

Background: AMPK is implicated as the mediator of AICAR action on liver metabolism.
Results: AICAR suppresses glucose production independent of AMPK. Regulation of mitochondrial function is AMPK-dependent.
Conclusion: Nucleotide monophosphates rely on AMPK to regulate energy metabolism but not to suppress glucose production.
Significance: Targeted AMPK activation will not lower glucose production in metabolic diseases but could improve hepatic energetics.
Keywords: AMP, AMP Kinase, AMP-activated Kinase (AMPK), Energy Metabolism, Liver Metabolism, AICAR, Fat Metabolism, In Vivo
Abstract
Metabolic stress, as well as several antidiabetic agents, increases hepatic nucleotide monophosphate (NMP) levels, activates AMP-activated protein kinase (AMPK), and suppresses glucose production. We tested the necessity of hepatic AMPK for the in vivo effects of an acute elevation in NMP on metabolism. 5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR; 8 mg·kg−1·min−1)-euglycemic clamps were performed to elicit an increase in NMP in wild type (α1α2lox/lox) and liver-specific AMPK knock-out mice (α1α2lox/lox + Albcre) in the presence of fixed glucose. Glucose kinetics were equivalent in 5-h fasted α1α2lox/lox and α1α2lox/lox + Albcre mice. AMPK was not required for AICAR-mediated suppression of glucose production and increased glucose disappearance. These results demonstrate that AMPK is unnecessary for normal 5-h fasting glucose kinetics and AICAR-mediated inhibition of glucose production. Moreover, plasma fatty acids and triglycerides also decreased independently of hepatic AMPK during AICAR administration. Although the glucoregulatory effects of AICAR were shown to be independent of AMPK, these studies provide in vivo support for the AMPK energy sensor paradigm. AICAR reduced hepatic energy charge by ∼20% in α1α2lox/lox, which was exacerbated by ∼2-fold in α1α2lox/lox + Albcre. This corresponded to a ∼6-fold rise in AMP/ATP in α1α2lox/lox + Albcre. Consistent with the effects on adenine nucleotides, maximal mitochondrial respiration was ∼30% lower in α1α2lox/lox + Albcre than α1α2lox/lox livers. Mitochondrial oxidative phosphorylation efficiency was reduced by 25%. In summary, these results demonstrate that the NMP capacity to inhibit glucose production in vivo is independent of liver AMPK. In contrast, AMPK promotes mitochondrial function and protects against a more precipitous fall in ATP during AICAR administration.
Introduction
The glucoregulatory system of the body coordinates glucose supply and demand with such precision that arterial glucose is maintained within narrow limits. Research has provided a clearer understanding of the hormonal and interorgan communication required for normoglycemia in vastly different physiological conditions (1). Indeed, disruption of this sensitive regulatory system has had a broad impact on public health (2). The alarming prevalence of diabetes and associated metabolic disorders makes the mechanistic understanding of glucose homeostasis imperative.
AMP-activated protein kinase (AMPK)2 is a conserved, heterotrimeric serine/threonine kinase composed of α, β, and γ subunits (3). AMPK senses a decrease in cellular energy and responds by triggering pathways that work to maintain energy balance (3). Furthermore, ADP and AMP bind the AMPK γ subunit, which primes the enzyme for αThr172 phosphorylation and protects against dephosphorylation. Phosphorylation increases AMPK activity, which is further stimulated by allosteric AMP activation (4). Removal of LKB1, the primary kinase responsible for AMPK phosphorylation in the liver, blunts AMPK activation and, interestingly, results in severe hyperglycemia (5). AMPK has been invoked as the major mediator of the suppressive effects of biguanides, 5-aminoimidasole-4-carboxamide-1-β-d-ribofuranoside (AICAR), and a number of endocrine factors on hepatic glucose production. However, recent studies in genetic models dispute the necessity of hepatic AMPK for the acute effects of some anti-glycemic agents (6–8). Adding yet another layer of complexity to the enigmatic role of AMPK in glycemic control, AMPK activation is highest in physiological conditions characterized by elevated glucagon action and hepatic glucose production (9–11).
AMPK activation correlates with endocrine and energetic states characterized by pro-oxidative, anti-lipogenic signaling in the liver (9–14). Evidence supports a role for AMPK in regulating fat utilization and mitochondrial function in the liver (15–17). Furthermore, AICAR (18–20) and metformin (18, 21) promote hepatic fat utilization and suppress lipogenic signaling. A major complication in determining the necessity of AMPK for the effects of glucagon, adiponectin, AICAR, and biguanides on hepatic metabolism is that they increase levels of nucleotide monophosphate (NMP) (6–10), which has multiple sites of action. To fully understand the mechanism of action of these hormones and pharmaceutical agents, it is necessary to distinguish the consequences of an increase in the NMP pool from the activation of AMPK on hepatic metabolism.
In the present studies, we tested the extent to which metabolic effects of increased hepatic NMP were dependent on AMPK. These experiments were carried out using an AICAR-euglycemic clamp in mice with a liver-specific removal of AMPK. The results demonstrate in vivo that an increase in hepatic NMP suppresses glucose production by a mechanism that is independent of hepatic AMPK. This finding challenges the paradigm that hepatic AMPK is required for acute glucoregulation by AICAR. However, these studies support the hypothesis that AMPK maintains hepatic adenylate energy balance in the liver during AICAR administration by regulating mitochondrial function.
EXPERIMENTAL PROCEDURES
Animal Models
All procedures were approved by Vanderbilt University Animal Care and Use Committee. To generate a liver-specific AMPK knock-out mouse, albumin-cre+ mice were crossed with mice containing floxed AMPKα1 and α2 subunits on a C57BL/6 background. Genotyping and Western blots were performed to confirm liver-specific deletion of AMPK, defined as α1α2lox/lox + Albcre or wild type, α1α2lox/lox. Mice were maintained on a 12:12 h light/dark cycle in a temperature- and humidity-controlled environment and given a chow diet (5.5% fat by weight; 5001 Purina laboratory rodent diet) with free access to water. On the day of the study, unrestrained 14–15 -week-old male mice were fasted 5 h prior to clamp or sacrifice.
Surgical Procedures and Body Composition
All clamps were performed in chronically catheterized mice as previously described (22). Briefly, right jugular vein and left common carotid artery catheters were surgically implanted 5–7 days prior to study. The mice were individually housed postsurgery; only mice within 10% of their presurgical weight were studied. An uncatheterized cohort of 14-week-old male α1α2lox/lox and α1α2lox/lox + Albcre mice were used to determine body composition using an mq10 nuclear resonance analyzer (Bruker Optics).
AICAR-Euglycemic and Saline Clamp
On the day of study, the mice were placed in bedded containers without food and water between 07:00 and 08:00 for a 5-h fast (t = −300 to 0 min). Infusion lines were connected to the indwelling catheters 90 min prior to infusions to minimize stress. AICAR (Toronto Research Chemicals) or saline clamps were performed similar to previously described insulin clamps (23) with minor modifications. At t = −90 min, a primed continuous infusion of [3-3H]glucose was administered to determine 5-h fasting and clamp glucose kinetics. Samples were taken at t = −15 and −5 min to determine basal blood and plasma parameters followed by the start of a continuous infusion of donor erythrocytes to prevent a fall in hematocrit during the clamp. At t = 0 min, AICAR or saline was delivered as a primed (40 mg·kg−1), continuous infusion (8 mg·kg−1·min−1) followed by a variable infusion of 50% dextrose to clamp blood glucose levels at 110 mg·dl−1. The AICAR infusion rate was selected to parallel the conditions of earlier clamps in rodents (24, 25), which afforded a more robust basis for comparing AMPK-dependent and -independent changes in metabolism. [3-3H]Glucose was mixed with the glucose infusate to minimize fluctuations in specific activity during the clamp steady state. Arterial glucose was monitored from a 5-μl blood sample (AccuCheck Advantage; Roche Diagnostics) every 10 min, and the glucose infusion rate (GIR) was adjusted to account for deviations from 110 mg·dl−1. Glucose kinetics and plasma metabolites were determined from arterial samples taken during the clamp steady state (t = 80–120 min). Blood samples were collected in EDTA-coated tubes and centrifuged, and the plasma layer was stored at −20 °C for subsequent analysis.
Plasma and Tissue Analyses
Immunoreactive insulin was measured using a double-antibody method (26). Plasma [3-3H]glucose radioactivity was determined by scintillation counting (22), and rates of endogenous glucose production (EndoRa) and disappearance (Rd) were calculated using modified Steele's non-steady-state equations (27). Lactate was measured fluorometrically. Free fatty acids and plasma/liver triglycerides were measured through enzyme-linked spectrophotometric assays (NEFA C kit, Wako Chemicals; and Triglycerides-GPO reagent set, Pointe Scientific).
At the end of the clamp, the mice were euthanized through cervical dislocation, and liver tissue was excised and freeze-clamped (<20 s) to prevent hypoxia driven changes in nucleotides. Hepatic adenine nucleotides were measured via HPLC through Vanderbilt's Hormone Assay and Analytical Services Core. ∼60 mg of frozen liver tissue was rapidly homogenized in 0.6 ml of ice-cold 0.4 m HClO4 containing 0.5 mm EGTA. Samples sat on ice for 3 min and were then centrifuged at 3200 × g at 4 °C for 5 min. The supernatant was neutralized with 0.5 m K2CO3. Samples sat on ice for another 3 min and were centrifuged at 3200 × g at 4 °C for 5 min, and the supernatants were saved for HPLC nucleotide analysis as previously described (10). Energy charge (EC) was calculated from the results as follows: EC = ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]).
For glycogen measurements, the liver was homogenized in 0.03 n HCl (0.1 mg of tissue/μl) and incubated at 80 °C for 10 min. A volume of the digest was applied to chromatography strips, dried, washed three times in 70% EtOH, and briefly rinsed with acetone. The chromatography strips were incubated in 5 ml of NaOAc-amyloglucosidase at 37 °C in a shaking water bath for 3 h. Glycogen concentration was determined from the isolate through an enzyme-linked colorimetric assay.
Immunoblotting
40–70 mg of liver tissue was homogenized in an extraction buffer (50 mm Tris, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 1% Triton X-100, pH 7.5) containing protease (Pierce) and phosphatase inhibitors. Homogenates were centrifuged for 20 min at 4500 × g at 4 °C, and the supernatants were assayed for protein concentration using the Bradford method. 5–25 μg of protein were separated in NuPAGE 4–12% (v/v) Bis-Tris gels (Invitrogen) and transferred to a PVDF membrane. The membranes were blocked in 5% milk (w/v) and probed for tAMPK, pAMPKT172, pACCS79, tACC, β-actin (Cell Signaling Technology), total OXPHOS, VDAC1, and GAPDH (Abcam). HRP-linked α-rabbit and α-mouse secondary antibodies were applied for ECL and visualization. Immunoblots for total OXPHOS were performed on mitochondrial extracts and normalized to VDAC1. ImageJ software was used for densitometry.
Mitochondria Isolation and Oxygen Consumption
Mitochondrial isolation was performed using standard homogenization and differential centrifugation methods as previously described (28). Briefly, liver tissue was weighed and subsequently minced in 5 ml of ice-cold mitochondrial extraction buffer containing 250 mm sucrose, 2 mm KH2PO4, 1 mm EGTA, and 20 mm Tris·HCl (pH 7.2). The minced tissue was washed twice and homogenized at 600 rpm with a Teflon Potter-Elvehjem pestle (6 up and down pulses). The homogenate was centrifuged for 10 min at 800 × g at 4 °C. The mitochondria-rich supernatant was then pelleted by centrifugation at 8000 × g at 4 °C for 10 min and gently resuspended in 0.5 ml of ice-cold extraction buffer. The mitochondrial sample received a second centrifugation at 8000 × g at 4 °C for 10 min, and the final pellet was resuspended in 200 μl of extraction buffer. Mitochondrial protein concentration was measured using the Bradford method. The freshly isolated liver mitochondria were used for oxygen consumption measurements. High resolution respirometry (Oroboros Instruments) was performed in duplicate at 37 °C in MiR05 (0.5 mm EGTA, 3 mm MgCl2·6H2O, 20 mm taurine, 10 mm KH2PO4, 20 mm HEPES, 1g/liter BSA, 60 mm potassium-lactobionate, 110 mm sucrose, pH 7.1, adjusted at 30 °C) as previously described (29). State 2 oxygen flux was assessed following titration of complex I substrates glutamate (10 mm) + malate (2 mm) + pyruvate (5 mm). State 3 oxygen consumption supported by complex I substrates was evaluated by a 5 mm ADP addition. State 4 was determined by titrating oligomycin (2 μg/ml). Carbonylcyanide-p-trifluoromethoxyphenyl-hydrazone (FCCP) was added in 0.05-μm steps to determine maximal electron transport system-mediated oxygen flux. An aliquot of freshly isolated mitochondria was frozen at −80 °C for immunoblot analysis.
Statistical Analysis
Comparisons were made using two-tailed t tests or two-way analysis of variance and Fisher's least significant difference test for specific differences. The data are expressed as means ± S.E. Significance was p ≤ 0.05.
RESULTS
Body Composition and 5-h Fasted Metabolites
A cohort of 14-week-old α1α2lox/lox and α1α2lox/lox + Albcre mice were assayed to determine whether the absence of hepatic AMPK impacted whole body weight and composition. There were no apparent differences in body composition (Table 1). 5-h fasted arterial glucose, plasma insulin, free fatty acids, lactate, and triglycerides (TGs) were not different between α1α2lox/lox and α1α2lox/lox + Albcre (Table 1).
TABLE 1.
Body composition and 5-h fasted metabolites of wild-type (α1α2lox/lox) and liver-AMPK knockout (α1α2lox/lox + Albcre) mice
Plasma metabolites were isolated from arterial blood samples taken from 5-h fasted α1α2lox/lox and α1α2lox/lox + Albcre mice. Body composition was performed on a separate cohort of α1α2lox/lox and α1α2lox/lox + Albcre mice. The data are expressed as means ± S.E. (n = 5–13 in each group).
| α1α2lox/lox | α1α2lox/lox + Albcre | |
|---|---|---|
| Body composition | ||
| Body weight (g) | 28.3 ± 1.0 | 28.2 ± 0.8 |
| Fat (%) | 9.3 ± 1.1 | 10.6 ± 0.5 |
| Muscle (%) | 87.6 ± 3.1 | 86.9 ± 0.5 |
| Plasma metabolites | ||
| Blood glucose | 118 ± 3 | 117 ± 4 |
| Insulin (ng/ml) | 1.9 ± 0.3 | 2.1 ± 0.4 |
| Triglycerides (mg/dl) | 95 ± 8 | 82 ± 7 |
| Lactate (mmol/liter) | 0.7 ± 0.1 | 0.6 ± 0.1 |
| Free fatty acids (mEq/liter) | 0.30 ± 0.03 | 0.38 ± 0.07 |
AICAR-mediated Inhibition of Glucose Production Is Independent of Acute AMPK Activation
In vivo and in vitro delivery of AICAR to hepatocytes inhibits glucose production (8, 24). To test whether AICAR requires AMPK for the inhibition of endogenous glucose production (EndoRa), AICAR was continuously infused at 8 mg·kg−1·min−1, and blood glucose was clamped at ∼110 mg·dl−1. During the experimental period, the GIR required to maintain euglycemia (Fig. 1A) was similar between genotypes in both saline and AICAR clamps (Fig. 1B); however, the sustained elevation in GIR was significantly higher with the infusion of AICAR than saline controls. EndoRa and glucose disappearance (Rd) were indistinguishable between genotypes prior to and during the AICAR or saline clamp (Fig. 1, C and D). AICAR inhibited EndoRa equivalently in α1α2lox/lox and α1α2lox/lox + Albcre mice (0.8 ± 1.4 and 1.1 ± 2.4 mg·kg−1·min−1). Likewise, AICAR induced an equal increment in Rd in α1α2lox/lox and α1α2lox/lox + Albcre mice (31.0 ± 1.2 and 32.8 ± 1.5 mg·kg−1·min−1). Plasma insulin levels were indistinguishable between the saline and AICAR clamp groups (Table 2). The data show that the inhibition of EndoRa by AICAR (Fig. 1, C and D) is disassociated from its capacity to activate AMPK (Fig. 2A) in vivo. AMPK deletion also provoked a reduction in liver glycogen (Table 2), which did not impact AICAR-mediated inhibition of EndoRa.
FIGURE 1.

Acute inhibition of glucose production by AICAR is independent of hepatic AMPK. A and B, arterial glucose (A) and GIR (B) during AICAR and saline clamps in α1α2lox/lox and α1α2lox/lox + Albcre mice. Mice were fasted 5 h prior to the primed (40 mg·kg−1) infusion of AICAR (8 mg·kg−1·min−1) or saline. Blood glucose was clamped at 110 mg·dl−1, and the time course is displayed to demonstrate experimental quality. 50% dextrose was infused to maintain euglycemia during the steady state of the clamp (t = 80–120 min). C and D, endogenous glucose production (EndoRa) and disappearance (Rd) during the basal period (5-h fasted) (C) and steady state of the AICAR clamp (D) in α1α2lox/lox (■) and α1α2lox/lox +Albcre (□) mice. GIR means from the experimental period were analyzed for significance. The data are expressed as means ± S.E., n = 5–12 in each group. *, p ≤ 0.05 versus saline.
TABLE 2.
Clamp metabolites
Plasma and liver metabolite concentrations during the saline or AICAR (8 mg·kg−1·min−1) euglycemic clamp from α1α2lox/lox and α1α2lox/lox + Albcre mice. Plasma metabolites were isolated from arterial blood samples drawn from externalized catheters in conscious, unstressed mice during the clamp steady state. Liver triglycerides and glycogen were measured from freeze-clamped tissue excised at the end of the clamp. Blood glucose means from the experimental period were analyzed for significance. The data are expressed as means ± S.E. (n = 4–7 in each group). ND, values lower than detectable range.
| α1α2lox/lox | α1α2lox/lox + Albcre | |
|---|---|---|
| Blood glucose (mg/dl) | ||
| Saline clamp | 109 ± 4 | 113 ± 4 |
| AICAR clamp | 111 ± 2 | 112 ± 5 |
| Insulin (ng/ml) | ||
| Saline clamp | 4.1 ± 0.5 | 3.0 ± 0.7 |
| AICAR clamp | 6.9 ± 1.5 | 4.5 ± 1.6 |
| Triglycerides (mg/dl) | ||
| Saline clamp | 41 ± 5 | 45 ± 5 |
| AICAR clamp | 17 ± 1a | 22 ± 4a |
| Lactate (mmol/liter) | ||
| Saline clamp | 0.8 ± 0.1 | 0.7 ± 0.1 |
| AICAR clamp | 5.0 ± 0.5a | 3.7 ± 0.3a,b |
| Free fatty acids (mEq/liter) | ||
| Saline clamp | 0.47 ± 0.06 | 0.48 ± 0.07 |
| AICAR clamp | NDa | 0.06 ± 0.02a |
| Liver triglycerides (mg/g of liver) | ||
| Saline clamp | 24.6 ± 0.8 | 29.1 ± 2.0 |
| AICAR clamp | 19.7 ± 1.5c | 26.7 ± 2.7b |
| Liver glycogen (mg/g of liver) | ||
| Saline clamp | 7.2 ± 2.2 | 3.1 ± 0.8 |
| AICAR clamp | 14.6 ± 3.4a | 2.1 ± 0.4b |
a p ≤ 0.05 versus saline.
b p ≤ 0.05 versus α1α2lox/lox.
c p = 0.08 versus saline.
FIGURE 2.
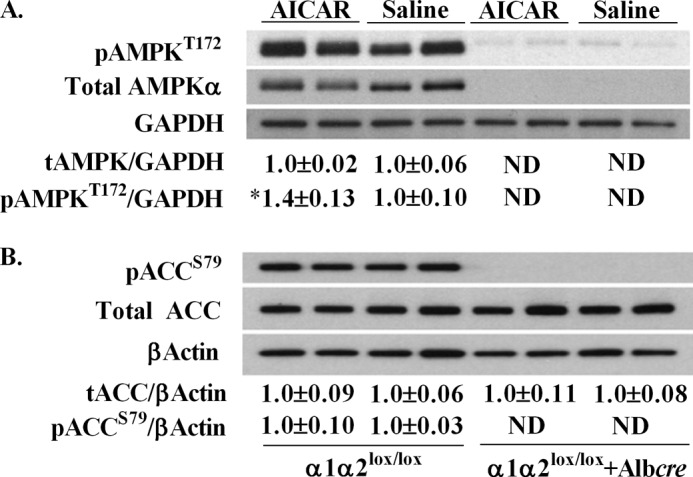
Effect of AICAR on liver AMPK activation state. Total, pAMPKT172 (A) and total, pACCS79 (B) from the livers of α1α2lox/lox and α1α2lox/lox + Albcre mice following the AICAR or saline clamp. The data are normalized to GAPDH or β-actin and expressed as means ± S.E. Values below the blots are arbitrary units normalized to α1α2lox/lox saline controls, n = 5–6 in each group. *, p ≤ 0.05 versus saline. ND, values lower than detectable range.
The 2-h AICAR infusion blunted plasma free fatty acid and plasma TG concentrations in α1α2lox/lox and α1α2lox/lox + Albcre mice (Table 2). Indeed, AICAR was so potent at clearing free fatty acids that circulating levels were undetectable in α1α2lox/lox mice (Table 2). AICAR elicited 6.6 ± 0.6- and 5.2 ± 0.5-fold increases in plasma lactate over saline in α1α2lox/lox and α1α2lox/lox + Albcre mice, respectively. The elevation in plasma lactate, however, was partially attenuated in α1α2lox/lox + Albcre mice (Table 2).
AMPK activation during energy stress is implicated in fat utilization in the liver (15). Liver TG levels in α1α2lox/lox and α1α2lox/lox + Albcre mice at the end of the saline infusion were not statistically different (24.6 ± 0.8 and 29.1 ± 2.0 mg/g of liver) (Table 2). The AICAR infusion in α1α2lox/lox mice reduced hepatic TG concentrations compared with saline controls (19.7 ± 1.5 versus 24.6 ± 0.8 mg/g of liver, p = 0.08). α1α2lox/lox + Albcre mice did not experience the same AICAR-mediated reduction as hepatic TG concentrations were significantly higher than α1α2lox/lox (26.7 ± 2.7 and 19.7 ± 1.5 mg/g of liver) (Table 2). Inhibitory phosphorylation of acetyl-CoA carboxylase (pACCS79) was similar between α1α2lox/lox saline and AICAR groups at the end of the clamp (Fig. 2B). Indeed, AICAR can promote carnitine palmitoyltransferase-1 activity independent of changes in malonyl-CoA (30). Thus, AICAR facilitates a decrease in liver TGs via hepatic AMPK.
AMPK Counters a Decrease in Hepatic Adenylate Energy Charge during an Acute AICAR Challenge in Vivo
The primed AICAR infusion resulted in comparable hepatic ZMP levels in the two genotypes (Fig. 3C). The conversion of AICAR to ZMP utilizes ATP (31), and ZMP has been demonstrated to inhibit complex I respiration in isolated mitochondria (16). Adenylate kinase maintains the equilibrium between ATP, ADP, and AMP; physiological and pharmacological perturbations in metabolism can alter this balance (6, 8–11). Thus, hepatic ATP, AMP, and ADP were measured to investigate how an acute AICAR infusion perturbs hepatic adenylate energy balance in a euglycemic, in vivo setting. Liver ATP levels were comparable in α1α2lox/lox and α1α2lox/lox + Albcre mice (1.7 ± 0.12 versus 1.5 ± 0.09 μmol·g−1) that received the saline infusion (Fig. 3A). The AMP/ATP ratio and EC were similar to previous measurements taken under postabsorptive conditions (11). The AICAR infusion resulted in a significant reduction in the total adenine nucleotide pool (3.3 ± 0.09 to 1.6 ± 0.02 μmol·g−1) in α1α2lox/lox and mice lacking hepatic AMPK (3.1 ± 0.06 to 1.4 ± 0.08μmol·g−1) (Fig. 3B). Hepatic AMP levels were equivalent (0.43 ± 0.04 versus 0.47 ± 0.03 μmol·g−1) in saline control groups. However, AICAR elicited a significant increase in AMP in α1α2lox/lox + Albcre over α1α2lox/lox mice (0.43 ± 0.02 versus 0.61 ± 0.09 μmol·g−1) (Fig. 3A). AICAR also elicited a reduction in ATP and ADP levels in both groups (Fig. 3A). The fall in ATP was exacerbated in the absence of hepatic AMPK. The deleterious effect of AICAR on the adenylate pool is exemplified by the magnitude of change in EC and the AMP/ATP ratio. α1α2lox/lox mice sustained a ∼20% reduction in EC, whereas α1α2lox/lox + Albcre mice incurred a nearly 2-fold greater reduction (Fig. 3D); the relative amount of available high energy adenylate phosphate was severely reduced as reflected by the ∼3- and ∼6-fold rise in the AMP/ATP ratio in α1α2lox/lox and α1α2lox/lox + Albcre mice, respectively (Fig. 3E). Thus, AMPK limits the fall in energy charge during an acute AICAR infusion in vivo. These data are consistent with the effects of metformin and AICAR in vitro (8).
FIGURE 3.

Liver AMPK deletion exacerbates AICAR effects on hepatic energy state. A and B, hepatic adenine nucleotides (A) and the total adenine nucleotide pool (TAN, B) were measured by HPLC from liver extracts taken from α1α2lox/lox and α1α2lox/lox + Albcre mice at the end of the AICAR or saline clamp. C, hepatic ZMP levels were also measured to assess the quality of AICAR delivery into the liver. D, energy charge was calculated by the following equation, ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]), to assess the energy state of the liver. E, the AMP/ATP ratio was provided for each clamp group. The data are expressed as means ± S.E., n = 5–6 in each group. *, p ≤ 0.05 versus saline; †, p ≤ 0.05 versus α1α2lox/lox.
Mitochondria from α1α2lox/lox + Albcre Mice Display Impairments in Mitochondrial Function
Livers from α1α2lox/lox + Albcre mice demonstrate an impaired capacity to utilize fat and maintain ATP levels during an acute AICAR challenge. Given the importance of oxidative phosphorylation in these interrelated processes, we hypothesized that hepatic mitochondria from α1α2lox/lox + Albcre mice would demonstrate impairments in integrative oxidative phosphorylation function compared with WT littermates. Polarographic oxygen consumption measurements were performed on mitochondria isolated from the livers of α1α2lox/lox and α1α2lox/lox + Albcre mice. The absence of hepatic AMPK had no effect on glutamate-, malate-, and pyruvate-supported basal oxygen consumption (state 2) (Fig. 4A). ADP-stimulated oxygen consumption (state 3), however, was attenuated in α1α2lox/lox + Albcre mice (Fig. 4A). The addition of the ATP-synthase inhibitor oligomycin demonstrated that nonphosphorylating oxygen consumption was no different between α1α2lox/lox and α1α2lox/lox + Albcre mice (Fig. 4A). Absolute FCCP-stimulated, uncoupled respiration was significantly higher in α1α2lox/lox mice (Fig. 4A) but equaled α1α2lox/lox + Albcre when normalized to state 3 and Leak respiration.3 Impaired state 3 respiration in α1α2lox/lox + Albcre mice led to a reduction in the respiratory control ratio—an index of mitochondrial efficiency—compared with α1α2lox/lox controls (Fig. 4B). These data indicate that hepatic AMPK supports efficient coupling of ADP phosphorylation and oxygen consumption. In addition, the removal of hepatic AMPK was sufficient to reduce the expression of mitochondrial complexes II and III (Fig. 4C). These results are consistent with the demonstration that hepatic AMPK activity is protective against a greater reduction in EC and elevation in the AMP/ATP ratio in response to AICAR.
FIGURE 4.

Oxygen flux data from mitochondria isolated from the livers of α1α2lox/lox and α1α2lox/lox + Albcre mice. A, mitochondrial respiration was measured in the presence of the OXPHOS substrates glutamate, malate, and pyruvate (State 2) in the presence of saturating ADP (State 3), oligomycin (Leak), and FCCP (electron transport system, ETS). B, the respiratory control ratio (RCR) is defined as the ratio of state 3 to state 2 oxygen consumption. C, mitochondrial complex expression in isolated mitochondria from the livers of α1α2lox/lox and α1α2lox/lox +Albcre mice was measured through Western blotting (normalized to VDAC1). The representative images are lanes obtained from different parts of the same immunoblot. The data are expressed as means ± S.E., n = 5–6 in each group. †, p ≤ 0.05 versus α1α2lox/lox; $, p > 0.05 versus α1α2lox/lox, absolute rates are significantly different but not when normalized to state 3 and leak respiration.
DISCUSSION
Compounds (endogenous and pharmaceutical) and conditions that increase hepatic AMPK activity have been demonstrated to mitigate hepatic processes central to the etiology of diabetes and obesity (6–8, 18–21, 32–38). Here we use genetic tools to distinguish between the AMPK-dependent and -independent effects of AICAR in vivo. Recent evidence challenges the role of AMPK as an indispensable arbiter for the anti-glycemic action of biguanides and adiponectin in hepatocytes (6–8). The studies herein define the regulatory role of energy state in terms of those metabolic effects that are mediated by hepatic AMPK and those that are not. This was accomplished by increasing the NMP concentration in the presence of a fixed glucose concentration using an AICAR-euglycemic clamp technique in the presence and absence of hepatic AMPK in vivo.
AMPK activation is broadly implicated in the transcriptional control of mediators of gluconeogenesis (17, 39–44). Cre-mediated removal of the AMPKα2 catalytic subunit from the liver results in substantial increases in fasting glucose and insulin (45). It has also been demonstrated that the overexpression of constitutively active AMPKα2 reduces fasting glucose with a concomitant attenuation of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase gene expression (17). Moreover, AICAR attenuates the gene expression of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase; this effect has been attributed to AMPK activation (40, 42, 44), but the requirement for this enzyme in acute control has recently been strongly contested in primary hepatocytes and metabolic tests in vivo (8). We demonstrate that the genetic removal of both AMPKα1 and α2 catalytic subunits from the liver has no impact on rates of endogenous glucose production or disappearance in the conscious, 5-h fasted mouse. During euglycemia, liver AMPK is neither required for the AICAR-mediated inhibition of EndoRa nor its stimulation of glucose disappearance. These data impart two important results regarding the role of AMPK in the acute glucoregulation: the genetic deletion of AMPK from the liver (a) does not affect 5-h fasting glucose kinetics and (b) is unnecessary for the AICAR-mediated suppression of EndoRa in the short-fasted mouse.
Although these studies highlight the power of AICAR to inhibit glucose production in vivo, the results do not discount the possibility that hepatic AMPK participates in the regulation of glucose flux under other conditions. The experiments were designed to study glucose flux in the short-fasted, postprandial mouse to limit the effects of fast duration on hepatic nucleotides and AMPK signaling. Applying physiological stressors that reduce energy state (i.e., acute, high intensity exercise) may uncover a role for hepatic AMPK in glucoregulation that is not evident when energy state is reduced using AICAR. These and other recent studies (6, 8, 10) emphasize the need to demarcate the in vivo effects of AMPK in physiology from pharmacology.
Recent literature on glycemic regulation by glucagon (10) and biguanides (6, 8) has reinvigorated the conversation surrounding AMP and energy charge as regulatory variables (46, 47). Glucagon-mediated flux through phosphoenolpyruvate carboxykinase appears to generate an autoregulatory feedback loop limiting the amount of ATP disposed to gluconeogenesis (10). These studies demonstrate that the unique plasticity of liver EC has an important function in response to a normal elevation in endocrine action. Indeed, glucagon provokes distinct changes in liver function that correspond to a relative decrease in available ATP (10), despite the stimulation of fat oxidation and suppression of triglyceride synthesis (48).
A therapeutic advantage of AMPK-activators like AICAR and biguanides is that they are not dependent on cataplerotic flux for an increase in the AMP/ATP ratio. Rather, metformin has been shown to inhibit mitochondrial complex I activity (49, 50), depress energy charge, and activate AMPK (8). AMP has the capacity to inhibit fructose 1,6-bisphosphatase (51) and adenylyl cyclase (6). ZMP, the AMP mimetic generated from AICAR, also inhibits the former (38) and interferes with complex I activity in isolated mitochondria (16). In well controlled conditions in which glucose is tightly regulated, the AICAR infusion induces a large drop in the total adenine nucleotide pool, EC, and an increase in the AMP/ATP ratio. The AICAR stimulated fall in ATP is even larger in the absence of hepatic AMPK, which manifests into a greater drop in EC and increase in AMP/ATP. In vivo (37, 52) and in vitro (8, 16) delivery of AICAR can disrupt the hepatic adenylate energy pool and induce a drop in ATP. AMPK clearly plays a protective role, maintaining energy state in the presence of the metabolic challenge induced by nucleotide phosphate disequilibrium.
Substrate utilization and energy production are functionally linked through oxidative phosphorylation. Physiological conditions characterized by increases in ATP demand also correspond to elevations in fat, amino acid, glycerol, and lactate utilization in the liver (53). The AICAR infusion provokes a fall in available energy. Liver AMPK knock-out impairs mitochondrial function and fat utilization, which exacerbates the effects of AICAR on hepatic energy state. Energy production from fat primarily results from the generation of reducing cofactors for oxidative phosphorylation. Additionally, AMPK promotes mitochondrial complex II and III expression, which serve as sites for the provision of reducing equivalents. During AICAR delivery, the efficient coupling of reduced cofactors with ATP production may be vital to preserving energy charge. Reductions in complex II and III expression and mitochondrial efficiency may trigger the larger increment in the AMP/ATP ratio observed in AMPK-deficient livers. It should be noted that impairments in hepatic energy homeostasis in α1α2lox/lox + Albcre mice only emerged with AICAR administration. No differences in adenine nucleotides, EC, or AMP/ATP were observed in short-fasted, euglycemic α1α2lox/lox and α1α2lox/lox + Albcre mice receiving the saline infusion. Thus, defects in substrate utilization and mitochondrial function in α1α2lox/lox + Albcre mice only affect liver energy state when linked with elevated metabolic stress.
Similar to the effects of metformin, ZMP interferes with complex I and impairs state 3 respiration (16). AICAR may promote AMPK-dependent and -independent fat transport into the mitochondria yet impede cofactor oxidation. As a result, liver energy charge may decline despite a relative abundance of substrate, NADH, and FADH2. A similar hypothesis has been proposed for the actions of metformin (54).
The deleterious effect of AICAR on EC may be compounded by the inhibition of hepatic glucose uptake. AICAR delivery into the portal vein renders the liver insensitive to net hepatic glucose uptake, even during hyperglycemia and hyperinsulinemia (52). In vitro work confirms that AICAR reduces glucokinase translocation and glycolysis in hepatocytes (55), in AMPK-dependent (56) and -independent (57) conditions. Our AICAR delivery rate was selected to permit a reasonable physiological comparison between our results and earlier AICAR-euglycemic clamps in rodents (24, 25). The resulting dose was sufficient to perturb hepatic adenylate energy balance, and we expect that the depletion of ATP could contribute to an inhibition of liver glycolysis (56, 57). AICAR also elevates plasma lactate in vivo (24, 37), which may result from increased production from muscle (58) and impaired uptake in the liver (38). Because AICAR induced an equal increment in glucose disappearance, it is reasonable to assume that muscle lactate production is unaffected by liver AMPK knock-out. The attenuated elevation in plasma lactate in α1α2lox/lox + Albcre mice may instead reflect a switch in substrate uptake and utilization in the liver during AICAR administration.
The metabolic challenge induced by AICAR inhibits both glucose flux in and out of the liver, effectively generating a condition in which the liver may become more reliant on fat oxidation. Ample molecular and physiological evidence connects AMPK activation with fat utilization in the liver. AICAR reduces hepatic TGs under various conditions in rodents (18–20) and increases fatty acid oxidation in hepatocytes (21, 30, 59). Knock-out of the AMPKα2 subunit in the liver (45) results in increased circulating fatty acids, TGs, and a decrease in β-hydroxybutyrate, whereas the short term overexpression of a constitutively active form of AMPKα2 reduces plasma TGs and increases β-hydroxybutyrate (17). Together, these results suggest that AMPK mediates fatty acid uptake and oxidation. Our data demonstrate that the acute effect of AICAR on hepatic TG utilization requires liver AMPK. pACCS79 is undetectable in α1α2lox/lox + Albcre mice and, thus, higher in AICAR-treated α1α2lox/lox mice (Fig. 2B), implicating increased carnitine palmitoyltransferase-1 activity as a potential mechanism for elevated fat oxidation in the liver. However, pACCS79 was not different between α1α2lox/lox saline and α1α2lox/lox AICAR. It is plausible that increased NMP drives fatty acid flux away from de novo lipogenesis and through carnitine palmitoyltransferase-1 (30), in an AMPK-dependent and pACCS79-independent manner.
Lastly, AMPK has been shown to regulate mediators of TG and cholesterol synthesis (14, 60–62). At the end of the AICAR infusion, higher liver TGs in α1α2lox/lox + Albcre mice could conceivably result from an impaired inhibition of TG synthesis or esterification. If this were the case, one might expect circulating TGs to follow a similar trend, yet circulating TGs were equivalent in α1α2lox/lox and α1α2lox/lox + Albcre mice. Evidence from these studies point to impairments in fat utilization as a source of elevated TGs in the livers of α1α2lox/lox + Albcre mice following AICAR administration.
Collectively, the data provide in vivo support for the energy sensor paradigm (63). AMPK acts as a sensor and safeguard of the hepatic adenylate energy pool during an acute challenge to the energy status of the liver. The absolute amounts of ATP, ADP, and AMP in the liver are dictated by changes in synthesis and breakdown, with adenylate kinase coordinating their relative balance. Genetic deletion of AMPK exacerbates the AICAR-mediated disturbance of the adenylate pool and a larger reduction in available ATP. The evidence suggests that AMPK works to promote fat utilization and mitochondrial function during a pharmacological increase in ATP consumption, as liver AMPK knock-out leaves liver TGs elevated. These data prove that AMPK is not required for the acute inhibition of endogenous glucose production during elevations in AMP, the AMP/ATP ratio, or ZMP in vivo. This and other recent research (6–8) have important implications for therapeutics designed to target the pathogenesis of diabetes and metabolic diseases.
Acknowledgments
We thank Susan Hajizadeh in the hormone assay and analytical services core at Vanderbilt for measuring plasma insulin. We also thank Jeffrey S. Bonner for careful review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants U24 DK59637 and R37 DK50277 (to D. H. W.) and DK20593 (the Diabetes Research and Training Center). This work was also supported by the Molecular Endocrinology Training Program at Vanderbilt.
C. M. Hasenour, D. E. Ridley, C. C. Hughey, F. D. James, E. P. Donahue, J. Shearer, B. Viollet, M. Foretz, and D. H. Wasserman, unpublished observation.
- AMPK
- AMP-activated protein kinase
- AICAR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- NMP
- nucleotide monophosphate
- FCCP
- carbonylcyanide-p-trifluoromethoxyphenylhydrazone
- EC
- energy charge
- GIR
- glucose infusion rate
- EndoRa
- endogenous glucose production
- Rd
- glucose disappearance
- TG
- triglyceride
- ACC
- acetyl-coA carboxylase.
REFERENCES
- 1. Wasserman D. H. (2009) Four grams of glucose. Am. J. Physiol. Endocrinol. Metab. 296, E11–E21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu F. B. (2011) Globalization of diabetes. The role of diet, lifestyle, and genes. Diabetes Care 34, 1249–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kahn B. B., Alquier T., Carling D., Hardie D. G. (2005) AMP-activated protein kinase. Ancient energy gauge provides clues into modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 4. Oakhill J. S., Scott J. W., Kemp B. E. (2012) AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol. Metab. 23, 125–132 [DOI] [PubMed] [Google Scholar]
- 5. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller R. A., Chu Q., Xie J., Foretz M., Viollet B., Birnbaum M. J. (2013) Biguanides suppress hepatic glucagon signaling by decreasing production of cyclic AMP. Nature 494, 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller R. A., Chu Q., Le Lay J., Scherer P. E., Ahima R. S., Kaestner K. H., Foretz M., Viollet B., Birnbaum M. J. (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J. Clin. Invest. 121, 2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berglund E. D., Lustig D. G., Baheza R. A., Hasenour C. M., Lee-Young R. S., Donahue E. P., Lynes S. E., Swift L. L., Charron M. J., Damon B. M., Wasserman D. H. (2011) Hepatic glucagon action is essential for exercise-induced reversal of mouse fatty liver. Diabetes 60, 2720–2729 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Berglund E. D., Lee-Young R. S., Lustig D. G., Lynes S. E., Donahue E. P., Camacho R. C., Meredith M. E., Magnuson M. A., Charron M. J., Wasserman D. H. (2009) Hepatic energy state is regulated by glucagon receptor signaling in mice. J. Clin. Invest. 119, 2412–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Camacho R. C., Donahue E. P., James F. D., Berglund E. D., Wasserman D. H. (2006) Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab. 290, E405–E408 [DOI] [PubMed] [Google Scholar]
- 12. Yamauchi T., Nio Y., Maki T., Kobayashi M., Takazawa T., Iwabu M., Okada-Iwabu M., Kawamoto S., Kubota N., Kubota T., Ito Y., Kamon J., Tsuchida A., Kumagai K., Kozono H., Hada Y., Ogata H., Tokuyama K., Tsunoda M., Ide T., Murakami K., Awazawa M., Takamoto I., Froguel P., Hara K., Tobe K., Nagai R., Ueki K., Kadowaki T. (2007) Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13, 332–339 [DOI] [PubMed] [Google Scholar]
- 13. Kimball S. R., Siegfried B. A., Jefferson L. S. (2004) Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J. Biol. Chem. 279, 54103–54109 [DOI] [PubMed] [Google Scholar]
- 14. Foretz M., Carling D., Guichard C., Ferré P., Foufelle F. (1998) AMP-activated protein kinase inhibits the glucose-activated expression of fatty acid synthase gene in rat hepatocytes. J. Biol. Chem. 273, 14767–14771 [DOI] [PubMed] [Google Scholar]
- 15. Hasenour C. M., Berglund E. D., Wasserman D. H. (2013) Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Mol. Cell. Endocrinol. 366, 152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guigas B., Taleux N., Foretz M., Detaille D., Andreelli F., Viollet B., Hue L. (2007) AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 404, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foretz M., Ancellin N., Andreelli F., Saintillan Y., Grondin P., Kahn A., Thorens B., Vaulont S., Viollet B. (2005) Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54, 1331–1339 [DOI] [PubMed] [Google Scholar]
- 18. Cool B., Zinker B., Chiou W., Kifle L., Cao N., Perham M., Dickinson R., Adler A., Gagne G., Iyengar R., Zhao G., Marsh K., Kym P., Jung P., Camp H. S., Frevert E. (2006) Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3, 403–416 [DOI] [PubMed] [Google Scholar]
- 19. Iglesias M. A., Ye J. M., Frangioudakis G., Saha A. K., Tomas E., Ruderman N. B., Cooney G. J., Kraegen E. W. (2002) AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat fed rats. Diabetes 51, 2886–2894 [DOI] [PubMed] [Google Scholar]
- 20. Song X. M., Fiedler M., Galuska D., Ryder J. W., Fernström M., Chibalin A. V., Wallberg-Henriksson H., Zierath J. R. (2002) 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia 45, 56–65 [DOI] [PubMed] [Google Scholar]
- 21. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ayala J. E., Bracy D. P., McGuinness O. P., Wasserman D. H. (2006) Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55, 390–397 [DOI] [PubMed] [Google Scholar]
- 23. Kang L., Ayala J. E., Lee-Young R. S., Zhang Z., James F. D., Neufer P. D., Pozzi A., Zutter M. M., Wasserman D. H. (2011) Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin α2β1 in mice. Diabetes 60, 416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bergeron R., Previs S. F., Cline G. W., Perret P., Russell R. R., 3rd, Young L. H., Shulman G. I. (2001) Effect of 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50, 1076–1082 [DOI] [PubMed] [Google Scholar]
- 25. Bergeron R., Russell R. R., 3rd, Young L. H., Ren J. M., Marcucci M., Lee A., Shulman G. I. (1999) Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am. J. Physiol. 276, E938–E944 [DOI] [PubMed] [Google Scholar]
- 26. Morgan C. R., Lazarow A. (1965) Immunoassay of pancreatic and plasma insulin following alloxan injection of rats. Diabetes 14, 669–671 [DOI] [PubMed] [Google Scholar]
- 27. Steele R., Wall J. S., De Bodo R. C., Altszuler N. (1956) Measurement of the size and turnover rate of body glucose pool by the isotope dilution method. Am. J. Physiol. 187, 15–24 [DOI] [PubMed] [Google Scholar]
- 28. Romestaing C., Piquet M. A., Letexier D., Rey B., Mourier A., Servais S., Belouze M., Rouleau V., Dautresme M., Ollivier I., Favier R., Rigoulet M., Duchamp C., Sibille B. (2008) Mitochondrial adaptations to steatohepatitis induced by a methionine- and choline-deficient diet. Am. J. Physiol. Endocrinol. Metab. 294, E110–E119 [DOI] [PubMed] [Google Scholar]
- 29. Hughey C. C., Hittel D. S., Johnsen V. L., Shearer J. (2011) Respirometric oxidative phosphorylation assessment in saponin-permeabilized cardiac fibers. J. Vis. Exp. 48, 2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Velasco G., Geelen M. J., Guzmán M. (1997) Control of hepatic fatty acid oxidation by 5′-AMP-activated protein kinase involves a malonyl-CoA-dependent and a malonyl-CoA-independent mechanism. Arch. Biochem. Biophys. 337, 169–175 [DOI] [PubMed] [Google Scholar]
- 31. Corton J. M., Gillespie J. G., Hawley S. A., Hardie D. G. (1995) 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 32. Holland W. L., Miller R. A., Wang Z. V., Sun K., Barth B. M., Bui H. H., Davis K. E., Bikman B. T., Halberg N., Rutkowski J. M., Wade M. R., Tenorio V. M., Kuo M. S., Brozinick J. T., Zhang B. B., Birnbaum M. J., Summers S. A., Scherer P. E. (2011) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 17, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim Y. D., Park K. G., Lee Y. S., Park Y. Y., Kim D. K., Nedumaran B., Jang W. G., Cho W. J., Ha J., Lee I. K., Lee C. H., Choi H. S. (2008) Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 57, 306–314 [DOI] [PubMed] [Google Scholar]
- 34. Iglesias M. A., Furler S. M., Cooney G. J., Kraegen E. W., Ye J. M. (2004) AMP-activated protein kinase activation by AICAR increases both muscle fatty acid and glucose uptake in white muscle of insulin-resistant rats in vivo. Diabetes 53, 1649–1654 [DOI] [PubMed] [Google Scholar]
- 35. Buhl E. S., Jessen N., Pold R., Ledet T., Flyvbjerg A., Pedersen S. B., Pedersen O., Schmitz O., Lund S. (2002) Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes 51, 2199–2206 [DOI] [PubMed] [Google Scholar]
- 36. Fryer L. G., Parbu-Patel A., Carling D. (2002) The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277, 25226–25232 [DOI] [PubMed] [Google Scholar]
- 37. Vincent M. F., Erion M. D., Gruber H. E., Van den Berghe G. (1996) Hypoglycaemic effect of AICAriboside in mice. Diabetologia 39, 1148–1155 [DOI] [PubMed] [Google Scholar]
- 38. Vincent M. F., Marangos P. J., Gruber H. E., Van den Berghe G. (1991) Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 40, 1259–1266 [DOI] [PubMed] [Google Scholar]
- 39. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., Shaw R. J. (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shirai T., Inoue E., Ishimi Y., Yamauchi J. (2011) AICAR response element binding protein (AREBP), a key modulator of hepatic glucose production regulated by AMPK in vivo. Biochem. Biophys. Res. Commun. 414, 287–291 [DOI] [PubMed] [Google Scholar]
- 41. Chanda D., Kim S. J., Lee I. K., Shong M., Choi H. S. (2008) Sodium arsenite induces orphan nuclear receptor SHP gene expression via AMP-activated protein kinase to inhibit gluconeogenic enzyme gene expression. Am. J. Physiol. Endocrinol. Metab. 295, E368–E379 [DOI] [PubMed] [Google Scholar]
- 42. Koo S. H., Flechner L., Qi L., Zhang X., Screaton R. A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., Takemori H., Montminy M. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 43. Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S., Yamashita S., Noda M., Kita S., Ueki K., Eto K., Akanuma Y., Froguel P., Foufelle F., Ferre P., Carling D., Kimura S., Nagai R., Kahn B. B., Kadowaki T. (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 44. Lochhead P. A., Salt I. P., Walker K. S., Hardie D. G., Sutherland C. (2000) 5-Aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49, 896–903 [DOI] [PubMed] [Google Scholar]
- 45. Andreelli F., Foretz M., Knauf C., Cani P. D., Perrin C., Iglesias M. A., Pillot B., Bado A., Tronche F., Mithieux G., Vaulont S., Burcelin R., Viollet B. (2006) Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147, 2432–2441 [DOI] [PubMed] [Google Scholar]
- 46. Krebs H. (1964) The Croonian Lecture, 1963. Gluconeogenesis. Proc. R. Soc. Lond. B. Biol. Sci. 159, 545–564 [DOI] [PubMed] [Google Scholar]
- 47. Atkinson D. E. (1968) The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7, 4030–4034 [DOI] [PubMed] [Google Scholar]
- 48. Longuet C., Sinclair E. M., Maida A., Baggio L. L., Maziarz M., Charron M. J., Drucker D. J. (2008) The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 8, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brunmair B., Staniek K., Gras F., Scharf N., Althaym A., Clara R., Roden M., Gnaiger E., Nohl H., Waldhäusl W., Fürnsinn C. (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I. A common mechanism contributing to their antidiabetic actions? Diabetes 53, 1052–1059 [DOI] [PubMed] [Google Scholar]
- 50. El-Mir M. Y., Nogueira V., Fontaine E., Avéret N., Rigoulet M., Leverve X. (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228 [DOI] [PubMed] [Google Scholar]
- 51. Taketa K., Pogell B. M. (1965) Allosteric inhibition of rat liver fructose 1,6-diphosphatase by adenosine 5′-monophosphate. J. Biol. Chem. 240, 651–662 [PubMed] [Google Scholar]
- 52. Pencek R. R., Shearer J., Camacho R. C., James F. D., Lacy D. B., Fueger P. T., Donahue E. P., Snead W., Wasserman D. H. (2005) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside causes acute hepatic insulin resistance in vivo. Diabetes 54, 355–360 [DOI] [PubMed] [Google Scholar]
- 53. Wasserman D. H., Cherrington A. D. (1991) Hepatic fuel metabolism during muscular work. Role and regulation. Am. J. Physiol. 260, E811–E824 [DOI] [PubMed] [Google Scholar]
- 54. Miller R. A., Birnbaum M. J. (2010) An energetic tale of AMPK-independent effects of metformin. J. Clin. Invest. 120, 2267–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vincent M. F., Bontemps F., Van den Berghe G. (1992) Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem. J. 281, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mukhtar M. H., Payne V. A., Arden C., Harbottle A., Khan S., Lange A. J., Agius L. (2008) Inhibition of glucokinase translocation by AMP-activated protein kinase is associated with phosphorylation of both GKRP and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R766–R774 [DOI] [PubMed] [Google Scholar]
- 57. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 58. Hunter R. W., Treebak J. T., Wojtaszewski J. F., Sakamoto K. (2011) Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60, 766–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. García-Villafranca J., Guillén A., Castro J. (2008) Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90, 460–466 [DOI] [PubMed] [Google Scholar]
- 60. Muoio D. M., Seefeld K., Witters L. A., Coleman R. A. (1999) AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle. Evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 338, 783–791 [PMC free article] [PubMed] [Google Scholar]
- 61. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., Cohen R. A., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Carling D., Zammit V. A., Hardie D. G. (1987) A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217–222 [DOI] [PubMed] [Google Scholar]
- 63. Hardie D. G., Hawley S. A. (2001) AMP-activated protein kinase. The energy charge hypothesis revisited. Bioessays 23, 1112–1119 [DOI] [PubMed] [Google Scholar]