

Background: The mechanistic role of DNA-induced structural changes in RecQ helicases is largely unexplored.
Results: DNA interaction of RecQ helicase depends on the nucleotide state of the enzyme and the presence of an intact HRDC domain.
Conclusion: We identified a structural transition of the RecQ-DNA complex that is linked to the mechanoenzymatic cycle.
Significance: This transition contributes to translocation along DNA and genome-maintaining activities.
Keywords: ATPases, DNA Repair, DNA-Protein Interaction, Molecular Motors, Protein Conformation, Recombination, Structure
Abstract
The allosteric communication between the ATP- and DNA-binding sites of RecQ helicases enables efficient coupling of ATP hydrolysis to translocation along single-stranded DNA (ssDNA) and, in turn, the restructuring of multistranded DNA substrates during genome maintenance processes. In this study, we used the tryptophan fluorescence signal of Escherichia coli RecQ helicase to decipher the kinetic mechanism of the interaction of the enzyme with ssDNA. Rapid kinetic experiments revealed that ssDNA binding occurs in a two-step mechanism in which the initial binding step is followed by a structural transition of the DNA-bound helicase. We found that the nucleotide state of RecQ greatly influences the kinetics of the detected structural transition, which leads to a high affinity DNA-clamped state in the presence of the nucleotide analog ADP-AlF4. The DNA binding mechanism is largely independent of ssDNA length, indicating the independent binding of RecQ molecules to ssDNA and the lack of significant DNA end effects. The structural transition of DNA-bound RecQ was not detected when the ssDNA binding capability of the helicase-RNase D C-terminal domain was abolished or the domain was deleted. The results shed light on the nature of conformational changes leading to processive ssDNA translocation and multistranded DNA processing by RecQ helicases.
Introduction
RecQ helicases, which are members of helicase superfamily 2 (SF2),3 are present in all cellular organisms. The versatile DNA-restructuring activities of these enzymes play key roles in homologous recombination (HR)-based DNA repair and genome maintenance processes (1–6). Escherichia coli RecQ helicase, the prototypic member of the family, plays roles in the regulation of HR-mediated DNA double-stranded break repair (7, 8) and the stabilization of stalled replication forks (9, 10). Mutations of three of the five human RecQ family members cause severe diseases, including Bloom (BLM), Werner (WRN), and Rothmund-Thompson syndrome (RTS), generally associated with genomic instability and high cancer predisposition (11).
The translocation of RecQ helicases along single-stranded DNA (ssDNA), which is the basis for the various DNA-restructuring activities of these enzymes, is fueled by ATP hydrolysis catalyzed within their ATPase active site. The detected tight linkage between ATP hydrolysis and ssDNA translocation (12–14) strongly suggests the existence of precise allosteric communication between the ATPase and DNA binding sites. However, little is known about this communication in RecQ helicases (15).
DNA-induced conformational changes were identified in various SF1, -2, and -6 helicases. DNA binding by E. coli UvrD (SF1) helicase is associated with a large rotation of the auxiliary DNA binding domain 2B relative to the rest of the protein (16, 17). DNA-induced structural changes were detected using biochemical methods in Rep and UvrD (SF1) (18), PriA (SF2) (19), and DnaB (SF6) (20, 21) helicases. The kinetic mechanism of DNA binding was determined for PriA and DnaB using signals from chemically modified DNA (22, 23). In both helicases, sequential multistep binding mechanisms were detected in which the initial binding step was followed by conformational changes occurring in the helicase-DNA complex.
X-ray structural studies have visualized nucleotide state-dependent changes in DNA-bound UvrD (SF1) (16) helicase. The coupling between nucleotide state and DNA binding has been investigated biochemically for PriA (SF2) (19) and DnaB (21) helicases. The interaction with DNA was shown to influence the conformational changes coupled to ATP hydrolysis in PcrA (SF1) (24) and RecD2 (SF1) helicases (25).
The DNA binding site of RecQ helicases has not been precisely mapped. However, it is generally accepted that the DNA interaction of these enzymes resides in multiple domains (26–28). RecQ helicases have a motor core comprising two tandem RecA-like domains, together forming the ATPase pocket. The Zn2+-binding domain (ZBD) and the winged helix domain (WHD) together constitute the RecQ C-terminal region. The helicase-RNase D C-terminal (HRDC) domain is connected to the other domains via a flexible linker. Proposed DNA-interacting elements of E. coli RecQ helicase were identified by Bernstein et al. (29), who crystallized a RecQ fragment comprising the RecA, ZBD, and WHD domains. An aromatic-rich region within the RecQ core (between conserved helicase motifs II and III) has been implicated in coupling nucleic acid binding, unwinding and ATP hydrolysis (30). Via its interaction with the γ-phosphate of the bound ATP molecule, the so-called arginine finger (in helicase motif VI) was shown to be essential in coupling ATP hydrolysis and DNA binding to the unwinding activity of human BLM (31). Alignment of the crystal structure of human RECQ1 with those of DNA-bound complexes of other helicases provided no indications for significant DNA-induced conformational changes (32, 33).
Besides its general role in protein structure stabilization, the ZBD has been implicated to contribute to DNA binding by RecQ helicases (34). The interactions of the WHD with the DNA substrate appear to be necessary for unwinding (26, 35, 36). Due to its flexible linkage to the helicase motor core, the HRDC domain could not be crystallized with the other RecQ domains. Thus, its structure has been determined in isolation from other parts of the molecule (35, 37, 38). This domain was found to be necessary for the processing of complex DNA structures in human BLM and E. coli RecQ (28, 39). Molecular dynamics studies suggested that the interdomain flexibility within E. coli RecQ leads to changes in the interactions at domain interfaces upon DNA binding (40). E. coli RecQ is generally thought to interact with ssDNA as a monomer (41, 42). However, there are implications for oligomerization and cooperativity during unwinding activity (43, 44).
Biochemical data on the coupling between nucleotide and DNA binding to RecQ helicases are scarce. A study of the DNA binding properties of E. coli RecQ DNA binding revealed only a weak effect of AMPPNP on the ssDNA affinity of the enzyme (45). Kinetic data on the DNA interaction or the coupling of nucleotide and DNA interactions of RecQ helicases are missing, except for the implication that in human BLM, ATP binding was largely unaffected by the presence of ssDNA (15).
Tryptophan (Trp) fluorescence has been frequently used to probe protein-nucleic acid interactions, with ssDNA-binding proteins (T4 gene 32 protein, SSB) and RecA as prominent examples (46–48). Nucleotide and DNA binding was found to affect the Trp fluorescence quenching properties of DnaB helicase, allowing the identification of different ligand-bound conformations of the enzyme (49). Studies utilizing fluorescence signals of protein Trp residues as well as those of fluorescently labeled DNA substrates revealed a two-step DNA binding process for Rep helicase, leading to dimerization of the enzyme (50, 51).
E. coli RecQ harbors four Trp residues located in the N-core RecA (Trp138 and Trp154), ZBD (Trp347), and WHD (Trp469) domains. In this study, we used Trp fluorescence signals in spectroscopic and rapid kinetic experiments to determine the kinetic mechanism of the interaction of the enzyme with ssDNA substrates.
EXPERIMENTAL PROCEDURES
Reagents and Experimental Conditions
Unless otherwise indicated, experiments were performed at 25 °C in SF50 buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 5 mm MgCl2). Unless otherwise stated, all materials were purchased from Sigma-Aldrich. ATP was from Roche Applied Science, and [γ-32P]ATP was from Institute of Isotopes Co. Ltd. (Budapest, Hungary). The RecQ-ADP-AlF4 complex was made by complementing a premixture of 3 μm RecQ and 1 mm ADP with 1 mm AlCl3 and 5 mm NaF, followed by incubation on ice for 3 h. In stopped-flow experiments using nucleotide-free RecQ, all reagents were preincubated with 0.02 unit/ml apyrase for 30 min at 25 °C to remove ATP contamination. Oligonucleotides and poly(dT) were obtained from Sigma-Aldrich. Heparin (Sigma H3393) and dextran sulfate (DxSO4) (Sigma D8906) were dissolved in sterile distilled water at 50 mg/ml and dialyzed against sterile distilled water and then SF50 buffer in molecular weight cut-off 3500 dialysis tubing (Serva 44183). DNA concentrations are expressed in terms of constituent nucleotide units (nt) throughout.
Cloning and Protein Expression
The coding regions of truncated RecQ constructs RecQ523 and RecQ414 were amplified by PCR using GoTaq polymerase, using the wild-type RecQ DNA construct as template (12). PCR products were digested and ligated into pTXB3 vector (New England Biolabs) between NotI and SapI (Fermentas) restriction sites. The RecQY555A DNA construct was prepared using QuikChange kit (Stratagene). All DNA constructs were verified by sequencing.
DNA constructs were transformed into E. coli BER2566 (NEB) cells. Induction, protein expression, and purification were performed as described previously for wild-type RecQ (12). Other protein constructs (RecQY555A, RecQ523, and RecQ414) were further purified on a MonoQ anion exchange column (GE Healthcare) (0–1 m NaCl, 50 mm Tris-HCl, pH 8.5, 1 mm DTT, 5 mm MgCl2, 10% glycerol, 1 ml/min for 50 min) instead of the heparin column.
Trp Fluorescence Spectral Measurements
Trp fluorescence was excited at 297 nm (with 1-nm bandwidth), and emission was detected at 310–420 nm (with 4-nm bandwidth) in a SPEX Fluoromax spectrofluorometer in SF50 buffer plus 10% glycerol. Spectra were corrected for the inner filter effect of DNA based on control experiments performed using N-acetyl-l-tryptophanamide instead of RecQ (applied at a concentration identical to that of RecQ tryptophans).
Stopped-flow Measurements
Stopped-flow experiments were performed in KinTek SF-2004 and BioLogic SFM 300 apparatuses. Trp fluorescence was excited at 297 nm (4-nm bandwidth), and emission was detected through a 340-nm interference filter (Comar). In order to obtain a sufficient number of data points throughout different time scales and ascertain their unbiased contribution to the fitted parameters, split linear or quasilogarithmic time base sampling was applied. Accordingly, the higher noise levels at shorter times resulted from the shorter signal integration times used in this regime.
Steady-state ATPase Assay
ATPase activities were measured using a pyruvate kinase/lactate dehydrogenase-linked (PK-LDH) assay (14 units/ml pyruvate kinase, 20 units/ml lactate dehydrogenase, 1 mm ATP, 1 mm phosphoenolpyruvate, 200 μm NADH in the presence of 50 μg/ml BSA in SF50 buffer). Time courses of NADH absorbance (ϵ340 nm = 6220 m−1 cm−1) were followed at 340 nm in a Shimadzu UV-2101PC photometer.
Data Analysis
Data analysis and fitting was performed using OriginLab version 8.0 (Microcal Corp.). Means ± S.E. are reported throughout.
The DNA concentration dependence of measured parameters was fitted by hyperbolic or quadratic binding equations (Equations 1 and 2, respectively).
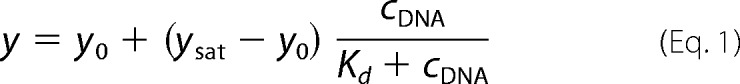 |
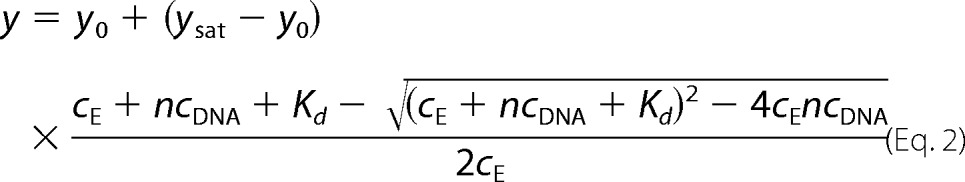 |
where y represents the measured value at a given DNA concentration; y0 and ysat are the values of the given parameter at zero and saturating DNA concentration, respectively; cE and cDNA are concentrations of RecQ and DNA (in nt), respectively; n is the inverse of the binding stoichiometry (mol of RecQ/mol of DNA in nt); and Kd is the (apparent) dissociation constant. The quadratic equation was used in cases where the tight interaction between RecQ and DNA allowed the separate determination of DNA binding stoichiometries (1/n) and Kd values.
RESULTS
DNA Binding by RecQ Leads to a Structural Transition Affected by the Nucleotide State of the Helicase
To mimic the nucleotide states adopted by RecQ helicase during the ATPase cycle, we used a series of nucleotides and nucleotide analogs, including ATP, ADP, AMPPNP (a non-hydrolyzable ATP analog), and ADP-AlF4 (a nucleotide analog thought to mimic an intermediate during the hydrolytic process). The addition of different nucleotides to RecQ caused different extents of quench (5–17%) and a 2–3-nm red shift in the emission spectrum of the protein (Fig. 1A and Table 1). The addition of dT54 ssDNA to nucleotide-free RecQ caused a large (62%) quench and a 4-nm blue shift (Fig. 1A and Table 1). Similar large extents of DNA-induced quench were seen also in the presence of nucleotides (Fig. 1A and Table 1). The DNA-induced blue shift was even larger in RecQ-nucleotide complexes than in nucleotide-free RecQ (by about 2 nm), indicating the formation of ternary complexes between the enzyme and the nucleotide and DNA ligands. Increasing the nucleotide and DNA concentrations specified in Fig. 1A did not cause further changes in Trp spectra, indicating that quasisaturating conditions were reached.
FIGURE 1.

DNA binding by RecQ and RecQ-nucleotide complexes. A, Trp fluorescence emission spectra of RecQ (3 μm), from top to bottom, in the absence of nucleotides and DNA and in ATP (1 mm), AMPPNP (200 μm), ADP (1 mm), and ADP-AlF4 (1 mm) and in 162 μm (nt) dT54 alone and in dT54 plus ATP, ADP, AMPPNP, and ADP-AlF4. B, Trp fluorescence stopped-flow time courses of dT54 binding to RecQ (0.5 μm) in the presence of 100 μm ADP. dT54 concentrations (nt) were (top to bottom) 13.5, 27, 54, 81, 108, and 378 μm. Trp fluorescence levels were normalized to that of the DNA-free state. Time courses were fitted by triple exponentials in all nucleotide states examined. (The slowest phase arose from photobleaching in each case; see “Results.”) C, comparison of fits comprising one (gray line) and two (black line) exponential DNA-binding phases (in addition to the slow photobleaching phase) to the trace recorded at 81 μm (nt) dT54 (cf. B). The inset shows the residuals of the fits (gray and black, respectively), indicating systematic deviation from data points when fitting a single binding phase (gray). D and E, dT54 concentration dependence of kobs values of the rapid (D) and slow (E) phases of dT54 binding by RecQ in the absence of nucleotide (squares) and in ADP (100 μm; circles), AMPPNP (100 μm; triangles), and ADP-AlF4 (500 μm; inverted triangles). The slopes and intercepts of linear fits to rapid phase kobs values (D) determined the forward (k1) and reverse (k−1) rate constants, respectively, of the first step of DNA binding (K1 in G). The upper bounds and intercepts of hyperbolic fits to slow phase kobs values (E) yielded the sum of the forward and reverse rate constants (k2 + k−2) and the reverse rate constant alone (k−2), respectively, of the subsequent isomerization step (K2 in G). F, dT54 concentration dependence of total (Atotal, main panel) and rapid phase (Arapid, inset) amplitudes of Trp fluorescence time courses upon dT54 binding (relative to the intensity of the DNA-free state) in different nucleotide states (symbols as in D). Quadratic fits to data sets were used to determine upper bounds and DNA binding stoichiometries (Table 1). G, proposed mechanistic scheme of DNA binding by RecQ and RecQ-nucleotide complexes (RecQ-N), involving an initial binding step (K1) followed by an isomerization (K2) representing a structural change occurring in the RecQ-DNA complex. Both steps are associated with a quench in Trp fluorescence (indicated by † and ††). K1 and the overall equilibrium constant of DNA binding (Keq) are defined as dissociation constants, whereas K2 is defined in the rightward direction. Data in this figure were recorded at 25 °C. Parameters derived from fits are shown in Table 1.
TABLE 1.
DNA binding properties of RecQ in different nucleotide states
Shown are 25 °C, dT54, 0.5 μm RecQ Trp fluorescence stopped-flow data (except for Trp fluorescence emission spectra). Nomenclature of parameters refers to Fig. 1G. All parameters with dimensions involving concentration are specified based on DNA concentrations expressed in constituent nucleotide units (nt).
| Parameter | Source data | Nucleotide-free | AMPPNP | ADP | ADP-AlF4 |
|---|---|---|---|---|---|
| Total DNA-induced quench (%)a | Trp spectrab | 65 ± 4 | 69 ± 4 (5 ± 1) | 62 ± 4 (11 ± 1) | 70 ± 3 (17 ± 2) |
| Amplitudes | 59 ± 1 | 61 ± 1 | 51 ± 2 | 62 ± 2 | |
| Contribution of slow phase (% of total quench) | Amplitudes | 49 ± 7 | 56 ± 3 | 14 ± 10 | 53 ± 2 |
| DNA-binding stoichiometry (nt/RecQ) | Amplitudes | 20 ± 3 | 14 ± 8 | 14 ± 3 | 40 ± 18 |
| k1 (μm−1 s−1) | Binding kinetics | 7.8 ± 1.7 | 5.0 ± 0.6 | 9.1 ± 0.6 | 0.76 ± 0.07 |
| k–1 (s−1) | Binding kinetics | 140 ± 110 | 88 ± 19 | 140 ± 20 | 10 ± 7 |
| K1 (μm) | k−1/k1 | 18 ± 18 | 18 ± 6 | 15 ± 3 | 13 ± 10 |
| k2 + k−2 (s−1) | Binding kinetics | 8.5 ± 3.0 | 3.2 ± 0.4 | 18 ± 5 | 2.0 ± 0.3 |
| k−2 (s−1) | Binding kinetics | 2.2 ± 3.3 | 0.30 ± 0.50 | 6.7 ± 2.7 | < 0.1 |
| DxSO4 chasingc | 4.1 ± 0.3 | 0.40 ± 0.03 | 12 ± 1 | < 0.01 | |
| Heparin chasingd | 4.2 ± 0.8 | 0.89 ± 0.03e | 10 ± 2 | < 0.01 | |
| K2 | k2/k−2 | 1.4 ± 0.2 | 5.0 ± 1.4 | 0.88 ± 0.11 | > 200 |
| Keq (μm)f | K1/(K2+1) | 7.4 ± 8.0 | 3.0 ± 1.7 | 8.0 ± 2.1 | < 0.065 |
| Amplitudes | < 5 | < 5 | < 5 | < 5 |
a Relative to the Trp fluorescence level of the corresponding DNA-free RecQ-nucleotide complex.
b Values in parentheses indicate the extent of nucleotide-induced quench in the absence of DNA.
c Determined at [DxSO4] = 0.1 mg/ml.
d Values extrapolated to zero heparin concentration (cf. Fig. 2D).
e Value for ATPγS: 3.3 ± 0.2 s−1.
f Overall equilibrium constant of DNA binding, defined as Keq = ([RecQ(.N)]eq × [DNA]eq)/([RecQ†.(N.)DNA]eq + [RecQ††.(N.)DNA]eq) (cf. Fig. 1G).
We investigated the kinetics of the interaction of RecQ with dT54 ssDNA by rapidly mixing RecQ-nucleotide complexes with increasing concentrations of dT54 in the stopped-flow apparatus. Importantly, DNA concentrations (and parameters derived therefrom) are expressed in terms of constituent nucleotide (nt) units throughout this work, (i) in order to facilitate comparison of results obtained with DNA substrates of different length and (ii) to provide parameters that are independent of uncertainties in the determined DNA binding stoichiometry of RecQ. Thus, the second-order binding rate constants and equilibrium dissociation constants specified in Tables 1–3 can be used to provide values pertinent to RecQ binding sites upon multiplication or division, respectively, by the applicable DNA binding stoichiometry (nt/RecQ).
TABLE 2.
DNA length dependence of the DNA binding properties of RecQ
Shown are 5 °C, 0.5 μm RecQ Trp fluorescence stopped-flow data. Nomenclature of parameters refers to Fig. 1G. All parameters with dimensions involving concentration are specified based on DNA concentrations expressed in constituent nucleotide units (nt).
| Parameter | Source data | dT18 | dT54 | dT90 | poly(dT) |
|---|---|---|---|---|---|
| Total DNA-induced quench (%, relative to DNA-free state) | Amplitudes | 31 ± 2 | 30 ± 2 | 42 ± 2 | 44 ± 2 |
| Contribution of slow phase (% of total quench) | Amplitudes | 9 ± 4 | 17 ± 4 | 23 ± 4 | 43 ± 5 |
| k1 (μm−1 s−1) | Binding kinetics | 2.8 ± 0.4 | 2.4 ± 0.4 | 2.5 ± 0.2 | 2.3 ± 0.7 |
| k−1 (s−1) | Binding kinetics | 35 ± 14 | 29 ± 11 | 21 ± 16 | < 10 |
| K1 (μm) | k−1/k1 | 13 ± 7 | 12 ± 7 | 8.4 ± 7.1 | < 5 |
| Amplitudes | 55 ± 27 | 49 ± 8 | 27 ± 8 | 23 ± 14 | |
| k2 + k−2 (s−1) | Binding kinetics | 15 ± 3 | 16 ± 4 | 17 ± 16 | > 20 |
| Keq (μm) | Amplitudes | 36 ± 16 | 16 ± 3a | 9.0 ± 2.0a | 8.0 ± 2.0a |
a Apparent Keq value limited by RecQ concentration and DNA binding stoichiometry (cf. Table 1), representing an upper bound for Keq.
TABLE 3.
DNA-activated ATPase activity and DNA binding properties of RecQ constructs
Shown are 5 °C, nucleotide-free RecQ construct (0.5 μm), Trp fluorescence stopped-flow data (except for ATPase parameters). Nomenclature of parameters refers to Fig. 1G. All parameters with dimensions involving concentration are specified based on DNA concentrations expressed in constituent nucleotide units (nt). ND, not determined.
| Parameter | Source data | RecQ | RecQY555A | RecQ523 | RecQ414 |
|---|---|---|---|---|---|
| ATPase kcat (s−1)a | PK-LDH assay | 32 ± 2 | 27 ± 1 | 59 ± 1 | 52 ± 2 |
| ATPase Kd,app (μm)a,b | PK-LDH assay | 0.068 ± 0.033 | 0.12 ± 0.02 | 0.21 ± 0.03 | 19 ± 2 |
| Total DNA-induced quench (%, relative to DNA-free state) | Amplitudes | 30 ± 2 | 20 ± 3 | 45 ± 4 | 13 ± 2 |
| Contribution of slow phase (% of total quench) | Amplitudes | 17 ± 4 | 0 | 0 | 0 |
| k1 (μm−1s−1) | Binding kinetics | 2.4 ± 0.4 | 1.7 ± 0.3 | 1.9 ± 0.2 | 0.4 ± 0.2 |
| k−1 (s−1) | Binding kinetics | 29 ± 11 | 170 ± 40 | 53 ± 27 | 280 ± 30 |
| K1 (μm)c | k−1/k1 | 12 ± 7 | 100 ± 40 | 28 ± 17 | 700 ± 410 |
| koff/k1c | 1.9 ± 0.9 | 18 ± 5 | 1.4 ± 0.4 | ND | |
| Amplitudes | 49 ± 8 | ND | 54 ± 14 | ND | |
| koffd | DxSO4 chasing | 4.5 ± 1.3 | 31 ± 3 | 2.7 ± 0.5 | ND |
| DxSO4 sensitivity (s−1(mg/ml)−1) | DxSO4 chasing | 18 ± 3 | 47 ± 5 | 0 | ND |
a 25 °C, 15 nm (wild-type RecQ) or 10 nm (other constructs) enzyme, dT54 data.
b Apparent dissociation constant of DNA binding during steady-state ATP hydrolysis (cf. Equations 1 and 2).
c In mutant constructs, K1 = Keq.
d koff derived from y intercept as in Fig. 5B.
The dT54 binding traces of RecQ and RecQ-nucleotide complexes were fitted by two exponentials (Fig. 1B and Table 1). A slower third phase of the traces was present also in DNA-free controls and was thus attributed to photobleaching. The biphasic nature of DNA binding was verified by the finding that fits comprising a single binding phase showed systematic deviation from the recorded transients (Fig. 1C). The observed rate constants (kobs) of the rapid and slow phases increased linearly and hyperbolically, respectively, with DNA concentration (Fig. 1, D and E). The total and rapid phase amplitudes of the traces (Atotal and Arapid, respectively) showed early saturation (Fig. 1F), reflecting tight DNA binding by RecQ and DNA binding stoichiometries (14–20 nt/RecQ except in ADP-AlF4; Table 1) consistent with those determined in earlier ATPase and ssDNA translocation studies by us (18 ± 2 nt/RecQ) (12) and others (10–34 nt/RecQ) (13, 14). Interestingly, the binding stoichiometries reflected a significantly larger occluded site size of RecQ on ssDNA in ADP-AlF4 than in other nucleotide states (Table 1), suggesting a change in the conformational dynamics of the protein.
The observed DNA-binding kinetic behavior of RecQ can be interpreted in the framework of a sequential two-step binding mechanism in which the initial collision step (K1) is followed by an isomerization event occurring in DNA-bound RecQ helicase (K2), representing a structural transition (Fig. 1G). Thus, the slopes and the intercepts of linear fits to the rapid phase kobs versus DNA concentration plots reflected the forward and reverse rate constants (k1 and k−1) of the initial collision step, respectively (Fig. 1D and Table 1). Importantly, with increasing DNA concentration, Atotal converged to significantly higher values than Arapid, reflecting that both steps of the DNA binding mechanism contribute to the observed quench in Trp fluorescence (Fig. 1, F and G, and Table 1). The upper bounds and the y intercepts of the hyperbolic DNA concentration dependence of slow phase kobs delineated (k2 + k−2) and k−2, respectively (Fig. 1E and Table 1). This way, all kinetic and equilibrium constants of the two-step DNA-binding mechanism could be determined (Fig. 1G and Table 1). The overall mechanism of RecQ DNA binding was found generally similar in all nucleotide states examined. The initial K1 process is slower in the ADP-AlF4-bound state than in other nucleotides, but its equilibrium constant is unchanged (Table 1). The K2 isomerization step is slowed down by nucleotide analogs occupying the γ-phosphate binding site (AMPPNP and ADP-AlF4) and slightly accelerated by ADP relative to the nucleotide-free state (Table 1). Importantly, the value of k−2 (reflected in the intercept of the slow phase kobs versus DNA concentration plots) was very low in ADP-AlF4, indicating that the K2 isomerization leads to very strong DNA binding in the presence of this nucleotide analog (Fig. 1E and Table 1). We therefore propose that the RecQ-ADP-AlF4 complex represents either an ATP-bound transition state or a posthydrolytic (ADP-Pi) state of the hydrolytic cycle, which is the most strongly DNA-bound “clamped” one during the mechanochemical cycle. This important finding was assessed further in DNA dissociation studies described below.
We monitored the dissociation of RecQ-nucleotide complexes from dT54 ssDNA in kinetic chasing experiments by rapidly mixing RecQ-nucleotide-dT54 complexes with commonly used DNA-mimicking protein traps (heparin or DxSO4) in the stopped-flow apparatus (Fig. 2). In DxSO4 chasing experiments, a rapid dissociation phase (reflecting k−1) was apparent (with rate constants around 100 s−1) (Fig. 2, A and B), but this phase was not seen in heparin chasing experiments, possibly due to the limited signal/noise ratio (Fig. 2C). The rate constants of the dominant slow dissociation phase were similar in the presence of DxSO4 and heparin and consistent with the k−2 values determined from DNA binding experiments (Fig. 1E and Table 1). We performed the heparin chasing experiments at a series of heparin concentrations (Fig. 2D). The amplitude of the DNA dissociation traces showed saturation at relatively low heparin concentrations, whereas the dissociation kobs values increased linearly with heparin concentration, indicating that the dissociation of the protein is accelerated by the trap (Fig. 2D). Values reported for heparin experiments in Table 1 are the y intercepts of kobs versus heparin concentration plots, representing heparin-free values. Relative to the nucleotide-free state of RecQ, ADP induced a slight acceleration of DNA dissociation, whereas AMPPNP slightly slowed down the process (Table 1). Importantly, no dissociation transient was seen in ADP-AlF4 at least until 30 s, corroborating the exceptionally strong DNA binding of RecQ in this state (Table 1).
FIGURE 2.

Effect of nucleotides on RecQ dissociation from DNA. A, normalized time courses of Trp fluorescence recorded upon “chasing” RecQ (0.5 μm) from dT54 (27 μm, nt) by rapidly mixing the RecQ-dT54 complex with DxSO4 (0.1 mg/ml) in the absence of nucleotide (apo; black dots), in ADP (100 μm; open dark gray symbols), AMPPNP (100 μm; gray dots), and ADP-AlF4 (0.5 mm; light gray dots). Lines show double exponential fits to data sets. B, comparison of single (gray line) and double (black line) exponential fits to the trace recorded in AMPPNP (cf. A). The inset shows the residuals of the fits (gray and black, respectively), indicating systematic deviation from data points when fitting a single phase (gray). C, kinetic “chasing” trace recoded in the presence of ADP as in A except that heparin (2.5 mg/ml) was used as a chaser instead of DxSO4, fitted by a single exponential (black line). Residuals shown in the inset indicated that the single-exponential fit was sufficient in the case of heparin chasing. D, heparin concentration dependence of kobs values (main panel) and amplitudes (inset) of transients recorded in the presence of ADP as in C. Lines show linear (main panel) and hyperbolic (inset) fits to the data sets (see “Results”). Data were recorded at 25 °C. Parameters derived from fits are shown in Table 1. AU, arbitrary units.
The Mechanism of DNA Binding Is Largely Independent of DNA Length
To further assess the detected two-step DNA binding mechanism, we monitored the DNA length dependence of DNA binding kinetics by rapidly mixing nucleotide-free RecQ with ssDNA molecules of different length (dT18, dT54, dT90 and poly(dT)) (Fig. 3). We performed these experiments at 5 °C to minimize amplitude loss in the first phase (cf. Fig. 3A) and allow better resolution of kinetic processes. This condition also allowed for a more precise determination of the apparent equilibrium constants of DNA binding from amplitude data (Fig. 3C and Table 2). The DNA concentration dependence of the observed rate constants of the two phases (krapid and kslow, respectively) appeared similar in the presence of DNA substrates of different length (Fig. 3B). The initial binding step (K1) did not appear to be strongly affected by ssDNA length; however, k−1 was lower, and the initial binding appeared to be stronger in poly(dT) than in shorter DNA substrates (Table 2). The contribution of the K2 isomerization process to the overall fluorescence change gradually increased with DNA length (Table 2). The overall kinetics of this step (k2 + k−2) was not greatly affected by DNA length (Table 2). The overall DNA affinity of RecQ gradually increased with DNA length, indicating influence from both steps (Table 2). The DNA length dependence of the overall DNA affinity of RecQ was in line with our earlier findings based on ATPase kinetic parameters (12).
FIGURE 3.

Interaction of RecQ with DNA molecules of different length. A, stopped-flow Trp fluorescence traces recorded upon rapidly mixing RecQ (0.5 μm) with dT18 (black), dT54 (dark gray), dT90 (gray), and poly(dT) (light gray; all DNA ligands applied at 81 μm, nt). Triple exponential fits are shown as lines. (The slowest phase arose from photobleaching; see “Results.”) Fluorescence levels were normalized to that of the DNA-free state. B, DNA concentration dependence of kobs values of the first (krapid; main panel) and second (kslow; inset) phases of transients recorded as in A (dT18 (squares), dT54 (circles), dT90 (triangles), and poly(dT) (inverted triangles)) alongside linear fits (lines). C, DNA concentration dependence of total (main panel) and rapid phase (inset) amplitudes of transients recorded as in A (symbols as in B) alongside hyperbolic fits (lines). Data were recorded at 5 °C. Parameters derived from fits are shown in Table 2.
Temperature Dependence of the Initial Binding and Isomerization Steps
The comparison of the results of Tables 1 and 2 (columns “Nucleotide-free” and “dT54”, listing data obtained with identical components at 25 and 5 °C, respectively) provides insights into the temperature dependence of the DNA binding process. The total quench in Trp fluorescence was markedly larger at higher temperature, mainly due to increase in the amplitude component originating from the K2 isomerization. The forward and reverse rate constants of the initial K1 binding step were both accelerated by 3–4-fold at increased temperature, leaving the K1 equilibrium constant essentially unchanged. The overall kinetics of the isomerization step (k2 + k−2) was also essentially unchanged, within experimental error. The overall DNA binding affinity (Keq) determined from amplitude data increased with temperature, suggesting an endothermic nature for DNA binding by RecQ. This increase was mostly dictated by the K2 isomerization process.
The Isomerization of Nucleotide-bound RecQ Is Detected Only in the Presence of an Intact HRDC Domain
To assess the contribution of the structural elements of RecQ helicase to the observed DNA binding processes, we compared the DNA interaction kinetics of wild-type RecQ with that of three mutant constructs. RecQY555A contains an amino acid substitution that was shown to practically abolish the ssDNA interaction of the HRDC domain (28). RecQ523 and RecQ414, terminated at the indicated amino acid positions, lack the HRDC and both the WHD and HRDC domains, respectively.
We performed Trp fluorescence stopped-flow measurements to monitor DNA binding to nucleotide-free RecQ constructs at 5 °C (Fig. 4A). The most striking feature of all mutants was the absence of the slow transient phase characteristic of the wild-type enzyme (Fig. 4, B and C). Thus, in the mutant proteins, DNA binding could be described by the K1 process alone (reflected in krapid observed rate constants; Fig. 4D). The k1 on-rate constant (reflected in the slopes of the plots in Fig. 4D) was slowed down in RecQ414 compared with the other constructs (Table 3). The k−1 off-rate (reflected in the intercepts of the plots in Fig. 4D) was accelerated in all mutants, most prominently in RecQ414. The amplitude of the DNA-induced quench in Trp fluorescence was variable among the investigated constructs (Fig. 4E and Table 3).
FIGURE 4.

DNA binding and DNA-activated ATPase activity of wild-type and mutant RecQ constructs. A, stopped-flow Trp fluorescence traces of the interaction of RecQ constructs (wild-type RecQ (black), RecQY555A (dark gray), RecQ523 (gray), and RecQ414 (light gray); all proteins applied at 0. 5 μm) with dT54 (81 μm, nt) at 5 °C. Fluorescence levels were normalized to that of the DNA-free state. Lines show triple (wild-type RecQ) or double exponential (all mutants) fits to the data sets. (The slowest phase arose from photobleaching in each case; see “Results.”) B and C, comparison of fits comprising one (B, gray line) and two (B, black line) exponential DNA-binding phases (in addition to the slow photobleaching phase) to the wild-type RecQ trace (cf. A). Residuals of the fits shown in the main panel of C (gray and black, respectively) indicated systematic deviation from data points when fitting a single binding phase (gray). The inset of C shows that, in the case of RecQ523, the residuals of fits comprising either one (gray) or two (black) binding phases showed no systematic deviation from data points (fitted lines were indistinguishable; cf. A). This finding applied to all mutant constructs. D and E, dT54 concentration dependence of rapid phase rate constants (krapid; D) and total amplitudes (Atotal; E) of transients recorded at 5 °C as in A (wild-type RecQ (black squares), RecQY555A (dark gray circles), RecQ523 (gray triangles), and RecQ414 (light gray inverted triangles)) alongside linear (D) and hyperbolic (E) fits. (RecQY555A and RecQ414 amplitude data could not be reasonably fitted due to noise in the low values.) F, dT54-activated ATPase activity of RecQ constructs (wild-type RecQ 15 nm, mutant constructs 10 nm; symbols as in D) as determined by a PK-LDH assay at 25 °C. Quadratic (wild-type RecQ, RecQY555A) and hyperbolic (RecQ523 and RecQ414) fits are shown as lines. Parameters derived from fits are shown in Table 3.
We further assessed the DNA binding properties of RecQ constructs in dT54-activated steady-state ATPase measurements performed at 25 °C. All mutants showed a marked acceleration of their ATPase activities by dT54 (Fig. 4F). As determined from the dT54 concentration dependence of the ATPase activities, the apparent DNA binding affinity of RecQY555A and RecQ523 was only moderately reduced compared with that of wild-type RecQ, whereas RecQ414 showed a marked (about 100-fold) weakening of the apparent DNA binding affinity (Fig. 4F and Table 3). (The dT54 concentration dependence of the ATPase activities was fitted by a quadratic equation (Equation 2) in the case of wild-type RecQ and RecQY555A, whereas hyperbolic fits (Equation 1) were applicable in the case of RecQ523 and RecQ414 due to the lower apparent DNA binding affinities of the latter two constructs.)
We monitored the dissociation of RecQ constructs from poly(dT) at 5 °C in DxSO4 chasing kinetic experiments (Fig. 5). The DxSO4 concentration dependence of kobs values of the dominant rapid phase of dissociation transients (Fig. 5B) indicated generally lower DxSO4-free dissociation rate constants (koff) than the k−1 values determined from DNA binding experiments (Fig. 4D) but showed a similar tendency among the constructs (i.e. the dissociation from DNA was accelerated several times in RecQY555A and was similar in RecQ523 to that in the wild-type enzyme) (Fig. 5B and Table 3). Importantly, no transient was seen in RecQ414 DxSO4 chasing experiments, but the fluorescence level of the traces was significantly elevated compared with that observed in DxSO4-free controls (Fig. 5A). Together with the high k−1 value determined in DNA binding experiments (Fig. 4D), this result indicates that the majority of RecQ414 molecules were not bound to DNA in the premixture in the applied conditions, and DxSO4 binding to DNA-free RecQ414 occurred in the dead time of the experiment (about 1 ms) (Table 3).
FIGURE 5.

Dissociation of wild-type and mutant RecQ constructs from DNA. A, Trp fluorescence stopped-flow transients recorded upon “chasing” RecQ constructs (0.5 μm; color coding as in Fig. 4A) from poly(dT) (25 μm, nt) by rapidly mixing RecQ-poly(dT) premixtures with DxSO4 (1 mg/ml). Fluorescence intensities were normalized to that of the DxSO4-free RecQ-poly(dT) premixture in the case of each construct. (The higher initial fluorescence level in RecQ414 (inset) indicates that the majority of RecQ414 molecules were not bound to DNA in the premixture, and DxSO4 binding to DNA-free RecQ414 occurred in the dead time of the experiment.) Double exponential fits are shown as lines. B, DxSO4 concentration dependence of observed rate constants (kobs; main panel) and amplitudes (inset) of the main (rapid) phase of DxSO4 chasing transients recorded as in A (symbols as in Fig. 4B). Linear fits to kobs data sets are shown as lines. Data were recorded at 5 °C. Parameters derived from fits are shown in Table 3. AU, arbitrary units.
DISCUSSION
Overall DNA Binding Properties Deduced from Trp Fluorescence Data
We found the Trp fluorescence emission of RecQ helicase to be sensitive to the interaction of the enzyme with ssDNA and mononucleotide substrates and analogs (Fig. 1A and Tables 1–3). The large quench occurring upon DNA binding, which was also sensitive to the nucleotide state of the RecQ-DNA complex (i.e. RecQ-nucleotide-DNA ternary complex formation), could be utilized in stopped-flow rapid kinetic experiments to determine the mechanism of DNA binding. In line with an earlier report (45), we found that RecQ bound to sites on oligo(dT) and poly(dT) substrates with a medium overall affinity (with equilibrium constants in the micromolar range), which was not greatly affected by ssDNA length above 54 nt (Tables 1 and 2). The DNA binding stoichiometry of RecQ, deduced from stopped-flow amplitudes (14–40 nt/RecQ in different nucleotide states; Table 1), was in line with the occluded sizes determined in earlier DNA binding and ATPase kinetic experiments (10–34 nt/RecQ) (12–14, 45).
Two-step DNA Binding Mechanism
Our rapid kinetic experiments revealed that the Trp fluorescence quench occurs in two major phases during the interaction of RecQ with ssDNA (Fig. 1, B and C). The DNA concentration dependence of the parameters of the DNA binding transients (Fig. 1, D and E) is consistent with a two-step mechanism comprising an initial binding (K1) and a subsequent isomerization (K2) step (Fig. 1G). A similar sequential three-step DNA binding mechanism was described for the DnaB hexamer (23). Two-step ssDNA binding behavior was observed earlier for T4 gene 32 ssDNA-binding protein, in which the first phase represented a diffusion-limited association, whereas the second phase arose from cooperative binding of free protein molecules to the preformed complex (46). In our studies, however, no signs of cooperativity were detected in RecQ binding to ssDNA.
The kinetic mechanism of RecQ binding to ssDNA was only moderately affected by ssDNA length (Table 2), suggesting that neither DNA end effects nor the potential binding of multiple RecQ molecules to the same DNA molecule contribute significantly to the observed phenomena. The increase in the amplitude of the slow phase of Trp fluorescence transients with ssDNA length suggests that the K2 isomerization may involve the interaction of the helicase with ssDNA segments outside the occupied primary binding site (Table 2). However, the dependence of the K2 process on the nucleotide state of RecQ clearly indicates that it is largely dependent on protein conformational changes occurring during the ATP hydrolytic cycle (see below). We observed significant temperature dependence for the rate constants, but not the equilibrium constant, of the initial binding (K1) step, whereas the kinetics of the isomerization (K2) step did not appear to be significantly temperature-dependent (Tables 1 and 2).
Detection of a DNA-clamped State; Implications for Translocation
The two-step DNA binding mechanism (Fig. 1G) was characteristic of RecQ in all nucleotide states examined (Table 1). The initial binding step (K1) showed similar kinetics in all nucleotide states except in ADP-AlF4, where it was slowed down, but its equilibrium constant was unchanged (Table 1). The most striking effect of the ADP-AlF4 nucleotide analog was the drastic slowing of the k−2 rate constant, resulting in a very high ssDNA affinity of RecQ in this state (Table 1). This kinetic difference was paralleled by a change in DNA binding stoichiometry, indicating a significantly larger occluded size for RecQ-ADP-AlF4 along ssDNA than in other nucleotides (Table 1). ADP-AlF4 has been proposed to mimic different ATPase intermediates in different enzymes (52, 53). Because the properties of the RecQ-ADP-AlF4 complex were markedly different from those of RecQ-AMPPNP representing a prehydrolysis complex (Table 1), we propose that the RecQ-ADP-AlF4 state can be regarded either as the transition state of ATP hydrolysis or a posthydrolytic ADP-Pi-bound state. Thus, our findings suggest that ATP hydrolysis is associated with a conformational change involving the closure of the DNA-binding region of RecQ. This structural change may be linked to a mechanical step along ssDNA.
The determined ssDNA binding parameters of RecQ and RecQ-nucleotide complexes provide further insights into the ATP hydrolysis-driven ssDNA translocation mechanism. Recently, we and others have shown that E. coli RecQ translocates along ssDNA with a tight mechanochemical coupling and moderate processivity (12–14). Our current results show that the mean DNA-bound lifetime of RecQ is short (≪1 s) in the nucleotide-free and ADP-bound states, whereas it is on the order of 1 s in AMPPNP (mimicking a prehydrolytic ATP-bound state) and is extremely long (>100 s) in the ADP-AlF4-bound state (Fig. 6A). The mean DNA-bound lifetime during ssDNA translocation (8.3 ± 0.7 s−1) (12) falls between those characteristic of the AMPPNP and ADP-AlF4 states. Similarly, the apparent dissociation equilibrium constant of the RecQ-DNA interaction during ssDNA-activated ATP hydrolysis lies between those in the AMPPNP and ADP-AlF4 states (Fig. 6B). Taken together, these data suggest that, at saturating ATP and DNA concentrations, the states mimicked by AMPPNP-bound and ADP-AlF4-bound RecQ are most abundant during steady-state ATP hydrolysis-driven ssDNA translocation. Thus, the rate-limiting step of the mechanochemical cycle may be linked to either ATP hydrolysis or phosphate release, depending on the precise nature of the ADP-AlF4-bound state. Further dissection of the enzymatic substeps of the RecQ ATPase kinetic cycle driving ssDNA translocation will be necessary to determine the transition rates between nucleotide states (i.e. those of ATP binding, hydrolysis, and product release) and the mechanistic basis of the described tight mechanochemical coupling.
FIGURE 6.

Comparison of DNA binding parameters of RecQ in different nucleotide states with those during ATP hydrolysis-driven ssDNA translocation. A, mean DNA-bound lifetime of RecQ in different nucleotide states (determined in this study), compared with that determined during ATP hydrolysis-driven ssDNA translocation (12) in identical physicochemical conditions. The mean DNA-bound lifetime of RecQ in a given nucleotide (N) state (〈τ〉) was calculated as 〈τ〉 = α1(1/k−1) + α2(1/k−2), where α1 and α2 are the fractional occupancies of the RecQ†.(N.)DNA and RecQ††.(N.)DNA states within the DNA-bound enzyme population, respectively (α1 = 1/(K2 + 1); α2 = K2/(K2 + 1); cf. Fig. 1G and Table 1). B, DNA-binding overall Kd (Keq) values of RecQ in different nucleotide states (cf. Table 1), compared with the apparent DNA binding Kd (Kd,app) values determined in steady-state ATPase experiments in identical physicochemical conditions (cf. Table 3). Arrows in the ADP-AlF4 columns indicate that only a lower bound for the DNA-bound lifetime (A) and an upper bound for the DNA-binding Kd (B) could be defined for this state. Error bars, S.E.
The effect of the nucleotide state on nucleic acid binding was observed in other helicases, including PriA (19), DnaB (21), and Rep (54). In the latter two cases, AMPPNP reduced the affinity of the helicase for ssDNA. Henn et al. (55) found that the low ssDNA affinity by ATP-bound DEAD box helicase DbpA undergoes significant strengthening upon a conformational change linked to ATP hydrolysis. This transition was shown to be indispensable for dsDNA unwinding.
Our results strongly indicate that the closed tandem RecA conformation of RecQ allows the other domains to form a “clamped,” very strongly DNA-bound structure. The formation of this state may be analogous to the DNA-induced closure of the conformation of E. coli UvrD helicase (16, 17). The cyclical change in the DNA affinity of RecQ (Fig. 6B) is presumably coordinated with the movements of the so-called aromatic loop containing the highly conserved sequence CXSQWGHDFR, which is essential for coupling ATP hydrolysis to DNA binding (30). Adjacent to the aromatic loop, the DEAH box motif is also expected to have a potential role in mediating conformational changes between these two regions, based on mutational studies in NS3 helicase (56). Conserved aromatic loops and residues of other helicases were implicated in hydrophobic stacking interactions necessary for translocation (16, 57, 58). Such interactions may play important roles also in RecQ family helicases. The strength of the DNA interaction must be coordinated with base stacking for effective translocation; a weak to strong DNA binding transition could be coupled to a shift in base stacking interactions. The motion of the RecA folds induced by changes in the nucleotide binding site may therefore coordinate the interaction with DNA, resulting in effective translocation cycles.
Implications for Domain Movements during DNA Binding
Our Trp fluorescence stopped-flow experiments revealed that the isomerization of DNA-bound RecQ (K2 in Fig. 1G) is not detectable if ssDNA binding by the HRDC domain is impaired (in RecQY555A) or the HRDC domain is deleted (in RecQ523) (Fig. 4 and Table 3). Importantly, these RecQ variants contain all four native tryptophans of the enzyme. The isomerization therefore appears to be associated with the binding of the HRDC domain to ssDNA. However, both above-mentioned mutant RecQ variants retained a relatively high ssDNA affinity, suggesting that the HRDC domain is not a dominant contributor to the ssDNA interaction of the whole RecQ enzyme molecule (Table 3). Interestingly, a RecQ variant lacking the HRDC domain showed markedly weakened binding to 3′-overhanged dsDNA in earlier electrophoretic mobility shift experiments, but the unwinding efficiency was unaffected by the truncation (28). There is no simple kinetic explanation for this discrepancy because the dissociation of RecQ523 from ssDNA was only moderately accelerated compared with that of wild-type RecQ in our experiments (Fig. 4D and Table 3).
RecQ414, in which both the WHD and HRDC domains have been deleted, showed a drastically impaired overall ssDNA interaction in all experiments, suggesting that the WHD has a significant contribution to the overall ssDNA affinity of RecQ. The high ATPase activity of this construct at saturating DNA concentrations indicated that the weakening of the DNA affinity does not originate from an impairment of the RecQ helicase core fold (Fig. 4F and Table 3).
Taken together, the present study provides evidence for a structural transition occurring in DNA-bound RecQ helicase that is sensitive to the nucleotide state of the enzyme. We detected a very strongly ssDNA-bound RecQ conformation, which is proposed to be important for mechanochemical coupling of ATP hydrolysis to ssDNA translocation. The detected structural transition appears to involve the binding of the HRDC domain to ssDNA. Together with earlier works on the RecQ ATPase and ssDNA translocation and dsDNA unwinding mechanism (12–14, 44, 59), mechanistic insight into the DNA interaction of RecQ enables a deeper understanding of the molecular processes leading to genome maintenance.
This work was supported by Human Frontier Science Program Grant RGY0072/2010 (to M. K.) and “Momentum” Program of the Hungarian Academy of Sciences Grant LP2011-006/2011 (to M. K.). This work was also supported by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of the TÁMOP 4.2.4.A/1-11-1-2012-0001 “National Excellence Program.”
- SF2
- SF1, and SF6, superfamily 2, 1, and 6, respectively
- AMPPNP
- adenosine 5′-(β,γ-imido)triphosphate
- PK-LDH
- pyruvate kinase/lactate dehydrogenase-linked
- HR
- homologous recombination
- HRDC
- helicase-RNase D C-terminal
- ZBD
- Zn2+-binding domain
- WHD
- winged helix domain
- SSB
- ssDNA-binding protein
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- DxSO4
- dextran sulfate
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Khakhar R. R., Cobb J. A., Bjergbaek L., Hickson I. D., Gasser S. M. (2003) RecQ helicases. Multiple roles in genome maintenance. Trends Cell Biol. 13, 493–501 [DOI] [PubMed] [Google Scholar]
- 2. Oh S. D., Lao J. P., Hwang P. Y., Taylor A. F., Smith G. R., Hunter N. (2007) BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 130, 259–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singleton M. R., Dillingham M. S., Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 4. Bohr V. A. (2008) Rising from the RecQ-age. The role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 33, 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bachrati C. Z., Hickson I. D. (2008) RecQ helicases. Guardian angels of the DNA replication fork. Chromosoma 117, 219–233 [DOI] [PubMed] [Google Scholar]
- 6. Wu L., Hickson I. D. (2006) DNA helicases required for homologous recombination and repair of damaged replication forks. Annu. Rev. Genet. 40, 279–306 [DOI] [PubMed] [Google Scholar]
- 7. Harmon F. G., Kowalczykowski S. C. (1998) RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 12, 1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanada K., Ukita T., Kohno Y., Saito K., Kato J., Ikeda H. (1997) RecQ DNA helicase is a suppressor of illegitimate recombination in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 94, 3860–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Handa N., Morimatsu K., Lovett S. T., Kowalczykowski S. C. (2009) Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23, 1234–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Courcelle J., Hanawalt P. C. (1999) RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 262, 543–551 [DOI] [PubMed] [Google Scholar]
- 11. Harrigan J. A., Bohr V. A. (2003) Human diseases deficient in RecQ helicases. Biochimie 85, 1185–1193 [DOI] [PubMed] [Google Scholar]
- 12. Sarlós K., Gyimesi M., Kovács M. (2012) RecQ helicase translocates along single-stranded DNA with a moderate processivity and tight mechanochemical coupling. Proc. Natl. Acad. Sci. U.S.A. 109, 9804–9809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rad B., Kowalczykowski S. C. (2012) Efficient coupling of ATP hydrolysis to translocation by RecQ helicase. Proc. Natl. Acad. Sci. U.S.A. 109, 1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rad B., Kowalczykowski S. C. (2012) Translocation of E. coli RecQ helicase on single-stranded DNA. Biochemistry 51, 2921–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gyimesi M., Sarlós K., Kovács M. (2010) Processive translocation mechanism of the human Bloom's syndrome helicase along single-stranded DNA. Nucleic Acids Res. 38, 4404–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J. Y., Yang W. (2006) UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell 127, 1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia H., Korolev S., Niedziela-Majka A., Maluf N. K., Gauss G. H., Myong S., Ha T., Waksman G., Lohman T. M. (2011) Rotations of the 2B sub-domain of E. coli UvrD helicase/translocase coupled to nucleotide and DNA binding. J. Mol. Biol. 411, 633–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao K., Lohman T. M. (1990) DNA and nucleotide-induced conformational changes in the Escherichia coli Rep and helicase II (UvrD) proteins. J. Biol. Chem. 265, 1067–1076 [PubMed] [Google Scholar]
- 19. Lucius A. L., Jezewska M. J., Bujalowski W. (2006) Allosteric interactions between the nucleotide-binding sites and the ssDNA-binding site in the PriA helicase-ssDNA complex. 3. Biochemistry 45, 7237–7255 [DOI] [PubMed] [Google Scholar]
- 20. Jezewska M. J., Kim U. S., Bujalowski W. (1996) Binding of Escherichia coli primary replicative helicase DnaB protein to single-stranded DNA. Long-range allosteric conformational changes within the protein hexamer. Biochemistry 35, 2129–2145 [DOI] [PubMed] [Google Scholar]
- 21. Jezewska M. J., Bujalowski W. (1996) Global conformational transitions in Escherichia coli primary replicative helicase DnaB protein induced by ATP, ADP, and single-stranded DNA binding. Multiple conformational states of the helicase hexamer. J. Biol. Chem. 271, 4261–4265 [DOI] [PubMed] [Google Scholar]
- 22. Galletto R., Jezewska M. J., Bujalowski W. (2004) Multistep sequential mechanism of Escherichia coli helicase PriA protein-ssDNA interactions. Kinetics and energetics of the active ssDNA-searching site of the enzyme. Biochemistry 43, 11002–11016 [DOI] [PubMed] [Google Scholar]
- 23. Bujalowski W., Jezewska M. J. (2000) Kinetic mechanism of the single-stranded DNA recognition by Escherichia coli replicative helicase DnaB protein. Application of the matrix projection operator technique to analyze stopped-flow kinetics. J. Mol. Biol. 295, 831–852 [DOI] [PubMed] [Google Scholar]
- 24. Soultanas P., Dillingham M. S., Velankar S. S., Wigley D. B. (1999) DNA binding mediates conformational changes and metal ion coordination in the active site of PcrA helicase. J. Mol. Biol. 290, 137–148 [DOI] [PubMed] [Google Scholar]
- 25. Toseland C. P., Webb M. R. (2013) ATPase Mechanism of the 5′-3′ DNA Helicase, RecD2. Evidence for a pre-hydrolysis conformation change. J. Biol. Chem. 288, 25183–25193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Kobbe C., Thomä N. H., Czyzewski B. K., Pavletich N. P., Bohr V. A. (2003) Werner syndrome protein contains three structure-specific DNA binding domains. J. Biol. Chem. 278, 52997–53006 [DOI] [PubMed] [Google Scholar]
- 27. Tadokoro T., Kulikowicz T., Dawut L., Croteau D. L., Bohr V. A. (2012) DNA binding residues in the RQC domain of Werner protein are critical for its catalytic activities. Aging 4, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernstein D. A., Keck J. L. (2005) Conferring substrate specificity to DNA helicases. Role of the RecQ HRDC domain. Structure 13, 1173–1182 [DOI] [PubMed] [Google Scholar]
- 29. Bernstein D. A., Zittel M. C., Keck J. L. (2003) High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 22, 4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zittel M. C., Keck J. L. (2005) Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ. Role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 33, 6982–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ren H., Dou S. X., Rigolet P., Yang Y., Wang P. Y., Amor-Gueret M., Xi X. G. (2007) The arginine finger of the Bloom syndrome protein. Its structural organization and its role in energy coupling. Nucleic Acids Res. 35, 6029–6041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pike A. C., Shrestha B., Popuri V., Burgess-Brown N., Muzzolini L., Costantini S., Vindigni A., Gileadi O. (2009) Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc. Natl. Acad. Sci. U.S.A. 106, 1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lucic B., Zhang Y., King O., Mendoza-Maldonado R., Berti M., Niesen F. H., Burgess-Brown N. A., Pike A. C., Cooper C. D., Gileadi O., Vindigni A. (2011) A prominent β-hairpin structure in the winged-helix domain of RECQ1 is required for DNA unwinding and oligomer formation. Nucleic Acids Res. 39, 1703–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J. L., Rigolet P., Dou S. X., Wang P. Y., Xi X. G. (2004) The zinc finger motif of Escherichia coli RecQ is implicated in both DNA binding and protein folding. J. Biol. Chem. 279, 42794–42802 [DOI] [PubMed] [Google Scholar]
- 35. Kitano K., Kim S. Y., Hakoshima T. (2010) Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Structure 18, 177–187 [DOI] [PubMed] [Google Scholar]
- 36. Hu J. S., Feng H., Zeng W., Lin G. X., Xi X. G. (2005) Solution structure of a multifunctional DNA- and protein-binding motif of human Werner syndrome protein. Proc. Natl. Acad. Sci. U.S.A. 102, 18379–18384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato A., Mishima M., Nagai A., Kim S. Y., Ito Y., Hakoshima T., Jee J. G., Kitano K. (2010) Solution structure of the HRDC domain of human Bloom syndrome protein BLM. J. Biochem. 148, 517–525 [DOI] [PubMed] [Google Scholar]
- 38. Liu S., Zhang W., Gao Z., Ming Q., Hou H., Lan W., Wu H., Cao C., Dong Y. (2013) NMR structure of the N-terminal-most HRDC1 domain of RecQ helicase from Deinococcus radiodurans. FEBS Lett. 587, 2635–2642 [DOI] [PubMed] [Google Scholar]
- 39. Wu L., Chan K. L., Ralf C., Bernstein D. A., Garcia P. L., Bohr V. A., Vindigni A., Janscak P., Keck J. L., Hickson I. D. (2005) The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 24, 2679–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pandey N., Govardhan S., Pathak R. K. (2011) Mobility in the structure of E. coli recQ helicase upon substrate binding as seen from molecular dynamics simulations. Bioinformation 7, 371–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu H. Q., Deprez E., Zhang A. H., Tauc P., Ladjimi M. M., Brochon J. C., Auclair C., Xi X. G. (2003) The Escherichia coli RecQ helicase functions as a monomer. J. Biol. Chem. 278, 34925–34933 [DOI] [PubMed] [Google Scholar]
- 42. Zhang X. D., Dou S. X., Xie P., Hu J. S., Wang P. Y., Xi X. G. (2006) Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 281, 12655–12663 [DOI] [PubMed] [Google Scholar]
- 43. Li N., Henry E., Guiot E., Rigolet P., Brochon J. C., Xi X. G., Deprez E. (2010) Multiple Escherichia coli RecQ helicase monomers cooperate to unwind long DNA substrates. A fluorescence cross-correlation spectroscopy study. J. Biol. Chem. 285, 6922–6936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harmon F. G., Kowalczykowski S. C. (2001) Biochemical characterization of the DNA helicase activity of the Escherichia coli RecQ helicase. J. Biol. Chem. 276, 232–243 [DOI] [PubMed] [Google Scholar]
- 45. Dou S. X., Wang P. Y., Xu H. Q., Xi X. G. (2004) The DNA binding properties of the Escherichia coli RecQ helicase. J. Biol. Chem. 279, 6354–6363 [DOI] [PubMed] [Google Scholar]
- 46. Lohman T. M., Kowalczykowski S. C. (1981) Kinetics and mechanism of the association of the bacteriophage T4 gene 32 (helix destabilizing) protein with single-stranded nucleic acids. Evidence for protein translocation. J. Mol. Biol. 152, 67–109 [DOI] [PubMed] [Google Scholar]
- 47. Kozlov A. G., Lohman T. M. (2002) Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry 41, 6032–6044 [DOI] [PubMed] [Google Scholar]
- 48. Stole E., Bryant F. R. (1997) The rate-determining step on the recA protein-catalyzed ssDNA-dependent ATP hydrolysis reaction pathway. Biochemistry 36, 3483–3490 [DOI] [PubMed] [Google Scholar]
- 49. Flowers S., Biswas E. E., Biswas S. B. (2003) Conformational dynamics of DnaB helicase upon DNA and nucleotide binding. Analysis by intrinsic tryptophan fluorescence quenching. Biochemistry 42, 1910–1921 [DOI] [PubMed] [Google Scholar]
- 50. Bjornson K. P., Moore K. J., Lohman T. M. (1996) Kinetic mechanism of DNA binding and DNA-induced dimerization of the Escherichia coli Rep helicase. Biochemistry 35, 2268–2282 [DOI] [PubMed] [Google Scholar]
- 51. Bjornson K. P., Hsieh J., Amaratunga M., Lohman T. M. (1998) Kinetic mechanism for the sequential binding of two single-stranded oligodeoxynucleotides to the Escherichia coli Rep helicase dimer. Biochemistry 37, 891–899 [DOI] [PubMed] [Google Scholar]
- 52. Kowalczykowski S. C., Krupp R. A. (1995) DNA-strand exchange promoted by RecA protein in the absence of ATP. Implications for the mechanism of energy transduction in protein-promoted nucleic acid transactions. Proc. Natl. Acad. Sci. U.S.A. 92, 3478–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geeves M. A., Holmes K. C. (1999) Structural mechanism of muscle contraction. Annu. Rev. Biochem. 68, 687–728 [DOI] [PubMed] [Google Scholar]
- 54. Wong I., Lohman T. M. (1992) Allosteric effects of nucleotide cofactors on Escherichia coli Rep helicase-DNA binding. Science 256, 350–355 [DOI] [PubMed] [Google Scholar]
- 55. Henn A., Cao W., Hackney D. D., De La Cruz E. M. (2008) The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J. Mol. Biol. 377, 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heilek G. M., Peterson M. G. (1997) A point mutation abolishes the helicase but not the nucleoside triphosphatase activity of hepatitis C virus NS3 protein. J. Virol. 71, 6264–6266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Korolev S., Hsieh J., Gauss G. H., Lohman T. M., Waksman G. (1997) Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell 90, 635–647 [DOI] [PubMed] [Google Scholar]
- 58. Velankar S. S., Soultanas P., Dillingham M. S., Subramanya H. S., Wigley D. B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97, 75–84 [DOI] [PubMed] [Google Scholar]
- 59. Pan B. Y., Dou S. X., Yang Y., Xu Y. N., Bugnard E., Ding X. Y., Zhang L., Wang P. Y., Li M., Xi X. G. (2010) Mutual inhibition of RecQ molecules in DNA unwinding. J. Biol. Chem. 285, 15884–15893 [DOI] [PMC free article] [PubMed] [Google Scholar]