

Background: The alginate epimerase AlgG converts mannuronate to its C5 epimer guluronate at the polymer level.
Results: The structure of Pseudomonas syringae AlgG has been determined, and the protein has been functionally characterized.
Conclusion: His319 acts as the catalytic base, whereas Arg345 neutralizes the negative charge of the carboxylate group during catalysis.
Significance: This is the first structural characterization of a periplasmic alginate epimerase.
Keywords: Biofilm, Crystal Structure, Enzyme Catalysis, Polysaccharide, Pseudomonas aeruginosa, Alginate, Epimerase
Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that forms chronic biofilm infections in the lungs of cystic fibrosis patients. A major component of the biofilm during these infections is the exopolysaccharide alginate, which is synthesized at the inner membrane as a homopolymer of 1–4-linked β-d-mannuronate. As the polymer passages through the periplasm, 22–44% of the mannuronate residues are converted to α-l-guluronate by the C5-epimerase AlgG to produce a polymer of alternating β-d-mannuronate and α-l-guluronate blocks and stretches of polymannuronate. To understand the molecular basis of alginate epimerization, the structure of Pseudomonas syringae AlgG has been determined at 2.1-Å resolution, and the protein was functionally characterized. The structure reveals that AlgG is a long right-handed parallel β-helix with an elaborate lid structure. Functional analysis of AlgG mutants suggests that His319 acts as the catalytic base and that Arg345 neutralizes the acidic group during the epimerase reaction. Water is the likely catalytic acid. Electrostatic surface potential and residue conservation analyses in conjunction with activity and substrate docking studies suggest that a conserved electropositive groove facilitates polymannuronate binding and contains at least nine substrate binding subsites. These subsites likely align the polymer in the correct register for catalysis to occur. The presence of multiple subsites, the electropositive groove, and the non-random distribution of guluronate in the alginate polymer suggest that AlgG is a processive enzyme. Moreover, comparison of AlgG and the extracellular alginate epimerase AlgE4 of Azotobacter vinelandii provides a structural rationale for the differences in their Ca2+ dependence.
Introduction
Alginate is an unbranched anionic polysaccharide that is produced by brown algae (Phaeophyceae), Pseudomonas spp., and Azotobacter genera (1–3). Alginate is initially formed as a 1–4-linked poly-β-d-mannuronate polymer at the inner membrane and is subsequently selectively modified as it passages through the periplasm. These modifications alter the properties of the polymer and provide significant benefits to the organism. For example, in alginate-producing bacteria, mannuronate (M)2 residues can be selectively acetylated at the C2 and/or C3 positions (4), a modification that helps Pseudomonas aeruginosa evade host defense mechanisms (5). In both brown algae and alginate-producing bacteria, unacetylated mannuronate can be epimerized to its C5 epimer, α-l-guluronate (G) (6, 7). Brown algae and Azotobacter vinelandii express more than one epimerase and are capable of producing alginate rich in guluronate blocks, which in the presence of Ca2+ form gels that are important for structural integrity and cyst formation in brown algae and A. vinelandii, respectively (4, 6–10). In contrast, Pseudomonas spp. contain a single periplasmic epimerase. The alginate produced by these bacteria do not contain guluronate blocks but rather polymannuronic acid (poly(M)) blocks and blocks of alternating MG sequence (MG blocks) (4, 8, 11). The importance of epimerization in Pseudomonas spp. alginate is not clear, but guluronate incorporation, like acetylation, makes alginate more viscous, which could contribute to the ability of P. aeruginosa to evade host immune defenses (12).
Polymer level epimerization of sugar molecules is a rare modification that has only been found to date in three polysaccharides: alginate and the glycosaminoglycans heparin/heparan sulfate and dermatan sulfate (6). Heparin/heparan sulfate and dermatan sulfate are components of the extracellular matrix of animal tissue (13). Because of their negative charge, these polymers interact with a number of proteins to fulfill their roles in cell signaling, coagulation, and wound healing (14, 15). Both glycosaminoglycans contain the uronic acid β-d-glucuronate, which is epimerized at its C5 position to α-l-iduronate (6). Epimerization of heparan sulfate is essential for prenatal development as mice lacking the C5-epimerase die shortly after birth due to lung failure (16). Although alginate and heparin/heparan sulfate/dermatan sulfate are made by different organisms, they share some striking similarities. (i) All three polysaccharides are linear, polyanionic polymers that contain uronic acids that undergo C5 epimerization at the polymer level, and (ii) each is believed to be synthesized by a large multiprotein complex, the alginate biosynthetic complex and the GAGosome in the case of glycosaminoglycan biosynthesis (17–19).
The proposed polysaccharide epimerase reaction mechanism is based on the β-elimination reaction of polysaccharide lyases (Fig. 1) (20). The general lyase β-elimination mechanism involves neutralization of the carboxylate group of the uronic acid by a positive charge, abstraction of the proton at the C5 position, and cleavage of the glycosidic bond with the formation of a double bond between C4 and C5. A proton is added to the leaving group, resulting in a new reducing end. In the epimerase reaction, a proton is added to the opposite face of the C5 carbon, forming the C5 epimer. Jerga et al. (21) found that a glycal intermediate is formed during the epimerization reaction. All alginate epimerases are predicted to adopt a β-helix fold (22), a prediction that is supported by the crystal structure of the catalytic domain of the extracellular epimerase AlgE4 of A. vinelandii (23). Despite the low overall sequence identity between bacterial periplasmic alginate epimerases (AlgG) and brown algae epimerases (∼15% identity), they all contain a putative active site DPHD sequence motif. A. vinelandii in addition to its periplasmic alginate epimerase also expresses seven extracellular alginate epimerases (AlgE1–7). These enzymes contain a slightly modified DPHE sequence motif as part of their active site and have been shown to be Ca2+-dependent (24, 25). The extracellular alginate epimerases of A. vinelandii share ∼70% sequence identity but have less than 10% sequence identity with the bacterial periplasmic alginate epimerases. The periplasmic alginate epimerase in alginate-producing Pseudomonas spp. and A. vinelandii is AlgG (26–29). These epimerases share ∼60% sequence identity and are Ca2+-independent with a pH optimum for activity of between pH 6 and 7.5 (27, 30, 31).
FIGURE 1.
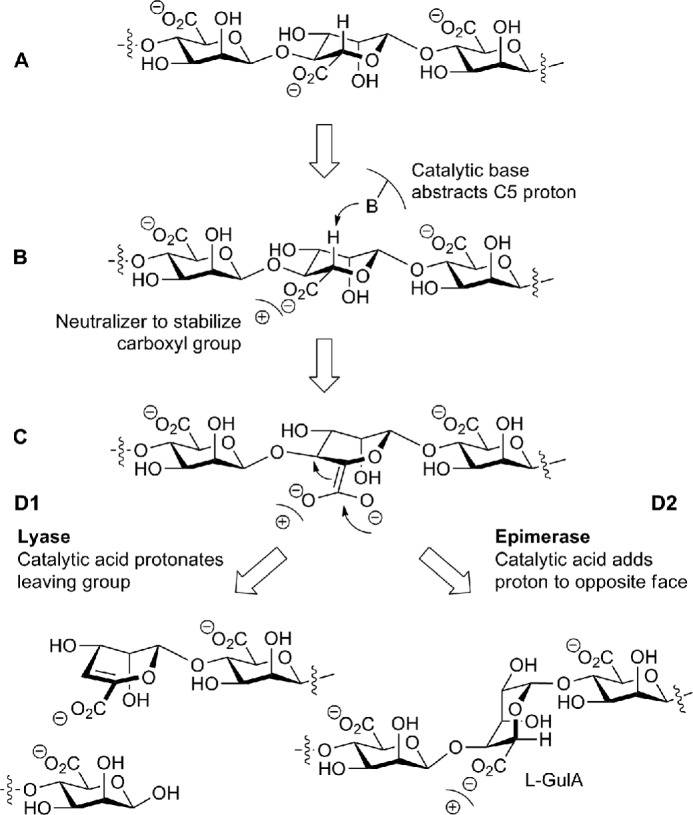
Lyase β-elimination and epimerase reaction mechanisms. A, a mannuronate residue within the mannuronate polymer is depicted. B, a positive charge stabilizes the carboxylate group while the catalytic base abstracts the C5 proton. C, an enolate group is formed. D1, a double bond between C4 and C5 is formed, the chain is cleaved, and the catalytic acid protonates the leaving group, forming a new reducing end. D2, a proton is added to the opposite face of the C5 by the catalytic acid, and the C5 epimer α-l-guluronate (l-GulA) is formed.
In this study, we present the first crystal structure of a periplasmic alginate epimerase. Our 2.1-Å structure of Pseudomonas syringae AlgG reveals that the protein, as expected, adopts a parallel β-helix fold with 11 complete coils and one incomplete coil. The β-helix is capped N-terminally by an unusually elaborate lid structure comprising a central helix flanked by two antiparallel β-sheets. Site-directed mutagenesis and functional analysis suggest that the catalytic mechanism utilizes His319 as the catalytic base, Arg345 neutralizes the charge on the uronic acid, and water acts as the catalytic acid. His319 and Arg345 are located in a conserved and electropositive groove that our analysis suggests contains at least nine saccharide binding subsites. This groove would not only facilitate polymannuronate binding but also suggests that AlgG is a processive enzyme. Furthermore, our comparison of AlgG and the extracellular alginate epimerase AlgE4 provides a rationale for the differences in Ca2+ dependence observed between the two classes of enzymes.
EXPERIMENTAL PROCEDURES
Cloning
The nucleotide sequence of algG from P. syringae pv. tomato strain DC3000 was obtained from the Pseudomonas Genome Database (32, 33) and used to design gene-specific primers. Residues 49–536 of AlgG were amplified from genomic DNA using the following forward and reverse primers: 5′-GCATCATATGGTCAAGGAGTTGCACCAGG-3′ and 5′-GCATAAGCTTTCATTCCCGCAGCTCGGTC-3′. (Italic font indicates that those nucleotides encode the protein.) The amplified PCR product was digested with NdeI and HindIII restriction endonucleases and cloned into a pET28a vector (Novagen) that had been linearized with the same enzymes. The resulting expression vector, pFW-AlgG49–536, encodes residues 49–536 of P. syringae AlgG fused to a cleavable N-terminal His6 tag (His6-AlgG49–536) for purification purposes.
The gene for Klebsiella pneumoniae Aly l-guluronate-specific lyase (alyA) was cloned in a similar fashion as described above using the following forward and reverse primers: 5′-GCATCATATGGCTGTGCGGCGCCGGGG-3′ and 5′-GCATAAGCTTACTATCGCTGCGCTCCGTGTGTCGC-3′ based on the sequence obtained from GenBankTM (accession number L19657.1). The alyA construct was amplified from the pRC5 plasmid (34). The resulting expression vector, pFW-G-Lyase21–307, encodes residues 21–307 fused to a cleavable N-terminal His6 tag (His6-G-Lyase21–307) for purification purposes.
Mutants of AlgG were constructed using the QuikChange® Lightning site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The fidelity of all constructs was verified by DNA sequencing (ACGT DNA Technologies Corp.).
Protein Expression and Purification
Escherichia coli BL21(DE) competent cells (Stratagene) transformed with pFW-AlgG49–536 or mutant AlgG were grown in 500 ml of terrific broth supplemented with 0.05 mg/ml kanamycin at 310 K. After the A600 of the cell culture reached 0.6, protein expression was induced with isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1.0 mm. The induced cells were incubated for 4 h at 310 K and harvested via centrifugation at 6,000 × g for 10 min at 277 K. The resulting cell pellet was stored at 193 K until required. His6-G-Lyase21–307 was expressed as described above for His6-AlgG49–536. For selenomethionine (SeMet) incorporation, the pFW-AlgG49–536 expression vector was transformed into E. coli B834 Met− competent cells (Novagen) and grown in 500 ml of selenomethionine-containing medium with 0.05 mg/ml kanamycin at 310 K using the protocol described by Lee et al. (35).
His6-AlgG49–536 was purified from the cell pellet of two 500-ml bacterial cultures after resuspension of the cell pellets in 40 ml of buffer A (50 mm MES, pH 6.4, 500 mm NaCl, 5% (v/v) glycerol, 2 mm MgSO4,10 mm imidazole) containing one tablet of SIGMAFAST protease inhibitor EDTA-free mixture (Sigma). The resuspended cells were lysed by sonication (Misonix Sonicator 3000), and cell debris was removed by centrifugation at 38,000 × g for 30 min at 277 K. The supernatant containing His6-AlgG49–536 was then loaded onto a 5-ml Ni2+-NTA Superflow cartridge (Qiagen) pre-equilibrated with buffer A. After washing the Ni2+-NTA column with at least 5 column volumes of buffer A, His6-AlgG49–536 was eluted with 5 column volumes of buffer B (50 mm MES, pH 6.4, 500 mm NaCl, 5% (v/v) glycerol, 2 mm MgSO4, 500 mm imidazole). The elution peak was pooled, concentrated, and run on a HiLoad 16/60 Superdex 200 prep grade gel filtration column (GE Healthcare) equilibrated with buffer C (25 mm MES, pH 6.4, 300 mm NaCl, 5% (v/v) glycerol, 2 mm MgSO4), and fractions corresponding to His6-AlgG49–536 were pooled. The purity of the protein was estimated using SDS-PAGE to be >95%. Mutant AlgG proteins were purified as described above for the wild-type protein. His6-G-Lyase21–307 was purified as described above using the following buffers: lysis/wash buffer (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 10 mm imidazole), Ni2+-NTA elution buffer (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 200 mm imidazole), and gel filtration buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl).
For structural studies, AlgG eluted from the Ni2+-NTA column was pooled and dialyzed twice in 1 liter of buffer D (50 mm MES, pH 6.4, 500 mm NaCl, 5% (v/v) glycerol, 2 mm MgSO4) at 277K for 1 h. Dialyzed His6-AlgG49–536 was treated with thrombin (Calbiochem; 1 unit of thrombin/3 mg of His6-AlgG49–536) for 12 h at 277 K to remove the histidine tag. Thrombin-treated His6-AlgG49–536 was loaded onto an open column filled with 3 ml of Ni2+-NTA-agarose (Qiagen) equilibrated with buffer A to separate untagged from tagged AlgG. The flow-through and the wash fraction (20 ml of buffer A) were pooled and concentrated by centrifugation (1240 × g at 277K) using a concentrator with a 30-kDa-molecular mass cutoff (Millipore) and run on a HiLoad 16/60 Superdex 200 prep grade gel filtration column (GE Healthcare) equilibrated with buffer C. Fractions corresponding to AlgG49–536 were pooled, methylated as described in Truebestein et al. (36), concentrated, and rerun on a HiLoad 16/60 Superdex 200 prep grade gel filtration column (GE Healthcare) equilibrated with buffer C. Fractions corresponding to methylated AlgG49–536 were pooled and concentrated to 4.4 mg/ml, and the protein was stored in buffer C at 277 K until required. SeMet-incorporated AlgG and the AlgG D317A mutant were purified as described above for the wild-type protein.
Crystallization, Data Collection, Structure Determination, and Analysis
Methylated AlgG49–536 concentrated to 4.4 mg/ml was subjected to crystallization trials using commercially available sparse matrix screens (Emerald BioSystems, Hampton Research, and Qiagen). Methylated AlgG49–536 was crystallized using the hanging drop vapor diffusion method at 293 K with 2 μl of protein and crystallization solution in a 1:1 ratio over a reservoir containing 0.2 ml of crystallization solution. Initial crystals appeared after 7 days in condition 78 of JCSG+ suite (Qiagen; 20% (w/v) PEG 3350, 0.24 m sodium malonate, pH 7.0) and diffracted to 9 Å. These crystals were harvested and used to make a 20× seed stock as described by Douglas Instruments Ltd. and stored at 253 K. Methylated AlgG49–536 was rescreened using the microseeding technique (1 μl of protein solution, 1 μl of crystallization solution, and 0.5 μl of 1× seed stock over 0.2 ml of crystallization solution). Crystals appeared after 2–3 months in 1.26 m (NH4)2SO4, 0.1 m HEPES, pH 7.5 (condition 15 of Wizard2, Emerald BioSystems) and diffracted to 2.1 Å. The crystals grew to maximum dimensions of 300 × 150 × 150 μm. Methylated selenomethionyl-incorporated AlgG and mutant AlgG were crystallized in the same way. Showers of methylated selenomethionyl-incorporated AlgG crystals appeared after 5 days and grew to maximum dimensions of 75 × 40 × 40 μm. Mutant D317A AlgG crystals appeared after 2–3 months and grew to maximum dimensions of 300 × 100 × 100 μm.
In preparation for data collection, the crystals were cryoprotected by soaking them for 10–30 s in crystallization solution supplemented with 20% (v/v) glycerol prior to vitrification by flash freezing and subsequently stored in liquid nitrogen. All data were collected at beam line X29 at the National Synchrotron Light Source (Brookhaven National Laboratory). For the native wild-type protein, 360 images of 1° Δφ oscillation on an Area Detector Systems Corp. Q315 charge-coupled device detector with a 250-mm crystal-to-detector distance with an exposure time of 0.4 s per image were collected. The data were processed, integrated, and scaled using the HKL-2000 program suite (37) (Table 1). SeMet single wavelength anomalous dispersion data consisting of 720 images of 0.5° Δφ oscillation on an Area Detector Systems Corp. Q315 charge-coupled device detector with a 350-mm crystal-to-detector distance with an exposure time of 0.4 s per image were also collected and processed, integrated, and scaled using the HKL-2000 program suite (37) (Table 1). The SeMet single wavelength anomalous dispersion data in conjunction with HKL2MAP (38) were used to locate 13 of 15 selenium sites. Density-modified phases were calculated using SOLVE/RESOLVE (39). The electron density map was interpretable, and the model (residues 69–492) was manually built using Coot (40), briefly refined using PHENIX.REFINE (41), and subsequently used as a search model using the PHENIX AutoMR wizard to determine the structure of native AlgG49–536 by molecular replacement. The AlgG D317A49–536 structure was determined using the PHENIX AutoMR wizard using the native structure as a search model and refined using PHENIX.REFINE.
TABLE 1.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell.
| AlgG SeMet | AlgG wild type | AlgG D317A | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.97920 | 1.075 | 1.075 |
| Temperature (K) | 100 | 100 | 100 |
| Space group | P3221 | P3221 | P3221 |
| Unit cell parameters (Å; °) | a = b = 126.4 Å, c = 97.3; α = β = 90 γ = 120 | a = b = 126.6, c = 97.7; α = β = 90 γ = 120 | a = b = 125.6, c = 98.5; α = β = 90 γ = 120 |
| Resolution (Å) | 50.0-2.5 (2.59-2.5) | 50.0-2.1 (2.18-2.1) | 50.0-2.3 (2.38-2.3) |
| Total no. of reflections | 699,311 | 1,242,960 | 958,215 |
| No. of unique reflections | 31,584 | 53,161 | 40,540 |
| Redundancy | 22.1 (22.3) | 23.4 (21.5) | 23.7 (19.0) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (99.7) |
| Average I/σ(I) | 28.8 (5.3) | 43.3 (6.8) | 31.9 (4.0) |
| Rmerge (%)a | 12.4 (64.0) | 8.8 (58.7) | 10.9 (72.4) |
| Refinement | |||
| Rwork/Rfree (%)b | 18.8/21.5 | 20.9/23.6 | |
| No. atoms | |||
| Protein | 3,354 | 3,344 | |
| Solvent | 171 | 29 | |
| Average B-factors (Å2) | |||
| Protein | 41.3 | 43.0 | |
| Water | 44.9 | 38.3 | |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.007 | 0.008 | |
| Bond angles (°) | 1.14 | 1.15 | |
| Ramachandran plotc | |||
| Total favored (%) | 95.8 | 96.0 | |
| Total allowed (%) | 100 | 100 | |
| Estimated coordinate error (Å)d | 0.20 | 0.31 | |
| PDB code | 4NK6 | 4NK8 | |
a Rmerge = ΣhklΣi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl) where Ii(hkl) and I(hkl) represent the diffraction intensity values of the individual measurements and the corresponding mean values, respectively.
b Rwork = Σ‖Fobs| − k|Fcalc‖/|Fobs| where Fobs and Fcalc are the observed and calculated structure factors, respectively. Rfree is the sum extended over a subset of reflections (5%) excluded from all stages of the refinement.
c As calculated using MolProbity (54).
d Maximum likelihood-based coordinate error as determined by PHENIX (41).
Structural alignments were performed in PyMOL (Schrödinger, LLC) or using the Dali pairwise comparison server (42). Structure figures were generated using PyMOL (Schrödinger, LLC). Quantitative electrostatics were calculated using the PDB2PQR (43) and APBS (44) software. Surface residue conservation was calculated by the ConSurf program (45) using a T-Coffee (46) alignment of AlgG sequences from Pseudomonas spp. and A. vinelandii (accession numbers NP_252235 (P. aeruginosa PAO1), NP_791068 (P. syringae pv. tomato strain DC3000), YP_258150 (Pseudomonas protegens Pf-5), YP_273381 (P. syringae pv. phaseolicola 1448A), YP_610004 (Pseudomonas entomophila L48), YP_002870646 (Pseudomonas fluorescens SBW25), ZP_18874249 (Pseudomonas chlororaphis subsp. aureofaciens 30-84), ZP_16384839 (Pseudomonas avellanae BPIC 631), ZP_10143451 (Pseudomonas synxantha BG33R), ADE09310 (Pseudomonas alkylphenolia), and CAA61231 (A. vinelandii)).
A β-d-mannuronate trisaccharide was modeled into the catalytic site of the wild-type AlgG structure using Coot (40). Subsequently, a dummy set of Fobs of the model of AlgG with the β-d-mannuronate trisaccharide was created at a resolution of 3 Å using PHENIX.FMODEL (41). This model was then energy-minimized using PHENIX.REFINE (41). The β-d-mannuronate trisaccharide was taken from the structure of extracellular alginate epimerase complexed with a mannuronate trisaccharide (Protein Data Bank code 2PYH) (23).
Epimerase Activity Assay
Deacetylated poly(M) was prepared from P. aeruginosa FRD462 as described in Chitnis and Ohman (26), lyophilized, and dissolved in water to a concentration of 20 mg/ml. A fixed time coupled assay for AlgG, modified from Jerga et al. (31), was used to determine the activity of wild-type AlgG and its mutant variants. In the first step, 4 mg of poly(M) and 500 μg of AlgG were mixed in 300 μl of buffer C and incubated at 310 K for 40 h. AlgG and its mutant variants were subsequently inactivated by heating the mixture to 373 K for 10 min. The mixture was then centrifuged at 10,000 × g for 2 min to separate the alginate from the denatured epimerase. In the second step, the volume of the alginate solution was adjusted to 1 ml with 100 mm Tris-HCl, pH 7.5, and 2.5 μg of K. pneumoniae Aly l-guluronate-specific lyase (His6-G-Lyase21–307) was added. The lyase reaction was terminated after 10 min at room temperature by heating the reaction mixture to 373 K for 10 min followed by centrifugation at 10,000 × g for 2 min. Unsaturated uronic acids, the product of the lyase reaction, were measured at 230 nm in a 1-cm-path length quartz cuvette in a Ultrospec 2100 UV/visible spectrometer (GE Healthcare) (47). The absorbance value of the K. pneumoniae Aly G-specific lyase on untreated poly(M) (control) was subtracted from the value from AlgG-treated poly(M). Relative activities for the AlgG mutants were calculated setting the value of the wild-type enzyme to 100%. All activity assays were performed in triplicate.
The folding of AlgG mutants that showed <5% of wild-type activity was assessed by circular dichroism spectroscopy. Circular dichroism spectra of purified AlgG (0.3 mg/ml) were recorded in 1 mm MES, pH 6.4, 10 mm NaCl at 293 K on a Jasco J-810 spectropolarimeter. The scans were done in triplicate.
Electrospray Ionization Mass Spectrometry (ESI-MS) Alginate Binding Assay
AlgG was dialyzed against aqueous 100 mm ammonium acetate, pH 7 using microconcentrators (Millipore Corp., Bedford, MA) with a molecular mass cutoff of 30 kDa and immediately used in the ESI-MS binding assay. A single chain Fv fragment (molecular mass, 26,539 Da) of the monoclonal antibody (mAb) Se155-4 was used as a reference protein to correct for nonspecific carbohydrate-protein binding during the ESI process.
Stock solutions of each of the individual alginate polymer ligands, tetramer through dodecamer (48), were prepared by dissolving the solid compounds in ultrafiltered water (Milli-Q, Millipore) at a concentration of 1 mm. Ligand solutions were stored at 253 K until needed.
The binding measurements were carried out on a Synapt G2 quadrupole-ion mobility separation-time of flight (Q-IMS-TOF) mass spectrometer (Waters, UK). A nanoflow ESI source was used. To perform nanoflow ESI, tips were produced from borosilicate tubes (1.0-mm outer diameter, 0.68-mm inner diameter) pulled to ∼5-μm outer diameter at one end using a P-1000 micropipette puller (Sutter Instruments, Novato, CA). A platinum wire was inserted into the nanoflow ESI tip, and a ∼1.0-kV voltage was applied to carry out the ESI. Mass spectra were obtained in positive ion mode using cesium iodide (concentration, 30 ng/μl) for calibration. A cone voltage of 35 V was used, and the source block temperature was maintained at 343 K. Ion transmission parameters (injection voltages) into the trap and transfer ion guides were maintained at 5 and 2 V, respectively. Argon was used in the trap and transfer ion guides at a pressure of 2.22 × 10−2 and 3.36 × 10−2 millibars, respectively. Data acquisition and processing were carried out using MassLynx (v4.1).
The determination of the association constant Ka value for a given protein-ligand (P-L) interaction using the direct ESI-MS assay is based on the measured ratio (R) of the total abundance (Ab) of ligand-bound to free protein ions for solutions of known initial concentrations of protein ([P]0) and ligand ([L]0). For a 1:1 P-L complex (Reaction 1), Ka is calculated using Equation 1.
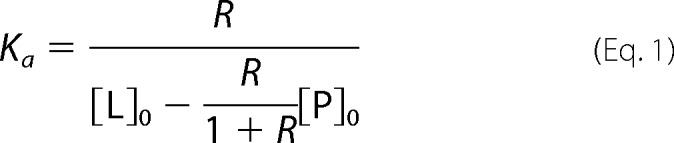 |
where R is given by Equation 2.
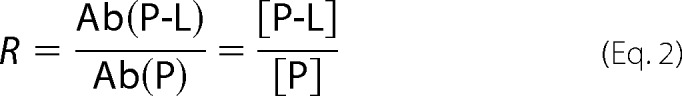 |
An underlying assumption is that P-L and P have similar ionization and detection efficiencies (i.e. similar ESI-MS response factors) such that the gas-phase abundance ratio is equal to the concentration ratio in solution. This assumption has been shown to be valid in cases where L is small compared with P such that P and P-L are similar in size and surface properties (49, 50). Nonspecific binding of free ligand to the free and bound protein can occur during the ESI process, leading to changes of their measured relative abundances. To account for nonspecific ligand binding, a non-binding protein (single chain Fv fragment  Pref) was added to the ESI-MS solution containing P and L. A corrected R value was obtained by the reference protein method using the following expression (51).
Pref) was added to the ESI-MS solution containing P and L. A corrected R value was obtained by the reference protein method using the following expression (51).
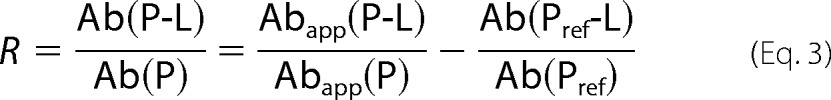 |
where Abapp(P) and Abapp(P-L) are apparent measured abundances of ions corresponding to P and P-L ions and Ab(Pref) and Ab(Pref-L) are the measured abundances of ions corresponding to free Pref and Pref bound nonspecifically to L.
Mass Spectrometry
Approximately 15 AlgG crystals were harvested, washed in well solution, dissolved in 50 μl of H2O, and analyzed by the in-solution tryptic digest LC/MS method (Advanced Protein Technology Centre, The Hospital for Sick Children).
RESULTS
AlgG Adopts a Right-handed Parallel β-Helix Fold
To better understand how alginate is modified as it passages through the periplasm prior to export, we have undertaken structural studies of the C5-epimerase AlgG. As our initial attempts to crystallize P. aeruginosa AlgG were unsuccessful, we generated a number of constructs for various AlgG homologues. Ultimately, a construct lacking the predicted type I export signal sequence, residues 1–36, and 12 additional N-terminal amino acids of P. syringae pv. tomato strain DC3000 AlgG, AlgG49–536, yielded high resolution diffraction quality crystals after the protein had been methylated. The crystals took 2–3 months to grow. The structure of P. syringae AlgG was determined using selenomethionine incorporation and the single wavelength anomalous dispersion technique and was refined against a native data set to 2.1-Å resolution (Table 1). AlgG crystallized in space group P3221 with unit cell dimensions of a = b = 126.6 Å, c = 97.7 Å, and γ = 120° with one molecule in the asymmetric unit. The final model was refined to an Rwork and Rfree of 18.8 and 21.5%, respectively (Table 1). Although the construct used produced protein that contains residues 49–536, the final refined model comprises residues 69–492 (Table 1). Analysis of the AlgG crystals by tryptic digest followed by LC/MS (data not shown) was able to detect a sequence starting at residue 128 and ending with residue 518, indicating that residues 493–518 were still part of the crystallized protein but could not be built due to the poor quality of the electron density. The available density suggests that this disordered C-terminal tail interacts with a symmetry-related protein molecule. Because of the limitation of the LC/MS technique, we could not determine whether the crystallized protein contained residues 49–69 or 518–536.
AlgG adopts a right-handed parallel β-helix fold, a fold first observed for Erwinia chrysanthemi pectate lyase C (52). AlgG is the first periplasmic alginate C5-epimerase whose structure has been determined and the second epimerase structure found to adopt a parallel β-helix fold. The parallel β-helix of AlgG comprises 11 complete coils and one unfinished coil (Fig. 2A). Each coil is made up of three β-sheets, PB1, PB2, and PB3, which are linked by turns T1 (between PB1 and PB2), T2 (between PB2 and PB3), and T3 (between PB3 and PB1) (Fig. 2A). As predicted by Douthit et al. (53), coils 4–10 form a carbohydrate-binding/sugar hydrolysis domain (22). Carbohydrate-binding/sugar hydrolysis domains comprise the central core of the β-helix fold and are characterized by internally repeating glycines and hydrophobic residues and frequently contain the active site (55). The features typically associated with a parallel β-helix fold are all present in AlgG. There are two internal asparagine ladders located at the T2 and T3 turns, confirming the predictions of Douthit et al. (53); several internal stacks of aliphatic residues located on the PB1, PB2, and PB3 sheets; and an N-terminal tail lacking regular secondary structure that is packed against the PB2 face (Fig. 2B). The hydrophobic core of the protein is capped N-terminally by a short helix, called the cap helix, comprising residues 142–148. This short cap helix is flanked by two antiparallel β-sheets, each of which consists of two β-strands that together form an elaborate lid over the N-terminal central core region (Fig. 2A). At the C terminus, the hydrophobic core of AlgG is packed against a symmetry-related molecule in a tail-to-tail arrangement. AlgG contains an additional 44 residues at the C terminus that could not be modeled in the current structure. The missing C terminus is predicted to contain a short β-strand, presumably completing the 12th and final coil, followed by a helix, which could potentially cap the hydrophobic core at the C terminus (56). Hence, it is likely that this tail-to-tail protein interaction is an artifact of crystal packing.
FIGURE 2.
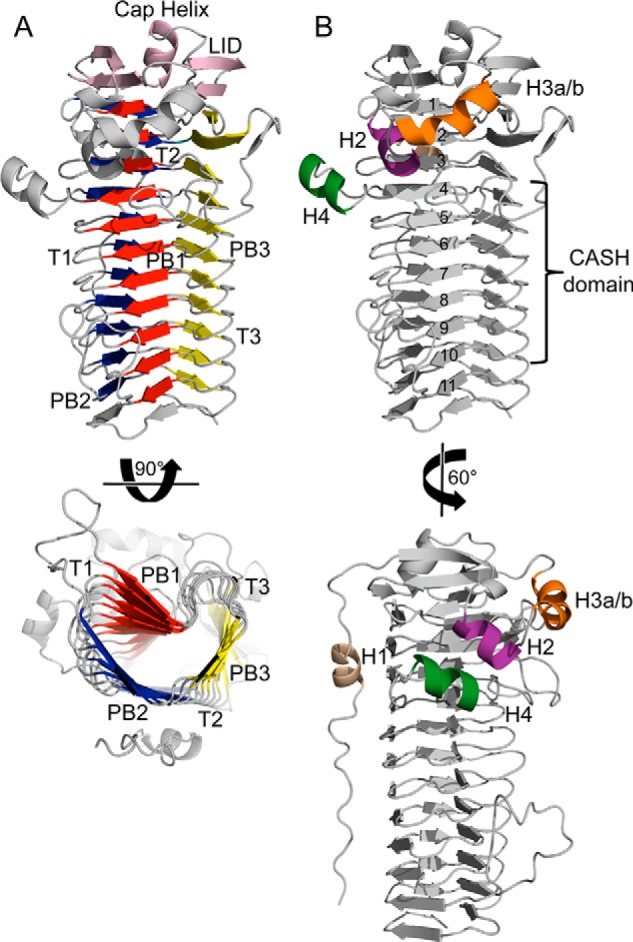
Overall structure of AlgG. A, schematic representation of AlgG. The parallel β-helix fold is made up of three parallel β-sheets, PB1 (red), PB2 (blue), and PB3 (yellow), which are connected via turns T1, T2, and T3. The parallel β-helix is capped N-terminally by an elaborate “lid” (pink). The lid consists of a short capping helix flanked by two antiparallel β-sheets. B, schematic representation of AlgG with coil numbers (1–11) and carbohydrate-binding/sugar hydrolysis (CASH) domain (22, 53). The N-terminal tail is packed against the PB2 surface and contains helix H1 (beige). AlgG contains three additional helices termed H2 (purple), H3a-H3b (orange), and H4 (dark green).
In addition to the N-terminal cap helix, there are four other helices present in the structure that we have termed the H1–H4 helices (Fig. 2B). Helix H1 (residues 83–89) is found within the mostly random coil N-terminal tail and packs against the PB2 surface (coils 2 and 3 of the β-helix). Helix H2 (residues 107–113) interacts with residues 266–272, helix H4, a part of the T1 loop of coil 4. Helix H3a/b (residues 116–126) belongs to the same extended loop as helix H2 and consists of a 310 helix (residues 116–118; helix H3a) and a short two-turn α-helix (residues 119–126; helix H3b).
His319 Is the Catalytic Base
The parallel β-helix fold found in AlgG is most commonly associated with polysaccharide lyases (PLs) belonging to families PL1, PL3, PL6, and PL9 (57, 58). Polysaccharide lyases and epimerases not only share the same fold but also have been suggested to have similar reaction mechanisms (Fig. 1) (20). As polysaccharide lyases have been extensively characterized, it is beneficial to use this knowledge to understand the catalytic mechanism of alginate C5-epimerases (57). Members of the PL1, PL3, PL6, and PL9 families utilize either Ca2+ or arginine to neutralize the negative charge on the carboxylate group of the uronic acid (Fig. 1) (57, 59, 60). Polysaccharide lyases with a β-helix fold typically have either an arginine or a lysine as the catalytic base, whereas the catalytic acid has been identified to be either arginine or a water molecule (57). Polysaccharide lyases of other folds use tyrosine or histidine as the catalytic base and tyrosine as the catalytic acid (57).
In vivo studies of P. aeruginosa AlgG revealed that mutagenesis of each of the residues in the conserved DPHD motif (residues 317–320 in P. syringae) abolished epimerase activity (53). This motif is part of the carbohydrate-binding/sugar hydrolysis domain and is proposed to be part of the active site (22, 53, 55). In our AlgG structure, the DPHD motif is composed of Asp317, Pro318, and His319, which are located on the concave PB1 surface, and Asp320, which is part of the adjacent T1 loop of coil 6 (Fig. 3, A and B). To determine whether these residues are important for epimerase activity in P. syringae, alanine point mutants of residues Asp317, His319, and Asp320 were made, and their ability to epimerize polymannuronate in vitro was tested. Pro318 was not mutated as its side chain is not surface-accessible but part of the β-helix core. Using an end point assay, mutation of His319 and Asp320 was found to completely ablate epimerase activity, whereas the D317A mutant retained ∼5% activity compared with wild-type levels, thus reinforcing the importance of the DPHD motif in catalysis (Fig. 4). CD spectroscopy of AlgG H319A and D320A indicates that the mutant proteins are folded (data not shown). Examination of the AlgG structure reveals that the side chain of His319 is in an unusual rotamer conformation (χ1 = −118°). This high energy conformation and its presence in the DPHD motif suggests that His319 plays a role in catalysis. His319 is the only rotamer outlier present in the AlgG structure. This conformation appears to be due to residues Tyr294, Asp317, and Asp320, which surround His319 and prevent it from adopting a preferred rotamer conformation. Asp320 Oδ1 is in hydrogen bond distance to His319 Nδ1 (Fig. 3B), and mutating Asp320 to alanine completely abolishes epimerase activity (Fig. 4). The His319-Asp320 interaction suggests that His319 may be deprotonated over the pH range observed for optimal AlgG activity (pH 6–7.5) (31), a hypothesis supported by calculations using the heuristic pKa prediction software PROPKA (61), which suggests that His319 has a pKa of 5.48. Because the formation of the His319-Asp320 interaction appears to be crucial for epimerase activity and a deprotonated His319 would be capable of abstracting a proton from the C5 of mannuronate, it would appear that His319 likely acts as the catalytic base in the epimerase reaction. Asp317 is also in close proximity to His319 but too far for a hydrogen bond interaction as the distance between Oδ1 of Asp317 and Nϵ2 of His319 is 3.7 Å. As we had found that the D317A AlgG mutant retains only ∼5% wild-type epimerase activity, to further probe whether Asp317 could play a role in the positioning of His319, we determined the structure of the D317A mutant to 2.3-Å resolution (Table 1). The D317A mutant structure is very similar to the wild-type AlgG structure with a root mean square deviation of 0.2 Å over 424 Cα atoms (42). The only residue that changed in the active site region of the AlgG D317A mutant structure is His319, which is no longer in a high energy conformation but adopts a more favorable side chain conformation, thus supporting our hypothesis that Asp317 helps to position His319 by forcing His319 into a high energy conformation to avoid a steric clash between Asp317 and His319 (Fig. 3B). The His319-Asp320 interaction is not formed in the AlgG D317A structure as His319 adopts the only possible low energy conformation that does not result in a steric clash. The loss of the His319-Asp320 interaction potentially explains why the D317A mutant only shows ∼5% epimerase activity compared with wild-type levels. The side chain of the third residue likely to be important for positioning His319 in wild-type AlgG, Tyr294, sits above and approximately perpendicular to His319 within van der Waals distance (shortest distance, ∼3.7 Å). Mutating Tyr294 to an alanine (Y294A) reduced AlgG epimerase activity to 24.5% of wild-type activity, whereas the Y294F mutant retained 66% activity (Fig. 4). These results suggest that Tyr294 may also contribute to the proper orientation of His319 and that the bulky side chain of Tyr294 limits the potential side chain conformations that His319 can adopt.
FIGURE 3.
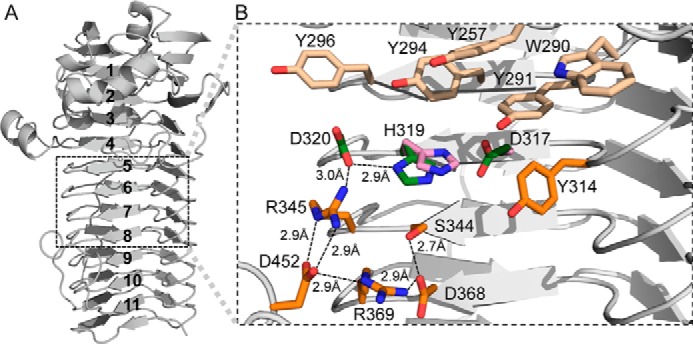
AlgG active site region. A, schematic representation of AlgG with coil numbers (1–11). The location of the active site on AlgG is boxed. B, a detailed view of the AlgG wild-type active site region with Asp317, His319, and Asp320 of the conserved DPHD motif (green), other active site residues (orange), and aromatics cluster above the active site (light orange) shown in stick representation. In the AlgG D317A structure, residue His319 (pink) has moved relative to other active site residues and no longer interacts with Asp320. His319 in the AlgG D317A mutant structure is e only residue whose orientation has changed relative to the active site of wild-type AlgG. Therefore, the other residues in the active site region of the D317A mutant have for clarity not been depicted.
FIGURE 4.

Epimerase activity of AlgG mutants. The relative activities of AlgG mutants compared with wild-type AlgG are shown. Wild-type epimerase activity was normalized to 100%. Results are the mean of at least three measurements ± S.E. (error bars).
Further examination of the wild-type structure reveals that Tyr294 is one of five conserved aromatic residues that lie directly above the active site (Fig. 3B). Mutation of the conserved aromatic residue Tyr296 to alanine reduced the epimerase activity to 66% of wild-type levels (Fig. 4). Tyr296 is within van der Waals distance of Asp320 and could potentially help position Asp320.
As it had previously been suggested for the calcium-dependent extracellular alginate epimerase AlgE4 from A. vinelandii (23) that Tyr149 (Tyr314 in AlgG P. syringae) was the catalytic base, we mutated Tyr314 to phenylalanine and tested this AlgG mutant for epimerase function. This single point mutation reduced the epimerase activity to ∼5% of wild-type levels (Fig. 4). CD spectroscopy confirmed that the Y314F mutant was folded (data not shown). The predicted pKa of Tyr314 is 13, suggesting that Tyr314 would be protonated between pH 6 and 7.5 and therefore unlikely to function as the catalytic base in the AlgG epimerase reaction (61), making it more likely that Tyr314 is involved in substrate recognition.
AlgG Uses Arg345 to Neutralize the Negative Charge on the Uronic Acid during the Epimerase Reaction
Parallel β-helix polysaccharide lyases use either Ca2+ or arginine to neutralize the negative charge of the uronic acid during catalysis (57). If alginate epimerases use a similar reaction mechanism and because the AlgG epimerization reaction was found to be Ca2+-independent, then it is likely that AlgG uses an arginine to neutralize the negative charge of the uronic acid (31). Examination of the AlgG structure suggests that the most likely candidate for this role is Arg345 (Fig. 3B). This arginine is in close proximity to the His319-Asp320 diad and is held in position by three salt bridges. The Nη1 of Arg345 interacts with the Oδ1 of Asp320, and Nϵ and Nη2 of Arg345 interact with Oδ1 and Oδ2 of Asp452, respectively (Fig. 3B). To probe the role that Arg345 may play in catalysis, site-directed mutants that replaced the arginine with alanine, glutamine, glutamate, or lysine were generated. The R345K mutant reduced the epimerase activity to 36% of the wild-type level, whereas both R345A and R345Q mutants exhibited less than 10% of wild-type activity (Fig. 4). No epimerase activity was detected in the R345E mutant (Fig. 4). CD spectroscopy confirmed that the Arg345 mutants, which showed less than 10% epimerase activity, were folded (data not shown). The retention of 36% activity of the R345K mutant suggests that the positive charge of lysine may be able, in part, to perform the same role as Arg345. In contrast, replacing Arg345 with the negatively charged glutamate completely abrogated epimerase activity, suggesting that the negative charge of glutamate might repel the negatively charged carboxylate group of mannuronate. The mutational results are consistent with the proposal that Arg345 is involved in neutralizing the negative charge of saccharide during catalysis.
His319, Asp320, and Arg345 are part of an extensive hydrogen bond and electrostatic interaction network of conserved residues that are arranged in a ringlike manner: His319-Asp320-Arg345-Asp452-Arg369-Asp368-Ser344 (Fig. 3B). The hydrogen bond-salt bridge ring is broken between His319 and Ser344 located just underneath His319. Two of these residues, Asp368 and Ser344 in P. syringae, were found to be important for epimerase activity in vivo in P. fluorescens (mutants in P. fluorescens are D361N and S337F) (62). To test whether these residues were important for epimerase activity in vitro and to probe the importance of this network, we mutated Asp368 to asparagine and Ser344 to alanine. CD spectroscopy confirmed both mutants to be folded (data not shown). AlgG D368N showed 5% of wild-type activity, confirming the in vivo observations and suggesting that Asp368 is important for epimerase function (Fig. 4). The S344A mutant retained 54% of wild-type activity (Fig. 4). The reason for the disparity between in vitro and in vivo results is explained by that fact that phenylalanine replaced serine in vivo, probably causing AlgG to become unstable, whereas the smaller alanine does not perturb folding. The D452A mutant has a less severe impact on AlgG epimerase activity, retaining 63% of wild-type levels (Fig. 4). The AlgG R369A mutant shows 23% epimerase activity compared with wild-type activity, indicating that it is important for proper epimerase function and might be involved in substrate binding (Fig. 4). Our analysis of the active site hydrogen bond network shows that several residues play a crucial role in epimerase activity.
A Conserved, Electropositive Groove on the Concave Face of AlgG Facilitates Alginate Binding
As the polymer is negatively charged, the alginate binding site is likely to have an overall positive electrostatic surface potential. Examination of the electrostatic surface properties of AlgG (Fig. 5A) indicates only one positively charged region on the enzyme located just below the putative catalytic site. This region is also highly conserved across all periplasmic AlgG alginate epimerases (Fig. 5B). We propose that this conserved region, which is ∼49 Å long, is an extended substrate binding site. Given that the lengths of the mannuronate trisaccharides found in the alginate lyase A1-III (63) and the alginate epimerase AlgE4 (23) structures are 15.5 and 14.7Å, respectively, the substrate binding site of AlgG could accommodate at least nine monomers. In vitro characterization of AlgG has found that the enzyme requires a minimum of nine mannuronate residues for catalysis to occur, suggesting that the enzyme contains several substrate binding subsites (31). Ligand binding studies of wild-type AlgG with mannuronate oligomers ranging in length from four to 12 residues show that the ligand affinity does not significantly increase in oligomers longer than a nonamer (Ka of 5.0 × 105 m−1) (Table 2). These data suggest that the AlgG substrate binding site contains at least nine subsites.
FIGURE 5.

Polymannuronate binding site. A, electrostatic potential surface representation of AlgG. The electrostatic properties of AlgG were determined with the APBS server. Positive surface potential is shown in blue, and negative surface potential is shown in red and contoured from −5 to +5 kT/e. B, surface representation of residue conservation of AlgG (dark magenta (high residue conservation) to dark cyan (variable residue conservation)). Residue conservation was produced with the ConSurf server using a T-Coffee alignment comprising AlgG sequences from Pseudomonas spp. and A. vinelandii. See “Experimental Procedures” for a list of sequences used. C, surface representation of AlgG with a modeled mannuronate trisaccharide in the putative poly(M) binding site. Residues His319, Asp320, and Arg345 are highlighted in green. D, stick representation of AlgG active and substrate binding site residues (orange) with modeled mannuronate trisaccharide (black).
TABLE 2.
Apparent association constants (Ka) for the binding of mannuronate oligomers to wild-type AlgG
| Oligomer length | Apparent Ka |
|---|---|
| m−1 | |
| 4-mer | (2.4 ± 0.4) × 104 |
| 5-mer | (2.2 ± 1.1) × 104 |
| 6-mer | (5.7 ± 0.6) × 104 |
| 7-mer | (2.2 ± 1.1) × 105 |
| 8-mer | (2.0 ± 1.0) × 105 |
| 9-mer | (5.0 ± 0.5) × 105 |
| 10-mer | (4.8 ± 0.3) × 105 |
| 11-mer | (6.3 ± 0.6) × 105 |
| 12-mer | (4.6 ± 2.5) × 105 |
As our current attempts to co-crystallize AlgG with mannuronate oligomers have been unsuccessful, we modeled a mannuronate trisaccharide into subsites +1 to −2 of the putative binding site (Fig. 5, C and D). The orientation of mannuronate in subsite +1 (site of catalysis) was based on our proposed catalytic mechanism. Fixing the position of the mannuronate in subsite +1 limits the possible orientations of mannuronates for subsites −1 and −2, which were placed using Coot. The resulting model was subsequently energy-minimized.
According to our substrate-bound model, His339 could hydrogen bond to the carboxylate group of the mannuronate residue in subsite −1. The conserved Lys338 does not interact with the modeled trisaccharide but is in the vicinity of the mannuronate residue in subsite −1. To examine the influence of His339 and Lys338 on the epimerase activity of AlgG, we mutated Lys338 and His339 to alanine. The K338A mutant has an epimerase activity of 87% of wild-type levels, whereas the H339A mutant retains only 49% epimerase activity. This suggests that His339 plays a bigger role in substrate binding than Lys338 and further validates our substrate-bound model.
Our model suggests that Arg415 likely interacts with the carboxylate group of mannuronate in subsite −2 (Fig. 5D). Interestingly, the corresponding residue in P. fluorescens AlgG, Arg408, is crucial for epimerase function in vivo as its mutation to cysteine results in a strain that only produces polymannuronate (62). To probe the role of Arg415, we introduced the equivalent mutation in P. syringae AlgG (R415C), confirmed it to be folded (data not shown), and found that it completely abolishes epimerization activity in our in vitro assay (Fig. 4). We did not anticipate that the mutation of a single residue within the alginate binding site ∼13 Å from the site of catalysis (His319) would cause a complete loss of function in AlgG. The loss of epimerase activity in the R145C mutant supports the in vivo data.
Tyr392 is also located in close proximity to the mannuronate in subsite −2 although not within hydrogen or π bonding distance in our model. Because aromatic residues are frequently implicated in protein-carbohydrate interactions, we mutated Tyr392 to phenylalanine and alanine and tested each mutant for epimerase function. Our analysis shows that both the Y392F mutant and the Y392A mutant have ∼65% of wild-type activity (Fig. 4). These results suggests that it is not the aromatic ring of the tyrosine residue but the hydroxyl group that may be important for AlgG epimerase function. The hydroxyl group of Tyr392 hydrogen bonds with Nη1 of Arg413. Hence, Tyr314 could be important for the correct positioning of Arg413.
Conservation of Active Site Residues of Periplasmic and Extracellular Alginate Epimerases Does Not Extend to the Polymer Binding Site
Although both periplasmic AlgG and the extracellular epimerase AlgE4 (Protein Data Bank code 2PYG) adopt the same parallel β-helix fold (23), these enzymes have distinctly different metal dependences. To gain insight into why AlgE4 requires Ca2+ for its enzymatic activity but AlgG does not, we have compared the two enzymes. Superimposition of the maximum number of coils of both structures resulted in their respective active sites being misaligned. This was probably due to the fact that their respective active sites are located on different coils. The active site of AlgG, Tyr314 and 317DPH, is located on coil 6, whereas the active site of AlgE4, Tyr149 and 152DPH, is located on coil 4. As initial inspection suggested that the active sites are very similar, a more meaningful comparison was obtained by aligning both structures using their active sites residues Tyr314-Tyr149, Asp317-Asp152, Pro318-Pro153, His319-His154, and Asp320-Glu155. When AlgG and AlgE4 are aligned in this manner, residues 294–482 of AlgG and 122–290 of AlgE4, the β-helix fold, superimpose well with a root mean square deviation of 0.753 Å over 543 backbone atoms (Fig. 6A). Both active sites have a similar organization (Fig. 6B). The Tyr294 of AlgG that we propose aids in orientation of AlgG His319 is equivalent to Phe122 in the AlgE4 structure (Fig. 6B). However, Asp320 in AlgG does not superimpose well with Glu155 of AlgE4. The Glu155 of AlgE4 was found to be critical for its epimerase activity (23). Interestingly, there is no AlgE4 residue equivalent to Arg345 of AlgG. In addition, examination of the active site region of AlgE4 shows a strong negatively charged surface potential as this active site lacks positively charged residues (Fig. 7, A and C). One of the negatively charged residues located in the active site region, Asp178, is crucial for the epimerase function of AlgE4 (23). This residue superimposes with Ser344 of AlgG (Fig. 6B). A closer look at the superimposition of the active sites of AlgG and AlgE4 reveals that Glu155 in AlgE4 partly overlaps with Arg345 in AlgG (Fig. 6B). Indeed, AlgE4 Glu155 and Asp178 surround the guanidinium group of Arg345 of AlgG in the superimposition. Because AlgE4 is Ca2+-dependent and the coordination of Ca2+ is achieved by negatively charged amino acids, it is feasible that residues Glu155 and Asp178 of AlgE4 are part of a putative Ca2+ binding site.
FIGURE 6.
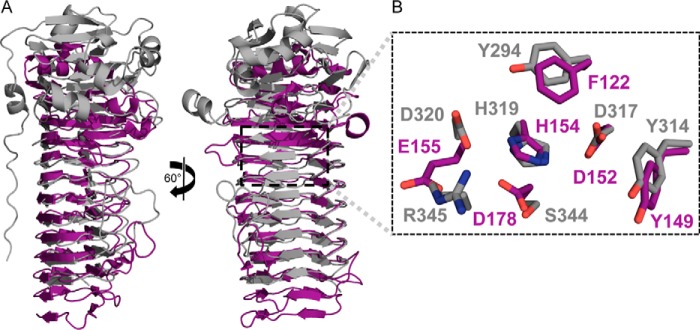
AlgG and AlgE4 superimposition. A, schematic representation of AlgG (gray) and AlgE4 (purple). AlgG and AlgE4 were structurally aligned according to their respective active sites. B, a detailed view of the superimposed active site residues of AlgG (gray) and AlgE4 (purple) are shown in stick representation.
FIGURE 7.
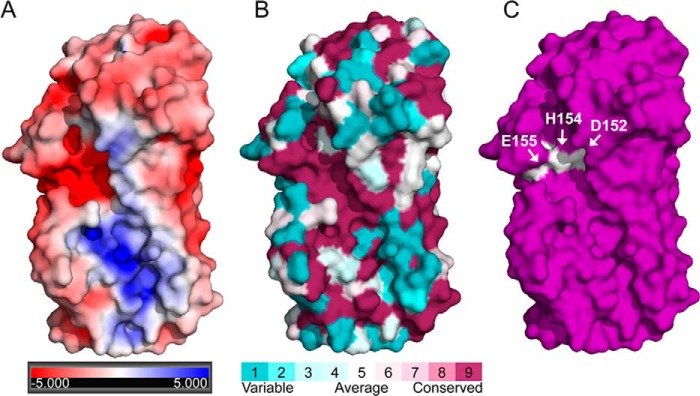
AlgE4 surface analysis. A, electrostatic potential surface representation of AlgE4. The electrostatic properties of AlgE4 were determined with the APBS server. Positive surface potential is shown in blue, and negative surface potential is shown in red and contoured from −5 to +5 kT/e. B, surface representation of residue conservation of AlgE4 (dark magenta (high residue conservation) to dark cyan (variable residue conservation)). Residue conservation was produced with the ConSurf server using a T-Coffee alignment comprising AlgE1–7 sequences from A. vinelandii. C, surface representation of AlgE4. Residues Asp152, His154, and Glu155 are highlighted in white.
The N-terminal cap regions of AlgG and AlgE4 do not superimpose as AlgG is capped by an elaborate lid, whereas AlgE4 is capped by a single N-terminal α-helix (Fig. 6A). Furthermore, the N-terminal tail of AlgG does not have an equivalent in AlgE4 (Fig. 6A). Interestingly, the packing of the molecules in the two structures is comparable as the C terminus of AlgE4 is capped by the C terminus of the second molecule in the asymmetric unit in a tail-to-tail fashion. This arrangement again appears to be due to crystal packing as full-length AlgE4 contains an 18-kDa regulatory domain at the C terminus, and this is not present in the construct used to determine the structure. Although the backbone atoms of the extended substrate binding site of both epimerases superimpose well, only a few residues in this region are conserved between the two proteins. One exception is Arg415 of AlgG that superimposes with AlgE4 Lys255, a residue that is 100% conserved within A. vinelandii AlgE proteins. This residue is also critical for AlgE4 function as mutating Lys255 to an alanine reduced AlgE4 epimerase activity to less than 10% of the wild-type level (23). Interestingly, the sequence conservation of the substrate binding site across the extracellular AlgE proteins is low, and the conserved area is smaller than the conserved substrate binding site of the periplasmic epimerases (Fig. 7, B and C).
DISCUSSION
To better understand how alginate is post-translationally modified as it passages through the periplasm, we determined the structure and functionally characterized the C5-epimerase AlgG. The protein adopts a parallel β-helix fold establishing that the periplasmic epimerase shares the same fold as the extracellular epimerase AlgE4 (23). This is in contrast to the glycosaminoglycan polysaccharide epimerases, heparin/heparan sulfate and dermatan sulfate epimerases, the only other known polymer level carbohydrate epimerases, that are predicted to adopt a α/α toroid fold (64, 65). Like the β-helix fold, the α/α toroid fold is also common in polysaccharide lyases, further emphasizing the strong connections between polysaccharide epimerases and lyases (57).
Because of their lack of sequence similarity and different characteristics, extracellular and periplasmic alginate epimerases were thought to have different mechanisms (6, 21, 31). Our data reveal that the active site architecture of both types of enzymes is very similar (Fig. 6B). The main difference in the active sites is that AlgE4 does not contain a residue equivalent to Arg345 found in AlgG but instead contains several acidic residues that we propose form the AlgE4 Ca2+ binding site. As found in most β-helix fold polysaccharide lyases, this Ca2+ would be in a position to neutralize the carboxylate group of the uronic acid during the reaction, thereby performing a role equivalent to that of Arg345. The coordination of calcium also helps explain why the active site region of AlgE4, which has a negative surface potential, is able to catalyze the epimerase reaction of the negatively charged polymannuronate. One reason why no Ca2+ was modeled in the active site of the AlgE4 structure is that calcium coordination may require the substrate, which is not present at this subsite in the AlgE4 structure. The A. vinelandii extracellular alginate epimerases also appear to use the histidine of their 100% conserved DPHE motif as the catalytic base. Although His154 does not interact with Glu155 in the structure of the extracellular alginate epimerase AlgE4, in a manner comparable with His319 and Asp320 in AlgG, the Nδ1 of His154 hydrogen bonds with the Oδ1 of Asp178. His154 is also predicted to have a pKa of 3.77 and therefore would be able to act as the catalytic base over the pH range of optimal AlgE4 activity (pH 6.5–7.0) (61, 66). The lack of sequence conservation in the substrate binding sites of the AlgG and AlgE4 enzymes and between periplasmic and extracellular alginate epimerases in general could reflect the fact that their substrates are slightly different and/or that the epimerases have adapted to their respective environments. Periplasmic AlgG catalyzes the epimerization of polymannuronate, whereas the extracellular epimerases only encounter the secreted alginate chain, which has already been modified by the periplasmic alginate epimerase and the acetylation machinery of A. vinelandii.
Our structure suggests that AlgG adheres to the first steps of the catalytic mechanism found in β-helix polysaccharide lyases and, although less commonly found in these polysaccharide lyases, appears to use arginine to neutralize the negative charge of the carboxylate group of the uronic acid (59, 60). β-Helix polysaccharide lyases, such as the extracellular pectate lyases, more frequently use Ca2+ to neutralize the negative charge (57). As calcium is used as a signaling molecule in bacteria, its concentration is tightly regulated in the bacterial cell (67). As reduced periplasmic Ca2+ concentration was found to promote biofilm formation in Pseudomonas (68), it is possible that periplasmic epimerases have adapted to this environment. Ca2+-dependent A. vinelandii AlgE alginate epimerases are secreted to the extracellular space where calcium is found in abundance (24, 25). To complete the epimerization reaction, a proton from a catalytic acid is required, and water is the most likely catalytic acid in the AlgG epimerase reaction. An A. vinelandii extracellular alginate epimerase has been found to incorporate tritium into guluronate residues during the epimerization reaction (69). Given the similarity of the active site architectures and proposed similarity of the reaction mechanisms, it is likely that in AlgG the proton, which is added to the opposite face of the sugar to complete the epimerization reaction, comes from the solvent. Water is also frequently used as the catalytic acid in the β-elimination reaction mechanism of β-helix polysaccharide lyases (57). Examination of the AlgG structure reveals that Tyr314 coordinates a water molecule, which is in the vicinity of the C5 position of the mannuronate in the +1 subsite and could be a potential candidate for the catalytic acid. The readdition of the proton to the opposite face of the sugar also explains why the reaction is irreversible (21). The C5 proton of the newly created guluronate points away from the active site, making it impossible for AlgG to convert it back to mannuronate. Flipping the alginate polymer so that the C5 proton of guluronate is pointing toward the catalytic base would also not allow the reverse reaction to take place as the epimerization reaction causes a substantial conformational change in the sugar with mannuronate adopting a 4C1 conformation, whereas guluronate typically adopts a 1C4 configuration. Hence, it is very unlikely that the +1 subsite could accommodate guluronate for the reverse epimerase reaction to occur. The main difference between the alginate epimerases reaction mechanism, including that of AlgG, and the reaction mechanism of β-helix polysaccharide lyases is the use of histidine rather than arginine or lysine as the catalytic base. This difference is reflected in the different pH optima of the enzymes with alginate epimerases and β-helix polysaccharide lyases having neutral and basic pH optima, respectively (66, 70–75). Taken together, our data suggest that AlgG has a slight variation of the otherwise conserved catalytic mechanism found in β-helix polysaccharide lyases. It is interesting to note that the polysaccharide epimerases, heparin/heparan sulfate and dermatan sulfate epimerases, follow the same catalytic mechanism as AlgG but are able to perform the reverse reaction as they contain two catalytic bases (76, 77). The fact that polysaccharide epimerases adopt folds that are prevalent in polysaccharide lyases and their similar reaction mechanisms suggest that polysaccharide epimerases are derived from polysaccharide lyases.
Our model of the mannuronate trisaccharide bound to AlgG suggests that for the epimerase reaction to occur the reducing end of the sugar needs to point toward the active site of AlgG. The same orientation is seen in the AlgE4-mannuronate structure (23). Alginate, cellulose, and poly-β-N-acetylglucosamine are synthase-dependent extracellular polysaccharides (78). The recent cellulose synthase study (79) modeled the nascent polymer with the reducing end as the first portion of the polymer chain to enter the periplasm. This modeling of the nascent chain was based on the finding that cellulose is polymerized at its non-reducing end (80). If the same polymerization mechanism takes place in the alginate system, the substrate binding site of AlgG, located below the active site where the trisaccharide is modeled, coordinates the nascent and unepimerized polymannuronate chain before the epimerase reaction occurs.
Although we only modeled a trisaccharide into the substrate binding site, AlgG has been shown to require longer substrates for catalysis to occur. Data from Jerga et al. (31) suggest that a minimum of nine mannuronate residues are required, which suggests that the substrate binding site of AlgG contains several subsites. The occurrence of subsites is a characteristic of processive enzymes, and many processive enzymes contain a groove to which the substrate binds (81). The catalytic and substrate binding sites of AlgG lie on a shallow concave surface of the parallel β-helix and form such a groove. AlgG has a low affinity for its substrate with a Ka of 5.0 × 105 m−1 for the nonamer (Table 2), additionally supporting the processivity hypothesis as tight binding would be detrimental to the sliding of the polymer. The sliding movement of the polymer could be a consequence of polymannuronate synthesis. The loss of epimerase activity in the AlgG R415C mutant suggests that the coordination of the carboxylate group of mannuronate in subsite −2 and hence the binding of the substrate in the correct register is crucial for the epimerase reaction to occur and that several subsites are indeed present on AlgG. The negatively charged carboxylate moieties of polymannuronate are offset by ∼180° in successive monomers due to the nature of the β-1–4 linkage. Because the alginate binding site of AlgG is lined with positively charged residues (Fig. 5, A and D), each mannuronate could potentially bind to any subsite, allowing polymannuronate to glide through the binding site. But maximally only every other mannuronate would be in the correct orientation to be converted to guluronate. This processive mode of action model would explain why there are only alternating sequences of M and G but no guluronate doublets found in Pseudomonad alginates (4, 8, 11, 26). In vitro AlgG was found to produce G blocks probably because the polymer and enzyme are free to dissociate in the in vitro assay (31). Another explanation for the mode of action of AlgG in vivo could be that a polymer where a mannuronate has been converted to a guluronate is a better substrate for the epimerase (preferred attack mode of action). In this case, AlgG should show a higher affinity for substrates containing guluronate residues than for pure mannuronate substrates if this preferred attack mode of action is followed. However, AlgG shows no preference for MG over MM substrates (21), thereby excluding a preferred attack mode of action. Thus, only a processive mode of action can explain the non-random distribution of guluronate in alginate (4, 82). The extracellular alginate epimerases AlgE4 and AlgE6 have been found to be processive, adding support to the suggestion that AlgG follows this mechanism (66, 82–84).
In this study, we have determined the first structure of a periplasmic alginate epimerase and have shown that it adopts a β-helix fold with an elaborate lid structure. Subsequent bioinformatics analysis, site-directed mutagenesis, enzymatic analysis, and substrate modeling have allowed us to propose a catalytic mechanism and mode of action that is able to explain the composition of mannuronate and guluronate found in alginate from Pseudomonas spp. Furthermore, comparing our structure of AlgG with the structure of the extracellular epimerase AlgE4 has enabled us to identify a probable Ca2+ binding site in AlgE4. Calcium is essential for catalysis in this enzyme and is believed to be involved in neutralizing the negative charge of mannuronate during the epimerase reaction of extracellular alginate epimerases.
Acknowledgments
We thank Ana Mirela Neculai for substantial contributions to the initial studies on P. aeruginosa AlgG; Laura Riley, Joel Weadge, John C. C. Whitney, and Perrin Baker for helpful discussions; and Dustin J. Little and Jason Koo for technical assistance. Beam line X29 at the National Synchrotron Light Source is supported by the United States Department of Energy and by National Center for Research Resources Grant P41RR012408 and National Institute of General Medical Sciences Grant P41GM103473 from the National Institutes of Health.
This work was supported in part by Canadian Institutes of Health Research Grant MT 13337 (to P. L. H.) and the Alberta Glycomics Centre (to E. N. K. and J. S. K.).
The atomic coordinates and structure factors (codes 4NK6 and 4NK8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- M
- β-d-mannuronate
- G
- α-l-guluronate
- poly(M)
- polymannuronic acid
- SeMet
- selenomethionine
- NTA
- nitrilotriacetic acid
- ESI
- electrospray ionization
- PL
- polysaccharide lyase.
REFERENCES
- 1. Linker A., Jones R. S. (1964) A polysaccharide resembling alginic acid from a Pseudomonas micro-organism. Nature 204, 187–188 [DOI] [PubMed] [Google Scholar]
- 2. Gorin P. A., Spencer J. F. (1966) Exocellular alginic acid from Azotobacter vinelandii. Can. J. Chem. 44, 993–998 [Google Scholar]
- 3. Haug A., Larsen B., Smidsrod O. (1974) Uronic acid sequence in alginate from different sources. Carbohydr. Res. 32, 217–225 [Google Scholar]
- 4. Skjåk-Braek G., Grasdalen H., Larsen B. (1986) Monomer sequence and acetylation pattern in some bacterial alginates. Carbohydr. Res. 154, 239–250 [DOI] [PubMed] [Google Scholar]
- 5. Pier G. B., Coleman F., Grout M., Franklin M., Ohman D. E. (2001) Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 69, 1895–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Valla S., Li J., Ertesvåg H., Barbeyron T., Lindahl U. (2001) Hexuronyl C5-epimerases in alginate and glycosaminoglycan biosynthesis. Biochimie 83, 819–830 [DOI] [PubMed] [Google Scholar]
- 7. Franklin M. J., Chitnis C. E., Gacesa P., Sonesson A., White D. C., Ohman D. E. (1994) Pseudomonas aeruginosa AlgG is a polymer level alginate C5-mannuronan epimerase. J. Bacteriol. 176, 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Skjåk-Braek G., Smidsrod O., Larsen B. (1986) Tailoring of alginates by enzymatic modification in vitro. Int. J. Biol. Macromol. 8, 330–336 [Google Scholar]
- 9. Nyvall P., Corre E., Boisset C., Barbeyron T., Rousvoal S., Scornet D., Kloareg B., Boyen C. (2003) Characterization of mannuronan C-5-epimerase genes from the brown alga Laminaria digitata. Plant Physiol. 133, 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tonon T., Rousvoal S., Roeder V., Boyen C. (2008) Expression profiling of the mannuronan C5-epimerase multigenic family in the brown alga Laminara digitata (Phaeophyceae) under biotic stress conditions. J. Phycol. 44, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 11. Sherbrock-Cox V., Russell N. J., Gacesa P. (1984) The purification and chemical characterisation of the alginate present in extracellular material produced by mucoid strains of Pseudomonas aeruginosa. Carbohydr. Res. 135, 147–154 [DOI] [PubMed] [Google Scholar]
- 12. Simpson J. A., Smith S. E., Dean R. T. (1988) Alginate inhibition of the uptake of Pseudomonas aeruginosa by macrophages. J. Gen. Microbiol. 134, 29–36 [DOI] [PubMed] [Google Scholar]
- 13. Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. (2002) Molecular Biology of the Cell, 4th Ed., pp. 1091–1092, Garland Science, New York [Google Scholar]
- 14. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 15. Trowbridge J. M., Gallo R. L. (2002) Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology 12, 117R–125R [DOI] [PubMed] [Google Scholar]
- 16. Li J. P., Gong F., Hagner-McWhirter A., Forsberg E., Abrink M., Kisilevsky R., Zhang X., Lindahl U. (2003) Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking l-iduronic acid and in neonatal lethality. J. Biol. Chem. 278, 28363–28366 [DOI] [PubMed] [Google Scholar]
- 17. Presto J., Thuveson M., Carlsson P., Busse M., Wilén M., Eriksson I., Kusche-Gullberg M., Kjellén L. (2008) Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc. Natl. Acad. Sci. U.S.A. 105, 4751–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Esko J. D., Selleck S. B. (2002) Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 [DOI] [PubMed] [Google Scholar]
- 19. Franklin M. J., Nivens D. E., Weadge J. T., Howell P. L. (2011) Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gacesa P. (1987) Alginate-modifying enzymes. A proposed unified mechanism of action for the lyases and epimerases. FEBS Lett. 212, 199–202 [Google Scholar]
- 21. Jerga A., Stanley M. D., Tipton P. A. (2006) Chemical mechanism and specificity of the C5-mannuronan epimerase reaction. Biochemistry 45, 9138–9144 [DOI] [PubMed] [Google Scholar]
- 22. Zdobnov E. M., Apweiler R. (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 [DOI] [PubMed] [Google Scholar]
- 23. Rozeboom H. J., Bjerkan T. M., Kalk K. H., Ertesvåg H., Holtan S., Aachmann F. L., Valla S., Dijkstra B. W. (2008) Structural and mutational characterization of the catalytic A-module of the mannuronan C-5-epimerase AlgE4 from Azotobacter vinelandii. J. Biol. Chem. 283, 23819–23828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ertesvåg H., Høidal H. K., Hals I. K., Rian A., Doseth B., Valla S. (1995) A family of modular type mannuronan C-5-epimerase genes controls alginate structure in Azotobacter vinelandii. Mol. Microbiol. 16, 719–731 [DOI] [PubMed] [Google Scholar]
- 25. Svanem B. I., Skjåk-Braek G., Ertesvåg H., Valla S. (1999) Cloning and expression of three new Azotobacter vinelandii genes closely related to a previously described gene family encoding mannuronan C-5-epimerases. J. Bacteriol. 181, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chitnis C. E., Ohman D. E. (1990) Cloning of Pseudomonas aeruginosa algG, which controls alginate structure. J. Bacteriol. 172, 2894–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rehm B. H., Ertesvåg H., Valla S. (1996) A new Azotobacter vinelandii mannuronan C-5-epimerase gene (algG) is part of an alg gene cluster physically organized in a manner similar to that in Pseudomonas aeruginosa. J. Bacteriol. 178, 5884–5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peñaloza-Vázquez A., Kidambi S. P., Chakrabarty A. M., Bender C. L. (1997) Characterization of the alginate biosynthetic gene cluster in Pseudomonas syringae pv. syringae. J. Bacteriol. 179, 4464–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morea A., Mathee K., Franklin M. J., Giacomini A., O'Regan M., Ohman D. E. (2001) Characterization of algG encoding C5-epimerase in the alginate biosynthetic gene cluster of Pseudomonas fluorescens. Gene 278, 107–114 [DOI] [PubMed] [Google Scholar]
- 30. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 31. Jerga A., Raychaudhuri A., Tipton P. A. (2006) Pseudomonas aeruginosa C5-mannuronan epimerase: steady-state kinetics and characterization of the product. Biochemistry 45, 552–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F. S., Hufnagle W. O., Kowalik D. J., Lagrou M., Garber R. L., Goltry L., Tolentino E., Westbrock-Wadman S., Yuan Y., Brody L. L., Coulter S. N., Folger K. R., Kas A., Larbig K., Lim R., Smith K., Spencer D., Wong G. K., Wu Z., Paulsen I. T., Reizer J., Saier M. H., Hancock R. E., Lory S., Olson M. V. (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406, 959–964 [DOI] [PubMed] [Google Scholar]
- 33. Winsor G. L., Lam D. K., Fleming L., Lo R., Whiteside M. D., Yu N. Y., Hancock R. E., Brinkman F. S. (2011) Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–D600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caswell R. C., Gacesa P., Lutrell K. E., Weightman A. J. (1989) Molecular cloning and heterologous expression of a Klebsiella pneumoniae gene encoding alginate lyase. Gene 75, 127–134 [DOI] [PubMed] [Google Scholar]
- 35. Lee J. E., Cornell K. A., Riscoe M. K., Howell P. L. (2001) Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure 9, 941–953 [DOI] [PubMed] [Google Scholar]
- 36. Truebestein L., Tennstaedt A., Mönig T., Krojer T., Canellas F., Kaiser M., Clausen T., Ehrmann M. (2011) Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 18, 386–388 [DOI] [PubMed] [Google Scholar]
- 37. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collection in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 38. Pape T., Schneider T. R. (2004) HKL2MAP: a graphical user interface for phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 39. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D. Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 43. Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 47. Preiss J., Ashwell G. (1962) Alginic acid metabolism in bacteria. I. Enzymatic formation of unsaturated oligosaccharides and 4-deoxy-l-erythro-5-hexoseulose uronic acid. J. Biol. Chem. 237, 309–316 [PubMed] [Google Scholar]
- 48. Walvoort M. T., van den Elst H., Plante O. J., Kröck L., Seeberger P. H., Overkleeft H. S., van der Marel G. A., Codée J. D. (2012) Automated solid-phase synthesis of β-mannuronic acid alginates. Angew. Chem. Int. Ed. Engl. 51, 4393–4396 [DOI] [PubMed] [Google Scholar]
- 49. Wang W., Kitova E. N., Klassen J. S. (2003) Influence of solution and gas phase processes on protein-carbohydrate binding affinities determined by nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 75, 4945–4955 [DOI] [PubMed] [Google Scholar]
- 50. Kitova E. N., El-Hawiet A., Schnier P. D., Klassen J. S. (2012) Reliable determinations of protein-ligand interactions by direct ESI-MS measurements. Are we there yet? J. Am. Soc. Mass Spectrom. 23, 431–441 [DOI] [PubMed] [Google Scholar]
- 51. Sun J., Kitova E. N., Wang W., Klassen J. S. (2006) Method for distinguishing specific from nonspecific protein-ligand complexes in nanoelectrospray ionization mass spectrometry. Anal. Chem. 78, 3010–3018 [DOI] [PubMed] [Google Scholar]
- 52. Yoder M. D., Keen N. T., Jurnak F. (1993) New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science 260, 1503–1507 [DOI] [PubMed] [Google Scholar]
- 53. Douthit S. A., Dlakic M., Ohman D. E., Franklin M. J. (2005) Epimerase active domain of Pseudomonas aeruginosa AlgG, a protein that contains a right-handed β-helix. J. Bacteriol. 187, 4573–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciccarelli F. D., Copley R. R., Doerks T., Russell R. B., Bork P. (2002) CASH—a β-helix domain widespread among carbohydrate-binding proteins. Trends Biochem. Sci. 27, 59–62 [DOI] [PubMed] [Google Scholar]
- 56. Buchan D. W., Ward S. M., Lobley A. E., Nugent T. C., Bryson K., Jones D. T. (2010) Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38, W563–W568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garron M. L., Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 [DOI] [PubMed] [Google Scholar]
- 58. Lombard V., Bernard T., Rancurel C., Brumer H., Coutinho P. M., Henrissat B. (2010) A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem. J. 432, 437–444 [DOI] [PubMed] [Google Scholar]
- 59. Vitali J., Schick B., Kester H. C., Visser J., Jurnak F. (1998) The tree-dimensional structure of Aspergillus niger pectin lyase B at 1.7-Å resolution. Plant Physiol. 116, 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mayans O., Scott M., Connerton I., Gravesen T., Benen J., Visser J., Pickersgill R., Jenkins J. (1997) Two crystal structures of pectin lyase A from Aspergillus reveal a pH driven conformational change and striking divergence in the substrate-binding clefts of pectin and pectate lyases. Structure 5, 677–689 [DOI] [PubMed] [Google Scholar]
- 61. Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 [DOI] [PubMed] [Google Scholar]
- 62. Gimmestad M., Sletta H., Ertesvåg H., Bakkevig K., Jain S., Suh S. J., Skjåk-Braek G., Ellingsen T. E., Ohman D. E., Valla S. (2003) The Pseudomonas fluorescens AlgG protein, but not its mannuronan C-5-epimerase activity, is needed for alginate polymer formation. J. Bacteriol. 185, 3515–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mikami B., Ban M., Suzuki S., Yoon H. J., Miyake O., Yamasaki M., Ogura K., Maruyama Y., Hashimoto W., Murata K. (2012) Induced-fit motion of a lid loop involved in catalysis in alginate lyase A1-III. Acta Crystallogr. D. Biol. Crystallogr. 68, 1207–1216 [DOI] [PubMed] [Google Scholar]
- 64. Pacheco B., Maccarana M., Goodlett D. R., Malmström A., Malmström L. (2009) Identification of the active site of DS-epimerase 1 and requirement of N-glycosylation for enzyme function. J. Biol. Chem. 284, 1741–1747 [DOI] [PubMed] [Google Scholar]
- 65. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 66. Høidal H. K., Ertesvåg H., Skjåk-Braek G., Stokke B. T., Valla S. (1999) The recombinant Azotobacter vinelandii mannuronan C-5-epimerase AlgE4 epimerizes alginate by a nonrandom attack mechanism. J. Biol. Chem. 274, 12316–12322 [DOI] [PubMed] [Google Scholar]
- 67. Dominguez D. C. (2004) Calcium signalling in bacteria. Mol. Microbiol. 54, 291–297 [DOI] [PubMed] [Google Scholar]
- 68. Boyd C. D., Chatterjee D., Sondermann H., O'Toole G. A. (2012) LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0–1, is a calcium-dependent protease. J. Bacteriol. 194, 4406–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Larsen B., Haug A. (1971) Biosynthesis of alginate. 3. Tritium incorporation with polymannuronic acid 5-epimerase from Azotobacter vinelandii. Carbohydr. Res. 20, 225–232 [DOI] [PubMed] [Google Scholar]
- 70. Scavetta R. D., Herron S. R., Hotchkiss A. T., Kita N., Keen N. T., Benen J. A., Kester H. C., Visser J., Jurnak F. (1999) Structure of a plant cell wall fragment complexed to pectate lyase C. Plant Cell 11, 1081–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Creze C., Castang S., Derivery E., Haser R., Hugouvieux-Cotte-Pattat N., Shevchik V. E., Gouet P. (2008) The crystal structure of pectate lyase peli from soft rot pathogen Erwinia chrysanthemi in complex with its substrate. J. Biol. Chem. 283, 18260–18268 [DOI] [PubMed] [Google Scholar]
- 72. Roy C., Kester H., Visser J., Shevchik V., Hugouvieux-Cotte-Pattat N., Robert-Baudouy J., Benen J. (1999) Modes of action of five different endopectate lyases from Erwinia chrysanthemi 3937. J. Bacteriol. 181, 3705–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ertesvåg H., Høidal H. K., Skjåk-Braek G., Valla S. (1998) The Azotobacter vinelandii mannuronan C-5-epimerase AlgE1 consists of two separate catalytic domains. J. Biol. Chem. 273, 30927–30932 [DOI] [PubMed] [Google Scholar]
- 74. Ramstadab M. V., Markussen S., Ellingsen T. E., Skjåk-Braek G., Levine D. W. (2001) Influence of environmental conditions on the activity of the recombinant mannuronan C-5-epimerase AlgE2. Enzyme Microb. Technol. 28, 57–69 [DOI] [PubMed] [Google Scholar]
- 75. Svanem B. I., Strand W. I., Ertesvag H., Skjåk-Braek G., Hartmann M., Barbeyron T., Valla S. (2001) The catalytic activities of the bifunctional Azotobacter vinelandii mannuronan C-5-epimerase and alginate lyase AlgE7 probably originate from the same active site in the enzyme. J. Biol. Chem. 276, 31542–31550 [DOI] [PubMed] [Google Scholar]
- 76. Hannesson H. H., Hagner-McWhirter A., Tiedemann K., Lindahl U., Malmström A. (1996) Biosynthesis of dermatan sulphate. Defructosylated Escherichia coli K4 capsular polysaccharide as a substrate for the D-glucuronyl C-5 epimerase, and an indication of a two-base reaction mechanism. Biochem. J. 313, 589–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hagner-Mcwhirter A., Lindahl U., Li J. (2000) Biosynthesis of heparin/heparan sulphate: mechanism of epimerization of glucuronyl C-5. Biochem. J. 347, 69–75 [PMC free article] [PubMed] [Google Scholar]
- 78. Whitney J. C., Howell P. L. (2013) Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol. 21, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morgan J. L., Strumillo J., Zimmer J. (2013) Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature 493, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Koyama M., Helbert W., Imai T., Sugiyama J., Henrissat B. (1997) Parallel-up structure evidences the molecular directionality during biosynthesis of bacterial cellulose. Proc. Natl. Acad. Sci. U.S.A. 94, 9091–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Breyer W. A., Matthews B. W. (2001) A structural basis for processivity. Protein Sci. 10, 1699–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Campa C., Holtan S., Nilsen N., Bjerkan T. M., Stokke B. T., Skjåk-Braek G. (2004) Biochemical analysis of the processive mechanism for epimerization of alginate by mannuronan C-5 epimerase AlgE4. Biochem. J. 381, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Holtan S., Bruheim P., Skjåk-Braek G. (2006) Mode of action and subsite studies of the guluronan block-forming mannuronan C-5 epimerases AlgE1 and AlgE6. Biochem. J. 395, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sletmoen M., Skjåk-Braek G., Stokke B. T. (2004) Single-molecular pair unbinding studies of mannuronan C-5 epimerase AlgE4 and its polymer substrate. Biomacromolecules 5, 1288–1295 [DOI] [PubMed] [Google Scholar]