because KLF15 expression has also been shown to be significantly reduced in rodent and human heart failure (21) with recovery of expression after mechanical unloading (Fig. 1D), these findings collectively raise the intriguing possibility that myocardial substrate flux and electrical activity might be subject to coordinate control by KLF15 as a form of “metabo-electrical coupling.”
Previous studies have identified key members of the nuclear receptor superfamily (e.g. PPARs and ERRα) as direct regulators of cardiac lipid metabolism (12, 15). We note that many of the lipid metabolic genes regulated by KLF15 are also known to be PPARα targets in the heart (12). Furthermore, we find that KLF15-deficient hearts have defects in lipid utilization as prominent as those observed with complete PPARα deficiency (15). Importantly, these defects in lipid metabolism and target gene expression seen with KLF15 deficiency occur with minimal alteration in expression of PPARs, ERRs, or several other important metabolic regulators (Fig. 4D). We postulate that KLF15 and nuclear receptors, such as PPARα, together exert coordinated control over a subset of important shared targets. Our observation that KLF15 binds and recruits p300 to promoters supports a transactivation model in which KLF15 occupancy favors an open chromatin configuration for the regulatory regions of its direct targets in the lipid flux pathway. It is also possible that the ability of KLF15 to influence intracellular lipid abundance may secondarily affect the bioavailability of endogenous PPAR ligands (13). Studies exploring potential cross-talk between KLFs and nuclear receptors in cardiac metabolism are an area of ongoing investigation by our group. Interestingly, KLF homologs and PPARα-like nuclear receptor genes in C. elegans have recently been shown to both play important roles in lipid metabolism (58, 59), raising the intriguing possibility that coordinated control of metabolic flux by these two ancient transcription factor families may have been present as early in evolution as the phylum Nematoda. We speculate that functional interactions between KLFs and nuclear receptor networks will emerge as an important theme in our understanding of metabolic plasticity in the myocardium as well as across a broad range of tissues.
Supplementary Material
Acknowledgments
We thank Daniel P. Kelly and Teresa C. Leone (Sanford-Burnham Medical Research Institute) for critical input and for providing the PDK4 reporter construct. We also thank Nanda Sambandam and Carla Weinheimer (Washington University Mouse Cardiovascular Core) for assistance with IWH studies and Carla Harris at the Vanderbilt University Mouse Metabolic Phenotyping Center for lipid analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants T32HL105338 and F32HL110538 (to D. A. P.), HL086614 and DK093821 (to S. M. H.), HL072952 and HL084154 (to M. K. J.), CA043703 and HL074237 (to C. L. H.), HL095742 and HL101272 (to P. C. S.), CA132977 (to J. Z.), and UL1RR024143 (National Center for Research Resources) (to K. M. A. and T. T.). This work was also supported by American Heart Association Grant 12SDG12070077 (to X. L.), the Hartwell Foundation (to S. M. H.), German Research Foundation Fellowship Grant AK137/1-1 (to K. M. A.), the Genome and Transcriptome Sequencing Core at Case Western Reserve University (to X. B.), and the Visconsi Research Scholar Fund (to S. M. H.).

This article contains supplemental Table 1.
- FA
- fatty acid
- TG
- triglyceride
- PPAR
- peroxisome proliferator-activated receptor
- ERR
- estrogen-related receptor
- KLF
- Kruppel-like factor
- NRVM
- Neonatal rat ventricular myocyte(s)
- LVAD
- left ventricular assisted device
- qPCR
- quantitative PCR
- IP
- immunoprecipitation
- IWH
- isolated working heart.
REFERENCES
- 1. Lopaschuk G. D., Ussher J. R., Folmes C. D., Jaswal J. S., Stanley W. C. (2010) Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258 [DOI] [PubMed] [Google Scholar]
- 2. Goldberg I. J., Trent C. M., Schulze P. C. (2012) Lipid metabolism and toxicity in the heart. Cell Metab. 15, 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertrand L., Horman S., Beauloye C., Vanoverschelde J. L. (2008) Insulin signalling in the heart. Cardiovasc. Res. 79, 238–248 [DOI] [PubMed] [Google Scholar]
- 4. Kolwicz S. C., Jr., Tian R. (2009) Metabolic therapy at the crossroad. How to optimize myocardial substrate utilization? Trends Cardiovasc. Med. 19, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neubauer S. (2007) The failing heart. An engine out of fuel. N. Engl. J. Med. 356, 1140–1151 [DOI] [PubMed] [Google Scholar]
- 6. Chokshi A., Drosatos K., Cheema F. H., Ji R., Khawaja T., Yu S., Kato T., Khan R., Takayama H., Knöll R., Milting H., Chung C. S., Jorde U., Naka Y., Mancini D. M., Goldberg I. J., Schulze P. C. (2012) Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 125, 2844–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Razeghi P., Young M. E., Alcorn J. L., Moravec C. S., Frazier O. H., Taegtmeyer H. (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104, 2923–2931 [DOI] [PubMed] [Google Scholar]
- 8. Ingwall J. S. (2009) On the control of metabolic remodeling in mitochondria of the failing heart. Circ. Heart Fail. 2, 275–277 [DOI] [PubMed] [Google Scholar]
- 9. Kolwicz S. C., Jr., Olson D. P., Marney L. C., Garcia-Menendez L., Synovec R. E., Tian R. (2012) Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 111, 728–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta A., Akki A., Wang Y., Leppo M. K., Chacko V. P., Foster D. B., Caceres V., Shi S., Kirk J. A., Su J., Lai S., Paolocci N., Steenbergen C., Gerstenblith G., Weiss R. G. (2012) Creatine kinase-mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J. Clin. Invest. 122, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Banke N. H., Wende A. R., Leone T. C., O'Donnell J. M., Abel E. D., Kelly D. P., Lewandowski E. D. (2010) Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARα. Circ. Res. 107, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huss J. M., Kelly D. P. (2004) Nuclear receptor signaling and cardiac energetics. Circ. Res. 95, 568–578 [DOI] [PubMed] [Google Scholar]
- 13. Haemmerle G., Moustafa T., Woelkart G., Büttner S., Schmidt A., van de Weijer T., Hesselink M., Jaeger D., Kienesberger P. C., Zierler K., Schreiber R., Eichmann T., Kolb D., Kotzbeck P., Schweiger M., Kumari M., Eder S., Schoiswohl G., Wongsiriroj N., Pollak N. M., Radner F. P., Preiss-Landl K., Kolbe T., Rülicke T., Pieske B., Trauner M., Lass A., Zimmermann R., Hoefler G., Cinti S., Kershaw E. E., Schrauwen P., Madeo F., Mayer B., Zechner R. (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Berlo J. H., Maillet M., Molkentin J. D. (2013) Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Invest. 123, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campbell F. M., Kozak R., Wagner A., Altarejos J. Y., Dyck J. R., Belke D. D., Severson D. L., Kelly D. P., Lopaschuk G. D. (2002) A role for peroxisome proliferator-activated receptor alpha (PPARα) in the control of cardiac malonyl-CoA levels. Reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARα are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J. Biol. Chem. 277, 4098–4103 [DOI] [PubMed] [Google Scholar]
- 16. Cheng L., Ding G., Qin Q., Huang Y., Lewis W., He N., Evans R. M., Schneider M. D., Brako F. A., Xiao Y., Chen Y. E., Yang Q. (2004) Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250 [DOI] [PubMed] [Google Scholar]
- 17. Battiprolu P. K., Hojayev B., Jiang N., Wang Z. V., Luo X., Iglewski M., Shelton J. M., Gerard R. D., Rothermel B. A., Gillette T. G., Lavandero S., Hill J. A. (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller I. J., Bieker J. J. (1993) A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell. Biol. 13, 2776–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bieker J. J. (2001) Kruppel-like factors. Three fingers in many pies. J. Biol. Chem. 276, 34355–34358 [DOI] [PubMed] [Google Scholar]
- 20. Fisch S., Gray S., Heymans S., Haldar S. M., Wang B., Pfister O., Cui L., Kumar A., Lin Z., Sen-Banerjee S., Das H., Petersen C. A., Mende U., Burleigh B. A., Zhu Y., Pinto Y., Liao R., Jain M. K. (2007) Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 104, 7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haldar S. M., Lu Y., Jeyaraj D., Kawanami D., Cui Y., Eapen S. J., Hao C., Li Y., Doughman Y. Q., Watanabe M., Shimizu K., Kuivaniemi H., Sadoshima J., Margulies K. B., Cappola T. P., Jain M. K. (2010) Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci. Transl. Med. 2, 26ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang B., Haldar S. M., Lu Y., Ibrahim O. A., Fisch S., Gray S., Leask A., Jain M. K. (2008) The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J. Mol. Cell Cardiol. 45, 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gray S., Feinberg M. W., Hull S., Kuo C. T., Watanabe M., Sen-Banerjee S., DePina A., Haspel R., Jain M. K. (2002) The Kruppel-like factor KLF15 regulates the insulin-sensitive glucose transporter GLUT4. J. Biol. Chem. 277, 34322–34328 [DOI] [PubMed] [Google Scholar]
- 24. Gray S., Wang B., Orihuela Y., Hong E. G., Fisch S., Haldar S., Cline G. W., Kim J. K., Peroni O. D., Kahn B. B., Jain M. K. (2007) Regulation of gluconeogenesis by Kruppel-like factor 15. Cell Metab. 5, 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeyaraj D., Scheer F. A., Ripperger J. A., Haldar S. M., Lu Y., Prosdocimo D. A., Eapen S. J., Eapen B. L., Cui Y., Mahabeleshwar G. H., Lee H. G., Smith M. A., Casadesus G., Mintz E. M., Sun H., Wang Y., Ramsey K. M., Bass J., Shea S. A., Albrecht U., Jain M. K. (2012) Klf15 orchestrates circadian nitrogen homeostasis. Cell Metab. 15, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haldar S. M., Jeyaraj D., Anand P., Zhu H., Lu Y., Prosdocimo D. A., Eapen B., Kawanami D., Okutsu M., Brotto L., Fujioka H., Kerner J., Rosca M. G., McGuinness O. P., Snow R. J., Russell A. P., Gerber A. N., Bai X., Yan Z., Nosek T. M., Brotto M., Hoppel C. L., Jain M. K. (2012) Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proc. Natl. Acad. Sci. U.S.A. 109, 6739–6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeyaraj D., Haldar S. M., Wan X., McCauley M. D., Ripperger J. A., Hu K., Lu Y., Eapen B. L., Sharma N., Ficker E., Cutler M. J., Gulick J., Sanbe A., Robbins J., Demolombe S., Kondratov R. V., Shea S. A., Albrecht U., Wehrens X. H., Rosenbaum D. S., Jain M. K. (2012) Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 483, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poulin D. L., Kung A. L., DeCaprio J. A. (2004) p53 targets simian virus 40 large T antigen for acetylation by CBP. J. Virol. 78, 8245–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Urvalek A. M., Wang X., Lu H., Zhao J. (2010) KLF8 recruits the p300 and PCAF co-activators to its amino terminal activation domain to activate transcription. Cell Cycle 9, 601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wende A. R., Huss J. M., Schaeffer P. J., Giguère V., Kelly D. P. (2005) PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα. A mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol. 25, 10684–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lehman J. J., Boudina S., Banke N. H., Sambandam N., Han X., Young D. M., Leone T. C., Gross R. W., Lewandowski E. D., Abel E. D., Kelly D. P. (2008) The transcriptional coactivator PGC-1α is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Heart Circ. Physiol. 295, H185–H196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belke D. D., Larsen T. S., Lopaschuk G. D., Severson D. L. (1999) Glucose and fatty acid metabolism in the isolated working mouse heart. Am. J. Physiol. 277, R1210–R1217 [DOI] [PubMed] [Google Scholar]
- 33. Palmer J. W., Tandler B., Hoppel C. L. (1977) Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739 [PubMed] [Google Scholar]
- 34. Puchowicz M. A., Varnes M. E., Cohen B. H., Friedman N. R., Kerr D. S., Hoppel C. L. (2004) Oxidative phosphorylation analysis. Assessing the integrated functional activity of human skeletal muscle mitochondria. Case studies. Mitochondrion 4, 377–385 [DOI] [PubMed] [Google Scholar]
- 35. Fujioka H., Tandler B., Hoppel C. L. (2012) Mitochondrial division in rat cardiomyocytes. An electron microscope study. Anat. Rec. 295, 1455–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phuc Le P., Friedman J. R., Schug J., Brestelli J. E., Parker J. B., Bochkis I. M., Kaestner K. H. (2005) Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet. 1, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 38. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate. A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 [Google Scholar]
- 39. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools. Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 41. Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P. H., Rybkin I. I., Shelton J. M., Manieri M., Cinti S., Schoen F. J., Bassel-Duby R., Rosenzweig A., Ingwall J. S., Spiegelman B. M. (2005) Transcriptional coactivator PGC-1 α controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271 [DOI] [PubMed] [Google Scholar]
- 42. Zhao G., Jeoung N. H., Burgess S. C., Rosaaen-Stowe K. A., Inagaki T., Latif S., Shelton J. M., McAnally J., Bassel-Duby R., Harris R. A., Richardson J. A., Kliewer S. A. (2008) Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 294, H936–H943 [DOI] [PubMed] [Google Scholar]
- 43. Chiu H. C., Kovacs A., Blanton R. M., Han X., Courtois M., Weinheimer C. J., Yamada K. A., Brunet S., Xu H., Nerbonne J. M., Welch M. J., Fettig N. M., Sharp T. L., Sambandam N., Olson K. M., Ory D. S., Schaffer J. E. (2005) Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 96, 225–233 [DOI] [PubMed] [Google Scholar]
- 44. Kim J. K., Gimeno R. E., Higashimori T., Kim H. J., Choi H., Punreddy S., Mozell R. L., Tan G., Stricker-Krongrad A., Hirsch D. J., Fillmore J. J., Liu Z. X., Dong J., Cline G., Stahl A., Lodish H. F., Shulman G. I. (2004) Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J. Clin. Invest. 113, 756–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frohnert B. I., Hui T. Y., Bernlohr D. A. (1999) Identification of a functional peroxisome proliferator-responsive element in the murine fatty acid transport protein gene. J. Biol. Chem. 274, 3970–3977 [DOI] [PubMed] [Google Scholar]
- 46. Mas C., Lussier-Price M., Soni S., Morse T., Arseneault G., Di Lello P., Lafrance-Vanasse J., Bieker J. J., Omichinski J. G. (2011) Structural and functional characterization of an atypical activation domain in erythroid Kruppel-like factor (EKLF). Proc. Natl. Acad. Sci. U.S.A. 108, 10484–10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morimoto T., Sunagawa Y., Kawamura T., Takaya T., Wada H., Nagasawa A., Komeda M., Fujita M., Shimatsu A., Kita T., Hasegawa K. (2008) The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J. Clin. Invest. 118, 868–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miyamoto S., Kawamura T., Morimoto T., Ono K., Wada H., Kawase Y., Matsumori A., Nishio R., Kita T., Hasegawa K. (2006) Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation 113, 679–690 [DOI] [PubMed] [Google Scholar]
- 49. Takaya T., Kawamura T., Morimoto T., Ono K., Kita T., Shimatsu A., Hasegawa K. (2008) Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J. Biol. Chem. 283, 9828–9835 [DOI] [PubMed] [Google Scholar]
- 50. Wei J. Q., Shehadeh L. A., Mitrani J. M., Pessanha M., Slepak T. I., Webster K. A., Bishopric N. H. (2008) Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 118, 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Randle P. J., Sugden P. H., Kerbey A. L., Radcliffe P. M., Hutson N. J. (1978) Regulation of pyruvate oxidation and the conservation of glucose. Biochem. Soc. Symp. 47–67 [PubMed] [Google Scholar]
- 52. Masuno K., Haldar S. M., Jeyaraj D., Mailloux C. M., Huang X., Panettieri R. A., Jr., Jain M. K., Gerber A. N. (2011) Expression profiling identifies Klf15 as a glucocorticoid target that regulates airway hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 45, 642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shimizu N., Yoshikawa N., Ito N., Maruyama T., Suzuki Y., Takeda S., Nakae J., Tagata Y., Nishitani S., Takehana K., Sano M., Fukuda K., Suematsu M., Morimoto C., Tanaka H. (2011) Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 13, 170–182 [DOI] [PubMed] [Google Scholar]
- 54. Mallipattu S. K., Liu R., Zheng F., Narla G., Ma'ayan A., Dikman S., Jain M. K., Saleem M., D'Agati V., Klotman P., Chuang P. Y., He J. C. (2012) Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J. Biol. Chem. 287, 19122–19135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Teshigawara K., Ogawa W., Mori T., Matsuki Y., Watanabe E., Hiramatsu R., Inoue H., Miyake K., Sakaue H., Kasuga M. (2005) Role of Kruppel-like factor 15 in PEPCK gene expression in the liver. Biochem. Biophys. Res. Commun. 327, 920–926 [DOI] [PubMed] [Google Scholar]
- 56. Newgard C. B. (2012) Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 15, 606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barth A. S., Aiba T., Halperin V., DiSilvestre D., Chakir K., Colantuoni C., Tunin R. S., Dimaano V. L., Yu W., Abraham T. P., Kass D. A., Tomaselli G. F. (2009) Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ. Cardiovasc. Genet. 2, 371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang J., Bakheet R., Parhar R. S., Huang C. H., Hussain M. M., Pan X., Siddiqui S. S., Hashmi S. (2011) Regulation of fat storage and reproduction by Kruppel-like transcription factor KLF3 and fat-associated genes in Caenorhabditis elegans. J. Mol. Biol. 411, 537–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van Gilst M. R., Hadjivassiliou H., Jolly A., Yamamoto K. R. (2005) Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 3, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.