

Abstract
Variation in the gene DTNBP1 encoding the protein dysbindin-1 is often associated with elevated risk for schizophrenia and with cognitive deficits prominent in that disorder. Because impaired function of the hippocampus is thought to play a role in these memory deficits and because NMDAR-dependent synaptic plasticity in this region is a proposed biological substrate for some hippocampal-dependent memory functions in schizophrenia, we hypothesized that reduced dysbindin-1 expression would lead to impairments in NMDAR-dependent synaptic plasticity and in contextual fear conditioning. Acute slices from male mice carrying 0, 1 or 2 null mutant alleles of the Dtnbp1 gene were prepared, and field recordings from the CA1 striatum radiatum were obtained before and after tetanization of Schaffer collaterals of CA3 pyramidal cells. Mice homozygous for the null mutation in Dtnbp1 exhibited significantly reduced NMDAR-dependent synaptic potentiation compared to wild type mice, an effect that could be rescued by bath application of the NMDA receptor co-agonist glycine (10 μM). Behavioral testing in adult mice revealed deficits in hippocampal memory processes. Homozygous null mice exhibited lower conditional freezing, without a change in the response to shock itself, indicative of a learning and memory deficit. Taken together, these results indicate that a loss of dysbindin-1 impairs hippocampal plasticity which may, in part, explain the role dysbindin-1 plays in the cognitive impairments of schizophrenia.
Keywords: Schizophrenia, Rodent, Genetic Model, Electrophysiology, Behavior
Introduction
Schizophrenia is a relatively common neuropsychiatric disorder characterized not only by positive and negative symptoms, but also by debilitating cognitive deficits significantly limiting psychosocial function (Green et al., 2000; Lesh et al., 2011). The disorder is highly heritable, and a number of promising candidate susceptibility genes have emerged in the last decade (Ayalew et al., 2012; Gejman et al., 2010). A firm understanding of the biological mechanisms by which these genetic risk factors cause cognitive deficits in schizophrenia could identify molecular targets for treating those deficits, which are largely refractory to current medications (Fumagalli et al., 2009; Goff et al., 2011; Tcheremissine et al., 2012).
Of the putative schizophrenia risk genes, the gene encoding dysbindin-1 (dystrobrevin-binding protein-1 or DTNBP1 (Guo et al., 2009; Talbot, 2009b) has been the target of considerable study. DTNBP1 lies within the chromosome 6p24-22 susceptibility locus (Straub et al., 2002; Straub et al., 1995). After discovery of this susceptibility locus through linkage analyses, twenty studies have reported associations between schizophrenia and DTNBP1 SNPs or haplotypes, though the specific risk variants identified have been inconsistent across studies/populations (Ayalew et al., 2012; Talbot, 2009b; Voisey et al., 2010). Although the association of DTNBP1 variants with schizophrenia has not met the criteria for significance (p<10−8) in genome-wide association studies (GWAS), a more recent analysis based on GWAS and other genetic and gene expression studies in humans and animals concluded that DTNBP1 is indeed among the top susceptibility genes for schizophrenia (Ayalew et al., 2012). Indeed, DTNBP1 risk SNPs are more common in a core subset of schizophrenia cases distinguished by earlier adult onset, more chronic course, and more prominent cognitive deficits and clinical symptoms (Wessman et al., 2009).
Consistent with a role for DTNBP1 in schizophrenia, abnormal dysbindin-1 gene and/or protein expression has been reported in the dorsolateral prefrontal cortex (DLPFC) and hippocampal formation (Talbot et al., 2004; Talbot et al., 2011; Tang et al., 2009a; Weickert et al., 2008; Weickert et al., 2004). While the existence and direction of the gene expression abnormalities in the DLPFC is controversial (Fung et al., 2011; Tang et al., 2009a; Weickert et al., 2004), dysbindin-1 protein reductions in schizophrenia are consistently seen in the DLPFC and hippocampal formation (Talbot et al., 2004; Talbot et al., 2011; Tang et al., 2009a).
Dysbindin-1 is expressed fairly ubiquitously throughout the brain in dopaminergic and glutamatergic neurons, including pre- and postsynaptic elements of glutamatergic cells (Talbot et al., 2006; Talbot et al., 2011; Talbot, 2009b). Amongst its functions, dysbindin-1 is involved in the control of pre-synaptic release of glutamate (Chen et al., 2008; Jentsch et al., 2009; Numakawa et al., 2004; Saggu et al., 2013) and is hypothesized to regulate synaptic NMDA receptor expression and function (Harrison and Weinberger, 2005; Karlsgodt et al., 2011). Such changes in glutamatergic transmission could potentially account for memory impairments on tasks such as the Morris water maze, Barnes circular maze, and the t-maze in mice with deletion mutations in Dtnbp1 resulting in loss of dysbindin-1 mRNA and protein, (Bhardwaj et al., 2009; Cox et al., 2009; Jentsch et al., 2009; Takao et al., 2008).
Given evidence that hippocampal dysfunction mediates some aspects of cognitive loss in schizophrenia (Boyer et al., 2007; Harrison, 2004) and that NMDAR-dependent plasticity of glutamatergic synapses is a proposed synaptic mechanism for memory-related processes(Bear, 1996; Citri and Malenka, 2008), we hypothesized that genetic loss of dysbindin-1 expression would compromise NMDA receptor-dependent synaptic plasticity in the hippocampus. We further hypothesized that positive allosteric modulation of the NMDA receptor complex would lessen these synaptic plasticity deficits. To test these hypotheses, we have performed field recordings of hippocampal field CA3 Schaffer collateral projections in mice carrying 1 or 2 null protein alleles of the Dtnbp1 gene (Li et al., 2003) and their wild-type littermates. Finally, we hypothesized that loss of dysbindin-1 would impair behavioral measures of hippocampal dependent learning. Due to a number of studies examining more conventional hippocampal dependent learning tasks, we hypothesized that dysbindin-1 deficient mice would also show impairments in contextual fear conditioning.
Methods
(More extensive methods are available in the supplementary materials)
Animals
The sandy mice used in these experiments were maintained on two backgrounds, DBA/2J (Figure 1&2) and C57Bl/6J (all other figures); in either case, the mice carry a 38.129 kb deletion in the Dtnbp1 gene, excising exons 6 and 7 that code for 52 amino acids in the coiled coil domain of dysbindin-1 (Li et al., 2003), resulting in partial dysbindin-1 reductions in heterozygous animals and total loss of the protein in homozygous animals (Li et al., 2003; Talbot, 2009a). Genotyping was performed according to methods described in Jentsch et al. (Jentsch et al., 2009). Experiments were performed at three different locations: the Medical University of South Carolina, the University of California at Los Angeles, and the University of Pennsylvania. The majority of slice electrophysiology experiments were performed at the Medical University of South Carolina on 35–50 day old male wild-type (dys +/+), heterozygous (dys +/−), and homozygous mutant (dys −/−) littermates. Studied performed at the University of Pennsylvania (Figure 3E–F) used male 90–120 days old dys +/+ and dys −/− mice on the C57Bl/6J background (n=4 per group). Behavioral studies leading to figures 5 and 6 were performed at the University of California at Los Angeles on male dys +/+, dys +/− and dys −/− mice on the C57Bl/6J background (n=16 per group). All experimental protocols were approved by the relevant Institutional Animal Care and Use Committees and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
Figure 1.

Synaptic plasticity in dysbindin deficient mice following tetanization. A) Field recordings were made from acute hippocampal slices of P35–50 dys +/+, and dys −/− SDY mice on the DBA/2J background. ANOVA revealed a significant interaction between time and genotype (p=0.04). B) Representative traces taken from dys +/+ and dys −/− field recording before and 55 minutes after tetanization. C) Post Hoc analysis showed significant differences (p=0.03) between dys +/+ and dys −/− in the average normalized differences in the first 5 minutes (STP). D) Post Hoc analysis showed significant and between dys +/+ and dys −/− (p=0.02) and between dys +/− and dys −/− (p=0.02) in the last 10 minutes (LTP) of each recording.
Figure 2.

Synaptic plasticity in dysbindin-1 deficient mice in the presence of 10μM glycine. A) Field recordings were made with bath-applied glycine (10μM). A two-way repeated measures ANOVA revealed a significant interaction between genotype and time (p=0.02). Unlike control conditions lacking glycine, Tukey’s HSD post-hoc analysis did not reveal any significant differences between genotypes. B) Average normalized differences for STP are plotted. Comparisons were made within genotype between treated and untreated recordings. No significant differences were found. C) Average normalized differences for LTP are plotted. Comparisons were made within genotype between treated and untreated recordings. Independent sample t-tests confirmed a significant increase in dys −/− LTP in the presence of glycine (p=0.03).
Figure 3.

Synaptic plasticity in dysbindin deficient mice following tetanization on the C57Bl/6J background. A) The experiment present in Figure 1 was repeated on the C57Bl/6J background on the DBA/2J background with dys +/+ and dys −/− mice. ANOVA revealed a significant interaction between genotype and time (p=0.03). B) A student’s t-test showed a significant differences between dys +/+ and dys −/− in the average normalized differences for the first 5 minutes (STP) of each recording (p=0.03). C) A student’s t-test revealed significant differences between dys +/+ and dys −/− in the average normalized differences for the last 10 minutes (LTP) of each recording (p=0.02). D) The basic finding presented in figure one was repeated independently at the University of Pennsylvania. Following tetanization (50–60 min), significantly less STP (p=0.004) and E) LTP (p=0.047) were observed in dys −/− compared to dys +/+ mice.
Figure 5.

Freezing during shock association acquisition. A) There were no differences between groups during the acquisition trial (pre shock). B) Following the foot shock, dys −/− mice showed significantly less freezing compared to dys +/+ mice.
Figure 6.
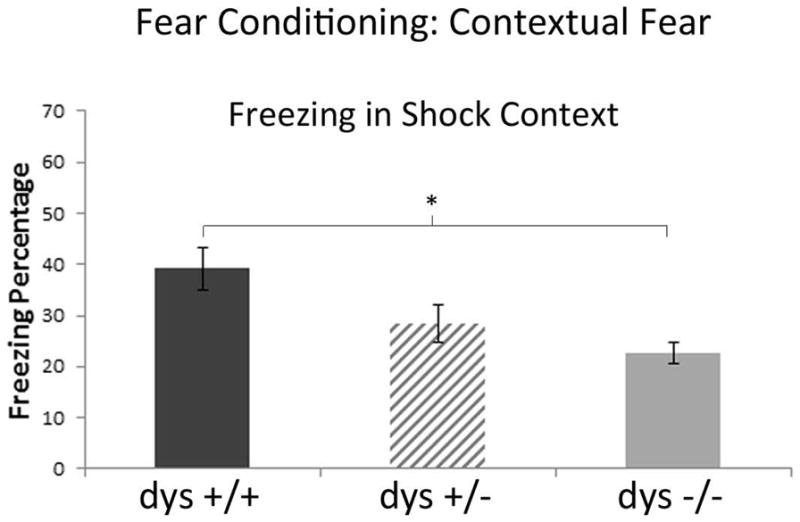
Freezing during contextual fear test. Mice were placed back into the context associated with shock (A) 24 hours after the acquisition. Significantly less freezing was observed in dys −/− mice compared to dys +/+ mice.
Electrophysiology
MUSC – Field Recordings
Mice were deeply anesthetized and the brain was rapidly removed and dissected into an ice-cold high-sucrose and cut in 300 μm horizontal sections. Slices were placed in an incubation chamber filled with room temperature artificial cerebrospinal fluid (ACSF) for 15–30 minutes. All solutions were constantly oxygenated with 95% O2/5% CO2.
Slices were next transferred to a recording chamber perfused with recording ACSF at room temperature. Stimulating electrodes were pulled from theta glass (WPI TST150-6) with an overall tip diameter of ~ 5 μm. Recording and stimulating electrodes were filled with recording ACSF. The recording electrode was placed in the stratum radiatum of hippocampal field CA1, and the stimulating electrode was placed 200–400 μm away from the recording electrode, stimulating Schaffer collateral projections originating from CA3. Field recordings were amplified using an Axopatch 200B amplifier. Data was acquired, stored and analyzed using Axograph X (www.axograph.com).
Square pulse stimulations were applied using a stimulator and a photoelectrically isolated unit. A paired-pulse protocol was delivered (0.2 msec pulses of ~25 μA with a 50 ms inter-stimulus interval) every 30 s until baseline responses stabilized (~30 min). An input/output curve (1–150 μA) was taken following stabilization, and baseline recordings were obtained for 10 min at half maximal stimulation (20–40 μA). Recordings that showed greater than 10% change across the baseline in either the stimulation artifact or the field excitatory postsynaptic potential (fEPSP) were excluded from analyses. Following baseline acquisition, long-term potentiation (LTP) was induced with either tetanization or theta burst stimulation (TBS). The tetanization protocol (0.2 msec pulses at 100 Hz for 1sec) was applied 3 times, 20 s apart. The TBS protocol (0.2 msec pulses at 100Hz for 40ms) was applied 4 times, 200 ms apart. In pharmacological experiments, glycine (10 μM) was dissolved in ACSF, bath applied before recording began, and maintained throughout the experiment.
UPENN - Field Recordings
Mice were anesthetized with isoflurane and decapitated; their brains were removed and were blocked in ice-cold ACSF. Hippocampal slices were cut at 350 μm with a vibrating tissue slicer. After preparation, slices were transferred into standard ACSF. Slices were transferred to a static interface chamber and warmed for 30 min and then maintained at room temperature for up to 6 h. A slice was transferred to an interface recording chamber for each experiment and superfused with warm (32 to 34 °C) oxygenated ACSF. Schaffer collateral stimulation was performed by an electrode placed near the border of CA1 and CA3 in stratum radiatum. Baseline stimulation was delivered every 30 seconds, tetanic stimulation was 100Hz for one second and was set to produce 30 to 40% of maximal fEPSP amplitude. To record field responses, a patch electrode filled with oxygenated ACSF and an Axoclamp 2D amplifier (Molecular Devices, Sunnyvale, CA) was used.
Behavior
Contextual-fear conditioning
All behavioral testing was assessed with Med Associates equipment (St Albans, VT). The fear-conditioning experiments were conducted in two distinctive contexts (A and B) with variable sensory features.
Fear was measured as mean percent time spent freezing to a tone or to a conditioned context (percentage of total time possible). Freezing is defined as complete cessation of movement and was measured using the Med Associates Near Infrared Video fear-conditioning system.
Habituation
Mice were habituated for 7 consecutive days. During this time they were transferred from the vivarium to the holding room for 1 h. For the first 15 min, they were left alone in the room. Over the next few minutes, each mouse was picked up by their tail, handled for at least 10 s, and then placed into a clean and empty 500 mL glass beaker.
Acquisition—Day 1
The mice were transferred from the vivarium into a quiet anteroom. They were individually placed in the context A chamber, and testing began immediately. There was a 4-min baseline followed by a 30-s tone played at 70 dB (2800 Hz). During the final 2 s of the tone, a 0.5 mA foot shock was delivered to the mice. This was followed by a 2 min post-shock period with no further programmed stimuli.
Context Fear Test—Day 2
The mice were taken from the vivarium and put in a quiet anteroom. They were then transferred to the context A chamber for a period of 8 min. No tones or shocks were presented.
Fear Generalization Test—Day 3
The mice were taken from the vivarium and placed in a quiet anteroom. From there, they were placed into the context B chambers for 8 min in the absence of tone and shock.
Statistical Analyses
Electrophysiology
Statistical analyses were performed in Predictive Analytics Software (PASW) Statistics 18 (http://www.spss.com/). In the primary analysis, two-way repeated measures ANOVAs were performed, with time being a within-subjects factor and genotype being a between-subjects factor. Degrees of freedom (df) were corrected in by the Greenhouse-Geisser method in cases where the assumption of sphericity was violated. Tukey’s HSD post hoc analysis was performed when significant time x genotype interactions were found. Analysis was performed on recordings from each slice.
Further analysis was performed in order to examine early and late components of synaptic plasticity. Average responses for each recording were taken for the first five and last ten minutes. One way ANOVA and Tukey’s HSD post hoc or Student’s t-test were used when appropriate.
Behavior
One-way ANOVAs were used to compare the percentage of time spent freezing in each context (A and B) between genotypes. The shock burst was measured as the maximum burst of activity captured by the Med Associates program. During the acquisition phase, freezing before and after the foot shock was measured. During the context fear test, the overall percentage of time spent freezing was measured for each mouse. During the fear generalization test, the overall percentage of time spent freezing was measured for each mouse.
Results
Electrophysiology
We first measured evoked synaptic plasticity under control conditions in DBA/2J dys +/+ (9 animals/19 recordings), dys +/− (11 animals/20 recordings), and dys −/− (10 animals/20 recordings) slices from sandy mice on the DBA/2J background (Figure 1). Under baseline conditions, no differences were observed in fEPSP slope between groups (dys +/+ =596.07±54.64 mV/s; dys +/− =605.15±76.02 mV/s; dys −/− =573.33±70.89 mV/s). Tetanization, however, induced a lasting change in the maximum slope of the fEPSP (Figure 1A). Representative traces of field recordings from a dys +/+ mouse are presented in Figure 1B. Statistical analysis revealed a significant genotype x time interaction. (F[8,224]=2.05, p=0.04). A significant effect of genotype on fEPSP slope was also found (F[2,56]=4.54, p<0.01). Post hoc comparisons showed a decrease in post-tetanization evoked field responses in the dys −/− vs. dys +/+ mice (p=0.01), but not between the latter and dys +/− mice. No differences were observed in baseline excitability as measured by I/O curve (data not shown).
Given the observed genotype x time interaction, we compared averaged responses recorded during the first 5 min (short term plasticity: STP) and the final 10 min of LTP in the three genotypic groups. ANOVA revealed significant differences in both STP (F[2,56]=4, p=0.03) and LTP (F[2,56]=4, p=0.02). Post hoc test of the STP time bin revealed significant differences between dys +/+ and dys −/−, but not between dys +/+ and dys +/−, mice (p=0.03; mean percent change from baseline ± SEM for dys +/+ =68.26±17.77%; dys +/− =58.08±16.41%; dys −/− =54.20±14.97%; Figure 1C). Moreover, a significant decrease in LTP in dys −/− vs dys +/+ mice (p=0.02) and between dys +/− and dys −/− mice (p=0.02) was also found; dys +/+ =21.37±13.30%; dys +/− =19.95±10.96%; dys −/− =12.21±5.5%; Figure 1D).
We next assessed if the impairments in synaptic plasticity observed in mutant mice could be rescued by positive allosteric modulation of NMDA receptor function via bath application of the NMDA receptor co-agonist glycine (10 μM; Figure 2A) on a C57Bl/6J background. As reported above, lasting synaptic plasticity was induced, and a significant genotype x time interaction was observed (F[6,157]=2.6, p=0.02). However, in the presence of glycine, there were no differences among the genotypes (F[2,49]=0.45, p=0.64) as confirmed by post hoc analyses (dys +/+ Gly vs. dys +/− Gly p=0.81; dys +/+ Gly vs. dys −/− Gly p=0.62; dys +/− Gly vs. dys −/− Gly p=0.95). Comparisons were made within genotype between treated and untreated recordings, revealing a rescue of LTP in dys −/− mice (t[19.2]=2.29, p=0.034; dys −/− =12.21±5.54%; dys −/− Gly =22.81±16.25%; Figure 2C).
These data show that complete but not partial loss of dysbindin-1 is associated with decreased long-term synaptic plasticity, and that this decrease can be recovered by augmentation of NMDA function. Considering previously published results (Tang et a., 2009) directly contradict those presented here, we performed additional studies in order to further confirm our results and attempt to explain the discrepancies.
We repeated the previous experiment in sandy mice on the C57Bl/6J background to determine if the effects observed above are strain-specific (Figure 3A–C). Given the absence of significant effects in the heterozygous sandy mice on the DBA2/J background, we studied only dys +/+ (6 animals/17 recordings) and dys −/− mice (10 animals/20 recordings). ANOVA revealed a genotype x time interaction similar to that seen in the mice on the DBA/2J background (F[2,82]=3.35, p=0.03; Figure 3A). T-tests confirmed that dys −/− mice on the C57Bl6/J background showed reduced STP (t[35]=2.13, p=0.04; dys +/+ =69.08±35.43%; dys −/− =44.03±35.96% Figure 3B) and LTP (t[35]=2.32, p=0.03; dys +/+ =27.30±20.25%; dys −/− =13.23±16.68% Figure 3C) when compared to dys +/+ mice. Combined with the studies of sandy mice on the DBA/2J background, these results show a clear decrease in synaptic plasticity in dysbindin-1-null mice independent of genetic background.
For further confirmation a small independent study (n=4 in each group) was also performed at the University of Pennsylvania examining synaptic plasticity in dys +/+ and dys −/− mice on the C57Bl/6J background (Figure 3D–E). Using a single stimulation tetanization on older mice (90–120 days) revealed a dramatic reduction in STP (t[6]=2.45, p=0.004; dys +/+.=68.33±5.93%; dys −/− =32.44±5.65%; Figure 3D) and LTP (t[3]=3.18, p=0.047; dys +/+ =163.78±52.59%; dys −/− = −11.57±11.66%; Figure 3E).
Since a previous study reported that LTP induced by theta burst stimulation (TBS) is increased, rather than decreased, in CA1 of sandy homozygotes on a C57Bl/6J background (Tang et al., 2009b), we also tested such mice and littermate controls using the TBS stimulation protocol. We could not replicate those findings. In our studies, both STP and LTP were induced following TBS in dys +/+ and dys −/−, but no significant group differences were observed (Figure 4A–C).
Figure 4.
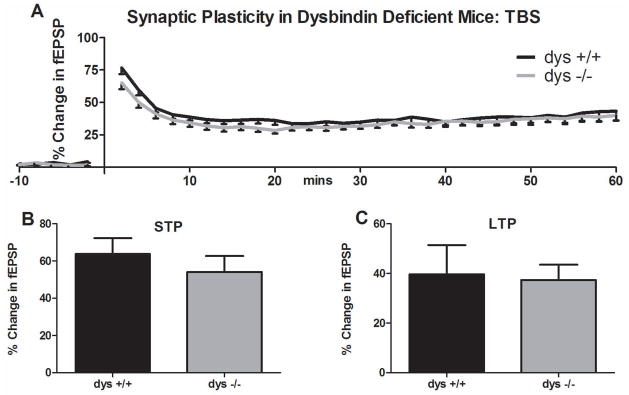
Synaptic plasticity in dysbindin deficient mice following theta burst stimulation (TBS). A) Time course of the percent change in fEPSP before and after TBS. No differences were observed in B) STP or C) LTP magnitude between groups during the last 10 minutes following TBS stimulation.
Taken together, these experiments performed on the DBA/2J and show a clear decrease in synaptic plasticity in response to tetanic stimulation in dysbindin-1-null mice independent of genetic background which can be recovered through augmentation of NMDA function. However, LTP was not decreased in dysbindin-1 null mice following TBS stimulation.
Contextual Fear Conditioning
For the acquisition phase, a one way ANOVA examined baseline freezing during the four minutes prior to the CS-US pairing (tone-shock). There was no significant differences between groups (F[1, 32]=0.085, p>.05) (Figure 5A). Subsequently, to measure sensitivity of animals to the foot shock, ANOVA was used to examine the shock-evoked activity burst between genotypes. The absence of a shock x genotype interaction (F[2,45]=1.29; p<.05) (data not shown) indicated that all groups responded to the shock with an equivalent unconditional response locomotor activity. Post-shock freezing (conditional response) was measured as it is indicative of hippocampal-dependent short-term memory for the shock event; ANOVA revealed a main effect of genotype (F[2,45]=37.44; p>.05; Figure 5B). Simple comparisons revealed that dys +/+ mice froze significantly more than dys −/− (M=4.82; p<.05).
Twenty-four hours later, the mice were exposed again to context A in the absence of tone and shock. ANOVA revealed a main effect of genotype (F[2,45)=5.98; p<.01; Figure 6). Simple post hoc analyses revealed that dys +/+ mice froze significantly more (mean=39.15%) than both dys +/− (M=28.35%; p<.05) and dys −/− (M=22.61%; p<.01). In contrast, in a test of fear generalization, the mice were placed in context B for 8 minutes, 48 hours after the shock paradigm. ANOVA revealed no main effect of genotype in the new context (F[2,45]=1.786; p>.05) (data not shown).
Discussion
Our current findings are consistent with a growing literature implicating dysbindin-1 in regulating glutamatergic (Chen et al., 2008; Jentsch et al., 2009; Numakawa et al., 2004; Saggu et al., 2013; Tang et al., 2009b), and dopaminergic (Hattori et al., 2008; Iizuka et al., 2007; Ji et al., 2009; Lutkenhoff et al., 2012; Murotani et al., 2007; Nagai et al., 2010; Papaleo et al., 2012) neurotransmitter systems affecting synaptic plasticity and associated cognitive processes known to be dysfunctional in schizophrenia. The data presented here specifically shed light on a putative mechanism by which dysbindin-1 deficits in schizophrenia may contribute to impaired NMDAR-dependent hippocampal synaptic plasticity and related behaviors. Synaptic plasticity likely plays a role in the development of positive and negative symptoms, as well as cognitive deficits (Javitt and Zukin, 1991; Jentsch and Roth, 1999; Kantrowitz and Javitt, 2009b). Proponents of the dysconnection theory of schizophrenia have argued that impaired plasticity may lead to dysfunctional connectivity between and within brain regions, thereby contributing to symptom expression (Stephan et al., 2006; Stephan et al., 2009). In fact, cognitive deficits are inarguably a core feature of schizophrenia (Elvevag and Goldberg, 2000; Lewis and Gonzalez-Burgos, 2008), with the degree of cognitive impairment being one of the best predictors of outcome for affected individuals (Green, 1996). While long-term memory deficits do not fully explain the loss of cognitive function in schizophrenia, the underlying cellular and molecular mechanisms behind synaptic plasticity, such as reduced NMDA function, are likely critical to the etiology of the disease state.
NMDA hypofunction is likely a critical component in schizophrenia etiology and cognitive dysfunction (Kantrowitz and Javitt, 2009a; Kantrowitz and Javitt, 2009b). NMDA antagonists and dopamine agonists are well known for their psychotomimetic properties, producing schizophrenia-like psychosis in unaffected individuals and exacerbating symptoms in patients with schizophrenia (Krystal et al., 1994). While dopamine agonists produce positive symptoms, NMDA antagonism also mimics some of the negative symptoms and cognitive deficits. Conversely, upregulation of NMDA function by modulation of the NMDA co-agonist glycine (Thomson, 1989) has proven effective clinically in the treatment of individuals with schizophrenia (Goff et al., 1999; Heresco-Levy et al., 2002; Heresco-Levy et al., 1999; Krystal et al., 1994; Tsai et al., 2006). Thus, regulation of NMDA function may be a critical component of future pharmacological therapies of schizophrenia (Krystal et al., 2009). Indeed, glycine treatment was found to restore LTP in dys −/− mice (Figure 2), suggesting a means for overcoming NMDA receptor hypofunction in the absence of dysbindin-1.
A recent publication examining NMDA receptor expression and synaptic plasticity in the hippocampus of dysbindin-1 null mutant mice reported increased LTP in dysbindin-1 deficient mice, inconsistent with the data presented here (Tang et al., 2009b). The primary methodological difference between that study and ours is the method used to induce LTP: Tang and colleagues utilized TBS while our study used tetanic stimulation. Both protocols involve the delivery of stimuli at 100 Hz, but tetanization applies a constant stimulation for ~1 s while TBS alternates several high frequency stimulations with quiescent inter-stimulation periods. Both protocols involve the activation of post-synaptic NMDA currents. TBS induces similar levels of plasticity with lower numbers of stimulation through a priming effect, activating feed-forward disinhibition through GABAB autoreceptors. Specifically, the initial burst activates presynaptic GABAB receptors, and the following burst exerts less inhibition, leading to enhanced excitation and NMDA receptor activation (Davies et al., 1991; Mott and Lewis, 1991; Pacelli et al., 1989; Staubli et al., 1999). While it was originally claimed that TBS is the more potent and physiological method of inducing LTP (Larson and Lynch, 1986), this has been contested more recently (Grover et al., 2009; Hernandez et al., 2005). Moreover, in intact C57Bl/6 mice, tetanization, but not TBS, induces LTP in CA1 (Buschler et al., 2012) arguing for the validity of tetanization procedure for inducing synaptic plasticity in CA1 of dysbindin-1 deficient mice. Indeed, we were unable to demonstrate increases in LTP in dys −/− mice with the TBS protocol. This may be especially so in the case of dysbindin-1 mutant mice, which exhibit deficits in inhibitory neuron function (Carlson et al., 2011; Ji et al., 2009) confounding results when using TBS given its effects on GABAB receptors noted above.
Here we also present evidence for impairments in contextual fear-conditioning, a form of learning which under normal conditions depends upon hippocampal processing (Maren et al., 2013). It has been well demonstrated that hippocampal lesion shortly after learning reduces contextual fear (Anagnostaras et al., 1999; Kim and Fanselow, 1992; Kim and Jung, 2006; Maren et al., 1997; Phillips and LeDoux, 1992). In contrast, hippocampal lesions performed before (Frankland et al., 1998; Maren et al., 1997) or long after (30+ days) (Anagnostaras et al., 1999; Kim and Fanselow, 1992; Maren et al., 1997) do not necessarily disrupt contextual fear conditioning. While dysbindin-1 deficiency did not affect the unlearned response to the noxious shock, namely the shock-induced activity burst, it did reduce post-shock freezing measured both within the condition session (Figure 5B) and in an extinction test the following day (Figure 6). Freezing in both conditions are at least in part learned responses to the shock and reflect memory processes (Fanselow, 1980). The reductions in contextual fear conditioning reported here may relate to reduced NMDA-dependent hippocampal synaptic plasticity in dysbindin-1 deficient mice. There is growing evidence supporting the relationship between hippocampal synaptic plasticity and contextual fear conditioning (Abeliovich et al., 1993; Bourtchuladze et al., 1994; Huerta et al., 2000; Liu et al., 2004). However, it is important to recognize that demonstrating a causal relationship between synaptic plasticity and memory processes has proven difficult and the role long-term potentiation in the hippocampus plays in establishing contextual fear conditioning requires further examination.
The results presented here provide the first evidence that dysbindin-1 deficiency reduces NMDA dependent synaptic plasticity. We demonstrate that this deficit in LTP can be recovered following NMDA enhancement. Finally, we demonstrated impairment in contextual fear conditioning. Together these results indicate that dysbindin-1 plays a critical role in synaptic plasticity and NMDA function. Reduced NMDA function and synaptic plasticity may in turn explain the impairments in spatial working memory found in dysbindin-1 deficient mice (Cox et al., 2009; Jentsch et al., 2009; Takao et al., 2008) and offers a mechanism by which dysbindin-1 may affect cognitive function in schizophrenic patients.
Supplementary Material
Acknowledgments
Grant Sponsor: NIMH; Grant Number: RL1 MH-83269; R01 MH-072880; P50 MH-064045;
Grant Sponsor: NIDCR; Grant Number: UL1 DE-019580
Grant Sponsor: NINDS; Grant Number: PL1 NS-062410
These studies were carried out as part of the UCLA Consortium for Neuropsychiatric Phenomics; they were funded, in part, by UL1-DE019580, PL1-NS062410 and RL1-083269. The author has performed the experiments presented here with contributions made by many colleagues including Judson Chandler, Heather Trantham-Davidson, Patrick Mulholland, Natasha New, and Amanda Grigg.
References
- Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993;75(7):1263–71. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Maren S, Fanselow MS. Temporally graded retrograde amnesia of contextual fear after hippocampal damage in rats: within-subjects examination. J Neurosci. 1999;19(3):1106–14. doi: 10.1523/JNEUROSCI.19-03-01106.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear Mark F. A synaptic basis for memory storage in the cerebral cortex. Proceedings of the National Academy of Sciences. 1996;93(24):13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj SK, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava LK. Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res. 2009;197(2):435–41. doi: 10.1016/j.bbr.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79(1):59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Boyer P, Phillips JL, Rousseau FL, Ilivitsky S. Hippocampal abnormalities and memory deficits: new evidence of a strong pathophysiological link in schizophrenia. Brain Res Rev. 2007;54(1):92–112. doi: 10.1016/j.brainresrev.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Buschler A, Goh JJ, Manahan-Vaughan D. Frequency dependency of NMDA receptor-dependent synaptic plasticity in the hippocampal CA1 region of freely behaving mice. Hippocampus. 2012;22(12):2238–48. doi: 10.1002/hipo.22041. [DOI] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, Phung QH, Gur RE, Arnold SE, Siegel SJ. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci U S A. 2011;108(43):E962–70. doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Feng YQ, Hao CJ, Guo XL, He X, Zhou ZY, Guo N, Huang HP, Xiong W, Zheng H, et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J Cell Biol. 2008;181(5):791–801. doi: 10.1083/jcb.200711021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33(1):18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Cox MM, Tucker AM, Tang J, Talbot K, Richer DC, Yeh L, Arnold SE. Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57BL/6J genetic background. Genes Brain Behav. 2009;8(4):390–7. doi: 10.1111/j.1601-183X.2009.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABA autoreceptors regulate the induction of LTP. Nature. 1991;349(6310):609–11. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14(1):1–21. [PubMed] [Google Scholar]
- Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15(4):177–82. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Cestari V, Filipkowski RK, McDonald RJ, Silva AJ. The dorsal hippocampus is essential for context discrimination but not for contextual conditioning. Behav Neurosci. 1998;112(4):863–74. doi: 10.1037//0735-7044.112.4.863. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Frasca A, Racagni G, Riva MA. Cognitive effects of second-generation antipsychotics: current insights into neurochemical mechanisms. CNS Drugs. 2009;23(7):603–14. doi: 10.2165/00023210-200923070-00005. [DOI] [PubMed] [Google Scholar]
- Fung SJ, Sivagnanasundaram S, Weickert CS. Lack of change in markers of presynaptic terminal abundance alongside subtle reductions in markers of presynaptic terminal plasticity in prefrontal cortex of schizophrenia patients. Biol Psychiatry. 2011;69(1):71–9. doi: 10.1016/j.biopsych.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Duan J. The role of genetics in the etiology of schizophrenia. Psychiatr Clin North Am. 2010;33(1):35–66. doi: 10.1016/j.psc.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245–53. doi: 10.1016/j.pbb.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. A Placebo-Controlled Trial of D-Cycloserine Added to Conventional Neuroleptics in Patients With Schizophrenia. Arch Gen Psychiatry. 1999;56(1):21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153(3):321–30. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26(1):119–36. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- Grover LM, Kim E, Cooke JD, Holmes WR. LTP in hippocampal area CA1 is induced by burst stimulation over a broad frequency range centered around delta. Learn Mem. 2009;16(1):69–81. doi: 10.1101/lm.1179109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo AY, Sun J, Riley BP, Thiselton DL, Kendler KS, Zhao Z. The dystrobrevin-binding protein 1 gene: features and networks. Mol Psychiatry. 2009;14(1):18–29. doi: 10.1038/mp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology. 2004;174(1):151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10(1):40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hattori S, Murotani T, Matsuzaki S, Ishizuka T, Kumamoto N, Takeda M, Tohyama M, Yamatodani A, Kunugi H, Hashimoto R. Behavioral abnormalities and dopamine reductions in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Biochem Biophys Res Commun. 2008;373(2):298–302. doi: 10.1016/j.bbrc.2008.06.016. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Ermilov M, Shimoni J, Shapira B, Silipo G, Javitt DC. Placebo-Controlled Trial of D-Cycloserine Added to Conventional Neuroleptics, Olanzapine, or Risperidone in Schizophrenia. Am J Psychiatry. 2002;159(3):480–482. doi: 10.1176/appi.ajp.159.3.480. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry. 1999;56(1):29–36. doi: 10.1001/archpsyc.56.1.29. [DOI] [PubMed] [Google Scholar]
- Hernandez RV, Navarro MM, Rodriguez WA, Martinez JL, Jr, LeBaron RG. Differences in the magnitude of long-term potentiation produced by theta burst and high frequency stimulation protocols matched in stimulus number. Brain Res Brain Res Protoc. 2005;15(1):6–13. doi: 10.1016/j.brainresprot.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Sun LD, Wilson MA, Tonegawa S. Formation of temporal memory requires NMDA receptors within CA1 pyramidal neurons. Neuron. 2000;25(2):473–80. doi: 10.1016/s0896-6273(00)80909-5. [DOI] [PubMed] [Google Scholar]
- Iizuka Y, Sei Y, Weinberger DR, Straub RE. Evidence that the BLOC-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J Neurosci. 2007;27(45):12390–5. doi: 10.1523/JNEUROSCI.1689-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20(3):201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Trantham-Davidson H, Jairl C, Tinsley M, Cannon TD, Lavin A. Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology. 2009;34(12):2601–8. doi: 10.1038/npp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Yang F, Papaleo F, Wang HX, Gao WJ, Weinberger DR, Lu B. Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc Natl Acad Sci U S A. 2009;106(46):19593–8. doi: 10.1073/pnas.0904289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrowitz J, Javitt D. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Javitt D, Kantorowitz J, editors. Handbook of Neurochemistry and Molecular Neurobiology. New York: Springer US; 2009a. pp. 39–89. [Google Scholar]
- Kantrowitz J, Javitt DC. Glutamatergic Approaches to the Conceptualization and Treatment of Schizophrenia. In: Lajtha A, Javitt D, Kantrowitz J, editors. Handbook of Neurochemistry and Molecular Neurobiology. 3. Springer US; 2009b. pp. 39–89. [Google Scholar]
- Karlsgodt KH, Robleto K, Trantham-Davidson H, Jairl C, Cannon TD, Lavin A, Jentsch JD. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol Psychiatry. 2011;69(1):28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256(5057):675–7. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev. 2006;30(2):188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Tolin DF, Sanacora G, Castner SA, Williams GV, Aikins DE, Hoffman RE, D’Souza DC. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug Discov Today. 2009;14(13–14):690–7. doi: 10.1016/j.drudis.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232(4753):985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316–38. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33(1):141–65. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35(1):84–9. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu IY, Lyons WE, Mamounas LA, Thompson RF. Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J Neurosci. 2004;24(36):7958–63. doi: 10.1523/JNEUROSCI.1948-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhoff E, Karlsgodt KH, Gutman B, Stein JL, Thompson PM, Cannon TD, Jentsch JD. Structural and functional neuroimaging phenotypes in dysbindin mutant mice. Neuroimage. 2012;62(1):120–9. doi: 10.1016/j.neuroimage.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res. 1997;88(2):261–74. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14(6):417–28. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott D, Lewis D. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252(5013):1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- Murotani T, Ishizuka T, Hattori S, Hashimoto R, Matsuzaki S, Yamatodani A. High dopamine turnover in the brains of Sandy mice. Neurosci Lett. 2007;421(1):47–51. doi: 10.1016/j.neulet.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kitahara Y, Shiraki A, Hikita T, Taya S, Kaibuchi K, Yamada K. Dysfunction of dopamine release in the prefrontal cortex of dysbindin deficient sandy mice: an in vivo microdialysis study. Neurosci Lett. 2010;470(2):134–8. doi: 10.1016/j.neulet.2009.12.071. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13(21):2699–708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- Pacelli GJ, Su W, Kelso SR. Activity-induced depression of synaptic inhibition during LTP-inducing patterned stimulation. Brain Res. 1989;486(1):26–32. doi: 10.1016/0006-8993(89)91273-0. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN, Weinberger DR. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol Psychiatry. 2012;17(1):85–98. doi: 10.1038/mp.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106(2):274–85. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Saggu S, Cannon TD, Jentsch JD, Lavin A. Schizophrenia Research. 2013. Potential molecular mechanisms for decreased synaptic glutamate release in dysbindin-1 mutant mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Scafidi J, Chun D. GABAB Receptor Antagonism: Facilitatory Effects on Memory Parallel Those on LTP Induced by TBS but Not HFS. J Neurosci. 1999;19(11):4609–4615. doi: 10.1523/JNEUROSCI.19-11-04609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59(10):929–39. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35(3):509–27. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337–48. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D, et al. A potential vulnerability locus for schizophrenia on chromosome 6p24-22: evidence for genetic heterogeneity. Nat Genet. 1995;11(3):287–93. doi: 10.1038/ng1195-287. [DOI] [PubMed] [Google Scholar]
- Takao K, Toyama K, Nakanishi K, Hattori S, Takamura H, Takeda M, Miyakawa T, Hashimoto R. Impaired long-term memory retention and working memory in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Mol Brain. 2008;1(1):11. doi: 10.1186/1756-6606-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K. The sandy (sdy) mouse: a dysbindin-1 mutant relevant to schizophrenia research. Prog Brain Res. 2009a;179:87–94. doi: 10.1016/S0079-6123(09)17910-4. [DOI] [PubMed] [Google Scholar]
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA, Kamins J, Hahn CG, Blake DJ, Arnold SE. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet. 2006;15(20):3041–54. doi: 10.1093/hmg/ddl246. [DOI] [PubMed] [Google Scholar]
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, Hahn CG, Siegel SJ, Trojanowski JQ, Gur RE, et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest. 2004;113(9):1353–63. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Louneva N, Cohen JW, Kazi H, Blake DJ, Arnold SE. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS One. 2011;6(3):e16886. doi: 10.1371/journal.pone.0016886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Ong WY, Blake DJ, Tang J, Louneva N, Carlson GC. Dysbindin-1 and its protein family with special attention to the potential role of dysbindin-1 in neuronal functions and the pathophysiology of schizophrenia. In: Kantorowitz DJaJ., editor. Handbook of Neurochemistry and Molecular Neurobiology. 3. New York: Springer US; 2009b. pp. 107–241. [Google Scholar]
- Tang J, LeGros RP, Louneva N, Yeh L, Cohen JW, Hahn CG, Blake DJ, Arnold SE, Talbot K. Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum Mol Genet. 2009a;18(20):3851–63. doi: 10.1093/hmg/ddp329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TT, Yang F, Chen BS, Lu Y, Ji Y, Roche KW, Lu B. Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc Natl Acad Sci U S A. 2009b doi: 10.1073/pnas.0910499106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcheremissine OV, Castro MA, Gardner DR. Targeting cognitive deficits in schizophrenia: a review of the development of a new class of medicines from the perspective of community mental health researchers. Expert Opin Investig Drugs. 2012;21(1):7–14. doi: 10.1517/13543784.2012.634798. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Glycine modulation of the NMDA receptor/channel complex. Trends in Neurosciences. 1989;12(9):349–353. doi: 10.1016/0166-2236(89)90042-8. [DOI] [PubMed] [Google Scholar]
- Tsai GE, Yang P, Chang YC, Chong MY. D-alanine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 2006;59(3):230–4. doi: 10.1016/j.biopsych.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Voisey J, Swagell CD, Hughes IP, Lawford BR, Young RM, Morris CP. Analysis of HapMap tag-SNPs in dysbindin (DTNBP1) reveals evidence of consistent association with schizophrenia. Eur Psychiatry. 2010;25(6):314–9. doi: 10.1016/j.eurpsy.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Rothmond DA, Hyde TM, Kleinman JE, Straub RE. Reduced DTNBP1 (dysbindin-1) mRNA in the hippocampal formation of schizophrenia patients. Schizophr Res. 2008;98(1–3):105–10. doi: 10.1016/j.schres.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. 2004;61(6):544–55. doi: 10.1001/archpsyc.61.6.544. [DOI] [PubMed] [Google Scholar]
- Wessman J, Paunio T, Tuulio-Henriksson A, Koivisto M, Partonen T, Suvisaari J, Turunen JA, Wedenoja J, Hennah W, Pietilainen OP, et al. Mixture model clustering of phenotype features reveals evidence for association of DTNBP1 to a specific subtype of schizophrenia. Biol Psychiatry. 2009;66(11):990–6. doi: 10.1016/j.biopsych.2009.05.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.