

ABSTRACT
Multiple cAMP phosphodiesterase (PDE) isoforms play divergent roles in cardiac homeostasis but the molecular basis for their non-redundant function remains poorly understood. Here, we report a novel role for the PDE4B isoform in β-adrenergic (βAR) signaling in the heart. Genetic ablation of PDE4B disrupted βAR-induced cAMP transients, as measured by FRET sensors, at the sarcolemma but not in the bulk cytosol of cardiomyocytes. This effect was further restricted to a subsarcolemmal compartment because PDE4B regulates β1AR-, but not β2AR- or PGE2-induced responses. The spatially restricted function of PDE4B was confirmed by its selective effects on PKA-mediated phosphorylation patterns. PDE4B limited the PKA-mediated phosphorylation of key players in excitation–contraction coupling that reside in the sarcolemmal compartment, including L-type Ca2+ channels and ryanodine receptors, but not phosphorylation of distal cytosolic proteins. β1AR- but not β2AR-ligation induced PKA-dependent activation of PDE4B and interruption of this negative feedback with PKA inhibitors increased sarcolemmal cAMP. Thus, PDE4B mediates a crucial PKA-dependent feedback that controls β1AR-dependent cAMP signals in a restricted subsarcolemmal domain. Disruption of this feedback augments local cAMP/PKA signals, leading to an increased intracellular Ca2+ level and contraction rate.
KEY WORDS: Cyclic nucleotide phosphodiesterase, PDE, cAMP, β-adrenergic receptors, Compartmentalization, Cardiac myocytes
INTRODUCTION
The cyclic adenosine monophosphate (cAMP) pathway is crucial for the sympathetic regulation of heart function. Upon β-adrenergic (βAR) stimulation, increased cAMP production activates protein kinase A (PKA), which in turn phosphorylates and regulates key proteins involved in excitation–contraction coupling (ECC), such as sarcolemmal Ca2+ channels (increasing the L-type Ca2+ current ICa,L), sarcoplasmic reticulum (SR) Ca2+ release channels [modifying ryanodin receptor (RyR2) gating], phospholamban (increasing SR Ca2+ pump rate via the sarcoplasmic reticulum Ca2+ ATPase SERCA) and contractile proteins (cardiac troponin I and myosin binding protein-C) (Bers, 2002). In parallel, activation of EPACs (exchange proteins directly activated by cAMP) causes branching of the cAMP signaling pathway to regulate additional components, most notably the Ca2+/calmodulin-dependent protein kinase II (Grimm and Brown, 2010). Given their crucial role in the control of cardiac function, cAMP signals are tightly regulated in their intensity, and temporal and spatial dimensions (Brunton et al., 1979; Mika et al., 2012).
It is now well established that cyclic nucleotide phosphodiesterases (PDEs) play an important role in the regulation of cAMP signaling in the heart. By degrading and inactivating cAMP, PDEs are thought to participate in the compartmentalization of the second messenger and in the generation of microdomains of signaling. Five PDE families, PDE1, PDE2, PDE3, PDE4 and PDE8, are predominantly responsible for cAMP degradation in the heart (Conti and Beavo, 2007; Patrucco et al., 2010). Of these, PDE4 isoforms are crucial in shaping global cAMP signals arising from β1AR and β2AR stimulation in cardiac myocytes in rodents and in humans (Mika et al., 2013; Molina et al., 2012). The PDE4 family is encoded by four genes (PDE4A, PDE4B, PDE4C and PDE4D); however, only PDE4A, PDE4B and PDE4D are expressed in cardiac myocytes (Richter et al., 2005; Richter et al., 2011). These three genes encode multiple isoforms generated through alternative splicing and use of alternative promoters/transcription start sites (Bender and Beavo, 2006; Jin and Conti, 2002).
Most of our understanding on the role of individual PDE4 subtypes in the heart is limited to PDE4D. Ablation of this gene in mice leads to PKA hyperphosphorylation of RyR2 and increased sensitivity to exercise-induced arrhythmias (Lehnart et al., 2005). PDE4D is also localized to other compartments of the cardiomyocyte, including the sarcomeric region, via anchoring to myomegalin (Verde et al., 2001), an A-kinase anchoring protein (AKAP) (Uys et al., 2011), the perinuclear region by an association with mAKAP (also known as AKAP6) (Dodge et al., 2001) and to the sarcolemmal region, where PDE4D3 is recruited to the cardiac K+ channel via AKAP9 (Terrenoire et al., 2009). PDE4D also associates with the PLB–SERCA complex and regulates the SERCA pump activity in the mouse heart (Beca et al., 2011). PDE4D5, PDE4D8 and PDE4D9 have been shown to associate with βARs either directly or indirectly by binding to β-arrestin (Baillie et al., 2003; De Arcangelis et al., 2009; Richter et al., 2008; Richter et al., 2013). These complex modes of interaction of βAR subtypes with PDEs shape divergent cAMP signals leading to specific physiological responses. More recently, the PDE4B isoform was identified as a component of the L-type Ca2+ channel (LTCC) complex and as the major PDE isoform regulating ICa,L current during βAR stimulation (Leroy et al., 2011). In this study, we show that PDE4B is crucial for the regulation of cardiac ECC because its ablation increases the propensity of arrhythmias. However, the molecular mechanisms by which PDE4B affects cAMP and channel function were not addressed.
Here, we investigated how PDE4B affects cAMP homeostasis in different compartments of cardiomyocytes. Using distinct approaches, our findings demonstrate a restricted function for PDE4B in a submembrane compartment of cardiomyocytes. We identified a local feedback regulation whereby β1AR stimulation activates PKA, which in turn phosphorylates and activates PDE4B, thereby desensitizing local cAMP signals. This negative feedback is required for maintaining the local cAMP steady state, which is necessary for fine tuning of ECC. Disruption of this feedback alters cAMP dynamics at the sarcolemma and increases phosphorylation of a subset of substrates involved in ECC, including CaV1.2 and RyR2, leading to an increase in intracellular Ca2+ level and an increased chronotropy.
RESULTS
PDE4B functions in a confined compartment near the sarcolemma of cardiac mycocytes
To investigate the function of PDE4B in cardiac myocytes, real-time cAMP measurements using FRET-based probes localized either in the cytosol (Epac2-CYTO) or at the plasma membrane (Epac2-PM) (Fig. 1A) were performed using neonatal cardiomyocytes (NCMs) derived from wild-type and PDE4B-knockout (PDE4BKO) mice. PDE4B ablation has no effect on βAR-dependent cAMP accumulation in the cytosol (Fig. 1B), but produces significant changes in the sarcolemmal compartment probed with the Epac2-PM sensor (Fig. 1C). Three parameters of the cAMP transient were altered in PDE4BKO compared to wild-type NCMs: maximal cAMP accumulation was increased, the time required for half maximal stimulation was also increased and the rate of decay of cAMP levels from maximum was significantly reduced in the absence of PDE4B (Fig. 1C). The differences in response to isoproterenol (Iso) between wild-type and PDE4BKO were abolished by inhibition of PDE4 with rolipram (Fig. 1D). This finding confirms that the cAMP phenotype is indeed caused by PDE4 ablation and the overall cAMP synthetic capacity is not altered in PDE4BKO cells.
Fig. 1.

PDE4B controls subsarcolemmal but not cytosolic cAMP. (A) YFP fluorescence in neonatal cardiac myocytes expressing either Epac2-CYTO (top) or Epac2-PM (bottom). (B–D) Wild-type and PDE4BKO myocytes expressing Epac2-CYTO (B) or Epac2-PM (C,D) were treated for the indicated times with Iso (10 nM) alone, or Iso (10 nM) and Rol (10 µM) followed by IBMX (100 µM) and Fsk (50 µM). The average ratio R/R0 (R = CFP/FRET) is reported and the number of cells analyzed (n) includes data from at least four experiments. Data were analyzed using a two-way ANOVA. Error bars correspond to the s.e.m. Rol, rolipram; Fsk, forskolin.
The above data provide initial evidence that chronic removal of PDE4B affects exclusively a membrane compartment. The possibility that there were compensatory changes in the pattern of PDE4 expression was excluded by monitoring the expression of PDE4A and PDE4D in the PDE4BKO myocytes. Consistent with previous observations, no changes in PDE4A or PDE4D expression could be detected (supplementary material Fig. S1). Similarly, no change in rolipram-insensitive PDE activity was detected in the hearts of PDE4BKO mice, indicating that no adaptive changes in PDEs other than PDE4 are induced (data not shown). To confirm that PDE4B functions in a restricted compartment and that the above data are not the result of long-term compensation for the absence of PDE4B, we used an alternative strategy to probe local PDE4B function. PDE4B-selective compounds have been developed to improve the efficacy and safety of PDE4 inhibition (Donnell et al., 2010; Kranz et al., 2009). We therefore used a PDE4B-selective inhibitor (supplementary material Fig. S2) to probe the consequences of acute PDE4B inhibition on cytosolic and subsarcolemmal cAMP in wild-type NCMs (Fig. 2A,B). This PDE4B-selective inhibitor caused an increase in basal and βAR-stimulated cAMP accumulation in the sarcolemmal compartment (Fig. 2B), which was quantitatively similar to that detected in the PDE4BKO. Moreover the cAMP response to Iso in the cytosol was not affected by the compound, with no statistical difference detected when compared to control (Fig. 2A). Thus, the acute inhibition of PDE4B faithfully recapitulates the PDE4BKO phenotype.
Fig. 2.
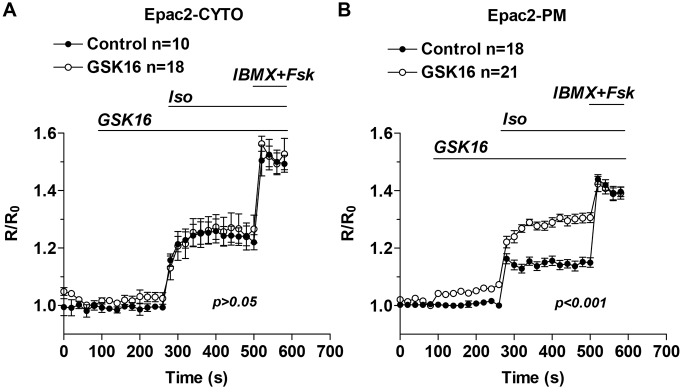
PDE4B-selective inhibition promotes subsarcolemmal but not cytosolic cAMP accumulation. (A,B) Wild-type myocytes expressing Epac2-CYTO (A) or Epac2-PM (B) were treated for the indicated times with a PDE4B-selective inhibitor (GSK16, 100 nM) and Iso (10 nM) followed by IBMX (100 µM) and Fsk (50 µM). The average ratio R/R0 (R = CFP/FRET) is reported and the number of cells analyzed (n) includes data from three experiments. Data were analyzed using a two-way ANOVA. Error bars correspond to the s.e.m. Fsk, forskolin.
Loss of PDE4B decreases cAMP decay at the plasma membrane
The above measurements at the membrane of NCMs indicate that cAMP concentration in PDE4BKO cells is maintained at a steady state for at least 10 min, suggesting that there is either a local decrease in cAMP hydrolysis or increased cAMP synthesis. To further distinguish between these two possibilities, Iso-mediated stimulation of cAMP synthesis was blocked by using an excess of the βAR antagonist, propranolol (1 µM) (Fig. 3A,B) to determine cAMP decay. Assuming complete blockade of adenylyl cyclase activation, the rate of cAMP decay should be dependent on PDE activity in the compartment. Under these experimental conditions, the t1/2 of cAMP decay was reduced by 50% in the PDE4BKO cells (69.8±8.5 s vs 122.5±23.4 s) confirming a decrease in PDE activity in this compartment (Fig. 3C). The fact that cAMP continues to decrease even in the absence of PDE4B indicates that (1) other PDEs are also involved in cAMP degradation in this compartment, and/or (2) that cAMP diffuses out of this compartment.
Fig. 3.
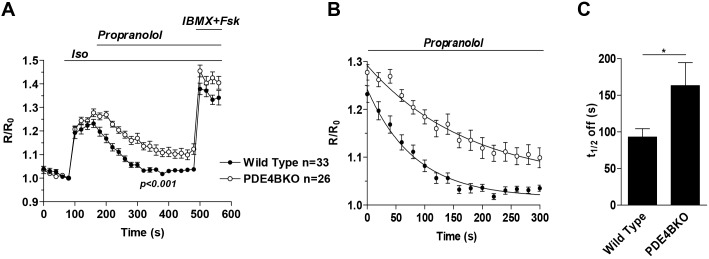
Rate of cAMP decay at the plasma membrane of wild-type and PDE4BKO myocytes. (A) Wild-type and PDE4BKO myocytes expressing Epac2-PM were stimulated for the indicated times with Iso (10 nM) followed 80 s later by treatment with propranolol (1 µM). At the end of the incubation IBMX (100 µM) plus Fsk (50 µM) was added to measure the maximal response of the cell. The average ratio R/R0 (R = CFP/FRET) is reported and the number of cells analyzed (n) includes data from three or more experiments. Data were analyzed using a two-way ANOVA. (B,C) Data were fitted with an exponential decay equation (B) and the average t1/2 off was calculated for the two genotypes (C). Data were analyzed using an unpaired Student's t-test; *P<0.05. The number of cells analyzed (n) includes data from at least three experiments. Error bars correspond to the s.e.m. Fsk, forskolin.
PDE4B controls β1AR but not β2AR or EPR responses in cardiac myocytes
The above findings indicate that there is a localized function of PDE4B in the proximity of the sarcolemma in cardiac myocytes. However, the plasma membrane compartment of these cells is not homogeneous but rather divided into subcompartments, including caveolae and non-caveolar lipid rafts, which contain distinct sets of receptors and signaling proteins (Insel et al., 2005). To further characterize the membrane subcompartment where PDE4B is active, we determined whether PDE4B ablation affects the signaling of different G-protein-coupled receptors (GPCRs). NCMs were preincubated with antagonists to block β1AR (CGP-20712A, 1 µM) or β2AR (ICI-118551, 1 µM) and cells were then stimulated with 10 nM Iso. Under these conditions, PDE4B ablation affects β1AR responses (Fig. 4A) but not cAMP signals induced by β2AR stimulation (Fig. 4B). Ablation of PDE4B also does not affect the cAMP transients induced by activation of the prostaglandin E receptors (EPRs) by PGE2 (100 nM) (Fig. 4C). Taken together, our data show that PDE4B controls a restricted pool of subsarcolemmal cAMP exclusively associated with β1AR activation.
Fig. 4.

PDE4B controls β1AR , but not β2AR or EPR responses in cardiac myocytes. Wild-type and PDE4BKO myocytes expressing Epac2-PM were preincubated with 1 µM ICI-118551 (ICI, β2 antagonist) (A) or 1 µM CGP-20712A (CGP, β1 antagonist) (B) for 5 min, then stimulated with Iso (10 nM) followed by IBMX (100 µM) plus Fsk (50 µM). (C) Wild-type and PDE4BKO myocytes were stimulated with PGE2 (100 nM) followed by IBMX (100 µM) plus Fsk (50 µM). The average ratio R/R0 (R = CFP/FRET) is reported and the number of cells analyzed (n) includes data from at least four experiments. Data were analyzed using a two-way ANOVA. Error bars represent the s.e.m. Fsk, forskolin.
PDE4B controls PKA activity at the sarcolemma but not in other compartments
As PDE4B regulates cAMP signaling in a restricted subsarcolemmal compartment, we wished to determine whether signaling events downstream of cAMP, namely activation of PKA and phosphorylation of its downstream targets, would be similarly restricted. To this end, we measured the phosphorylation level of several PKA substrates located in different subcellular regions of the NCM in wild-type and PDE4BKO cells. PKA phosphorylation of the LTCC subunit CaV1.2 as well as the ryanodine receptor (RyR2) and vasodilator-stimulated phosphoprotein (VASP), a component of the membrane actin cytoskeleton, was greatly increased in the PDE4BKO cardiomyocytes stimulated with Iso (10 nM) (Fig. 5A–C). Conversely, phosphorylation of phospholamban (PLB), which controls the activity of the SERCA pump, was the same in wild-type and PDE4BKO cells (Fig. 5D). Similarly, PKA phosphorylation of two substrates associated with the sarcomere, myosin-binding protein C (MyBP-C) and troponin I (TnI), was not affected by PDE4B ablation (Fig. 5E,F).
Fig. 5.

PDE4B controls PKA activity in a subsarcolemmal compartment, but not global PKA activity. Detergent extracts prepared from wild-type and PDE4BKO myocytes treated for 10 min with or without Iso (10 nM) were probed for phospho-CaV1.2 (P-CaV1.2, Ser1928) (A), phospho-ryanodin receptor 2 (P-RyR2, Ser2808) (B), phospho-VASP (P-VASP, Ser157) (C), phospho-phospholamban (P-PLB, Ser16) (D), phospho-myosine-binding protein C (P-MyBP-C, Ser282) (E) and phospho-troponin I (P-TnI, Ser23/Ser24) (F). Calsequestrin (CSQ) was used as a loading control. A representative western blot of three performed is included for each substrate. For quantification, the ratios of the immunoblot intensity for the phosphorylated proteins over that for CSQ were normalized to untreated (without Iso) wild-type cells and expressed as mean±s.e.m. *P<0.05; **P<0.01; ns, not significant (Student's unpaired t-test).
In wild-type NCMs, acute and selective inhibition of PDE4B promoted β1AR-, but not β2-induced PKA-mediated phosphorylation of CaV1.2, RyR2 and VASP, whereas PKA-mediated phosphorylation of PLB, MyBP-C and TnI were not altered (supplementary material Fig. S3). These discrete effects on PKA-mediated phosphorylations confirm that PDE4B predominantly functions to control a sequestered cAMP pool in the vicinity of the sarcolemma and that elevated cAMP levels in this compartment following PDE4B ablation do not spill over into other compartments including those containing the myofilament proteins and PLB.
PDE4B mediates a PKA-dependent feedback regulation of β1AR-signaling
At least five distinct protein variants, PDE4B1 to PDE4B5, are generated from the PDE4B gene. Western blots of NCM extracts with PDE4B-specific antibodies indicate the presence of a single prominent immunoreactive band with an apparent molecular mass of 95 kDa (Fig. 6A). This band corresponds to the PDE4B3 variant (supplementary material Fig. S4). Consistent with this view, immunoprecipitation with specific anti-PDE4B3 antibodies, but not with anti-PDE4B1 antibodies, led to recovery of a 95-kDa band in the immunoprecipitation pellet (supplementary material Fig. S4). This variant is a so-called long PDE4B form that contains a PKA phosphorylation consensus site at its N-terminus, and phosphorylation at this site mediates activation of the recombinant PDE4B protein (MacKenzie et al., 2002). Total extracts from NCMs were subjected to immunoprecipitation with PDE4B or control IgG antibodies. Fig. 6B confirms the specificity of the PDE4B antibody used for immunoprecipitation. No significant immunoprecipitation activity was detected when the experiment was repeated in PDE4BKO myocytes (Fig. 6B). Selective stimulation of β1AR, but not stimulation of β2AR or EPR, induced a more than 2-fold increase in cardiomyocyte PDE4B activity (Fig. 6C). This β1AR-dependent increase in PDE4B activity is reversed when the cells were pretreated with the PKA inhibitor H89 (Fig. 6C). Taken together, these data demonstrate that PDE4B expressed in NCMs is selectively phosphorylated and activated by PKA after stimulation of β1AR. Moreover, they provide independent confirmation that PDE4B is involved in the regulation of β1AR but not β2AR signaling; being activated only when the former receptor is occupied.
Fig. 6.
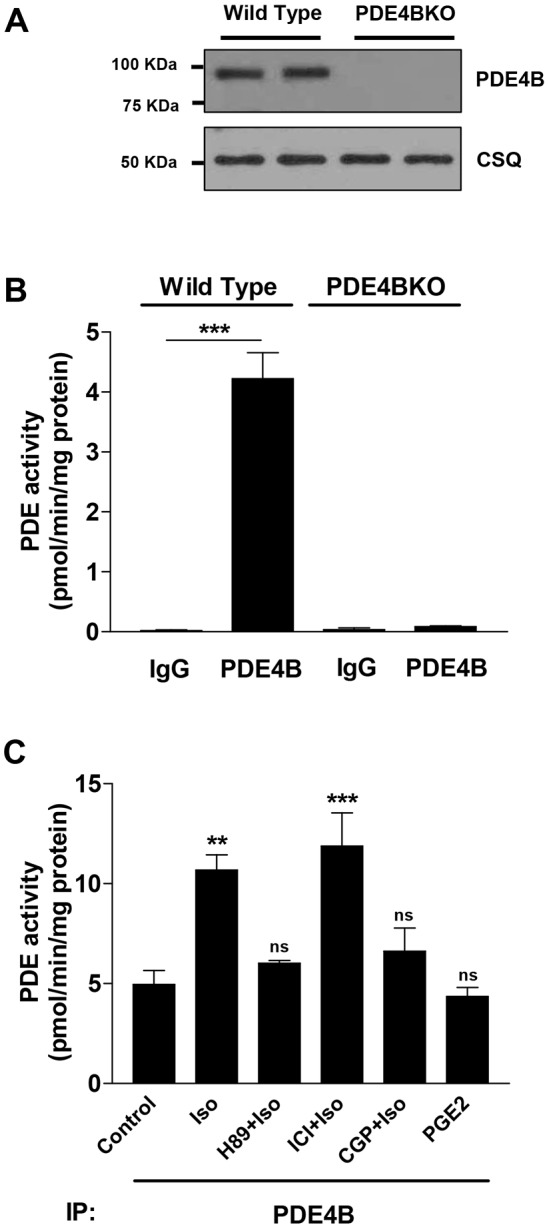
Phosphorylation and activation of PDE4B in cardiac myocytes. (A) Detergent extracts prepared from wild-type and PDE4BKO NCMs were probed by western blotting with anti-PDE4B antibodies. Calsequestrin (CSQ) was used as a loading control. (B) Total extracts from wild-type and PDE4BKO NCMs were subjected to immunoprecipitation (IP) with anti-PDE4B antibodies or anti-IgG antibodies. Shown is the PDE activity recovered in the immunoprecipitation pellets. (C) Wild-type myocytes were preincubated with mock, PKA inhibitor (H89, 20 µM, 1 h), ICI-118551 (ICI, β2 antagonist, 1 µM, 5 min) or CGP-20712A (CGP, β1 antagonist, 1 µM, 5 min), then stimulated with Iso (100 nM) or PGE2 (100 nM) for 10 min. Total extracts were then subjected to immunoprecipitation with anti-PDE4B antibodies and the PDE activity recovered in the immunoprecipitation pellets was measured. For B and C, data represent the mean±s.e.m. of experiments performed three times. **P<0.01; ***P<0.001; ns, not significant (Student's unpaired t-test).
PKA-mediated activation of PDE4B is crucial for cAMP signaling at the plasma membrane
To determine the consequences of the PKA-mediated phosphorylation and activation of PDE4B in the control of cAMP accumulation at the plasma membrane, FRET measurements of cAMP in this compartment were performed in the presence or absence of the PKA inhibitor H89 (20 µM). At this concentration, this inhibitor completely blocked the β1AR-dependent activation of the endogenous PDE4B. Inhibition of PKA triggered a significant increase of Iso-stimulated cAMP levels at the plasma membrane of wild-type myocytes (Fig. 7A). Conversely, the effect of H89 was dramatically reduced in PDE4BKO cells (Fig. 7B) suggesting that PDE4B activation is the main mechanism of PKA-mediated feedback regulation of cAMP signaling in this submembrane compartment. In the cytosol, pretreatment with H89 induced comparable increases in cAMP accumulation in wild-type cells (Fig. 7C) and PDE4BKO cells (Fig. 7D), suggesting that a different PDE mediates the PKA effects in the cytosol.
Fig. 7.

PDE4B phosphorylation by PKA contributes to the pattern of cAMP accumulation. Wild-type (A,C) and PDE4BKO (B,D) myocytes expressing Epac2-PM (A,B) or Epac2-CYTO (C,D) were pretreated for 1 h with or without H89 (20 µM) before stimulation for the indicated times with Iso (10 nM) and IBMX (100 µM) plus Fsk (50 µM). The average ratio R/R0 (R = CFP/FRET) is reported and the number of cells analyzed (n) includes data from four or more experiments. Data were analyzed using a two-way ANOVA. Error bars correspond to the s.e.m. Fsk, forskolin.
PDE4B ablation increases intracellular Ca2+ levels and myocyte contraction rate after βAR stimulation
It is well established that stimulation of β1AR signaling increases heart rate (Devic et al., 2001; Xiang et al., 2005) and that this effect is due in part to increased PKA-mediated phosphorylation and activation of CaV1.2 and RyR2 (Bers, 2008; Shan et al., 2010). The fact that PDE4B controls the PKA-mediated phosphorylation of LTCC and RyR2 prompted us to compare the intracellular Ca2+ levels and the contraction rate of wild-type and PDE4BKO NCMs. To this end, we measured Fura-2 ratios and the spontaneous beating rate after βAR stimulation with 10 nM Iso (Fig. 8). We found that Iso-induced intracellular Ca2+ levels in PDE4BKO cells are significantly increased compared to wild-type myocytes (Fig. 8A). The contraction rate measured in PDE4BKO cells tends to be higher than in wild-type cells under basal conditions (although not significant) and is significantly increased after Iso stimulation (Fig. 8B). This finding is consistent with the hypothesis that PDE4B ablation evokes an increase in the βAR-stimulated contraction rate of cardiac myocytes, likely via increased PKA-mediated phosphorylation of ECC proteins such as CaV1.2 and/or RyR2.
Fig. 8.
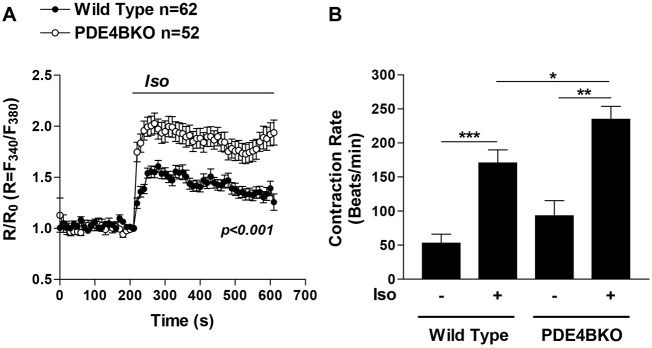
PDE4B regulates intracellular Ca2+ level and contraction rate in βAR-stimulated cardiac myocytes. (A) Wild-type and PDE4BKO myocytes loaded with Fura-2 AM (5 µM) were treated for the indicated times with Iso (10 nM). The average ratio R/R0 (R = F340/F380) is reported and the number of cells analyzed (n) includes data from three experiments. Data were analyzed using a two-way ANOVA. Error bars correspond to the s.e.m. (B) The contraction rate of wild-type and PDE4BKO myocytes was measured before and 5 min after βAR stimulation with 10 nM Iso. The bar graphs represent the mean ± s.e.m. of five experiments. *P<0.05, **P<0.01, ***P<0.001 (unpaired Student's t-test).
DISCUSSION
The data described above demonstrate a local PDE feedback regulation in cardiac myocytes whereby β1AR signals activate PDE4B3 via PKA-mediated phosphorylation. PKA-activated PDE4B, in turn, reduces the local steady-state cAMP concentration in a confined subsarcolemmal domain. The functional consequences of interrupting this feedback include increased PKA-mediated phosphorylation of Cav1.2 and RyR2, which is necessary for fine-tuning of ECC, a finding consistent with the cardiac phenotype of PDE4BKO mice (Leroy et al., 2011). Functional data on intracellular Ca2+ levels and contraction rate confirm the altered cAMP/PKA signaling in PDE4BKO myocytes. These conclusions are based on measurements of βAR-induced cAMP accumulation at the plasma membrane of PDE4BKO myocytes, the effects of acute PDE4B-selective inhibition in wild-type myocytes, the β1AR-dependent activation of PDE4B and the altered PKA-mediated phosphorylation of some, but not all substrates involved in Ca2+ homeostasis and contraction. Further proof for a restricted function of PDE4B is our finding that this enzyme is not regulated by β2AR signals and does not control β2AR-dependent cAMP accumulation at the plasma membrane. Thus, PDE4B serves a highly specialized and localized function to control β1AR-dependent ECC in cardiac myocytes.
The major PDE4B isoform expressed in neonatal and adult mouse cardiac myocytes is a so-called long form that contains a conserved consensus site for PKA phosphorylation (Conti et al., 2003). Here, we show that during βAR stimulation, PKA phosphorylates and activates this PDE4B isoform. Its mobility in SDS/PAGE gels is identical to the PDE4B isoforms identified in the adult mouse, rat and human heart (Abi-Gerges et al., 2009; Richter et al., 2011). Thus, any regulation of this isoform is likely conserved and relevant to human cardiac physiology.
Being a substrate for PKA phosphorylation, activation of different PDE4s by itself might be used to explore the selectivity of receptor signaling as well as define its spatial propagation. We have previously shown that β1AR and β2AR activate different subsets of PDE4D variants. β1AR ligation preferentially activates PDE4D8 localized in the vicinity of the receptor, whereas β2AR activates PDE4D9 and PDE4D5 (Richter et al., 2008). This concept is reinforced and extended with the demonstration that β1AR, but not β2AR, signals to activate PDE4B in neonatal cardiac myocytes. Previously, we have shown that PDE4B co-immunoprecipitates with the LTCC in adult mouse ventricular myocytes suggesting that this isoform is tethered in a macromolecular complex with the channel at the plasma membrane and is located in the proximity of the T-tubules (Leroy et al., 2011). By contrast, PDE4D5 preferentially associates with β-arrestin (Lynch et al., 2005), and PDE4D9 co-immunoprecipitates with the β2AR (De Arcangelis et al., 2009). These distinct interactions and regulations of individual PDE4 variants by the two β-adrenergic receptors demonstrate that cAMP hydrolysis during catecholamine stimulation is not uniformly activated throughout the cell. Instead, dissipation of the cAMP signal in different subdomains occurs at different rates depending on the PDE4 subtype activated by different receptors.
β1AR-dependent cAMP signals in cardiac myocytes are far reaching and prolonged, whereas β2AR signals are transient and limited to restricted subdomains (Nikolaev et al., 2006). This difference might explain the finding that PDE4B is activated by β1AR but not β2AR activation. Localized β2AR signals might not reach and activate PDE4B, which in turn does not affect β2AR-dependent cAMP accumulation. We have previously reported that the chronotropic effects of β2AR stimulation are not affected in PDE4BKO neonatal cardiac myocytes, reinforcing the present observation of the lack of functional interaction between PDE4B and β2AR. One additional explanation of our finding is that the two signaling components are located in two separate membrane domains. β2AR has been localized to lipid rafts where adenylate cyclase (AC)5, AC6 and AKAP150 (Gorelik et al., 2013; Xiang et al., 2002) are located, but also in T-tubules (Nikolaev et al., 2010). Our data suggest that PDE4B is located instead in the vicinity of the β1AR and in close proximity to a pool of PKA that regulates LTCC and RyR2, all components present in the dyad of neonatal and adult cardiac myocytes (Bers, 2008). It is possible that β1AR and β2AR are mutually exclusive in their localization in the dyad space and that PDE4B is present exclusively in the domain where β1AR is present. The presence of different pools of LTCC coupled to β1AR or β2AR have been proposed (Nichols et al., 2010) but the mechanisms underlying this sequestration are at present unknown.
Surprisingly, PDE4B ablation does not lead to spillover of cAMP to other compartments that are distant from the membrane, as one would expect if this PDE constitutes the primary barrier to cAMP diffusion. Absence of detectable changes in PKA phosphorylation in domains close to the myofilaments where MyBP-C and TnI are phosphorylated or close to the PLB–SERCA complex, are consistent with this view. After removal of PDE4B, this compartment also remains inaccessible to either β2AR or PGE2 signals, again suggesting the presence of a barrier not dependent on PDE4B. Such hindered diffusion might include physical barriers generated by the network of SR membranes or the actin cytoskeletal meshwork present below the membrane. More importantly, the presence of cAMP-binding proteins that slow the diffusion rate may be crucial to stabilizing this compartment. It is also possible that the pool of cAMP affected by PDE4B, although functionally important, is small compared to the total cAMP and that spill over to a much larger pool would be difficult to detect. A role of other PDEs such as PDE4D in limiting diffusion should also be considered.
In summary, our findings reveal a highly specialized function of PDE4B in βAR signaling of cardiac myocytes. The regulation of this enzyme is likely relevant to the pathophysiology of heart failure. We demonstrate here that ablation of PDE4B leads to aberrant phosphorylation of RyR2 and LTCC phosphorylation (but not of PLB), affecting Ca2+ handling, similar to that observed in heart failure (Chen et al., 2002; Lehnart et al., 2005; Schröder et al., 1998). Importantly, a decrease in PDE4B expression and activity is detected during cardiac hypertrophy (Abi-Gerges et al., 2009). Thus, PDE4B dysregulation might be a molecular adaptive change that contributes to arrhythmias in the failing heart.
MATERIALS AND METHODS
Reagents
βAR ligands Isoproterenol (Iso), ICI-118551 (ICI), CGP-20712A (CGP) and Propranolol, the pan-PDE inhibitor IBMX (3-Isobutyl-1-methylxanthine) and the PDE4-selective inhibitor Rolipram were from Sigma-Aldrich (St. Louis, MO, USA). The adenylyl cyclase activator Forskolin (Fsk) was from Tocris Bioscience (Bristol, UK). The PKA inhibitor H89 and the EP2/4 agonist prostaglandin E2 (PGE2) were from Cayman Chemicals (Ann Arbor, MI, USA). The selective PDE4B inhibitor [3-[[5-(3-fluorophenyl)-2-pyrimidinyl]amino]4-ethoxy-phenyl]-4-morpholinyl-methanone (GSK16) was synthesized by Grünenthal GmbH (Aachen, Germany), according to a poster presentation by Mitchell C. et al. (GlaxoSmithKline) at the 46th International Conference on Medicinal Chemistry (RICT 2010, Reims, France). The Fura-2 AM Ca2+ indicator was from Invitrogen (Grand Island, NY, USA). Antibodies against the following proteins were used in this study: phospho-phospholamban (P-PLB, Ser16) and cardiac phospho-troponin I (P-TnI, Ser23/24), both from Cell Signaling Technology (Danvers, MA, USA), cardiac phospho-myosin binding protein C (P-MyBP-C, Ser282), from Alexis Biochemicals (Farmingdale, NY, USA) and calsequestrin (CSQ), from Affinity BioReagents (Golden, CO, USA). L-type Ca2+ channel pore forming subunit (Cav1.2) phosphorylation at Ser1928 was detected using the anti-CH3-P antibody (De Jongh et al., 1996; Hulme et al., 2006). The anti-phospho-ryanodine receptor (P-RyR2, Ser2808) antibody was a kind gift from Andrew Marks (Columbia University, New York, USA) (Wehrens et al., 2004). Pan-selective antibodies against PDE4A (AC55) and PDE4D (M3S1) were as described previously (Richter et al., 2005). Rabbit antibody 113-4, raised against the C-terminus of PDE4B, was used to detect PDE4B. Antibodies against PDE4B1 and PDE4B3 were from Fabgennix (Frisco, TX, USA). Horseradish-peroxidase-conjugated anti-rabbit IgG was from Amersham (Pittsburg, PA, USA).
Isolation of neonatal cardiac myocytes and adenoviral infection
Neonatal cardiac myocytes (NCMs) were isolated from the excised hearts of 1–2-day-old neonatal mice as described previously (Richter et al., 2008). They were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% Nu Serum IV (BD Biosciences, San Jose, CA, USA), 5% fetal bovine serum, 1 mM glutamine, 30 µg/ml penicillin, 100 µg/ml streptomycin, 3 mM Hepes and 1× ITS media supplement (Gibco, Grand Island, NY, USA) on plates precoated with 10 µg/ml laminin (Invitrogen). All cells were cultured at 37°C and under a 5% CO2 atmosphere. All experiments were carried out on day 3 of culture. All animal studies were performed according to the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of the University of California San Francisco. For expression of the FRET sensors Epac2-CYTO or Epac2-PM, NCMs cultured on laminin-coated glass coverslips for 48 h were infected with the respective adenoviruses at a multiplicity of infection of 500.
Measurement of local cAMP levels using the Epac2-CYTO and Epac2-PM FRET sensors
For FRET (fluorescence resonance energy transfer) microscopy, glass coverslips were placed in a modified Sykes-Moore Chamber and kept in 500 µl of Locke's medium (5 mM HEPES pH 7.4, 154 mM NaCl, 5.6 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 3.6 mM NaHCO3, 5 mM glucose, 0.05% BSA) at 37°C. Images were acquired with a Nikon TE2000 inverted fluorescence microscope using a 60× fluorescence objective. CFP (donor) fluorescence was viewed by exciting at 430–455 nm and measuring emission at 470–490 nm. YFP (acceptor) fluorescence was viewed by exciting at 500–520 nm and measuring emission at 535–565 nm. FRET was viewed by exciting at 430–455 nm (donor excitation) and measuring fluorescence at 535–565 nm (acceptor emission). Images were acquired every 20 s. Background and bleedthrough were subtracted from FRET images to obtain corrected FRET images using MetaMorph software (Molecular Devices, Sunnyvale, CA, USA) and average FRET intensity was measured directly in the corrected FRET images.
Preparation of protein extracts
Isolated NCMs were homogenized in an ice-cold buffer containing 150 mM NaCl, 20 mM HEPES (pH 7.4), 2 mM EDTA and 0.2 mM EGTA, supplemented with 10% glycerol, 0.2% Triton X-100, 1 µmol/l microcystin-LR (Cayman Chemicals) and Complete Protease Inhibitor Tablets (Roche Diagnostics, Pleasanton, CA, USA). Lysates were rotated at 4°C for 30 min followed by a 10-min centrifugation at 20,000 g and 4°C. Supernatants were either used directly for western blotting or subjected to immunoprecipitation (see below).
Immunoprecipitation of PDE4B from cell lysates
Immunoprecipitation of PDE4B was performed as described previously (Richter et al., 2011). In brief, cell and tissue extracts, prepared as described in ‘Preparation of protein extracts’, were precleared for 1 h using 50 µl protein-G–Sepharose (Invitrogen), followed by a second centrifugation at 20,000 g and 4°C for 10 min. Precleared detergent extracts were then immunoprecipitated using 10 µg of PDE4B antibody or 10 µg of control rabbit IgG for 2 h at 4°C.
PDE assay
The hydrolysis of cAMP by PDEs was measured according to the method of Thompson and Appleman as described previously (Richter and Conti, 2002). In brief, samples were assayed in a 200-µl reaction mixture containing 40 mM Tris-HCl (pH 8.0), 1 mM MgCl2, 1.4 mM β-mercaptoethanol, 1 µM cAMP, 0.75 mg/ml BSA and 0.1 µCi of [3H]cAMP for 20 min at 33°C. The reaction was terminated by heat inactivation in a boiling water bath for 1 min. The PDE reaction product 5′-AMP was then hydrolyzed by incubation of the assay mixture with 50 µg Crotalus atrox snake venom for 20 min at 33°C, and the resulting adenosine was separated by anion exchange chromatography using 1 ml AG1-X8 resin (Bio-Rad) and quantified by scintillation counting.
Measurement of intracellular Ca2+ level
NCMs cultured on laminin-coated glass coverslips for 48 h, were loaded with Fura-2 AM (5 µM, 30 min) then washed in Locke's medium. The coverslips were placed in a modified Sykes-Moore Chamber and kept in 500 µl of Locke's medium at 37°C. Images were acquired with a Nikon TE2000 inverted fluorescence microscope using a 20× objective. Fura-2 ratio (measured at 512 nm upon excitation at 340 nm and 380 nm) was recorded every 10 s using the MetaFluor software.
Measurement of myocyte contraction rate
Myocytes were cultured on laminin-coated cell culture dishes to obtain a uniformly beating syncytium. After 48 h in culture, the medium was replaced with serum-free medium, and experiments were carried out on day 3 of culture. To measure the myocyte contraction rate, the culture dish was placed on an inverted microscope for 5 min to equilibrate. Stacks of images were then stream acquired at 1-min intervals for 5 min before, and for another 10 min after addition of Iso (10 nM). The resulting images were analyzed with MetaMorph Object-Track to determine beat frequency.
Supplementary Material
Acknowledgments
We are indebted to Thomas C. Rich (University of South Alabama, Mobile, AL) for critical advice and to George S. Baillie (University of Glasgow, Glasgow, UK) for providing the PDE4C2 construct.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
D.M. and W.R. performed the experiments and analyzed the data. D.M., W.R. and M.C. designed the study. D.M., W.R., R.E.W., W.A.C. and M.C. interpreted the data and prepared the manuscript.
Funding
This work was supported by grants from the National Institutes of Health [grant numbers HL0927088 to M.C., HL107960 to W.R.]; and Fondation Leducq [grant number 06CVD02 cycAMP to M.C.]. D.M. is a recipient of a postdoctoral fellowship from the American Heart Association. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.140251/-/DC1
References
- Abi-Gerges A., Richter W., Lefebvre F., Mateo P., Varin A., Heymes C., Samuel J. L., Lugnier C., Conti M., Fischmeister R. et al. (2009). Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res. 105, 784–792 10.1161/CIRCRESAHA.109.197947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie G. S., Sood A., McPhee I., Gall I., Perry S. J., Lefkowitz R. J., Houslay M. D. (2003). beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA 100, 940–945 10.1073/pnas.262787199 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Beca S., Helli P. B., Simpson J. A., Zhao D., Farman G. P., Jones P. P., Tian X., Wilson L. S., Ahmad F., Chen S. R. et al. (2011). Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ. Res. 109, 1024–1030 10.1161/CIRCRESAHA.111.250464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T., Beavo J. A. (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520 10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- Bers D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- Bers D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 10.1146/annurev.physiol.70.113006.100455 [DOI] [PubMed] [Google Scholar]
- Brunton L. L., Hayes J. S., Mayer S. E. (1979). Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase. Nature 280, 78–80 10.1038/280078a0 [DOI] [PubMed] [Google Scholar]
- Chen X., Piacentino V., III, Furukawa S., Goldman B., Margulies K. B., Houser S. R. (2002). L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91, 517–524 10.1161/01.RES.0000033988.13062.7C [DOI] [PubMed] [Google Scholar]
- Conti M., Beavo J. (2007). Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76, 481–511 10.1146/annurev.biochem.76.060305.150444 [DOI] [PubMed] [Google Scholar]
- Conti M., Richter W., Mehats C., Livera G., Park J. Y., Jin C. (2003). Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 278, 5493–5496 10.1074/jbc.R200029200 [DOI] [PubMed] [Google Scholar]
- De Arcangelis V., Liu R., Soto D., Xiang Y. (2009). Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J. Biol. Chem. 284, 33824–33832 10.1074/jbc.M109.020388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jongh K. S., Murphy B. J., Colvin A. A., Hell J. W., Takahashi M., Catterall W. A. (1996). Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35, 10392–10402 10.1021/bi953023c [DOI] [PubMed] [Google Scholar]
- Devic E., Xiang Y., Gould D., Kobilka B. (2001). Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol. Pharmacol. 60, 577–583 [PubMed] [Google Scholar]
- Dodge K. L., Khouangsathiene S., Kapiloff M. S., Mouton R., Hill E. V., Houslay M. D., Langeberg L. K., Scott J. D. (2001). mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20, 1921–1930 10.1093/emboj/20.8.1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnell A. F., Dollings P. J., Butera J. A., Dietrich A. J., Lipinski K. K., Ghavami A., Hirst W. D. (2010). Identification of pyridazino[4,5-b]indolizines as selective PDE4B inhibitors. Bioorg. Med. Chem. Lett. 20, 2163–2167 10.1016/j.bmcl.2010.02.044 [DOI] [PubMed] [Google Scholar]
- Gorelik J., Wright P. T., Lyon A. R., Harding S. E. (2013). Spatial control of the βAR system in heart failure: the transverse tubule and beyond. Cardiovasc. Res. 98, 216–224 10.1093/cvr/cvt005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M., Brown J. H. (2010). Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 48, 322–330 10.1016/j.yjmcc.2009.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme J. T., Westenbroek R. E., Scheuer T., Catterall W. A. (2006). Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc. Natl. Acad. Sci. USA 103, 16574–16579 10.1073/pnas.0607294103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel P. A., Head B. P., Ostrom R. S., Patel H. H., Swaney J. S., Tang C. M., Roth D. M. (2005). Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann. N. Y. Acad. Sci. 1047, 166–172 10.1196/annals.1341.015 [DOI] [PubMed] [Google Scholar]
- Jin S. L., Conti M. (2002). Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc. Natl. Acad. Sci. USA 99, 7628–7633 10.1073/pnas.122041599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz M., Wall M., Evans B., Miah A., Ballantine S., Delves C., Dombroski B., Gross J., Schneck J., Villa J. P. et al. (2009). Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg. Med. Chem. 17, 5336–5341 10.1016/j.bmc.2009.03.061 [DOI] [PubMed] [Google Scholar]
- Lehnart S. E., Wehrens X. H., Reiken S., Warrier S., Belevych A. E., Harvey R. D., Richter W., Jin S. L., Conti M., Marks A. R. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123, 25–35 10.1016/j.cell.2005.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J., Richter W., Mika D., Castro L. R., Abi-Gerges A., Xie M., Scheitrum C., Lefebvre F., Schittl J., Mateo P. et al. (2011). Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 121, 2651–2661 10.1172/JCI44747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. J., Baillie G. S., Mohamed A., Li X., Maisonneuve C., Klussmann E., van Heeke G., Houslay M. D. (2005). RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 280, 33178–33189 10.1074/jbc.M414316200 [DOI] [PubMed] [Google Scholar]
- MacKenzie S. J., Baillie G. S., McPhee I., MacKenzie C., Seamons R., McSorley T., Millen J., Beard M. B., van Heeke G., Houslay M. D. (2002). Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 136, 421–433 10.1038/sj.bjp.0704743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika D., Leroy J., Vandecasteele G., Fischmeister R. (2012). PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 52, 323–329 10.1016/j.yjmcc.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Mika D., Bobin P., Pomérance M., Lechêne P., Westenbroek R. E., Catterall W. A., Vandecasteele G., Leroy J., Fischmeister R. (2013). Differential regulation of cardiac excitation-contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc. Res. 100, 336–346 10.1093/cvr/cvt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina C. E., Leroy J., Richter W., Xie M., Scheitrum C., Lee I. O., Maack C., Rucker-Martin C., Donzeau-Gouge P., Verde I. et al. (2012). Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J. Am. Coll. Cardiol. 59, 2182–2190 10.1016/j.jacc.2012.01.060 [DOI] [PubMed] [Google Scholar]
- Nichols C. B., Rossow C. F., Navedo M. F., Westenbroek R. E., Catterall W. A., Santana L. F., McKnight G. S. (2010). Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ. Res. 107, 747–756 10.1161/CIRCRESAHA.109.216127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev V. O., Bünemann M., Schmitteckert E., Lohse M. J., Engelhardt S. (2006). Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 99, 1084–1091 10.1161/01.RES.0000250046.69918.d5 [DOI] [PubMed] [Google Scholar]
- Nikolaev V. O., Moshkov A., Lyon A. R., Miragoli M., Novak P., Paur H., Lohse M. J., Korchev Y. E., Harding S. E., Gorelik J. (2010). Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657 10.1126/science.1185988 [DOI] [PubMed] [Google Scholar]
- Patrucco E., Albergine M. S., Santana L. F., Beavo J. A. (2010). Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 49, 330–333 10.1016/j.yjmcc.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Conti M. (2002). Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J. Biol. Chem. 277, 40212–40221 10.1074/jbc.M203585200 [DOI] [PubMed] [Google Scholar]
- Richter W., Jin S. L., Conti M. (2005). Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem. J. 388, 803–811 10.1042/BJ20050030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Day P., Agrawal R., Bruss M. D., Granier S., Wang Y. L., Rasmussen S. G., Horner K., Wang P., Lei T. et al. (2008). Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 27, 384–393 10.1038/sj.emboj.7601968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Xie M., Scheitrum C., Krall J., Movsesian M. A., Conti M. (2011). Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 106, 249–262 10.1007/s00395-010-0138-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Mika D., Blanchard E., Day P., Conti M. (2013). β1-adrenergic receptor antagonists signal via PDE4 translocation. EMBO Rep. 14, 276–283 10.1038/embor.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder F., Handrock R., Beuckelmann D. J., Hirt S., Hullin R., Priebe L., Schwinger R. H., Weil J., Herzig S. (1998). Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 98, 969–976 10.1161/01.CIR.98.10.969 [DOI] [PubMed] [Google Scholar]
- Shan J., Kushnir A., Betzenhauser M. J., Reiken S., Li J., Lehnart S. E., Lindegger N., Mongillo M., Mohler P. J., Marks A. R. (2010). Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Invest. 120, 4388–4398 10.1172/JCI32726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C., Houslay M. D., Baillie G. S., Kass R. S. (2009). The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 284, 9140–9146 10.1074/jbc.M805366200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uys G. M., Ramburan A., Loos B., Kinnear C. J., Korkie L. J., Mouton J., Riedemann J., Moolman-Smook J. C. (2011). Myomegalin is a novel A-kinase anchoring protein involved in the phosphorylation of cardiac myosin binding protein C. BMC Cell Biol. 12, 18 10.1186/1471-2121-12-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I., Pahlke G., Salanova M., Zhang G., Wang S., Coletti D., Onuffer J., Jin S. L., Conti M. (2001). Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 276, 11189–11198 10.1074/jbc.M006546200 [DOI] [PubMed] [Google Scholar]
- Wehrens X. H., Lehnart S. E., Reiken S. R., Marks A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70 10.1161/01.RES.0000125626.33738.E2 [DOI] [PubMed] [Google Scholar]
- Xiang Y., Rybin V. O., Steinberg S. F., Kobilka B. (2002). Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J. Biol. Chem. 277, 34280–34286 10.1074/jbc.M201644200 [DOI] [PubMed] [Google Scholar]
- Xiang Y., Naro F., Zoudilova M., Jin S. L., Conti M., Kobilka B. (2005). Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc. Natl. Acad. Sci. USA 102, 909–914 10.1073/pnas.0405263102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.