

Background: HDAC inhibitors exert anti-inflammatory properties.
Results: ITF2357 shifts the balance from Th17 cells toward regulatory T cells via suppression of the IL-6R expression on naïve T cells.
Conclusion: The HDAC inhibitor ITF2357 modulates T cell polarization in experimental colitis.
Significance: Learning the mode of action of this compound class will serve to optimize future therapeutic strategies.
Keywords: Colitis, Histone Deacetylase Inhibitors, Inflammatory Bowel Disease, Macrophages, STAT3, IL-6R, Il-17, T Helper Cells, Promoter Acetylation
Abstract
Histone deacetylase (HDAC) inhibitors have been associated primarily with an anti-proliferative effect in vitro and in vivo. Recent data provide evidence for an anti-inflammatory potency of HDAC inhibitors in models of experimental colitis. Because the balance of T cell subpopulations is critical for the balance of the mucosal immune system, this study explores the regulatory potency of HDAC inhibitors on T cell polarization as a mechanistic explanation for the observed anti-inflammatory effects. Although HDAC inhibition suppressed the polarization toward the pro-inflammatory T helper 17 (Th17) cells, it enhanced forkhead box P3 (FoxP3)+ regulatory T cell polarization in vitro and in vivo at the site of inflammation in the lamina propria. This was paralleled by a down-regulation of the interleukin 6 receptor (IL-6R) on naïve CD4+ T cells on the mRNA as well as on the protein level and changes in the chromatin acetylation at the IL6R gene and its promoter. Downstream of the IL-6R, HDAC inhibition was followed by a decrease in STAT3 phosphorylation as well as retinoic acid receptor-related orphan receptor γT (RORγT) expression, thus identifying the IL-6/STAT3/IL-17 pathway as an important target of HDAC inhibitors. These results directly translated to experimental colitis, where IL-6R expression was suppressed in naïve T cells, paralleled by a significant reduction of Th17 cells in the lamina propria of ITF2357-treated animals, resulting in the amelioration of disease. This study indicates that, in experimental colitis, inhibition of HDAC exerts an anti-inflammatory potency by directing T helper cell polarization via targeting the IL-6 pathway.
Introduction
Epigenetic modifications represent an additional regulatory level for gene expression and, thus, comprise potential therapeutic targets for autoinflammatory diseases or cancer. This work focuses on the modification of histone acetylation that is under a physiological state regulated by the balance of histone acetyltransferases and histone deacetylases (HDACs).2 HDAC inhibitors have been primarily described for their antiproliferative potency, as shown in animal models for colon, mammary, prostate, and bladder cancer (1, 2). Subsequent clinical trials resulted in Food and Drug Administration approval of the HDAC inhibitor suberoylanilide hydroxamid acid (SAHA) for the treatment of cutaneous T cell lymphoma (3). Remarkably, these antiproliferative effects have been partially attributed to the inhibition of the nuclear factor-κB pathway (4, 5). At the same time, a strong anti-inflammatory capacity of this compound class was identified. Initial in vitro studies revealed a dose-dependent suppression of proinflammatory cytokine production by SAHA at concentrations 10- to 100-fold lower than required for apoptosis induction (6, 7). These results were complemented in vivo by investigating inflammatory models such as concanavalin A-induced hepatitis or LPS-induced shock in mice (7). Following these initial data, the anti-inflammatory potency could be confirmed in models of nephritis (8), arthritis (9), intestinal inflammation (6, 10), graft versus host disease (11), and asthma (12). In addition, the HDAC inhibitor ITF2357 has shown clinical efficacy in clinical trials for juvenile arthritis (13).
The maintenance of the mucosal homeostasis requires the balance of CD4+ T cell subsets within the compartment of the lamina propria. In particular, proinflammatory T helper 17 (Th17) cells and anti-inflammatory regulatory T (Treg) cells have been identified as critical factors that define the course and severity of intestinal inflammation (14, 15). This concept is supported by studies indicating a beneficial effect by targeting the Th17-inducing cytokine IL-23 in experimental colitis (16). This strategy has been proven successful in clinical trials for inflammatory bowel disease (17, 18). In addition, transfer of regulatory T cells resulted in the amelioration in various models of experimental colitis (19). Thus, for inflammatory bowel disease as well as for other chronic inflammatory conditions, a therapeutic approach targeting both T cell subsets would be intriguing.
For the in vitro generation of both Th17 and inducible Treg cells from naïve CD4+ cells, TGFβ has been identified as a mandatory cytokine (20, 21). Adding IL-6 to low doses of TGFβ results in the differentiation of Th17 cells in vitro (22–24), thus identifying IL-6 as the key cytokine defining the polarization of these two cell types. IL-6 is a pleiotropic cytokine and was first described to function as a B cell differentiation factor being produced by several cell types, such as macrophages, monocytes, and T cells (25). In acute inflammation such as sepsis, IL-6 is critical for the induction of acute phase reactions (26).
IL-6 deploys its effects through binding to the IL-6 receptor (IL-6R). The IL-6·IL-6R complex then signals through the ubiquitously expressed subunit gp130, leading to activation of the JAK/STAT pathway (classic signaling) (27). Remarkably, the expression of the membrane-bound IL-6R is restricted to naïve T cells and hepatocytes, thus emphasizing the significance of this cytokine for these cell types. However, many other cell types can be activated by IL-6. To enable signaling in cells not expressing the IL-6R, the soluble IL-6R forms a complex with IL-6 that binds to gp130 and, thereby, activates cells that would be naturally unresponsive to IL-6 (28).
The aim of this study was to define the effect of HDAC inhibition on CD4+ T cell polarization and to provide an explanation for the anti-inflammatory effects observed in vivo. In fact, we were able to link HDAC inhibition to decreased IL-6R expression, resulting in suppression of the associated signaling pathway and, ultimately, the suppression of an inflammatory T cell response.
MATERIALS AND METHODS
Reagents
PBS, RPMI 1640 medium, DMEM, penicillin/streptomycin, and sterile l-glutamine solution were from PAA Laboratories (Cölbe, Germany). Fetal calf serum was bought from Linaris (Bettingen, Germany), and LPS (ultrapure) was from InvivoGen (Toulouse, France). All other chemicals were obtained from Sigma-Aldrich (Taufkirchen, Germany). Cytokines were measured using the respective cytometric bead array Flex-Sets (BD Biosciences).
HDAC Inhibitors
ITF2357 was synthesized by the Chemical Department of Italfarmaco (Cinisello Balsamo, Italy) and processed as described previously (5). SAHA was provided by MSD (Haar, Germany). SAHA and ITF2357 were diluted in sterile distilled water and heated to 95 °C for complete dissolution before being added to the cultures. For use in animal models, SAHA or ITF2357 were freshly dissolved in sterile PBS, heated to 95 °C, and kept at room temperature.
Mice
Six- to eight-week-old female BALB/c mice were obtained from Harlan Winkelmann (Borchen, Germany). DO11.10 mice were obtained from the Bundesinstitut für Risikobewertung (Berlin, Germany). Animal protocols were approved by the regional animal study committee of Berlin (Germany).
Experimental Colitis
Colitis was induced by administering 4% dextran sulfate sodium (DSS; molecular mass 40 kDa; MP Biomedicals, Illkirch, France) dissolved in drinking water from day 1 to day 7, followed by 2 days of regular drinking water. ITF2357 and SAHA treatment were initiated simultaneously at the start of colitis induction via oral gavage. Final doses were 10 mg/kg body weight (ITF2357) and 50 mg/kg body weight (SAHA), administered in 200 μl of PBS once daily.
Assessment of Colitis Severity
Body weight and the presence of occult or gross blood per rectum were determined daily. Bleeding was scored as follows: 0, no blood using hemocare (Fresenius, Friedberg, Germany); 2 points, positive hemocare samples; and 4 points, gross bleeding. Mice were sacrificed by cervical dislocation. The entire colon was removed from the cecum to the anus, and colon length was measured as a marker of inflammation. Whole colon tissue (1 cm) was cut open longitudinally and cultured overnight in 1 ml of serum-free medium. Cytokine concentrations were assessed by ELISA and calculated as a ratio versus total protein concentration.
Cell Culture
Lymphocytes were isolated from spleens and mesenteric lymph nodes (MLN) using standard procedures. Lamina propria mononuclear cells (LPMC) were isolated from the colon as described previously (6). Naïve T cells were defined as CD4+ CD62L+. Purification (>98%) was achieved through MACS technology (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were incubated with ITF2357 for 1, 2, or 3 h and subsequently lysed for RNA isolation or precipitation. For the generation of Treg cells, naïve T cells were stimulated with plate-bound anti-CD3 (clone 2C11, 10 μg/ml) and anti-CD28 (clone 37.51, 3 μg/ml). For the polarization of naïve T cells to Th1, Th2, or Th17 cells, antigen-specific activation was performed. Naïve T cells were isolated from DO11.10 mice (transgenic for a T cell receptor specific for ovalbumin peptide OVA323–339 (29)) and cocultured with T cell-depleted splenocytes from congenic BALB/c mice in the presence of OVA323–339 (5 μg/ml). Polarizing conditions and achieved polarization (antibody concentrations of 10 μg/ml) were as follows: Th1 (IL-12 (30 ng/ml), anti-IL-4 (11B11); IFNγ >50%), Th2 (IL-4 (50 ng/ml), anti-IL12 (c17.8), anti-IFNγ: (XMG1.2); IL-4 >30%), Th17 (TGFβ (10 ng/ml), IL-6 (20 ng/ml), anti-IL4, anti-IFNγ; IL-17 >30%), and Treg cells (TGFβ, anti-IL-4, anti-IFNγ; FoxP3 >30%).
To generate murine bone marrow-derived macrophages, the cavities of femur and tibia bones of BALB/c mice were flushed with PBS. The single cell suspensions were cultured in high-glucose DMEM supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml macrophage colony-stimulating factor (Peprotech, Hamburg, Germany). After 24 h of incubation, non-adherent macrophage progenitor cells were isolated and incubated for another 6 days. Thereafter, non-adhesive cells were discarded. Only populations with >98% of CD11b+ F4/80+ cells were used for further analysis. Cells (106/ml) were then cultured in medium devoid of macrophage colony-stimulating factor and incubated with the indicated concentration of ITF2357 for 1 h before further processing. LPS (1 μg/ml) was added for 18 h as described below.
Flow Cytometry Analysis
For intracellular staining, cells were incubated for 4 h with 50 ng/ml phorbol myristate acetate and 500 ng/ml ionomycin, plus 2 mg/ml brefeldin A during the last 2 h. The cells were kept in a tissue culture incubator at 37 °C. Cells were fixed with 4% paraformaldehyde for 5 min at 37 °C and subsequently stained for cytokines in 0.5% saponin for 25 min at room temperature. Intranuclear FoxP3 as well as RORγT staining were performed using the FoxP3 staining kit from eBioscience (San Diego, CA) according to the manual. STAT3 staining was performed using 100% methanol as the permeabilization reagent. Surface staining was performed for 10 min on ice. Anti-CD4, anti-CD62L, anti-IFNγ, and anti-IL-4 were purchased from BD Biosciences. Anti-IL-17A, anti-FoxP3, and anti-phosphorylated STAT3 (anti-pSTAT3) were from eBioscience. Fixable live/dead dye (aqua) was purchased from Invitrogen and used as indicated.
Real-time RT-PCR
Total RNA was isolated using the RNAeasy kit (Qiagen, Hilden, Germany) and reverse-transcribed (high-capacity cDNA reverse transcription kit, Invitrogen). Quantitative PCR was performed using TaqMan Universal Master Mix, the TaqMan gene expression assay, and the StepOnePlus real-time PCR system (all from Applied Biosystems). Primers were purchased from TibMolBiol (Berlin, Germany): IL-6R, CCAggTgCCCTgTCAgTATT (forward) and TTgTCACCCTCCAggATCTC (reverse); IL-6R promoter, ACAATCTCTgggCTCgA (forward) and GGCTCTggCTgTTAAAgTAg (reverse); IL-6 promoter, TCgATgCTAAACgACgTCACA (forward) and CTCCAATgCTCAAgTCTT (reverse); and GAPDH, CATCCTgCACCACCAACTgC (forward) and ACgCCACAgCTTTCCAgAgg (reverse).
Western Blot Analysis
For pSTAT3, 106 cells were lysed in SDS loading buffer (Cell Signaling Technology, Beverly, MA), heated to 95 °C for 5 min, and loaded onto a 7% polyacrylamide gel. The proteins were transferred to a PVDF membrane and detected with ECL reagent (GE Healthcare). The following antibodies were applied: anti-STAT3 and anti-pSTAT3 (Cell Signaling Technology) and anti-β-actin and horseradish peroxidase-labeled rabbit anti-goat and goat anti-rabbit antibodies (Dako, Hamburg, Germany). Densitometric analysis was done using Fuji MultiGauge software (Fujifilm, Düsseldorf, Germany).
ChIP
Cells were lysed in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris (pH 8.1)) and fixed with 0.5% formaldehyde, followed by sonication. ChIP was performed using protein A MicroBeads (Miltenyi Biotec) and anti-acetyl-histone 3 antibody (Lys9, Cell Signaling Technology) according to the protocol of the manufacturer.
Statistical Analysis
The data are expressed as mean ± S.E. Statistical significance of differences between treatment and control groups were determined by factorial analysis of variance and a Bonferroni-Dunn procedure as a post hoc test using GraphPad Prism software (GraphPad Software, La Jolla, CA).
RESULTS
HDAC Inhibition Impacts CD4+ T Cell Polarization
We recently demonstrated an anti-inflammatory effect of HDAC inhibitor treatment in experimental colitis (6). To reveal the mechanism behind this anti-inflammatory effect on a cellular level at the site of inflammation, LPMCs were stimulated with concanavalin A in the presence or absence of increasing concentrations of the HDAC inhibitors ITF2357 or SAHA. HDAC inhibition dose-dependently decreased IFNγ synthesis down to 20% relative to control samples (Fig. 1A). This effect could be reproduced in purified CD4+ T cells, with the rate of inhibition ranging from 40–70% (Fig. 1B). Besides IFNγ, IL-6 synthesis was almost completely suppressed (up to 90%), whereas the release of the Th2 cytokine IL-4 remained unaffected (Fig. 1B).
FIGURE 1.

HDAC inhibition suppresses IFNγ and IL-6 production in vitro as well as in vivo. LPMCs (A) as well as CD4+ T cells (B) were isolated from healthy wild-type mice and were pretreated with increasing concentrations of the HDAC inhibitors SAHA or ITF2357 for 1 h. Cells were subsequently stimulated with concanavalin A (Con A) for 48 h, and IFNγ, IL-6, or IL-4 concentrations were determined in the supernatants. C, DSS colitis was induced as described under “Materials and Methods.” Mice were treated with either vehicle, SAHA (50 mg/kg body weight), or ITF2357 (10 mg/kg body weight) once daily. At the end of the experiment, colon culture was performed, and the concentration of IFNγ and IL-6 in the supernatant was analyzed. Error bars represent the mean ± S.E. (n = 6 (A), n = 8 (B), and n = 7 (C)). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
This suppression of IFNγ and IL-6 production mediated by HDAC inhibition on a cellular level could be translated to ex vivo colon cultures from colitic mice that were treated with HDAC inhibitors (Fig. 1C). Having shown that HDAC inhibition altered the quality of the T cell response, and considering the impact of this cell type in intestinal inflammation, we next asked whether HDAC inhibitors exhibit the potency to regulate T cell polarization. To address this question, naïve CD4+ T cells were polarized in the presence or absence of the HDAC inhibitor ITF2357 into four different T helper cell subsets, namely Th1, Th2, Th17, and induced Treg cells. No influence on the “classic” T cell polarization to either Th1 or Th2 cells was observed (data not shown). In contrast, the presence of 200 nm ITF2357 resulted in increased Treg cell generation and a profound suppression of Th17 cell polarization (Fig. 2).
FIGURE 2.

HDAC inhibition regulates T helper cell polarization in vitro. Naïve CD4+ T cells were isolated from DO.11.10 mice, and antigen-specific T cell stimulation was performed under polarizing conditions in the presence of increasing concentrations of the HDAC inhibitor ITF2357, as indicated. At day 7, intracellular staining was performed for IL-17 and IFNγ as well as FoxP3. A, representative staining for the indicated experimental groups. B, error bars represent the mean ± S.E. (n = 6). *, p < 0.05; **p < 0.01.
HDAC Inhibition in Vivo Regulates T Helper Cell Polarization at the Site of Inflammation
To evaluate whether the effect of HDAC inhibition on T helper cell subsets might contribute to the anti-inflammatory potency during intestinal inflammation, colitis was induced by DSS, and mice received either the HDAC inhibitor ITF2357 or vehicle during the entire experimental time course (Fig. 3G). At the end of the experiment, the ratio of CD4+/IL-17+ as well as CD4+/FoxP3+ T cells within the LPMCs and the MLNs was analyzed (Fig. 3, A–F). In healthy mice, no considerable changes in the experimental groups were detected. In contrast, in mice suffering from colitis, inhibition of HDAC was followed by a significant increase of Treg cells in the lamina propria as well as in the draining MLNs (Fig. 3, A–C). In line with the in vitro data, this increase in Treg cells was paralleled by a significant decrease of Th17 cells at both sites to levels of healthy control mice (Fig. 3, D–F).
FIGURE 3.

HDAC inhibition regulates T cell polarization in vivo. DSS colitis was induced as described under “Materials and Methods.” Mice were treated either with vehicle or ITF2357 (10 mg/kg body weight) once daily. Mononuclear cells were isolated from the lamina propria (LPMCs), and lymphocytes from the MLNs were prepared. Cells were gated on CD4+, and the percentage of FoxP3+ (A–C) or IL-17+ cells (D–F) was evaluated. A and D, representative staining. The numbers indicate the percentage of FoxP3-positive (left panels) or IL-17-positive (right panels) cells among the CD4+ T cells. Staining for LPMCs (B and E) and for MLNs (C and F) is shown. G, weight was determined daily and expressed as the percentage of initial body weight. The bleeding score was determined daily. At the end of the experiment, colon length was measured. All three groups were fed daily via oral gavage, either ITF2357 or vehicle (PBS), whereas the H2O group was given regular drinking water instead of DSS. Horizontal lines represent the mean (n = 8). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
HDAC Inhibition Suppresses IL-6R Expression and Downstream Signaling
When polarizing naïve CD4+ T cells with TGFβ, the presence of IL-6 determines the direction toward Th17 cells, whereas the absence of IL-6 results in inducible Treg cells. Thus, the ability of naïve CD4+ T cells treated with ITF2357 to react to IL-6 was analyzed. In the presence of ITF2357, naïve CD4+ T cells down-regulated IL-6R expression on the cell surface in a time- and dose-dependent manner, as shown in protein as well as mRNA levels (Fig. 4, A and B). Confirming the impact of the HDAC inhibitor, a hyperacetylation of histone 3 at the site of the IL-6R locus was detected, whereas the IL-6R promoter region was deacetylated after ITF2357 treatment. The IL-6 promoter region remained unaffected (Fig. 4C). In parallel to the in vitro findings, the IL-6R was down-regulated significantly in the naïve CD4+ T cells from MLNs of ITF2357-treated mice (Fig. 4D). This effect was observed in healthy as well as in diseased mice but appeared statistically stronger in the DSS experiments.
FIGURE 4.

ITF2357 down-regulates IL-6R expression on naïve CD4+ T cells. A, naïveCD4+ T cells were isolated from BALB/c mice and cultured in the presence of ITF2357 as indicated. Cells were harvested at the indicated time points, and naïve CD4+ T cells were analyzed for their IL-6R expression via flow cytometry. Left panel, representative histogram blots. Right panel, shift in the mean fluorescence intensity (MFI) in the presence of ITF2357 as indicated. B, naïve 4+ T cells were isolated from BALB/c mice and cultured in the presence of ITF2357 (200 nm). Cells were analyzed for their expression of IL-6R mRNA relative to untreated cells via quantitative PCR. C, after 3 h, these naïve CD4+ T cells were analyzed for their histone 3 acetylation at the IL-6R gene site via ChIP/quantitative PCR displayed as fold enrichment of the IL-6R gene (top panel), IL-6R promoter (center panel), and IL-6 promoter (bottom panel) in the histone 3 precipitate. D, DSS colitis was induced, and mice were treated with either vehicle (PBS) or ITF2357 (10 mg/kg body weight) once daily. Naïve CD4+ T cells from MLNs were analyzed for IL-6R expression. Left panel, representative histogram blots, including the gating on naïve CD4+ T cells. Right panel, mean fluorescence intensity (MFI) for IL-6R (two experiments with n = 5 for each group). Data are mean ± S.E. (n = 8 (A), n = 12 (B), n = 4 (C), n = 10 (D)). *, p < 0.05; **, p < 0.005.
To reveal whether or not the decrease in IL-6R expression affects downstream signaling, phosphorylation of STAT3 was evaluated (30). Fig. 5 indicates that the presence of the HDAC inhibitor ITF2357 resulted in a dose-dependent reduction of IL-6-induced STAT3 phosphorylation in naïve CD4+ T cells. These findings are complemented by additional signaling data indicating that RORγT expression downstream of STAT3 is also down-regulated in ITF2357-treated, Th17-polarized T cells (Fig. 6).
FIGURE 5.

ITF2357 reduces STAT3 phosphorylation on naïve CD4+ T cells. Naïve 4+ T cells were isolated from BALB/c mice, preincubated with ITF2357 for 1 h, and subsequently treated with IL-6 for 15 min. Cells were analyzed for STAT3 phosphorylation via Western blot analysis (A) and flow cytometry (B). The left panels show representative Western blot analyses or histogram plots. Error bars represent the mean ± S.E. (n = 3). *, p < 0.05.
FIGURE 6.

RORγT expression is diminished in ITF2357-treated, Th17-polarized T cells. Naïve CD4+ T cells were isolated from DO.11.10 mice, and antigen-specific T cell stimulation was performed under Th17-polarizing conditions in the presence of ITF2357 (200 mm). On day 5, intracellular staining was performed for RORγT. Exemplary cytometry data are shown as a dot blot (A) and histogram (B). The columns (C) represent percentages of RORγT-positive cells (two experiments, n = 12 each). Data are mean ± S.E. ***, p < 0.001.
HDAC Inhibition Modulates IL-6 and IL-6R Expression of Macrophages
Having shown that HDAC inhibition leads to a suppressed response to IL-6 in naïve CD4+ T cells and a subsequent redirection of T cell polarization, one has to consider the effect of HDAC inhibition on other IL-6-producing cell populations. Macrophages are known to release IL-6 as well as soluble IL-6R upon stimulation and represent a critical cell type within the lamina propria and for directing T cell polarization (31, 32). LPS stimulation of macrophages in the presence of ITF2357 resulted in a dose-dependent reduction of IL-6R and soluble IL-6R expression. Remarkably, the expression of gp130 remained unaffected (Fig. 7, A–C). In addition to receptor expression, IL-6 synthesis itself was strongly down-regulated by HDAC inhibition (Fig. 7D).
FIGURE 7.
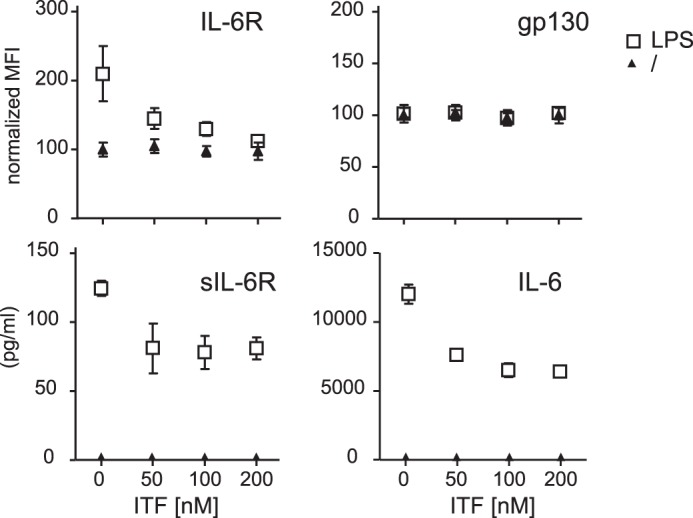
ITF2357 down-regulates IL-6R expression as well as IL-6 itself. Bone marrow-derived macrophages generated from BALB/c mice were preincubated with ITF2357 as indicated and stimulated with LPS (1 μg/ml) overnight. IL-6R (A) and gp130 (B) were analyzed via flow cytometry (mean fluorescence intensity (MFI) normalized to untreated cells). Soluble IL-6R (C) and IL-6 (D) were analyzed by ELISA. Data are mean ± S.E. (n = 3). *, p < 0.05.
DISCUSSION
Inhibition of HDAC results in the amelioration of colitis in a variety of models. However, the underlying mechanism remains largely to be defined. In these models, the severity, but even more the chronicity, of disease depends on the presence of CD4+ T cells and the balance of distinct subpopulations at the site of inflammation (33). This study serves to identify the regulatory capacity of HDAC inhibition on T cell polarization and, consecutively, on this critical balance of T cell subpopulations. A regulatory role of T cell function was indicated by the suppression of IL-6 and IFNγ synthesis in stimulated CD4+ T cells in the presence of an HDAC inhibitor. More important, HDAC inhibition enhanced Treg cell generation and suppressed polarization toward Th17 cells in vitro as well as in vivo at the site of inflammation. Because Th1 and Th2 polarization was unaffected, the IL-6 pathway came into focus as potential target because IL-6 represents the key cytokine distinguishing polarization toward Th17 cells from inducible Treg cells. The subsequent studies revealed a unique anti-inflammatory mode of action via regulation of the IL-6R and downstream signaling.
Genetic as well as environmental factors contribute to the dysregulation of the mucosal immune system in Crohn disease and ulcerative colitis (34). Although the balance of T helper cell subpopulations has been identified to play a critical role in inflammatory bowel disease, recent studies provide evidence that Th17 cells, in particular, add to the inflammatory process in the lamina propria (35, 36). Several studies identified IL-6 to be essential for the development of Th17 cells (22–24). Noguchi et al. (37) were able to demonstrate that blocking the IL-6 pathway through a specific IL-6R antibody resulted not only in the amelioration of experimental colitis but, furthermore, was paralleled by a significant decrease in Th17 cells in the lamina propria.
Treatment with pan-HDAC inhibitors ameliorates experimental colitis in mice and inhibits the production of proinflammatory cytokines at the site of inflammation (6). Via stimulation of LPMCs in the presence of HDAC inhibitors, the local effect of these compounds on effector cells could be proven. A similar dose-dependent suppression of IFNγ and IL-6 synthesis was observed when isolated CD4+ cells were stimulated in the presence of HDAC inhibitors. Both experiments suggest that the local T helper cell population within the lamina propria represents a critical target for HDAC inhibition. In the majority of experimental colitis models, Th1 as well as Th17 cells have been shown to represent a key cell population in the disease course (38). The absence of IL-17-producing T cells in the transfer colitis model almost completely abolished the characteristic inflammation (39). In contrast, the presence of Treg cells has been demonstrated to exert an ameliorating effect in experimental colitis (40, 41).
There are several options regarding how HDAC inhibition might modulate the T cell response toward an anti-inflammatory direction. Studying the effects of HDAC inhibition on T helper cell polarization, we were able to demonstrate, in vitro, that the release of Th1 cytokines was suppressed significantly by treatment with HDAC inhibitors (Fig. 1 and Refs. 6, 7, 42). Although the polarization toward either Th1 or Th2 cells was not affected by the presence of an HDAC inhibitor, the generation of Treg cells was enhanced significantly, and the polarization of Th17 cells was suppressed dose-dependently. In parallel to the in vitro findings, the analysis of T cell subsets in the lamina propria and MLN from mice with experimental colitis treated with ITF2357 showed a reduction in the frequency of Th17 cells within the CD4+ T cell fraction, and, vice versa, the Treg cell population was increased. Hence, the anti-inflammatory shift in the Treg/Th17 cell balance demonstrated in vitro could be confirmed directly in the animal model at the site of inflammation, delivering an explicit mode of action for these compounds regarding the anti-inflammatory effect exerted in a variety of inflammation models (6–12).
Complementary to our findings, Wayne Hancock and co-workers (10, 43) showed an HDAC inhibitor-dependent increase in Treg cells that mediated the anti-inflammatory effect. Because experimental colitis was associated with an increased local expression of HDAC9, HDAC9 knockout mice were subsequently investigated in models of experimental colitis where they proved to be protected (10, 43). For the IL-17-producing T cells, Koenen et al. (44) described a reduced in vitro differentiation of Treg to Th17 cells in the presence of pan-HDAC inhibitors. Furthermore, Bosisio et al. (45) suggest that the production of Th1- and Th17-polarizing cytokines by dendritic cells is suppressed by HDAC inhibition. Together with our data regarding the lack of Th1 polarizing cytokines and the decrease in Th17 cells, an explanation for the anti-inflammatory potency in various models of inflammation is provided.
Considering the effect of HDAC inhibition on IL-6 production and the critical role of IL-6 in the polarization of naïve CD4+ T cells toward Th17 cells, the suppression of IL-6 signaling could result in a consecutive decrease of Th17 cells. Thus, our following studies targeted the IL-6/IL-6R pathway. IL-6 exerts its function by binding to the IL-6R, starting a cascade, with STAT3 being one of the key signal transduction molecules downstream (30). HDAC inhibition not only resulted in a down-regulation of the IL-6R but, consecutively, lead to a dose-dependent reduction of IL-6-induced STAT3 phosphorylation in naïve CD4+ T cells. Modifications of this pathway lead directly to RORγT, the key transcription factor for IL17 production, and, therefore, to the polarization of Th17 cells (46). Consequently, RORγT is also down-regulated in ITF2357-treated cells. To prove a direct impact of the HDAC inhibitor on the IL-6R expression on the chromatin level, we assessed changes in the histone acetylation pattern on the respective loci. Although our ChIP data revealed a hyperacetylation for the IL-6R gene locus, which is mostly associated with an increased transcription of the respective region, the IL-6R promoter locus was, in fact, deacetylated, indicating reduced gene transcription and, thus, confirming the proposed mechanism.
Furthermore, our data demonstrate that, also in macrophages, the expression of the IL-6R is diminished by ITF2357, even in its soluble form. Given that the secretion of IL-6 itself is reduced in macrophages, one of the main sources of this cytokine in the lamina propria, we were able to describe a massive down-modulation of the IL-6 signaling pathway in our models.
In view of these data and the literature discussed, we propose the IL-6/STAT3/IL-17 pathway as critical in the anti-inflammatory effect achieved by HDAC inhibition. Other studies have described IL-6 as the determining factor in the polarization of Th17 from naïve CD4+ T cells, thus being crucial for obtaining the balance between Treg and Th17 cells (47). Because naïve CD4+ T cells are one of the few cell types in possession of the membrane-bound IL-6R, they are offering a possible target for the indicated regulation of T cell polarization (48). Complementary to the results presented here, it has been described that treatment of multiple myeloma cells with HDAC inhibitors resulted in down-modulation of IL-6R signaling (49, 50). Here, the authors emphasized the functional consequences achieved by down-modulation of the IL-6R because the response to IL-6 was equally reduced, as determined by STAT3 phosphorylation (50). Clinical studies are underway to evaluate HDAC inhibitors as a treatment option for relapsed multiple myeloma (51), adding to the variety of other substances targeting the IL-6R in pharmacotherapy for different diseases, such as rheumatoid arthritis or Crohn disease, with remarkable clinical effects (47, 52, 53). Hence, HDAC inhibitors might provide a novel therapeutic approach for the treatment of autoimmune diseases and other diseases by targeting the IL-6 receptor pathway and, thereby, influencing the critical balance of T cell subpopulations at the site of inflammation.
Acknowledgments
We thank Inka Freise for technical assistance and Ulrike Erben for critical reading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft Grant SI 749/5-3 and by the Helmholtz Alliance Preclinical Cancer Comprehensive Center.
- HDAC
- histone deacetylase
- SAHA
- suberoylanilide hydroxamid acid
- Th17 cell
- T helper 17 cell
- Treg cell
- regulatory T cell
- IL-6R
- IL-6 receptor
- DSS
- dextran sulfate sodium
- MLN
- mesenteric lymph node
- LPMC
- lamina propria mononuclear cell
- RORγT
- retinoic acid receptor-related orphan receptor γT.
REFERENCES
- 1. Kelly W. K., Marks P. A. (2005) Drug insight. Histone deacetylase inhibitors. Development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat. Clin. Pract. Oncol. 2, 150–157 [DOI] [PubMed] [Google Scholar]
- 2. Marks P., Rifkind R. A., Richon V. M., Breslow R., Miller T., Kelly W. K. (2001) Histone deacetylases and cancer. Causes and therapies. Nat. Rev. Cancer 1, 194–202 [DOI] [PubMed] [Google Scholar]
- 3. Marks P. A., Breslow R. (2007) Dimethyl sulfoxide to vorinostat. Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 [DOI] [PubMed] [Google Scholar]
- 4. Krämer O. H., Baus D., Knauer S. K., Stein S., Jäger E., Stauber R. H., Grez M., Pfitzner E., Heinzel T. (2006) Acetylation of Stat1 modulates NF-κB activity. Genes Dev. 20, 473–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glauben R., Batra A., Stroh T., Erben U., Fedke I., Lehr H. A., Leoni F., Mascagni P., Dinarello C. A., Zeitz M., Siegmund B. (2008) Histone deacetylases. Novel targets for prevention of colitis-associated cancer in mice. Gut 57, 613–622 [DOI] [PubMed] [Google Scholar]
- 6. Glauben R., Batra A., Fedke I., Zeitz M., Lehr H. A., Leoni F., Mascagni P., Fantuzzi G., Dinarello C. A., Siegmund B. (2006) Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 176, 5015–5022 [DOI] [PubMed] [Google Scholar]
- 7. Leoni F., Zaliani A., Bertolini G., Porro G., Pagani P., Pozzi P., Donà G., Fossati G., Sozzani S., Azam T., Bufler P., Fantuzzi G., Goncharov I., Kim S. H., Pomerantz B. J., Reznikov L. L., Siegmund B., Dinarello C. A., Mascagni P. (2002) The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. U.S.A. 99, 2995–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mishra N., Reilly C. M., Brown D. R., Ruiz P., Gilkeson G. S. (2003) Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111, 539–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishida K., Komiyama T., Miyazawa S., Shen Z. N., Furumatsu T., Doi H., Yoshida A., Yamana J., Yamamura M., Ninomiya Y., Inoue H., Asahara H. (2004) Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 50, 3365–3376 [DOI] [PubMed] [Google Scholar]
- 10. Tao R., de Zoeten E. F., Ozkaynak E., Chen C., Wang L., Porrett P. M., Li B., Turka L. A., Olson E. N., Greene M. I., Wells A. D., Hancock W. W. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13, 1299–1307 [DOI] [PubMed] [Google Scholar]
- 11. Reddy P., Sun Y., Toubai T., Duran-Struuck R., Clouthier S. G., Weisiger E., Maeda Y., Tawara I., Krijanovski O., Gatza E., Liu C., Malter C., Mascagni P., Dinarello C. A., Ferrara J. L. (2008) Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Invest. 118, 2562–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choi J. H., Oh S. W., Kang M. S., Kwon H. J., Oh G. T., Kim D. Y. (2005) Trichostatin A attenuates airway inflammation in mouse asthma model. Clin. Exp. Allergy 35, 89–96 [DOI] [PubMed] [Google Scholar]
- 13. Vojinovic J., Damjanov N., D'Urzo C., Furlan A., Susic G., Pasic S., Iagaru N., Stefan M., Dinarello C. A. (2011) Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 63, 1452–1458 [DOI] [PubMed] [Google Scholar]
- 14. Mucida D., Park Y., Kim G., Turovskaya O., Scott I., Kronenberg M., Cheroutre H. (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 [DOI] [PubMed] [Google Scholar]
- 15. Fantini M. C., Rizzo A., Fina D., Caruso R., Becker C., Neurath M. F., Macdonald T. T., Pallone F., Monteleone G. (2007) IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur. J. Immunol. 37, 3155–3163 [DOI] [PubMed] [Google Scholar]
- 16. Elson C. O., Cong Y., Weaver C. T., Schoeb T. R., McClanahan T. K., Fick R. B., Kastelein R. A. (2007) Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 [DOI] [PubMed] [Google Scholar]
- 17. Sands B. E., Jacobson E. W., Sylwestrowicz T., Younes Z., Dryden G., Fedorak R., Greenbloom S. (2010) Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn's disease. Inflamm. Bowel Dis. 16, 1209–1218 [DOI] [PubMed] [Google Scholar]
- 18. Sandborn W. J., Gasink C., Gao L. L., Blank M. A., Johanns J., Guzzo C., Sands B. E., Hanauer S. B., Targan S., Rutgeerts P., Ghosh S., de Villiers W. J., Panaccione R., Greenberg G., Schreiber S., Lichtiger S., Feagan B. G. (2012) Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N. Engl. J. Med. 367, 1519–1528 [DOI] [PubMed] [Google Scholar]
- 19. Izcue A., Coombes J. L., Powrie F. (2006) Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 212, 256–271 [DOI] [PubMed] [Google Scholar]
- 20. Chen W., Jin W., Hardegen N., Lei K. J., Li L., Marinos N., McGrady G., Wahl S. M. (2003) Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volpe E., Servant N., Zollinger R., Bogiatzi S. I., Hupé P., Barillot E., Soumelis V. (2008) A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 9, 650–657 [DOI] [PubMed] [Google Scholar]
- 22. Bettelli E., Carrier Y., Gao W., Korn T., Strom T. B., Oukka M., Weiner H. L., Kuchroo V. K. (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 [DOI] [PubMed] [Google Scholar]
- 23. Mangan P. R., Harrington L. E., O'Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T. (2006) Transforming growth factor-β induces development of the T(H)17 lineage. Nature 441, 231–234 [DOI] [PubMed] [Google Scholar]
- 24. Veldhoen M., Hocking R. J., Atkins C. J., Locksley R. M., Stockinger B. (2006) TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 [DOI] [PubMed] [Google Scholar]
- 25. Hirano T., Yasukawa K., Harada H., Taga T., Watanabe Y., Matsuda T., Kashiwamura S., Nakajima K., Koyama K., Iwamatsu A. (1986) Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 324, 73–76 [DOI] [PubMed] [Google Scholar]
- 26. Hideshima T., Mitsiades C., Tonon G., Richardson P. G., Anderson K. C. (2007) Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 7, 585–598 [DOI] [PubMed] [Google Scholar]
- 27. Hibi M., Murakami M., Saito M., Hirano T., Taga T., Kishimoto T. (1990) Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63, 1149–1157 [DOI] [PubMed] [Google Scholar]
- 28. Rose-John S., Heinrich P. C. (1994) Soluble receptors for cytokines and growth factors. Generation and biological function. Biochem. J. 300, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murphy K. M., Heimberger A. B., Loh D. Y. (1990) Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science 250, 1720–1723 [DOI] [PubMed] [Google Scholar]
- 30. Stephanou A., Isenberg D. A., Akira S., Kishimoto T., Latchman D. S. (1998) The nuclear factor interleukin-6 (NF-IL6) and signal transducer and activator of transcription-3 (STAT-3) signalling pathways co-operate to mediate the activation of the hsp90β gene by interleukin-6 but have opposite effects on its inducibility by heat shock. Biochem. J. 330, 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farache J., Zigmond E., Shakhar G., Jung S. (2013) Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense. Immunol. Cell Biol. 91, 232–239 [DOI] [PubMed] [Google Scholar]
- 32. Hosokawa T., Kusugami K., Ina K., Ando T., Shinoda M., Imada A., Ohsuga M., Sakai T., Matsuura T., Ito K., Kaneshiro K. (1999) Interleukin-6 and soluble interleukin-6 receptor in the colonic mucosa of inflammatory bowel disease. J. Gastroenterol. Hepatol. 14, 987–996 [DOI] [PubMed] [Google Scholar]
- 33. Powrie F. (2004) Immune regulation in the intestine. A balancing act between effector and regulatory T cell responses. Ann. N.Y. Acad. Sci. 1029, 132–141 [DOI] [PubMed] [Google Scholar]
- 34. Siegmund B., Zeitz M. (2009) Clinical aspects of inflammatory bowel disease. Eur. J. Immunol. 39, 2026–2030 [DOI] [PubMed] [Google Scholar]
- 35. Annunziato F., Cosmi L., Santarlasci V., Maggi L., Liotta F., Mazzinghi B., Parente E., Filì L., Ferri S., Frosali F., Giudici F., Romagnani P., Parronchi P., Tonelli F., Maggi E., Romagnani S. (2007) Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujino S., Andoh A., Bamba S., Ogawa A., Hata K., Araki Y., Bamba T., Fujiyama Y. (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noguchi D., Wakita D., Ohkuri T., Tajima M., Chamoto K., Kitamura H., Nishimura T. (2011) Blockade of IL-6-signaling inhibits the pathogenesis of CD4+ T cell-mediated lethal graft-versus-host reaction against minor histocompatibility antigen. Immunol. Lett. 136, 146–155 [DOI] [PubMed] [Google Scholar]
- 38. Dohi T., Fujihashi K. (2006) Type 1 and 2 T helper cell-mediated colitis. Curr. Opin. Gastroenterol. 22, 651–657 [DOI] [PubMed] [Google Scholar]
- 39. Leppkes M., Becker C., Ivanov I. I., Hirth S., Wirtz S., Neufert C., Pouly S., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Becher B., Littman D. R., Neurath M. F. (2009) RORγ-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136, 257–267 [DOI] [PubMed] [Google Scholar]
- 40. Mottet C., Uhlig H. H., Powrie F. (2003) Cutting edge. Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 [DOI] [PubMed] [Google Scholar]
- 41. Kjellev S., Lundsgaard D., Poulsen S. S., Markholst H. (2006) Reconstitution of Scid mice with CD4+CD25− T cells leads to rapid colitis. An improved model for pharmacologic testing. Int. Immunopharmacol. 6, 1341–1354 [DOI] [PubMed] [Google Scholar]
- 42. Leoni F., Fossati G., Lewis E. C., Lee J. K., Porro G., Pagani P., Modena D., Moras M. L., Pozzi P., Reznikov L. L., Siegmund B., Fantuzzi G., Dinarello C. A., Mascagni P. (2005) The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 11, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Zoeten E. F., Wang L., Sai H., Dillmann W. H., Hancock W. W. (2010) Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 138, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koenen H. J., Smeets R. L., Vink P. M., van Rijssen E., Boots A. M., Joosten I. (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112, 2340–2352 [DOI] [PubMed] [Google Scholar]
- 45. Bosisio D., Vulcano M., Del Prete A., Sironi M., Salvi V., Salogni L., Riboldi E., Leoni F., Dinarello C. A., Girolomoni G., Sozzani S. (2008) Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J. Leukocyte Biol. 84, 1540–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang X. O., Panopoulos A. D., Nurieva R., Chang S. H., Wang D., Watowich S. S., Dong C. (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282, 9358–9363 [DOI] [PubMed] [Google Scholar]
- 47. Ichiyama K., Yoshida H., Wakabayashi Y., Chinen T., Saeki K., Nakaya M., Takaesu G., Hori S., Yoshimura A., Kobayashi T. (2008) Foxp3 inhibits RORγt-mediated IL-17A mRNA transcription through direct interaction with RORγt. J. Biol. Chem. 283, 17003–17008 [DOI] [PubMed] [Google Scholar]
- 48. Jones G. W., McLoughlin R. M., Hammond V. J., Parker C. R., Williams J. D., Malhotra R., Scheller J., Williams A. S., Rose-John S., Topley N., Jones S. A. (2010) Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J. Immunol. 184, 2130–2139 [DOI] [PubMed] [Google Scholar]
- 49. Mitsiades C. S., Mitsiades N. S., McMullan C. J., Poulaki V., Shringarpure R., Hideshima T., Akiyama M., Chauhan D., Munshi N., Gu X., Bailey C., Joseph M., Libermann T. A., Richon V. M., Marks P. A., Anderson K. C. (2004) Transcriptional signature of histone deacetylase inhibition in multiple myeloma. Biological and clinical implications. Proc. Natl. Acad. Sci. U.S.A. 101, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Todoerti K., Barbui V., Pedrini O., Lionetti M., Fossati G., Mascagni P., Rambaldi A., Neri A., Introna M., Lombardi L., Golay J. (2010) Pleiotropic anti-myeloma activity of ITF2357. Inhibition of interleukin-6 receptor signaling and repression of miR-19a and miR-19b. Haematologica 95, 260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weber D. M., Graef T., Hussein M., Sobecks R. M., Schiller G. J., Lupinacci L., Hardwick J. S., Jagannath S. (2012) Phase I trial of vorinostat combined with bortezomib for the treatment of relapsing and/or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 12, 319–324 [DOI] [PubMed] [Google Scholar]
- 52. Ito H., Takazoe M., Fukuda Y., Hibi T., Kusugami K., Andoh A., Matsumoto T., Yamamura T., Azuma J., Nishimoto N., Yoshizaki K., Shimoyama T., Kishimoto T. (2004) A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 126, 989–996 [DOI] [PubMed] [Google Scholar]
- 53. Maini R. N., Taylor P. C., Szechinski J., Pavelka K., Bröll J., Balint G., Emery P., Raemen F., Petersen J., Smolen J., Thomson D., Kishimoto T., and CHARISMA Study Group (2006) Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 54, 2817–2829 [DOI] [PubMed] [Google Scholar]