

Abstract
Kaposis's sarcoma associated herpesvirus (KSHV) has been reported to infect, among others, monocytes and dendritic cells DCs impairing their function. However, the underlying mechanisms remain not completely elucidated yet. Here we show that DC exposure to active or UV-inactivated KSHV resulted in STAT3 phosphorylation. This effect, partially dependent on KSHV-engagement of DC-SIGN, induced a high release of IL-10, IL-6 and IL-23, cytokines that in turn might maintain STAT3 in a phosphorylated state. STAT3 activation also correlated with a block of autophagy in DCs, as indicated by LC3II reduction and p62 accumulation. The IL-10, IL-6 and IL-23 release and the autophagic block could be overcome by inhibiting STAT3 activation, highlighting the role of STAT3 in mediating such effects. In conclusion, here we show that STAT3 activation can be one of the molecular mechanisms leading to KSHV-mediated DC dysfunction, that might allow viral persistence and the onset of KSHV-associated malignancies.
Dendritic cells (DCs) are at crossroad between innate and adaptive immunity, being able to prime naive T lymphocytes to novel antigens and to initiate a specific immune response1,2. Kaposi's sarcoma-associated herpesvirus (KSHV) is a human gammaherpesvirus found in all forms of Kaposi's sarcoma (KS) and is also highly associated with lymphoproliferative disorders, such as primary effusion lymphomas (PEL) and multicentric Castleman's disease (MCD)3,4,5. As other members of Herpesvirus family6,7,8, KSHV is able to infect plasmacytoid DCs, myeloid DCs or their monocyte precursors9,10,11, leading to reduction of costimulatory molecules, altered cytokine release and impaired allostimulatory capacity11,12. Monocytes exposure to KSHV results also in down-regulation of chemokine receptors and consequently in reduced migration capacity in response to chemokines13. DC functional impairment has also been observed in vivo, in patients with KSHV-associated disease, such as Kaposi's Sarcoma14,15. To date, the molecular mechanisms underlying the KSHV-mediated immunosuppressive effects are not completely known. KSHV entry into myeloid DCs is mediated by dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN; CD209), although other receptor molecules seem to be involved10. Furthermore, KSHV binds heparan sulfates (HS) on the surface of monocytes and THP-1 monocytic cell line, activating molecules such as PI3K and NF-kB16. These signaling pathways are also involved in the modulation of DC function17,18,19,20 in addition to the signal transducer and activator of transcription 3 (STAT3), whose activation by immunosuppressive factors, released by tumor cells, leads to DC dysfunction21,22,23. Recently STAT3 activation by IL-10, in bystander macrophages/monocytes, in the course of HIV infection, has been reported to interfere also with the autophagic process in these cells24. STAT3 inhibitors, on the other hand, are reported to be potent autophagy inducers25.
Autophagy is characterized by the formation of double-membrane vesicles that, surrounding aggregated proteins or damaged organelles, fuse with lysosomes, leading to the degradation of their content26. It may allow cells to survive in stressful conditions, such as nutrient shortage and microbial infections27. Moreover, autophagy plays an essential role in pathogen clearance and MHC-presentation by antigen presenting cells (APC)28 and has been reported to be required also for monocyte differentiation into macrophages and DCs29,30. Given its important role in the immune response, it is not surprising that pathogens have developed strategies to interfere with the autophagy to avoid the immune control31.
The aim of our study was to investigate which molecular pathways could be activated by KSHV in human DCs. We showed that both active and UV-inactivated KSHV induced STAT3 phosphorylation in DCs. STAT3 activation was partially dependent on DC-SIGN engagement and correlated with a high release of IL-10, IL-6 and IL-23 cytokines. Moreover, a reduced IL-12 production upon LPS stimulation was observed. Finally, we found that, by activating STAT3, KSHV induced a block of autophagy, essential for the immune function of these cells.
Results
KSHV activates STAT3 in DCs
DCs, overnight serum-starved, were mock-treated or exposed to KSHV (~9 × 106 viral DNA copies/1 × 106 DCs) for 15 and 30 minutes at 37°C. STAT3 phosphorylation was then analyzed by western blot. We observed that STAT3 phosphorylation increased after 15 min of KSHV-exposure, in comparison to mock-treated DCs, and was sustained for 30 min (fig. 1a). We next investigated whether KSHV would also influence the activation of other molecules, such as NF-kB and AKT, involved in the regulation of DC function and activated by KSHV in other cell types16,32. Differently from STAT3, KSHV slightly influenced the phosphorylation of NF-kB p65 subunit and AKT after 15 or 30 minutes of viral exposure (fig. 1a). Next, to establish whether viral gene expression was required for STAT3 activation, we performed the same experiment with UV-inactivated KSHV. As shown in fig. 1b, UV-inactivated KSHV was still able to induce STAT3 phosphorylation in DCs, suggesting that viral binding/entry is sufficient for STAT3 activation and it occurs independently of viral gene expression. As for active KSHV, the UV-inactivated virus slightly modified the phosphorylation of NF-kB and AKT (fig. 1b).
Figure 1. Short exposure to KSHV induces STAT3 activation in human DCs.
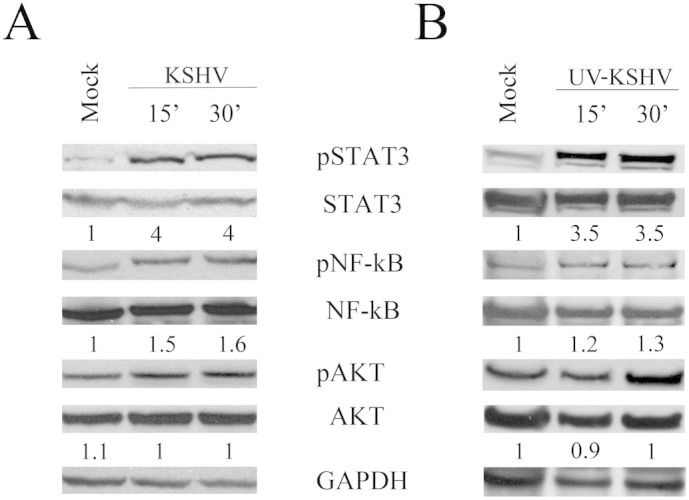
DCs were mock treated or exposed to (A) active or (B) UV-inactivated virus (UV-KSHV) for 15 and 30 min and STAT3, NF-kB p65 and AKT phosphorylation were analysed by western blot. Total STAT3, NF-kB p65, AKT are also shown. STAT3, NF-kB p65 and AKT phosphorylation and their fold of induction, based on densitometric analysis of the ratio of phospho/total molecules, are shown. GAPDH was included as protein loading control. 1 × 106 overnight serum-starved DCs/point were used in all experiments. One representative experiment out of three is shown.
STAT3 activation by KSHV is partially dependent on viral engagement of DC-SIGN
We then focused our study on STAT3 since it was highly phosphorylated in DCs exposed to active or UV-inactivated KSHV. We first evaluated if STAT3 activation could be induced by the engagement of KSHV with DC-SIGN, receptor molecule known to mediate KSHV binding and/or internalization into DCs10. For this purpose, we inhibited KSHV interaction with DC-SIGN by pre-incubating DCs with anti-DC-SIGN antibody (20 μg/ml for 1 hour at 4°C), according to a previous study10. The results obtained showed that anti-DC-SIGN pre-treatment led to ~60% reduction of KSHV-mediated STAT3 phosphorylation, based on densitometric analysis of phosphorylated vs total STAT3 protein ratio (fig. 2a), suggesting that STAT3 phosphorylation is partially mediated by viral interaction with DC-SIGN, although other molecules seem to be involved. Similar results were obtained with UV-inactivated virus (fig. 2b). Of note, the exposure to anti-DC-SIGN, in the absence of KSHV, did not affect STAT3 phosphorylation (fig. 2c). Finally, by inhibiting viral interaction with DC-SIGN, we strongly reduced KSHV entry into these cells, as measured by real-time DNA PCR (fig. 2d), confirming the importance of DC-SIGN as KSHV receptor on DC surface10.
Figure 2. STAT3 activation by KSHV is partially dependent on viral engagement of DC-SIGN.

DCs were mock-treated or pre-incubated with anti-DC-SIGN (20 μg/ml) for 1 hr at 4°C, before exposure for 15 min at 37°C to (A) active KSHV or (B) UV-KSHV. STAT3 phosphorylation and its fold of induction, based on densitometric analysis of the ratio of phospho/total STAT3, are shown. (C) DCs were incubated with anti-DC-SIGN antibody alone for 15 min at 37°C and STAT3 phosphorylation was analyzed by western blotting. Total STAT3 and GAPDH were included as control. 1 × 106/point overnight serum-starved DCs were used in all experiments. One representative experiment out of three is shown. (D) Real-time PCR showing KSHV DNA copy number/105 mock and KSHV-exposed DCs, pre-treated or not pre-treated with anti-DC-SIGN (20 μg/ml). Viral exposure was performed for 15 min at 37°C. Mean ± SD of three experiments is reported.
The KSHV-mediated STAT3 activation influences cytokine production in DCs
STAT3 phosphorylation has been reported to induce a release of cytokines33,34,35,36 that are able in turn to phosphorylate STAT3, by engaging their specific receptors37. Hence, we investigated if STAT3 activation by KHSV in DCs could influence the production of IL-10, IL-6 and IL-23. To this aim, serum-starved DCs were exposed to active or UV-inactivated KSHV and cultured overnight at 37°C in serum-free medium. Active and, to lesser extent, UV-inactivated KSHV induced a high release of IL-10, IL-6 and IL-23, in comparison to mock-treated DCs, as detected by ELISA assay (fig. 3). Moreover, DCs exposed to KSHV and then treated with LPS showed a reduced production of IL-12p70, the main cytokine responsible for TH1 polarization. Next, to demonstrate that the altered cytokines release was dependent on KSHV-mediated STAT3 activation, we pre-treated DCs with STAT3 inhibitor AG490, before exposure to KSHV19. As shown in fig. 3, AG490 pre-treatment strongly reduced the production of IL-10, IL-6 and IL-23. Conversely, the IL-12 production was restored by AG490 pretreatment. These results suggest that STAT3 activation is the main signaling that leads to an altered cytokine release by DCs exposed to KSHV.
Figure 3. STAT3 activation by KSHV alters DC cytokine release.

ELISA assay to measure the IL-10, IL-6 and IL-23 release by DCs or IL-12p70 release by LPS-treated DCs, exposed to (A) active or (B) UV-KSHV. Histograms (mean ± SD of three independent experiments) represent the amount of cytokines produced by mock and KSHV-exposed DCs, in the presence or in the absence of STAT3 inhibitor AG490 (50 μg/ml).
The KSHV-mediated STAT3 activation interferes with the autophagic flux in DCs
It has been reported that IL-10 activates STAT3 in bystander monocytes, in the course of HIV infection, and that STAT3 activation interferes with the autophagic process in these cells24. These observations prompted us to investigate whether KSHV-mediated STAT3 activation could interfere with the autophagic process in DCs. For this purpose, DCs were exposed to UV-inactivated KSHV, to rule out the known influence of viral encoded proteins on this process38,39. Mock or UV-inactivated KSHV exposed DCs were overnight serum-starved to induce autophagy, in the presence or in the absence of STAT3 inhibitor AG490. The autophagic flux was then evaluated by analyzing the expression levels of LC3I/II and p62, the two main autophagic markers, by western blot. As shown in fig. 4a, DCs, exposed to UV-inactivated KSHV, displayed a reduction of LC3II, concomitantly to the persistent STAT3 activation. The KSHV/STAT3-mediated increase of p62 expression level further indicates that an autophagic block was occurring in virus exposed DCs. The inhibition of STAT3 phosphorylation by AG490 pre-treatment (fig. 4a) was able to overcome the autophagic block, suggesting that it was mainly dependent on STAT3 activation by KSHV. Although STAT3 was activated after 15 minutes of viral exposure, the expression level of LC3II and p62 was slightly affected (fig. 4b), indicating that the interference with the autophagic process required longer viral exposure. More recently, STAT3 activation has been shown to inhibit autophagy by up-regulating Mcl-1, protein that, like BCL-2 and BCL-xL, is able to bind and sequester Beclin140,41. Hence, we evaluated if UV-KSHV autophagic block could be due to an up-regulation of Mcl-1 in DCs, dependent on STAT3 activation. We found that UV-KSHV induced an up-regulation of Mcl-1 in DCs, that was prevented by AG490 pre-treatment (fig. 4c). Furthermore, we analysed the expression level of BCL-2 and BCL-xL, two proteins belonging to the same family of Mcl-1 and also able to interact with Beclin1. The results shown in fig. 4c indicate that UV-KSHV slightly affected BCL-xL and BCL-2 and that Beclin1 itself was also not influenced, suggesting that KSHV mediated a specific Mcl-1 up-regulation through STAT3 that could be responsible for the interference with the autophagic process. To confirm the role of KSHV-mediated STAT3 activation in the autophagy inhibition, we knocked-down the STAT3 gene expression in DCs with specific siRNA (fig. 5a). According to the results obtained with AG490, the STAT3 silencing was able to prevent the increase of p62 and Mcl1 (fig. 5a) and the LC3II decrease (fig. 5b) induced by UV-KSHV in DCs. To confirm that the reduction of LC3II was due to an autophagic block, 3-Methyladenine (3-MA) was used as control to inhibit autophagy (fig. 5c).
Figure 4. STAT3 activation by KSHV correlates with an autophagy block in DCs.
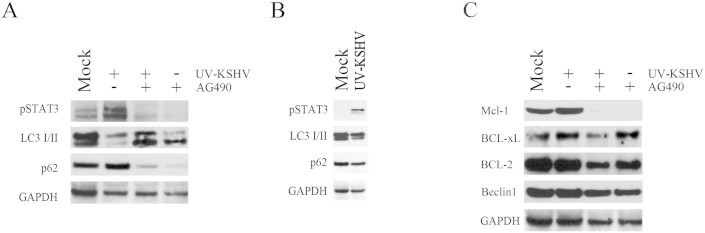
(A) Western blot analysis showing pSTAT3, LC3I/II and p62 expression level in DC mock or exposed to UV-KSHV for 15 min and cultured 24 hours in serum free conditions, in the presence or in the absence of STAT3 inhibitor AG490 (50 μg/ml). (B) Western blot analysis of pSTAT3, LC3I/II and p62 after 15 min of DC exposure to UV-KSHV (C) Western blot analysis of Mcl-1, BCL-xL, BCL-2 and Beclin1 expression in DCs mock or UV-KSHV-exposed, in the presence or in the absence of STAT3 inhibitor AG490 (50 μg/ml). GAPDH was included as protein loading control. 1 × 106 overnight serum-starved DCs/point were used in all experiments. Data, representative of three independent experiments, are reported.
Figure 5. STAT3 silencing prevents the KSHV-mediated autophagic block in DCs.
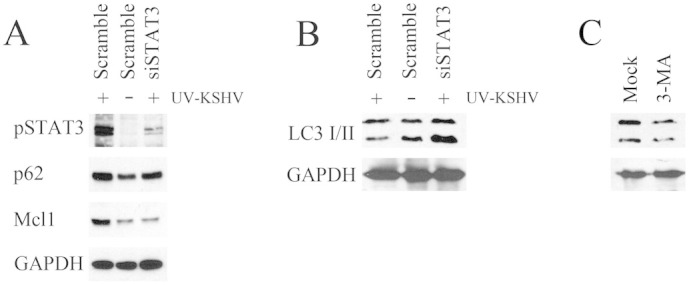
(A) Western blot analysis showing pSTAT3, p62 and Mcl1 expression and (B) LC3I/II in DC treated with control (scramble) or STAT3 specific siRNA exposed to UV-KSHV for 15 min and cultured 24 hours in serum free conditions. (C) LC3I/II was also evaluated in the presence of 3-MA autophagic blocker. GAPDH was included as protein loading control. 1 × 106 overnight serum-starved DCs/point were used in all experiments. Data, representative of three independent experiments, are reported.
Discussion
Based on our results, we suggest that STAT3 activation could be one of the molecular mechanisms underlying KSHV-mediated immunosuppression in human DCs. STAT3 has been previously reported to be activated by KSHV in endothelial cells42. To the best of our knowledge, this is the first report showing that STAT3 is also activated by KSHV in human DCs. The importance of this observation relays on the fact that STAT3 activation correlates with an immunosuppressive phenotype and function of DCs that is observed in the immune cells in tumor microenvironment and also in the peripheral blood of tumor bearing patients22,35,36, such as KS patients14,15. Here we show STAT3 activation in DCs occurs following KSHV-engagement of its DC receptor, DC-SIGN, and that it occurs independently of viral replication. We correlated STAT3 activation with a reduced production of IL12p70 in response to LPS stimulation and with an higher release of IL-10, IL-6 and IL-23. This cytokine pattern could skew the TH1/TH2 profile towards TH2 and/or TH17, promoting immunosuppression and inflammation. These cytokines might also, in turn, maintain STAT3 persistently phosphorylated, by binding on their specific receptors on DC surface, as previously reported in bystander monocytes in the course of HIV infection24. Finally, KSHV-induced STAT3 activation correlated with an interference with the autophagic process that could be overcome by the pre-treatment with AG490 or by STAT3 specific siRNA. We found that STAT3 activation correlated an up-regulation of Mcl-1 expression that was reversed by inhibiting STAT3 activation. Mcl-1 is one of the proteins able to bind and sequester Beclin1, hampering its essential role in autophagosome formation. Indeed, it has been recently shown that the reduction of STAT3 phosphorylation by Sorafenib treatment, in a hepatocarcinoma cell line, resulted in the down-regulation of Mcl-1 and disruption of Beclin1-Mcl-1 complex41. The Mcl-1 increased expression, induced by STAT3 activation by KSHV in DCs, could be a possible mechanism leading to the block of autophagy observed in this study.
The interference with the autophagic process is of pivotal importance for pathogens to impair antigen processing and presentation by DCs and therefore to avoid their own clearance. Of note, we observed that the autophagic block occurred after UV-inactivation of KSHV, excluding the involvement in such effect of vBCL2 and vFLIP viral proteins, that are reported to mediate such effect by binding Beclin1and Atg3, respectively38,39. Moreover, these data suggest that KSHV is able to mediate an immunosuppressive effect also by simply interacting with its receptor/s on the surface of DCs. In conclusion, by unveiling a new molecular pathway underlying KSHV-induced DC dysfunction, this study may help to find new therapeutic strategies to prevent and/or improve the outcome of KSHV-associated malignancies.
Methods
Cell lines, viral production, quantitation and UV-inactivation
BC3, a human B-cell line derived from PEL, carrying latent KSHV, was cultured in RPMI 1640 (SIGMA, R0883) supplemented with 10% fetal bovine serum (Invitrogen) in the presence of 5% CO2.
KSHV was obtained by inducing viral lytic cycle in BC3 cells treated with 0.3 mM Na-butyrate and TPA (10 ng/ml) for 96 hours, as described43. BC3 were then analyzed for the expression of K8.1A lytic antigen by IFA and the virus was collected when the percent of K8.1A positive cells was higher than 30%. The virus was then concentrated 200× in serum-free medium, in aliquots containing ~9 × 106 viral DNA copies, as measured by real-time DNA PCR (Nanogen Advanced Diagnostics, Milan, Italy), using primers amplifying the KSHV capsid protein gene, as previously described44. KSHV UV-inactivation was carried out at 1500 mJ in a UV cross-linker for 8 min.
Reagents and antibodies
TPA and Na-butyrate were purchased by SIGMA (cat.no P1585 and B5887, respectively). AG490 was purchased by Calbiochem (cat.no 658411). The following antibodies were used: mouse-anti-STAT3 and mouse anti-phosphoSTAT3 (pY705) (BD Transduction Laboratories, cat.no 610189 and 612356, respectively), rabbit anti-phospho-NF-kB p65 (Cell Signaling, cat.no S536), rabbit anti-NF-kB p65 (Santa Cruz, cat.no sc-109), mouse anti-GAPDH (Santa Cruz, cat.no sc-137179), mouse anti-DC-SIGN (Abcam, clone 120507), rabbit polyclonal anti-LC3 (Novus Biologicals, cat.no NB 100-222055), mouse monoclonal anti-p62 (BD Transduction Laboratories, cat.no 610833), rabbit polyclonal anti-BCL-xL (Cell Signalling, cat.no 5446), rabbit polyclonal anti-BCL-2 (Cell Signalling, cat. No 50E3) and rabbit polyclonal anti-Mcl-1 (Cell Signalling, cat.no D35A5).
Generation of dendritic cells and treatments
To generate monocyte-derived DCs, human peripheral blood mononuclear cells (PBMC), obtained from healthy donors (under informed consent), were isolated by Fycoll-Paque gradient centrifugation (Pharmacia, Uppsala, Sweden) from buffy coats. CD14+ monocytes were positively selected using anti-CD14 MAb-conjugated magnetic microbeads (Miltenyi Biotec, cod. no.130-050-301). Purified monocytes were cultured at a density of 106 cells/3 ml in 12-well plates for 6 days in RPMI 1640 containing 10% fetal calf serum (FCS), glutamine (300 μg/ml), 100 U/ml penicillin G, 100 μg/ml streptomycin and recombinant human granulocyte-macrophage colony stimulating factor (GM-CSF, 50 ng/ml) and interleukin 4 (IL-4, 20 ng/ml) (Miltenyi Biotec, cod. no 130-095-372 and 130-093-917, respectively) to generate DC (DC). Cytokines were replenished every other day with 10% fresh medium44. Depending on the experiments, over-night serum-starved DC were exposed to KSHV for different times at 37°C, or DCs were pre-incubated with anti-DC-SIGN monoclonal antibody (20 μg/ml) for 1 hour at 4°C and then exposed to KSHV or incubated with anti-DC-SIGN alone for 15 min at 37°C. To study the effect of KSHV-mediated STAT3 activation on autophagy, DCs were pretreated with STAT3 inhibitor, AG490 (50 μg/ml), or with DMSO as control, for 2 hours at 37°C and then exposed to KSHV for 15 min at 37°C. In some experiments STAT3 knock-down with specific siRNA (Santa Cruz) was performed before viral exposure, using INTERFERin (Polyplus) to transfect DCs. Cells were subsequently cultured overnight in serum-free medium. In some experiments, as control, 3-Methyladenine (3-MA) (5 mM) was used to inhibit autophagy.
Measurement of KSHV internalization by real-time DNA PCR
DCs untreated or exposed to KSHV for 15′ at 37°C, with or without pretreatment with anti-DC-SIGN, were washed twice in PBS and incubated with 0.25% trypsin-EDTA for 30′ at 37°C, to remove the non-internalized virus. The internalized KSHV was then quantified by real-time DNA PCR (Nanogen Advanced Diagnostics, Milan, Italy), using primers amplifying the KSHV capsid protein gene, as previously described44. Briefly, to standardize the assay, we used a plasmid containing part of ORF26. Serial dilutions of this plasmid ranging from 5 to 5 000 000 copies were used to characterize the linearity, precision, specificity, and sensitivity of the real-time DNA PCR. Amplification of human genomic β-globin DNA was also used to assess the absence of PCR-inhibitory substances45,46.
Western blotting
Cells (1 × 106) were washed twice with PBS and lysed in a modified RIPA buffer containing 150 mM NaCl, 1% NP-40, 50 mM Tris-HCl (pH 8), 0.5% deoxycholic acid, 0.1% SDS, 1% Triton X-100, protease and phosphatase inhibitors. The lysates were subjected to electrophoresis on 4–12% NuPage Bis-Tris gels (Life Technologies) and transferred to nitrocellulose membranes (Protran BA 85, Whatman). The membranes were blocked with 3% BSA (SIGMA, cod.no A4503) and probed with specific primary antibodies, overnight at 4°C. The membranes were washed and incubated with appropriated secondary antibody conjugated to horseradish peroxidase (Santa Cruz), used at 1:10.000. The membranes were washed and immunoreactivity was detected by enhanced chemiluminescence kit (Thermo Scientific, cod.no 32209).
ELISA assay
Serum-starved DCs mock were pretreated with STAT3 inhibitor, AG490 (50 μg/ml), or with DMSO as control, for 2 hours at 37°C and exposed to KSHV for 15 min at 37°C. Cells were then cultured for an additional 24 hours for IL-6, IL-10 and IL-23 production. For IL12p70 release DCs, during the 24 hours LPS (1 μg/ml, Salmonella abortus equi, SIGMA) was added to the cell culture. ELISA assays were performed using commercially available reagents and standards (for IL-6, IL-10 and IL-12 RayBiotech, Inc and for IL-23 Abcam ELISA kits were used). The supernatants were added in duplicate to appropriate pre-coated plates. After the plates were washed, horseradish peroxidase-conjugated detection antibody was added. The substrate used for color development was tetramethylbenzidine (TMB). The optical density was measured at 450 nm with a microplate reader (Multiskan Ex, Thermo Labsystem). The minimum detection dose is typically less than 1 pg/ml.
Statistical analyses
All data are represented by the mean ± standard error of at least three independent experiments.
Ethics statement
The study was approved by the ethical Committee of Policlinico Umberto I, Sapienza University, Rome, Italy.
Author Contributions
M.C. and R.S. conceived the experiments, R.S., G.D.G., R.G. and M.G. performed Western blot analysis, G.G. and A.C. performed Real Time PCR, L.C. performed ELISA assay, M.C., R.S. and A.F. interpreted results and wrote the paper. All authors reviewed and approved the manuscript.
Acknowledgments
This work was partially supported by grants from MIUR, Associazione Italiana per la ricerca sul Cancro (AIRC) (n. 10265), progetto strategico ISS 9ACF/1 of Ministero della salute and Pasteur Cenci-Bolognetti foundation. We thank Sandro Valia and Francesco Cosimi for the technical assistance. We also thank Gabriella D'Orazi for helpful discussion.
References
- Banchereau J. et al. Immunobiology of dendritic cells. Annu rev immunol 18, 767–811, 10.1146/annurev.immunol.18.1.767 (2000). [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A. & Sallusto F. Regulation of T cell immunity by dendritic cells. Cell 106, 263–266 (2001). [DOI] [PubMed] [Google Scholar]
- Cesarman E., Chang Y., Moore P. S., Said J. W. & Knowles D. M. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332, 1186–1191, 10.1056/NEJM199505043321802 (1995). [DOI] [PubMed] [Google Scholar]
- Soulier J. et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86, 1276–1280 (1995). [PubMed] [Google Scholar]
- Chang Y. et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869 (1994). [DOI] [PubMed] [Google Scholar]
- Grigoleit U. et al. Human cytomegalovirus induces a direct inhibitory effect on antigen presentation by monocyte-derived immature dendritic cells. Br J Haematol 119, 189–198 (2002). [DOI] [PubMed] [Google Scholar]
- Li L. et al. Epstein-Barr virus inhibits the development of dendritic cells by promoting apoptosis of their monocyte precursors in the presence of granulocyte macrophage-colony-stimulating factor and interleukin-4. Blood 99, 3725–3734 (2002). [DOI] [PubMed] [Google Scholar]
- Salio M., Cella M., Suter M. & Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol 29, 3245–3253 (1999). [DOI] [PubMed] [Google Scholar]
- West J. A., Gregory S. M., Sivaraman V., Su L. & Damania B. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J Virol 85, 895–904, 10.1128/JVI.01007-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappocciolo G. et al. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J Immunol 176, 1741–1749 (2006). [DOI] [PubMed] [Google Scholar]
- Cirone M. et al. Human herpesvirus 8 (HHV-8) inhibits monocyte differentiation into dendritic cells and impairs their immunostimulatory activity. Immunol lett 113, 40–46, 10.1016/j.imlet.2007.07.013 (2007). [DOI] [PubMed] [Google Scholar]
- Hensler H. R., Rappocciolo G., Rinaldo C. R. & Jenkins F. J. Cytokine production by human herpesvirus 8-infected dendritic cells. J Gen Virol 90, 79–83, 10.1099/vir.0.006239-0 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirone M. et al. HHV-8 reduces dendritic cell migration through down-regulation of cell-surface CCR6 and CCR7 and cytoskeleton reorganization. Virol J 9, 92, 10.1186/1743-422X-9-92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Bella S. et al. Quantitative and functional defects of dendritic cells in classic Kaposi's sarcoma. Clin Immunol 119, 317–329, 10.1016/j.clim.2006.01.011 (2006). [DOI] [PubMed] [Google Scholar]
- Stebbing J. et al. Disease-associated dendritic cells respond to disease-specific antigens through the common heat shock protein receptor. Blood 102, 1806–1814, 10.1182/blood-2003-03-0891 (2003). [DOI] [PubMed] [Google Scholar]
- Kerur N. et al. Characterization of entry and infection of monocytic THP-1 cells by Kaposi's sarcoma associated herpesvirus (KSHV): role of heparan sulfate, DC-SIGN, integrins and signaling. Virology 406, 103–116, 10.1016/j.virol.2010.07.012 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno M., Martino M., Sutherland C. L., Gold M. R. & Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med 188, 2175–2180 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparros E. et al. DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and modulates cytokine production. Blood 107, 3950–3958, 10.1182/blood-2005-03-1252 (2006). [DOI] [PubMed] [Google Scholar]
- Cirone M. et al. Dendritic cell differentiation blocked by primary effusion lymphoma-released factors is partially restored by inhibition of P38 MAPK. Int J Immunopathol Pharmacol 23, 1079–1086 (2010). [DOI] [PubMed] [Google Scholar]
- Yoshimura S., Bondeson J., Foxwell B. M., Brennan F. M. & Feldmann M. Effective antigen presentation by dendritic cells is NF-kappaB dependent: coordinate regulation of MHC, co-stimulatory molecules and cytokines. Int Immunol 13, 675–683 (2001). [DOI] [PubMed] [Google Scholar]
- Nefedova Y. et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol 175, 4338–4346 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefedova Y. et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172, 464–474 (2004). [DOI] [PubMed] [Google Scholar]
- Melillo J. A. et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol 184, 2638–2645, 10.4049/jimmunol.0902960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Grol J. et al. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PloS One 5, e11733, 10.1371/journal.pone.0011733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell 48, 667–680, 10.1016/j.molcel.2012.09.013 (2012). [DOI] [PubMed] [Google Scholar]
- He C. & Klionsky D. J. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43, 67–93, 10.1146/annurev-genet-102808-114910 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Marino G. & Levine B. Autophagy and the integrated stress response. Mol Cell 40, 280–293, 10.1016/j.molcel.2010.09.023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Lee S. & Jung J. U. When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin Immunopathol 32, 323–341, 10.1007/s00281-010-0226-8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquel A. et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 119, 4527–4531, 10.1182/blood-2011-11-392167 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang Y., Morgan M. J., Chen K., Choksi S. & Liu Z. G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 119, 2895–2905, 10.1182/blood-2011-08-372383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D. & Munz C. Innate and adaptive immunity through autophagy. Immunity 27, 11–21, 10.1016/j.immuni.2007.07.004 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira D. E., Ballon G. & Cesarman E. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol 18, 248–257, 10.1016/j.tim.2010.04.001 (2010). [DOI] [PubMed] [Google Scholar]
- Brocks C. P. et al. Functional alteration of myeloid dendritic cells through head and neck cancer. Anticancer Res 27, 817–824 (2007). [PubMed] [Google Scholar]
- Buisson S. et al. Monocyte-derived dendritic cells from HIV type 1-infected individuals show reduced ability to stimulate T cells and have altered production of interleukin (IL)-12 and IL-10. J Infect Dis 199, 1862–1871, 10.1086/599122 (2009). [DOI] [PubMed] [Google Scholar]
- Bharadwaj U., Li M., Zhang R., Chen C. & Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 67, 5479–5488, 10.1158/0008-5472.CAN-06-3963 (2007). [DOI] [PubMed] [Google Scholar]
- Kortylewski M. et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15, 114–123, 10.1016/j.ccr.2008.12.018 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. F. et al. Receptors for interleukin (IL)-10 and IL-6-type cytokines use similar signaling mechanisms for inducing transcription through IL-6 response elements. J Biol Chem 271, 13968–13975 (1996). [DOI] [PubMed] [Google Scholar]
- Pattingre S. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939, 10.1016/j.cell.2005.07.002 (2005). [DOI] [PubMed] [Google Scholar]
- Lee J. S. et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 11, 1355–1362, 10.1038/ncb1980 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M. et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 30, 395–407, 10.1038/emboj.2010.327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai W. T. et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 4, e485, 10.1038/cddis.2013.18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjabi A. S., Carroll P. A., Chen L. & Lagunoff M. Persistent activation of STAT3 by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J Virol 81, 2449–2458, 10.1128/JVI.01769-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli R. et al. Identification and characterization of the product encoded by ORF69 of Kaposi's sarcoma-associated herpesvirus. J Virol 82, 4562–4572, 10.1128/JVI.02400-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigoni B. et al. Human herpesvirus 8 is present in the lymphoid system of healthy persons and can reactivate in the course of AIDS. J Infect Dis 173, 542–549 (1996). [DOI] [PubMed] [Google Scholar]
- White I. E. & Campbell T. B. Quantitation of cell-free and cell-associated Kaposi's sarcoma-associated herpesvirus DNA by real-time PCR. J Clin Microbiol 38, 1992–1995 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile G. et al. Human herpesvirus 8 DNA in serum during seroconversion in allogeneic bone marrow transplant recipients. J Natl Cancer Inst 97, 1008–1011, 10.1093/jnci/dji177 (2005). [DOI] [PubMed] [Google Scholar]