

Background: Regulation and function of ERK5 in adipose depots is not known.
Results: Deletion of ERK5 in adipose depots increases adiposity and adipokine secretion. Reduced phosphorylation and potentiated NFATc4 nuclear localization are found. ERK5 also modulates PKA activation.
Conclusion: ERK5 impinges on NFATc4 nucleo-cytoplasmic shuttling and modulates PKA activation in adipocytes.
Significance: These results expand the repertoire of ERK5 physiological function and downstream targets.
Keywords: Adipose tissue, MAP Kinases (MAPKs), Metabolic Regulation, NFAT Transcription Factor, Protein Kinase A (PKA)
Abstract
Increased adiposity due to energy imbalance is a critical factor of the epidemic crisis of obesity and type II diabetes. In addition to the obvious role in energy storage, regulatory factors are secreted from adipose depots to control appetite and cellular homeostasis. Complex signaling cross-talks within adipocyte are also evident due to the metabolic and immune nature of adipose depots. Here, we uncover a role of extracellular signal-regulated kinase 5 (ERK5) in adipocyte signaling. We find that deletion of ERK5 in adipose depots (adipo-ERK5−/−) increases adiposity, in part, due to increased food intake. Dysregulated secretion of adipokines, leptin resistance, and impaired glucose handling are also found in adipo-ERK5−/− mice. Mechanistically, we show that ERK5 impinges on transcription factor NFATc4. Decreased phosphorylation at the conserved gate-keeping Ser residues and increased nuclear localization of NFATc4 are found in adipo-ERK5−/− mice. We also find attenuated PKA activation in adipo-ERK5−/− mice. In response to stimulation of β-adrenergic G-protein-coupled receptor, we find decreased NFATc4 phosphorylation and impaired PKA activation in adipo-ERK5−/− mice. Reduced cAMP accumulation and increased phosphodiesterase activity are also found. Together, these results demonstrate integration of ERK5 with NFATc4 nucleo-cytoplasmic shuttling and PKA activation in adipocyte signaling.
Introduction
Obesity correlates with the epidemic crisis of type II diabetes in the United States and in many other countries. Increased adiposity due to energy imbalance is a critical factor of obesity/diabetes. Adipose tissue has been traditionally considered as the major fat and energy storage at the obese state. Recent advances have indicated that adipose depots are active endocrine tissues and secret regulatory factors to control appetite and cellular homeostasis.
A variety of bioactive secretory proteins, including complements, growth factors, chemokines, and cytokines (generally termed as adipokines) are released from adipose tissue to regulate the growth and maintenance of adipose tissue (1–4). Adipokines are also required for the vascular homeostasis and inflammation through intercellular communications with stromal vascular cells and infiltrating monocytes/macrophages, respectively. Given the metabolic and immune nature of adipose depots, complex signaling cross-talks within adipocyte are also evident (5–12). These include sympathetic/parasympathetic activation of β-adrenergic signaling and immune signaling in response to metabolic dysregulations. Therefore, understanding of the signaling networks in adipocyte is important to combat obesity and its subsequent pathogenesis.
Mitogen-activated protein kinase (MAPK) is a group of Ser/Thr protein kinases activated by mitogen and stress (13–18). The MAPK cascades regulate diverse physiological processes, including cell growth, proliferation, differentiation, and death. The MAPK cascade is composed of mitogen-activated protein kinase kinase kinase (MAP3K), which phosphorylates and activates mitogen-activated protein kinase kinase (MAP2K). MAP2K further phosphorylates and leads to activation of MAPK. Extracellular signal-regulated kinase 5 (ERK5) is a member of the MAPK family and is activated by upstream MAP2K MEK5, which is activated by MAP3K, MEKK2, or MEKK3 (16, 19–21). ERK5 is also known as BMK1 (big mitogen-activated protein kinase 1) because it is more than twice the size of other members of the MAPK family (22, 23). The difference in size is, in part, due to the presence of a transcription activation domain in ERK5.
Little is known about the role of ERK5 in adipose tissues, in part, due to embryonic lethality of Erk5−/− mice during development (24–27). Erk5−/− embryo was found dead at around embryonic day 10.5 due to defects in angiogenesis and vascular formation. Endothelium-specific deletion of ERK5 led to vasculature deformation and embryonic lethality (27), phenocopying the observations found in systemic Erk5−/− embryos (24–27). Cardiomyocyte-specific Erk5−/− mice, however, are fertile and exhibited normal heart development (19). These data indicated that ERK5 in endothelial cells is the major contributor for the defects in vasculature and subsequent heart deformation found in systemic Erk5−/− mice in utero.
Recent studies indicate that neuron-specific deletion of ERK5 (using Nestin-Cre) affected development of the olfactory bulb and impaired odor discrimination (28). Furthermore, conditional deletion of ERK5 (using Nestin-CreER) altered adult neurogenesis (29). Additional tissue-specific Erk5−/− mice have also been generated, including hepatocyte-specific Erk5−/− mice (using albumin-Cre), mammary epithelium-specific Erk5−/− mice (using mouse mammary tumor virus (MMTV)-Cre), and T cell-specific Erk5−/− mice (using CD4- or Vav-Cre) (19, 30). Unfortunately and surprisingly, no obvious phenotype has been reported from these tissue-specific Erk5−/− mice despite the wide tissue expression of ERK5. Hence, the physiological role of peripheral ERK5, except for its role in vasculature, remains elusive.
The lack of knowledge of physiological substrates of ERK5 further complicates our understanding of ERK5 function in vivo. Connexin 43, p90 RSK, and MEF2C can be phosphorylated by ERK5 (31–34). Phosphorylation of RSK by ERK5 may mediate a cross-talk for the role of ERK5 in NFκB activation (35–37). The ERK5-MEF2C axis may be important for the skeletal muscle cell differentiation and neurotrophin signaling (32, 38, 39). The ERK5 signaling is also integrated into Akt (40) and PKA signaling (35, 41, 42). In addition, ERK5 suppresses the expression of cyclin-dependent protein kinase inhibitors and regulates cell cycle re-entry (43). Our recent findings that ERK5 regulates rephosphorylation of NFATc4 further expand the repertoire of physiological substrates of ERK5 (44). We propose that ERK5 contributes to the rephosphorylation upon transcription termination of NFATc4.
The transcription factor NFAT family was first identified as an important regulator for IL-2 gene expression (45). It was once hypothesized that NFAT function was restricted to the immune system. However, characterization of NFAT cDNAs indicates that NFAT is involved in numerous biological processes (reviewed in Refs. 46–49). We previously reported that Nfatc2−/− Nfatc4−/− mice are lean and resistant to diet-induced obesity via a non-cell-autonomous mechanism (50). In analogous to its role in cytokine expression in immune cells, we have demonstrated that NFAT also regulates adipokine transcription (50). The physiological relevance of ERK5 on NFAT or on other ERK5 targets in adipose depots is not known.
Here, we uncover a role of ERK5 in adipocyte signaling. We find that deletion of ERK5 in adipose depots increased adiposity, in part, due to increased food intake. Dysregulated secretion of adipokines, leptin resistance, and impaired glucose handling are also found in adipo-ERK5−/− mice. Mechanistically, we show that ERK5 impinges on NFATc4. ERK5 also modulates PKA activation. Together, these results demonstrate integration of ERK5 with NFATc4 nucleo-cytoplasmic shuttling and PKA activation in adipocyte signaling.
EXPERIMENTAL PROCEDURES
Mice
Animal experiments were performed in accordance with guidelines of the Albert Einstein College of Medicine Institute of Animal Studies. Erk5fl/fl mice and adiponectin-Cre transgenic mice were generated as described previously (40, 51). Erk5fl/fl mice were backcrossed with C57BL/6 mice at least 10 times before use. Deletion of ERK5 in adipose depots (adipo-ERK5−/−) was achieved by crossing Erk5fl/fl mice and adiponectin-Cre transgenic mice. Control mice were either Erk+/+ or Erkfl/fl/+ in the background of adiponectin-Cre. All adipo-ERK5−/− and control mice were in the background of homozygous adiponectin-Cre to achieve sufficient deletion as described previously (51). Eight-week-old mice were used for experiments except as indicated. Mice were fed ad libitum with regular chow (Research Diets, Inc.) except when fasted for experiments.
Reagents
Antibodies used were as follows: ERK5 (Upstate Biotechnology, 07-039), phospho-PKA substrate antibodies (Cell Signaling, 9621), hormone-sensitive lipase (Cell Signaling, 4107), phospho-hormone-sensitive lipase (Cell Signaling, 4126), NFATc4 (Santa Cruz Biotechnology, Inc., sc13036), phospho-NFATc4 (Santa Cruz Biotechnology, sc135770), STAT3 (Santa Cruz Biotechnology, sc482), and phospho-STAT3 (Santa Cruz Biotechnology, sc8059). Tubulin antibody (clone E7) was obtained from the monoclonal antibody facility at the University of Iowa. Glucose was measured from mouse tail vein using a glucometer (TrueTest System). Adipokine and insulin measurement was performed by using multiplex mouse adipokine assays and an insulin radioimmunoassay kit from Millipore and LINCO Research, respectively. ERK5-specific inhibitor BIX2189 was obtained from SelleckChem. Phosphodiesterase activity was determined by using the Bridge-It cAMP-PDE assay kits from Mediomics.
Cell Culture
Mouse embryonic fibroblasts (MEFs)2 were prepared from embryonic day 13.5 embryos after trypsin digestion as described previously (50). MEFs were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were harvested in Triton lysis buffer (20 mm Tris, pH 7.4, 134 mm NaCl, 2 mm EDTA, 25 mm β-glycerophosphate, 2 mm NaPPi, 10% glycerol, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine, and 10 μg/ml leupeptin). Fractionation of adipocytes from stromal vascular cells was performed as described previously (52). Briefly, epididymal white adipose depots were collected, minced, and digested with collagenase (1.5 mg/ml) at 37 °C for 1.5 h. After passage through a 100-μm filter to remove undigested tissues, collected cells were centrifuged at 450 × g for 5 min to prepare the stromal vascular fraction from the bottom sediment and adipocytes from the top floating layer.
Metabolic Tests
Adipo-ERK5−/− and control male mice (8 weeks old) were fasted for 4–6 h before intraperitoneal injection with a bolus of insulin (0.75 unit/kg) for insulin tolerance tests (50). Serum glucose was measured from tail vein using a glucometer (TrueTest System) at 0, 15, 30, 45, 60, 90, and 120 min after the insulin injection. For glucose tolerance tests, mice were orally gavaged with a bolus of glucose solution (2.5 g/kg). Serum glucose from the tail vein was measured with a glucometer at 0, 15, 30, 45, 60, 90, and 120 min after the gavage. To determine oxygen consumption (VO2), carbon dioxide production (VCO2), energy expenditure, and locomotor activity, mice were individually housed and were allowed to acclimate to the respiratory chambers for 2 days before the gas exchange measurements began. Data on gas exchanges were recorded for 2–3 days. Indirect calorimetry was performed with a computer-controlled open circuit calorimetry system (Oxymax, Columbus Instruments) composed of eight respiratory chambers. Oxygen consumption, carbon dioxide production, energy expenditure, and locomotor activity were measured from a total of eight adipo-ERK5−/− mice and eight control mice in two independent experiments. Body composition (adiposity and lean mass) of adipo-ERK5−/− and control mice were determined by using NMR-MRI-based technology from EchoMRI. Hypothalamic stimulation by leptin in adipo-ERK5−/− mice and control mice was carried out as described previously (53). Briefly, overnight-fasted adipo-ERK5−/− mice and control mice were injected with recombinant murine leptin (2 mg/kg) for 45 min before harvesting the hypothalamus to detect the levels of STAT3. The levels of phospho-STAT3 were determined by immunoprecipitation and immunoblotting.
β-Adrenergic Receptor Activation
To determine β-adrenergic receptor/PKA activation ex vivo, epididymal adipose depots (white adipose tissues (WAT)) were harvested, minced, and challenged with CL316,243 (1 μm) or isoproterenol (1 μm) for the times indicated. Tissues were homogenized and extracted in Triton lysis buffer. Extracts prepared were subjected to immunoblotting analysis using phospho-PKA motif antibodies. Expression of tubulin in tissue extracts was used for normalization. Cyclic AMP was measured from minced adipose depots after isoproterenol stimulation (5 min) using a colorimetric enzyme immunoassay kit (Arbor Assays, K019).
Immunofluorescence Analysis and Immunohistochemistry
Paraffin-embedded sections of epididymal adipose depots from adipo-ERK5−/− and control male mice were deparaffinized in xylene and rehydrated through a series of ethanol (100, 100, 95, 70, and 50%) and double-distilled water. Antigen retrieval was performed by heating the sections in a microwave oven in sodium citrate buffer (0.3% sodium citrate, 0.4% citric acid, pH 6.0). After rinsing in PBS, the sections were blocked in 5% goat serum for 30 min and incubated overnight with primary antibody at 4 °C. The sections were washed three times in PBS and incubated with secondary antibody for 1 h. After washing in PBS three times, the sections were mounted with DAPI-fluor mounting solution, and images were taken with a Leica AOBS SP2 confocal microscope with a ×63 objective lens. For immunoperoxidase staining, paraffin-embedded sections were deparaffinized in xylene and rehydrated with ethanol as described above. Sections were quenched in methanol containing 0.3% H2O2, rinsed with Tris-buffered saline (TBS), and blocked for 1 h in TBS containing 1% BSA, 5% serum, and 1% Triton-X-100. Sections were incubated with primary antibody (1:500) for 48 h at 4 °C. After rinsing in TBS, sections were left in labeled polymer-HRP anti-rabbit secondary antibody (DAKO EnVision+ System-HRP-DAB) for 2 h at room temperature. After washing three times in TBS, sections were stained with DAB for 10 min. Remaining DAB solution was rinsed off with TBS, and sections were dehydrated with ethanol and examined using a Zeiss axiovert microscope with a ×20 objective lens.
Statistical Analysis
Data are shown as mean ± S.E. Student's t test was performed, and values of p < 0.05 were considered to be significant.
RESULTS
Reduced Expression of ERK5 in Adipose Depots of Adipo-ERK5−/− Mice
To investigate the role of ERK5 in adipocyte signaling, we deleted ERK5 in adipose tissues. Reduced expression of ERK5 in white adipose tissues was found in adipo-ERK5−/− mice (Fig. 1A). As expected, expression of ERK5 in heart, liver, and skeletal muscle was similar in control and adipo-ERK5−/− mice (Fig. 1A). Quantitative RT-PCR also showed significant reduction of ERK5 mRNA in white adipose tissues of the adipo-ERK5−/− mice (Fig. 1B). The remaining expression of ERK5 in adipose depots of the adipo-ERK5−/− mice could be derived from non-adipocytes, such as endothelial cells and infiltrating immune cells.
FIGURE 1.

Reduced expression of ERK5 in adipo-ERK5−/− mice. A, expression of ERK5 protein in epididymal fat pads (WAT), heart, liver, and skeletal muscle of adipo-ERK5−/− and control mice. B, expression of ERK5 and actin in epididymal fat pads of adipo-ERK5−/− and control mice was quantitated by RT-quantitative PCR. Relative expression of ERK5 and actin was normalized to the expression of cyclophilin and shown. C, detection of ERK5 expression in adipose depots of adipo-ERK5−/− and control mice by immunohistochemistry. *, crownlike morphology of adipocytes in adipo-ERK5−/− mice. D, adipocytes and stromal vascular cells were separated by density centrifugation. The levels of ERK5 in the isolated adipocytes and stromal vascular cells were determined. *, p < 0.05. Error bars, S.E.
To ascertain reduced expression of ERK5 in adipocytes of the adipo-ERK5−/− mice, we performed immunochemistry to detect ERK5 (Fig. 1C). Furthermore, we performed density centrifugation to separate adipocytes from stromal vascular cells and determined the levels of ERK5 expression by immunoblots (Fig. 1D). Immunohistochemistry revealed reduced expression of ERK5 in adipocytes of the adipo-ERK5−/− mice (Fig. 1C). Immunochemistry also showed the presence of infiltrating immune cells, which expressed high levels of ERK5, in the adipose depots of the adipo-ERK5−/− mice (Fig. 1C). In addition, we found reduced expression of ERK5 in the isolated adipocytes of the adipo-ERK5−/− mice (Fig. 1D). The levels of ERK5 in the stromal vascular fractions of the adipo-ERK5−/− mice, however, were potentiated, supporting the presence of infiltrating immune cells in the adipose depots of the adipo-ERK5−/− mice (Fig. 1C). These data indicate reduced expression of ERK5 in adipocytes of the adipo-ERK5−/− mice. The presence of infiltrating immune cells and crownlike morphology suggest inflammation and possible metabolic changes upon deletion of ERK5 in adipocytes.
Increased Adiposity in Adipo-ERK5−/− Mice
To assess metabolic changes in adipo-ERK5−/− mice, age-matched adipo-ERK5−/− mice and control littermates were fed ad libitum with regular chow or a high fat diet for 20 weeks. Adipo-ERK5−/− mice exhibited increase in body weight gain, as compared with control littermates, as early as 10 weeks old (Fig. 2A). Increase in body weight gain was more evident in adipo-ERK5−/− mice fed with high fat diet (Fig. 2B). Increased body weight in adipo-ERK5−/− mice was, in part, due to increased adipose content because the lean mass is indistinguishable in control and adipo-ERK5−/− mice (Fig. 2C). These data demonstrate that ERK5 plays a role in adipose tissues.
FIGURE 2.

Increased adiposity in adipo-ERK5−/− mice. A and B, adipo-ERK5−/− and control mice were fed ad libitum with regular chow (A) or high fat diet (B). Body weight of the mice was determined for 20 weeks and shown. C, 12-week-old adipo-ERK5−/− and control mice were subjected to EchoMRI to determine fat mass and lean mass. *, p < 0.05; #, p < 0.005. Error bars, S.E.
Altered Metabolic Parameters in Adipo-ERK5−/− Mice
Next, we determined the metabolic rate of the adipo-ERK5−/− mice and control littermates (Fig. 3). Neither oxygen consumption nor carbon dioxide production was markedly different between the adipo-ERK5−/− and control mice (Fig. 3, A and B). Hence, similar respiratory quotient and energy expenditure were found in adipo-ERK5−/− and control mice (Fig. 3, C–E).
FIGURE 3.

Altered metabolic parameters in adipo-ERK5−/− mice. Oxygen consumption (VO2) (A), carbon dioxide production (VCO2) (B), respiratory quotient (RER) (C and D), energy expenditure (Heat) (E), food intake (F), meal number (G), and meal size (H) of 12-week-old adipo-ERK5−/− and control mice (n = 8) were determined by an indirect open circuit calorimeter system. Locomotor activity (I and J) of the adipo-ERK5−/− and control mice were also shown. *, p < 0.05; #, p < 0.005. Error bars, S.E.
Food intake of the adipo-ERK5−/− mice, however, was elevated as compared with the control littermates (Fig. 3F). Increased meal number was also found in the adipo-ERK5−/− mice (Fig. 3G). The meal size among the adipo-ERK5−/− and control mice, however, was similar (Fig. 3H). We also observed increased locomotor activity in the adipo-ERK5−/− mice (Fig. 3D), especially in the dark phase (Fig. 3E). Together, these data indicate that increased adiposity may be, in part, due to increased food intake of the adipo-ERK5−/− mice.
Changes in Adipokine Levels in Adipo-ERK5−/− Mice
Given the endocrine nature of adipose tissues, adipokines are released to control appetites and cellular homeostasis (1–4). Because the adipo-ERK5−/− mice showed infiltration by immune cells (Fig. 1) and increased food intake (Fig. 3), we asked whether the level of adipokines was altered. At 20 weeks old, the serum level of leptin, adiponectin, and resistin was elevated in the adipo-ERK5−/− mice (Fig. 4A). Increased fasting levels of glucose and insulin were also found in the adipo-ERK5−/− mice. Adipo-ERK5−/− mice fed with a high fat diet further showed altered adipokine levels (Fig. 4A). The level of inflammatory marker PAI-1, however, was also increased in the adipo-ERK5−/− mice. Changes in the adipokine levels in the 20-week-old adipo-ERK5−/− mice may be a consequence of the elevated inflammatory status.
FIGURE 4.

Changes in adipokine levels in adipo-ERK5−/− mice. The levels of adiponectin, leptin, resistin, and PAI-1 in serum of 20-year-old (A) and 12-year-old (B) adipo-ERK5−/− and control mice (n = 4–6) were determined by mouse adipokine multiplex assays. Fasting levels of glucose and insulin in adipo-ERK5−/− and control mice were also shown. Effects of a high fat diet (HFD) on adipokine levels were also determined. *, p < 0.05; #, p < 0.005. Error bars, S.E.
To investigate whether ERK5 plays a role in adipokine regulation, we determined the adipokine levels of 12-week-old adipo-ERK5−/− mice. At 12 weeks old, the level of PAI-1 in the adipo-ERK5−/− and control mice was similar (Fig. 4B). The level of insulin and resistin was also similar in the adipo-ERK5−/− and control mice. Leptin and adiponectin levels, however, remained elevated in the 12-week-old adipo-ERK5−/− mice. Adipo-ERK5−/− mice also exhibited increased fasting glucose level at 12 weeks old. These data indicate that dysregulated leptin and adiponectin secretion are probably early changes to account for hyperglycemia in the adipo-ERK5−/− mice. Dysregulated insulin and resistin, along with increased PAI-1 secretion, however, might be consequences due to hyperadiponectinemia and/or hyperleptinemia.
Impaired Glucose Handling and Reduced Insulin Sensitivity in Adipo-ERK5−/− Mice
Increased level of fasting glucose suggested that glucose handling and insulin sensitivity might be compromised in the adipo-ERK5−/− mice. We performed glucose tolerance tests and insulin tolerance tests to examine the glucose handling and insulin sensitivity, respectively, in the adipo-ERK5−/− and control mice (Fig. 5). Oral injection of a bolus of glucose to control mice increased serum glucose levels, which were declined and leveled off with time (Fig. 5A). In the adipo-ERK5−/− mice, the level of glucose, however, remained elevated in the early phase (up to ∼45 min) before reduction at a later time (Fig. 5A). The area under the curve further supported significant changes between control and adipo-ERK5−/− mice in glucose tolerance tests. These data indicate that adipo-ERK5−/− mice exhibit impairment, especially in the early phase, in glucose handling.
FIGURE 5.

Impaired glucose handling and reduced insulin sensitivity in adipo-ERK5−/− mice. Eight-week-old adipo-ERK5−/− (n = 7) and control mice (n = 9) were subjected to glucose tolerance tests (A) and insulin tolerance tests (B). The glucose levels were measured at the indicated times and presented. The area under the curve was also determined. *, p < 0.05; #, p < 0.005. Error bars, S.E.
In the insulin tolerance test, injection of a bolus of insulin reduced serum glucose levels in control mice as expected (Fig. 5B). In the adipo-ERK5−/− mice, insulin sensitivity was attenuated in the early time points (Fig. 5B). At later times, the level of glucose in the adipo-ERK5−/− and control mice was similar. The area under the curve further supported significant changes between control and adipo-ERK5−/− mice in insulin tolerance tests. These data indicate that the adipo-ERK5−/− mice show similar attenuation in the early phase of insulin sensitivity.
Leptin Resistance in Adipo-ERK5−/− Mice
Impaired glucose handling and reduced insulin sensitivity (Fig. 5) corroborates with the increased body weight (Fig. 2) and the presence of infiltrating immune cells (Fig. 1) in the adipo-ERK5−/− mice. Increased food intake (Fig. 3F) further suggests that the satiety signal of the adipo-ERK5−/− mice is probably impaired, and this contributes to the metabolic dysregulations. The satiety signal is centrally controlled, in part, by the action of leptin in the hypothalamus (54–57). Adipocyte-derived leptin activates JAK2, which phosphorylates STAT3, in the hypothalamus to suppress feeding. Indeed, ablation of leptin or leptin receptor in mice leads to hyperphagia and obesity (58–60).
Hyperleptinemia and subsequent leptin resistance are well correlated with obesity and insulin insensitivity (54–57). Because the level of leptin in the adipo-ERK5−/− mice is elevated (Fig. 4), we hypothesized that the adipo-ERK5−/− mice may develop leptin resistance. To test this hypothesis, we examined STAT3 activation in the hypothalamus upon leptin stimulation. As expected, leptin stimulation led to a ∼2-fold increase in STAT3 phosphorylation in the hypothalamus of the control mice (Fig. 6). In the adipo-ERK5−/− mice, even in the absence of leptin stimulation, the levels of STAT3 phosphorylation in the hypothalamus were increased ∼2.5-fold as compared with untreated control (Fig. 6). Leptin stimulation, however, did not further significantly increase phosphorylation of STAT3 in the hypothalamus of the adipo-ERK5−/− mice. These data indicate that hyperleptinemia and subsequent leptin resistance contribute to the obese phenotype and metabolic dysregulations in the adipo-ERK5−/− mice.
FIGURE 6.

Leptin resistance in adipo-ERK5−/− mice. A, recombinant murine leptin (2 mg/kg) or saline was administered to overnight-fasted, 12-week-old adipo-ERK5−/− and control mice via intraperitoneal injection. Levels of phospo-STAT3 were determined by immunoprecipitations (IP) and immunoblottings (IB). Levels of STAT3 were also shown. B, relative level of STAT3 phosphorylation was determined by densitometry and shown. *, p < 0.05; NS, not significant.
Integration of ERK5 and NFATc4 in Adipocyte Signaling
Changes in adipokine expression in the adipo-ERK5−/− mice suggested a role of ERK5 in adipocyte signaling. Previously, we showed that transcription factors NFATc2 and NFATc4 contribute to adipokine gene expression (50). We also showed that ERK5 phosphorylates Ser168 and Ser170 of endogenous NFATc4 (44). Phosphorylation of Ser168 and Ser170 on NFATc4 by ERK5 provides a priming effect for subsequent phosphorylation mediated by CK1α protein kinase. Similar conserved gate-keeping Ser residues are found in other NFAT members for the control NFAT subcellular distribution, which is opposed by the calcineurin phosphatase. Dysregulated phosphorylation and altered nucleo-cytoplasmic distribution of NFATc4 will affect adipokine expression in the adipo-ERK5−/− mice.
Next, we tested whether phosphorylation of NFATc4 at Ser168 and Ser170 was affected in the adipo-ERK5−/− mice using a phospho-specific antibody. Immunoblotting analyses showed phosphorylation of Ser168 and Ser170 of NFATc4 in control mice (Fig. 7A). Decreased phosphorylation of NFATc4 at Ser168 and Ser170, however, was found in the adipo-ERK5−/− mice (Fig. 7A). In addition, phosphorylation of NFATc4 at Ser168 and Ser170 upon stimulation of β-adrenergic receptors was also impaired in the adipo-ERK5−/− mice (Fig. 7B). These data indicate that ERK5 phosphorylates Ser168 and Ser170 of NFATc4.
FIGURE 7.
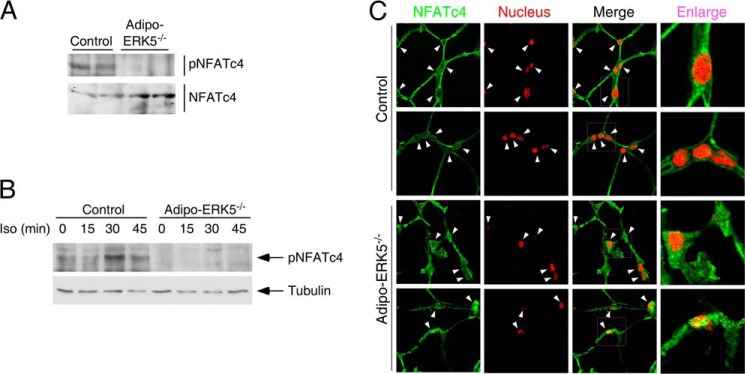
Integration of ERK5 and NFATc4 in adipocyte signaling. A, tissue extracts from epididymal fat pads of 8-week-old adipo-ERK5−/− and control mice were examined to determine the levels of NFATc4 phosphorylation. B, WAT isolated from adipo-ERK5−/− and control mice (n = 3) were challenged with β-adrenergic receptor agonist isoproterenol (Iso; 1 μm) for the times indicated. Tissue extracts prepared were used to determine the levels of NFATc4 phosphorylation. C, paraffin sections of epididymal fat pads of 8-week-old adipo-ERK5−/− and control mice were stained with rabbit polyclonal NFATc4 antibody (green). DNA in nuclei was visualized using DAPI and represented in the red channel. The presence or the lack of NFATc4 in the nucleus is indicated by arrowheads. Error bars, S.E.
Dephosphorylation promotes nuclear localization of NFAT. Next, we determined subcellular distribution of NFATc4 in adipo-ERK5−/− and control mice. Confocal microscopy showed that NFATc4 was mainly resided in the cytosol with little staining in the nucleus in control mice (Fig. 7C). In adipo-ERK5−/− mice, the presence of NFATc4 in the nucleus was evident (Fig. 7C). These data indicate that ablation of ERK5 in adipose tissues reduced phosphorylation and promoted nuclear localization of NFATc4.
Integration of ERK5 and PKA Activation in Adipocyte Signaling
We also examined whether ERK5 impinges on the PKA signaling axis, given the critical role of PKA in adipocyte functions (61, 62). In addition, previous studies showed that ERK5 and cAMP-specific phosphodiesterase-4D (PDE4D) form a scaffolding complex with the PKA-anchoring protein mAKAP (41, 42), which binds PKA. Phosphorylation of PDE4D by PKA activates whereas ERK5 kinase inhibits PDE4D activity. Similar to ERK5, ERK2 also phosphorylates and inhibits PDE4D (63, 64). Dysregulation of PDEs would lead to aberrant turnover of cAMP, affect the duration and amplitude of PKA activation, and contribute to the role of ERK5 in adipocyte functions.
We found that the levels of phosphorylation of PKA substrates in adipo-ERK5−/− mice were reduced as compared with control mice (Fig. 8A). To further ascertain the cross-talk of ERK5 on the PKA signaling axis, we examined the activation of the β-adrenergic receptor pathway in isolated epididymal fat depots in adipo-ERK5−/− and control mice. Ex vivo analysis on the isolated fat depots indicated that β-adrenergic receptor agonists, including isoproterenol (Fig. 8B) and CL316,243 (Fig. 8C), led to an increase in phosphorylation of PKA substrates. Adipose depots isolated from the adipo-ERK5−/− mice, however, showed attenuated phosphorylation of PKA substrates upon stimulation with β-adrenergic receptor agonists (Fig. 8, B and C). Similar reduction in phosphorylation of hormone-sensitive lipase by PKA was also found in the adipo-ERK5−/− mice upon stimulation with isoproterenol (Fig. 8B). These data indicate that ERK5 impinges on the PKA signaling axis in adipose tissues.
FIGURE 8.

Integration of ERK5 and PKA activation in adipocyte signaling. A, tissue extracts from epididymal fat pads of 8-week-old adipo-ERK5−/− and control mice were examined to determine the levels of activation of PKA by immunoblotting analysis using phospho-PKA motif antibodies (Phospho-PKA substrates). B and C, WAT isolated from adipo-ERK5−/− and control mice (n = 3) were challenged with β-adrenergic receptor agonist isoproterenol (Iso; 1 μm) (B) or CL316,243 (CL; 1 μm) (C) for the times indicated. Tissue extracts prepared were used to determine activation of PKA by immunoblotting analysis using phospho-PKA motif antibodies (Phospho-PKA substrates). The effect of isoproterenol stimulation on phosphorylation of hormone-sensitive lipase (HSL) was also shown (B). D, primary embryonic fibroblasts (MEFs) were challenged with β-adrenergic receptor agonist isoproterenol (10 μm) for the times indicated, in the absence (DMSO) and presence (BIX2189) of MEK5 inhibitor (BIX2189; 10 μm). Activation of PKA was determined by using phospho-PKA motif antibodies (Phospho-PKA substrates). E, Mek5+/+ (Control) and Mek5−/− fibroblasts were challenged with β-adrenergic receptor agonist isoproterenol (10 μm) for the times indicated. Activation of PKA was determined by using phospho-PKA motif antibodies (Phospho-PKA substrates). F, WAT isolated from adipo-ERK5−/− and control mice were challenged with isoproterenol (1 μm) for 5 min. Levels of cAMP and protein concentration were determined by ELISA and colorimetric assays, respectively. Relative levels of cAMP production are shown (G). Tissue extracts from epididymal fat pads of 8-week-old adipo-ERK5−/− and control mice were examined to determine the levels of phosphodiesterase activity. *, p < 0.05. Error bars, S.E.
We also examined the effect of ERK5 inhibition in activation of the β-adrenergic receptor pathway. Primary MEFs were pretreated or not with MEK5-specific inhibitor BIX2189 and then stimulation with isoproterenol (Fig. 8D). The effect of ERK5 inhibition in activation of β-adrenergic receptor pathway was also examined in fibroblasts isolated from MEK5 null mice (Fig. 8E). In the absence of MEK5, activation of ERK5 is impaired (65). Similar to deletion of ERK5 in adipose depots, either chemical inhibition of MEK5 (Fig. 8D) or deletion of MEK5 (Fig. 8E) attenuated the level of phosphorylation of PKA substrates upon stimulation with isoproterenol. These data confirm that ERK5 regulates the PKA signaling axis.
Activation of PKA is mediated by second messenger cAMP (41, 66–69). We therefore determined the levels of cAMP upon stimulation with isoproterenol. Ex vivo analysis on the isolated fat depots indicated that isoproterenol led to an increase in cAMP (Fig. 8F). Adipose depots isolated from the adipo-ERK5−/− mice, however, showed reduced accumulation of cAMP (Fig. 8F). These data indicate that ERK5 impinges on the PKA signaling axis in adipose tissues via controlling the levels of cAMP.
Given that ERK5 and PDE4D form a scaffolding complex with mAKAP and ERK5 inhibits PDE4D activity (41, 42), we determined PDE activity from epididymal fat depots in adipo-ERK5−/− and control mice (Fig. 8G). Adipose depots isolated from the adipo-ERK5−/− mice showed increased PDE activity as compared with control mice (Fig. 8G). These data support a previous model in which ERK5 inhibits PDE activity and affects PKA function.
DISCUSSION
In this report, we demonstrate that ERK5 contributes to adipocyte signaling. Impaired adipocyte signaling in the adipo-ERK5−/− mice may account for the dysregulated adipokine secretion. In particular, we find increased adiponectin secretion in the adipo-ERK5−/− mice. In addition to adiponectin, we also find increased levels of leptin in the adipo-ERK5−/− mice. Levels of resistin, another adipokine, are also elevated in the adipo-ERK5−/− mice. Adiponectin regulates energy expenditure in peripheral tissues, such as muscle and liver (70), whereas leptin plays a central role as a satiety signal (54). Hyperleptinemia and subsequent leptin resistance, however, are well correlated with obesity and insulin insensitivity (54–57). Our recent findings also indicate that resistin causes hypothalamic leptin resistance.3 In addition, hyperadiponectinemia correlates with dysregulated insulin signaling (71–74). Thus, increased levels of adipokines, including adiponectin, leptin, and resistin, and subsequent adiponectin dysregulation and/or leptin resistance could account for metabolic dysfunctions found in the adipo-ERK5−/− mice. Indeed, we found that the adipo-ERK5−/− mice showed leptin resistance (Fig. 6). Perhaps activation of AMP-activated protein kinase, a downstream effector of adiponectin receptors, is also dysregulated in the adipo-ERK5−/− mice due to hyperadiponectinemia.
Increased secretion of leptin, resistin, and adiponectin in the adipo-ERK5−/− mice may be due to the effect of ERK5 in regulating adipocyte growth and differentiation. ERK5 suppresses the expression of cyclin-dependent protein kinase inhibitors, such as p21 and p27 (43). The loss of ERK5, therefore, delays cell cycle re-entry and may inadvertently promote cell differentiation. Notably, cell cycle arrest is a prerequisite for adipocyte differentiation (75, 76). Hence, increased expression of leptin, resistin, and adiponectin in the adipo-ERK5−/− mice may be a consequence of an increased number/mass of adipocytes due to aberrant differentiation.
Alternatively, ERK5 may impinge on transcription factors that control adipokine gene expression. Among the known ERK5 effectors, we previously showed that ERK5 opposes calcineurin-mediated NFATc4 nuclear localization (44). In particular, ERK5 is an endogenous kinase regulating rephosphorylation of NFATc4. Indeed, the duration of nuclear NFATc4 is potentiated upon ablation of ERK5. In addition, NFAT plays a role in adiponectin, leptin, and resistin expression (11, 50). In adipo-ERK5−/− mice, we also observed decreased phosphorylation and increased nuclear localization of NFATc4. These data support the model that ERK5 modulates NFATc4 nucleo-cytoplasmic shuttling.
Once in the nucleus, NFAT interacts with distinct NFAT partners to positively and negatively regulate gene transcription. Indeed, NFAT interacts with Fos-Jun and plays an important role in cytokine gene expression in immune cells (77). Analogously, we demonstrated that NFAT interacts with CCAAT/enhancer-binding protein and regulates adipokine gene expression in adipocytes (78). NFAT can also form homodimers to mediate specific gene transcription (79, 80). Formation of hetero- and homo-NFAT nuclear complexes with distinctive partners, along with the differential role of different NFAT kinases, provide multiple mechanisms to positively and negatively regulate gene transcription. Nonetheless, increased NFATc4 nuclear localization in adipo-ERK5−/− mice will probably affect NFATc4-mediated gene expression. Perhaps nuclear NFATc4 contributes to the expression of adiponectin, resistin, and leptin in the adipo-ERK5−/− mice.
In addition to NFATc4, we also show that ERK5 impinges on PKA activation. Our findings that ERK5 negatively regulates PKA activation are parallel to the previous models in which ERK2 phosphorylates and inhibits PDE4 (63, 64). Previous studies showed that ERK2 phosphorylates Ser579 of PDE4D3. An effect on PDE4D enzyme activity due to phosphorylation-induced conformational changes is one proposed mechanism (63, 64, 81). Recently, we showed that PDE4D3 protein stability is phosphorylation-dependent (82). We showed that Ser616 and Ser618 of PDE4D3 are phosphorylated by GSK3β and CK1α, whereas dephosphorylation is carried out by calcineurin. Given that Ser579 is located close to Ser616 and Ser618, which we have identified as a critical phosphodegron for PDE4D3, it is tempting to speculate that phosphorylation at Ser579 of PDE4D3 provides an additional means to control PDE4D3. Notably, phosphorylation at multiple sites has been shown to facilitate phospho-dependent degradation (83, 84). Perhaps ERK phosphorylation coordinates subsequent GSK3β/CK1α phosphorylation, including phosphorylation-induced conformational changes, and modulates PDE4D protein stability. This model is in parallel to the role of JNK MAPK in facilitating GSK3-mediated phospho-dependent degradation of Mcl-1. The priming action of ERK5 to facilitate subsequent phosphorylation mediated by CK1α has also been found in NFATc4 rephosphorylation (44).
The negative role of ERK5 in PDE4D regulation is also found when examining the mAKAP scaffolding complex (41, 42). In this model, mAKAP functions as a scaffolding platform and recruits ERK5, PDE4D, PKA, and calcineurin to control cAMP signaling. The positive role of PKA in PDE4D regulation is opposed by ERK5 phosphorylation. ERK5, therefore, potentiates cAMP-mediated responses by inhibiting PDE4D. Alternatively, ERK5 provides a termination signal to nullify the activation of PDE4D by PKA. Nonetheless, our data support the model in which ERK5 impinges on PKA activation, possibly via modulating PDE4D function.
Our findings that ERK5 regulates NFATc4 nucleo-cytoplasmic shuttling and PKA activation, which are also integrated by calcineurin-mediated dephosphorylation, present an intriguing example of opposing actions of kinases and phosphatases (Fig. 9). The opposing actions of ERK5 and calcineurin control the duration of NFATc4 in the nucleus and provide a long term regulation via gene transcription. For PKA activation, current models indicate that the opposing actions of ERK5 and calcineurin regulate enzyme activity and protein stability, respectively. The ultimate effect of ERK5 and calcineurin will modulate the levels of second messenger cAMP, which controls a wide range of effectors (62, 69, 85). These include cAMP-dependent protein kinase (PKA), cAMP-regulated guanine nucleotide exchange factors (Epac), and cAMP-gated ion channels. It is likely that additional substrates are co-regulated by ERK5 and calcineurin in similar opposing actions. Identification of these common substrates will provide new understanding of the physiological role of ERK5 and calcineurin.
FIGURE 9.
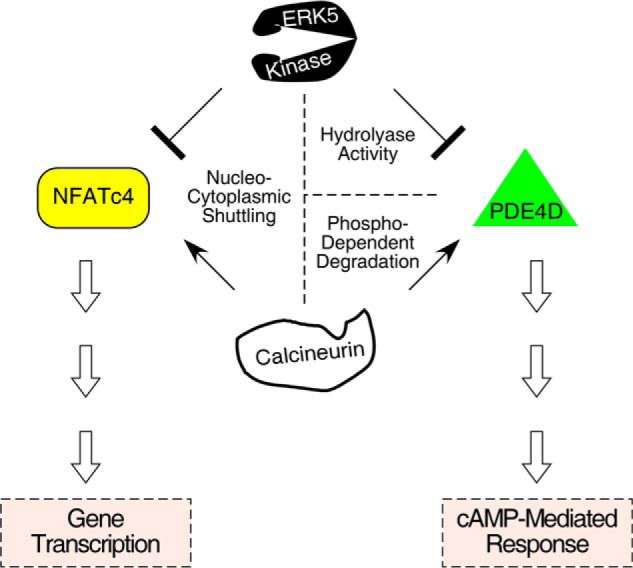
Opposing actions of ERK5 and calcineurin on NFATc4 and PDE4D. Conserved gate-keeping Ser residues are dephosphorylated by calcineurin phosphatase to promote nuclear localization of NFATc4. Nuclear NFATc4 then modulates gene transcription. Rephosphorylation of the conserved gate-keeping Ser residues of nuclear NFATc4 is carried out by ERK5. The opposing actions of ERK5 and calcineurin, therefore, regulate nucleo-cytoplasmic shuttling and subsequent NFATc4-mediated gene transcription. For PDE4D, dephosphorylation mediated by calcineurin stabilizes protein expression and increases cAMP hydrolysis. Hence, phospho-dependent degradation of PDE4D, which is triggered by GSK3β/CK1α phosphorylation, is attenuated upon calcineurin activation. Hydrolyase activity of PDE4D, however, is inhibited by ERK5 phosphorylation and thus decreases cAMP hydrolysis. Therefore, cAMP-mediated responses are opposed by calcineurin and ERK5 by regulating protein stability and enzyme activity of PDE4D, respectively.
In conclusion, we have uncovered interplays of ERK5 in adipocyte signaling. ERK5 impinges on NFATc4-mediated gene transcription, which may account for the changes in adipokine expression. ERK5 also regulates the cAMP signaling pathway by affecting PKA activation via PDE4D. Our data thus provide new physiological insights into the role of ERK5 and its downstream targets.
Acknowledgments
We thank Dr. Cathy Tournier for the Erk5fl/fl mice. We also thank Drs. Hyok-Joon Kwon and Sudarshana Purkayastha for technical advice. Furthermore, we thank members of our laboratories for suggestions and for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK0904881 (to C.-W. C.) and P01-DK088761, R01-DK55758, and R01-DK099110 (to P. E. S.).
P. E. Scherer, unpublished observations.
- MEF
- mouse embryo fibroblast
- WAT
- white adipose tissues
- PDE
- phosphodiesterase
- DAB
- 3,3′-diaminobenzidine.
REFERENCES
- 1. Kim S., Moustaid-Moussa N. (2000) Secretory, endocrine and autocrine/paracrine function of the adipocyte. J. Nutr. 130, 3110S–3115S [DOI] [PubMed] [Google Scholar]
- 2. Steppan C. M., Lazar M. A. (2002) Resistin and obesity-associated insulin resistance. Trends Endocrinol. Metab. 13, 18–23 [DOI] [PubMed] [Google Scholar]
- 3. Rangwala S. M., Lazar M. A. (2000) Transcriptional control of adipogenesis. Annu. Rev. Nutr. 20, 535–559 [DOI] [PubMed] [Google Scholar]
- 4. Rosen E. D., Walkey C. J., Puigserver P., Spiegelman B. M. (2000) Transcriptional regulation of adipogenesis. Genes Dev. 14, 1293–1307 [PubMed] [Google Scholar]
- 5. Collins S., Cao W., Robidoux J. (2004) Learning new tricks from old dogs. β-Adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol. Endocrinol. 18, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 6. Robidoux J., Martin T. L., Collins S. (2004) β-Adrenergic receptors and regulation of energy expenditure. A family affair. Annu. Rev. Pharmacol. Toxicol. 44, 297–323 [DOI] [PubMed] [Google Scholar]
- 7. Niswender K. D., Schwartz M. W. (2003) Insulin and leptin revisited. Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol. 24, 1–10 [DOI] [PubMed] [Google Scholar]
- 8. Lazar M. A. (2007) Resistin- and obesity-associated metabolic diseases. Horm. Metab. Res. 39, 710–716 [DOI] [PubMed] [Google Scholar]
- 9. de Luca C., Olefsky J. M. (2008) Inflammation and insulin resistance. FEBS Lett. 582, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calabro P., Yeh E. T. (2007) Obesity, inflammation, and vascular disease. The role of the adipose tissue as an endocrine organ. Subcell. Biochem. 42, 63–91 [PubMed] [Google Scholar]
- 11. Kim H. B., Kong M., Kim T. M., Suh Y. H., Kim W. H., Lim J. H., Song J. H., Jung M. H. (2006) NFATc4 and ATF3 negatively regulate adiponectin gene expression in 3T3-L1 adipocytes. Diabetes 55, 1342–1352 [DOI] [PubMed] [Google Scholar]
- 12. Tilg H., Moschen A. R. (2006) Adipocytokines. Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6, 772–783 [DOI] [PubMed] [Google Scholar]
- 13. Roberts O. L., Holmes K., Müller J., Cross D. A., Cross M. J. (2009) ERK5 and the regulation of endothelial cell function. Biochem. Soc. Trans. 37, 1254–1259 [DOI] [PubMed] [Google Scholar]
- 14. Rincón M., Davis R. J. (2009) Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 228, 212–224 [DOI] [PubMed] [Google Scholar]
- 15. Kyriakis J. M., Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 [DOI] [PubMed] [Google Scholar]
- 16. Raman M., Chen W., Cobb M. H. (2007) Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112 [DOI] [PubMed] [Google Scholar]
- 17. Johnson G. L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 [DOI] [PubMed] [Google Scholar]
- 18. Morrison D. K., Davis R. J. (2003) Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118 [DOI] [PubMed] [Google Scholar]
- 19. Hayashi M., Lee J. D. (2004) Role of the BMK1/ERK5 signaling pathway. Lessons from knockout mice. J. Mol. Med. 82, 800–808 [DOI] [PubMed] [Google Scholar]
- 20. Wang X., Tournier C. (2006) Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 18, 753–760 [DOI] [PubMed] [Google Scholar]
- 21. Nishimoto S., Nishida E. (2006) MAPK signalling. ERK5 versus ERK1/2. EMBO Rep. 7, 782–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou G., Bao Z. Q., Dixon J. E. (1995) Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 270, 12665–12669 [DOI] [PubMed] [Google Scholar]
- 23. Lee J. D., Ulevitch R. J., Han J. (1995) Primary structure of BMK1. A new mammalian MAP kinase. Biochem. Biophys. Res. Commun. 213, 715–724 [DOI] [PubMed] [Google Scholar]
- 24. Regan C. P., Li W., Boucher D. M., Spatz S., Su M. S., Kuida K. (2002) Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. U.S.A. 99, 9248–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sohn S. J., Sarvis B. K., Cado D., Winoto A. (2002) ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 277, 43344–43351 [DOI] [PubMed] [Google Scholar]
- 26. Yan L., Carr J., Ashby P. R., Murry-Tait V., Thompson C., Arthur J. S. (2003) Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayashi M., Kim S. W., Imanaka-Yoshida K., Yoshida T., Abel E. D., Eliceiri B., Yang Y., Ulevitch R. J., Lee J. D. (2004) Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Invest. 113, 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou J., Pan Y. W., Wang Z., Chang S. Y., Wang W., Wang X., Tournier C., Storm D. R., Xia Z. (2012) Targeted deletion of ERK5 MAP kinase in the developing nervous system impairs development of GABAergic interneurons in the main olfactory bulb and behavioral discrimination between structurally similar odorants. J. Neurosci. 32, 4118–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan Y. W., Chan G. C., Kuo C. T., Storm D. R., Xia Z. (2012) Inhibition of adult neurogenesis by inducible and targeted deletion of ERK5 mitogen-activated protein kinase specifically in adult neurogenic regions impairs contextual fear extinction and remote fear memory. J. Neurosci. 32, 6444–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ananieva O., Macdonald A., Wang X., McCoy C. E., McIlrath J., Tournier C., Arthur J. S. (2008) ERK5 regulation in naive T-cell activation and survival. Eur. J. Immunol. 38, 2534–2547 [DOI] [PubMed] [Google Scholar]
- 31. English J. M., Pearson G., Baer R., Cobb M. H. (1998) Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J. Biol. Chem. 273, 3854–3860 [DOI] [PubMed] [Google Scholar]
- 32. Kato Y., Kravchenko V. V., Tapping R. I., Han J., Ulevitch R. J., Lee J. D. (1997) BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 16, 7054–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ranganathan A., Pearson G. W., Chrestensen C. A., Sturgill T. W., Cobb M. H. (2006) The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch. Biochem. Biophys. 449, 8–16 [DOI] [PubMed] [Google Scholar]
- 34. Cameron S. J., Malik S., Akaike M., Lerner-Marmarosh N., Yan C., Lee J. D., Abe J., Yang J. (2003) Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase 1/ERK5 but not ERK1/2 kinase activation. J. Biol. Chem. 278, 18682–18688 [DOI] [PubMed] [Google Scholar]
- 35. Pearson G. W., Earnest S., Cobb M. H. (2006) Cyclic AMP selectively uncouples mitogen-activated protein kinase cascades from activating signals. Mol. Cell. Biol. 26, 3039–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cude K., Wang Y., Choi H. J., Hsuan S. L., Zhang H., Wang C. Y., Xia Z. (2007) Regulation of the G2-M cell cycle progression by the ERK5-NFκB signaling pathway. J. Cell Biol. 177, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garaude J., Cherni S., Kaminski S., Delepine E., Chable-Bessia C., Benkirane M., Borges J., Pandiella A., Iñiguez M. A., Fresno M., Hipskind R. A., Villalba M. (2006) ERK5 activates NF-κB in leukemic T cells and is essential for their growth in vivo. J. Immunol. 177, 7607–7617 [DOI] [PubMed] [Google Scholar]
- 38. Liu L., Cavanaugh J. E., Wang Y., Sakagami H., Mao Z., Xia Z. (2003) ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 8532–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cavanaugh J. E., Ham J., Hetman M., Poser S., Yan C., Xia Z. (2001) Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J. Neurosci. 21, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X., Finegan K. G., Robinson A. C., Knowles L., Khosravi-Far R., Hinchliffe K. A., Boot-Handford R. P., Tournier C. (2006) Activation of extracellular signal-regulated protein kinase 5 downregulates FasL upon osmotic stress. Cell Death Differ. 13, 2099–2108 [DOI] [PubMed] [Google Scholar]
- 41. Dodge-Kafka K. L., Kapiloff M. S. (2006) The mAKAP signaling complex. Integration of cAMP, calcium, and MAP kinase signaling pathways. Eur. J. Cell Biol. 85, 593–602 [DOI] [PubMed] [Google Scholar]
- 42. Dodge-Kafka K. L., Soughayer J., Pare G. C., Carlisle Michel J. J., Langeberg L. K., Kapiloff M. S., Scott J. D. (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437, 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Perez-Madrigal D., Finegan K. G., Paramo B., Tournier C. (2012) The extracellular-regulated protein kinase 5 (ERK5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (CDKs). Cell. Signal. 24, 2360–2368 [DOI] [PubMed] [Google Scholar]
- 44. Yang T. T., Yu R. Y., Agadir A., Gao G. J., Campos-Gonzalez R., Tournier C., Chow C. W. (2008) Integration of protein kinases mTOR and extracellular signal-regulated kinase 5 in regulating nucleocytoplasmic localization of NFATc4. Mol. Cell. Biol. 28, 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Durand D. B., Shaw J. P., Bush M. R., Replogle R. E., Belagaje R., Crabtree G. R. (1988) Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol. Cell. Biol. 8, 1715–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hogan P. G., Chen L., Nardone J., Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 [DOI] [PubMed] [Google Scholar]
- 47. Rao A., Avni O. (2000) Molecular aspects of T-cell differentiation. Br. Med. Bull. 56, 969–984 [DOI] [PubMed] [Google Scholar]
- 48. Olson E. N., Williams R. S. (2000) Remodeling muscles with calcineurin. BioEssays 22, 510–519 [DOI] [PubMed] [Google Scholar]
- 49. Graef I. A., Chen F., Crabtree G. R. (2001) NFAT signaling in vertebrate development. Curr. Opin. Genet. Dev. 11, 505–512 [DOI] [PubMed] [Google Scholar]
- 50. Yang T. T., Suk H. Y., Yang X., Olabisi O., Yu R. Y., Durand J., Jelicks L. A., Kim J. Y., Scherer P. E., Wang Y., Feng Y., Rossetti L., Graef I. A., Crabtree G. R., Chow C. W. (2006) Role of transcription factor NFAT in glucose and insulin homeostasis. Mol. Cell. Biol. 26, 7372–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Z. V., Deng Y., Wang Q. A., Sun K., Scherer P. E. (2010) Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo K., McMinn J. E., Ludwig T., Yu Y. H., Yang G., Chen L., Loh D., Li C., Chua S., Jr., Zhang Y. (2007) Disruption of peripheral leptin signaling in mice results in hyperleptinemia without associated metabolic abnormalities. Endocrinology 148, 3987–3997 [DOI] [PubMed] [Google Scholar]
- 54. Myers M. G., Cowley M. A., Münzberg H. (2008) Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 70, 537–556 [DOI] [PubMed] [Google Scholar]
- 55. Myers M. G., Jr., Leibel R. L., Seeley R. J., Schwartz M. W. (2010) Obesity and leptin resistance. Distinguishing cause from effect. Trends Endocrinol. Metab. 21, 643–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thaler J. P., Schwartz M. W. (2010) Minireview. Inflammation and obesity pathogenesis. The hypothalamus heats up. Endocrinology 151, 4109–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Enriori P. J., Evans A. E., Sinnayah P., Cowley M. A. (2006) Leptin resistance and obesity. Obesity 14, 254S–258S [DOI] [PubMed] [Google Scholar]
- 58. Lee G. H., Proenca R., Montez J. M., Carroll K. M., Darvishzadeh J. G., Lee J. I., Friedman J. M. (1996) Abnormal splicing of the leptin receptor in diabetic mice. Nature 379, 632–635 [DOI] [PubMed] [Google Scholar]
- 59. Tartaglia L. A., Dembski M., Weng X., Deng N., Culpepper J., Devos R., Richards G. J., Campfield L. A., Clark F. T., Deeds J., Muir C., Sanker S., Moriarty A., Moore K. J., Smutko J. S., Mays G. G., Wool E. A., Monroe C. A., Tepper R. I. (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 60. Coleman D. L. (1978) Obese and diabetes. Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14, 141–148 [DOI] [PubMed] [Google Scholar]
- 61. Holm C. (2003) Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 31, 1120–1124 [DOI] [PubMed] [Google Scholar]
- 62. Ahmad F., Degerman E., Manganiello V. C. (2012) Cyclic nucleotide phosphodiesterase 3 signaling complexes. Horm. Metab. Res. 44, 776–785 [DOI] [PubMed] [Google Scholar]
- 63. Hoffmann R., Baillie G. S., MacKenzie S. J., Yarwood S. J., Houslay M. D. (1999) The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 18, 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. MacKenzie S. J., Baillie G. S., McPhee I., Bolger G. B., Houslay M. D. (2000) ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. The involvement of COOH-terminal docking sites and NH2-terminal UCR regions. J. Biol. Chem. 275, 16609–16617 [DOI] [PubMed] [Google Scholar]
- 65. Wang X., Merritt A. J., Seyfried J., Guo C., Papadakis E. S., Finegan K. G., Kayahara M., Dixon J., Boot-Handford R. P., Cartwright E. J., Mayer U., Tournier C. (2005) Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell. Biol. 25, 336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Houslay M. D., Adams D. R. (2003) PDE4 cAMP phosphodiesterases. Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 370, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baillie G. S., Houslay M. D. (2005) Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr. Opin. Cell Biol. 17, 129–134 [DOI] [PubMed] [Google Scholar]
- 68. McConnachie G., Langeberg L. K., Scott J. D. (2006) AKAP signaling complexes. Getting to the heart of the matter. Trends Mol. Med. 12, 317–323 [DOI] [PubMed] [Google Scholar]
- 69. Szaszák M., Christian F., Rosenthal W., Klussmann E. (2008) Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell. Signal. 20, 590–601 [DOI] [PubMed] [Google Scholar]
- 70. Dyck D. (2009) Adipokines as regulators of muscle metabolism and insulin sensitivity. Appl. Physiol. Nutr. Metab. 34, 396–402 [DOI] [PubMed] [Google Scholar]
- 71. Kim C.-H., Pennisi P., Zhao H., Yakar S., Kaufman J. B., Iganaki K., Shiloach J., Scherer P. E., Quon M. J., LeRoith D. (2006) MKR mice are resistant to the metabolic actions of both insulin and adiponectin. Discordance between insulin resistance and adiponectin responsiveness. Am. J. Physiol. Endocrinol. Metab. 291, E298–E305 [DOI] [PubMed] [Google Scholar]
- 72. Kim J. J., Kido Y., Scherer P. E., White M. F., Accili D. (2007) Analysis of compensatory β-cell response in mice with combined mutations of Insr and Irs2. Am. J. Physiol. Endocrinol. Metab. 292, E1694–E1701 [DOI] [PubMed] [Google Scholar]
- 73. Lin H. V., Kim J.-Y., Pocai A., Rossetti L., Shapiro L., Scherer P. E., Accili D. (2007) Adiponectin resistance exacerbates insulin resistance in insulin receptor transgenic/knockout mice. Diabetes 56, 1969–1976 [DOI] [PubMed] [Google Scholar]
- 74. Semple R. K., Cochran E. K., Soos M. A., Burling K. A., Savage D. B., Gorden P., O'Rahilly S. (2008) Plasma adiponectin as a marker of insulin receptor dysfunction. Diabetes Care 31, 977–979 [DOI] [PubMed] [Google Scholar]
- 75. Altiok S., Xu M., Spiegelman B. M. (1997) PPARγ induces cell cycle withdrawal. Inhibition of E2F/DP DNA- binding activity via down-regulation of PP2A. Genes Dev. 11, 1987–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gregoire F. M., Smas C. M., Sul H. S. (1998) Understanding adipocyte differentiation. Physiol. Rev. 78, 783–809 [DOI] [PubMed] [Google Scholar]
- 77. Macián F., López-Rodriguez C., Rao A. (2001) Partners in transcription. NFAT and AP-1. Oncogene 20, 2476–2489 [DOI] [PubMed] [Google Scholar]
- 78. Yang T. T., Chow C. W. (2003) Transcription cooperation by NFAT·C/EBP composite enhancer complex. J. Biol. Chem. 278, 15874–15885 [DOI] [PubMed] [Google Scholar]
- 79. Soto-Nieves N., Puga I., Abe B. T., Bandyopadhyay S., Baine I., Rao A., Macian F. (2009) Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J. Exp. Med. 206, 867–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jin L., Sliz P., Chen L., Macián F., Rao A., Hogan P. G., Harrison S. C. (2003) An asymmetric NFAT1 dimer on a pseudo-palindromic κB-like DNA site. Nat. Struct. Biol. 10, 807–811 [DOI] [PubMed] [Google Scholar]
- 81. Liu H., Maurice D. H. (1999) Phosphorylation-mediated activation and translocation of the cyclic AMP-specific phosphodiesterase PDE4D3 by cyclic AMP-dependent protein kinase and mitogen-activated protein kinases. A potential mechanism allowing for the coordinated regulation of PDE4D activity and targeting. J. Biol. Chem. 274, 10557–10565 [DOI] [PubMed] [Google Scholar]
- 82. Zhu H., Suk H. Y., Yu R. Y., Brancho D., Olabisi O., Yang T. T., Yang X., Zhang J., Moussaif M., Durand J. L., Jelicks L. A., Kim J. Y., Scherer P. E., Frank P. G., Lisanti M. P., Calvert J. W., Duranski M. R., Lefer D. J., Huston E., Baillie G. S., Houslay M. D., Molkentin J. D., Jin J., Chow C. W. (2010) Evolutionarily conserved role of calcineurin in phosphodegron-dependent degradation of phosphodiesterase 4D. Mol. Cell. Biol. 30, 4379–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ardley H. C., Robinson P. A. (2005) E3 ubiquitin ligases. Essays Biochem. 41, 15–30 [DOI] [PubMed] [Google Scholar]
- 84. Hunter T. (2007) The age of crosstalk. Phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 85. Baillie G. S. (2009) Compartmentalized signalling. Spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 276, 1790–1799 [DOI] [PubMed] [Google Scholar]