

Abstract
Protein ubiquitination is an important post-translational modification (PTM) that regulates almost every aspect of cellular function and many cell signaling pathways in eukaryotes. Alterations of protein ubiquitination have been linked to many diseases, such as cancer, neurodegenerative diseases, cardiovascular diseases, immunological disorders, and inflammatory diseases. In order to understand the roles of protein ubiquitination in these diseases and in cell signaling pathways, it is necessary to identify ubiquitinated proteins and their modification sites. However, due to the nature of protein ubiquitination, it is challenging to identify the exact modification sites under physiological conditions. Recently, ubiquitin remnant profiling, an immunoprecipitation approach, which utilizes monoclonal antibodies to specifically enrich for peptides derived from the ubiquitinated portion of proteins and mass spectrometry (MS) for their identification, was developed to determine ubiquitination events from cell lysates. This approach has now been widely applied to profile protein ubiquitination in several cellular contexts. In this review, we discuss MS-based methods for the identification of protein ubiquitination sites, analyze their advantages and disadvantages, and discuss their application for proteomic analysis of ubiquitination.
Introduction
Protein ubiquitination is a post-translational modification (PTM) involving in multiple enzymes in eukaryotic cells (Hershko and Ciechanover, 1998). The amino acid sequence of ubiquitin is highly conserved across different organisms (Fig. 1A). During ubiquitination, this 76-amino acid polypeptide is first activated by a ubiquitin-activating enzyme, E1, and then transferred to a ubiquitin-conjugating enzyme, E2. In the final step, a ubiquitin ligase, E3, covalently links the C-terminus of the ubiquitin, which was conjugated to the E2, to the ε-amino group of a lysine residue of its substrates by forming an isopeptide linkage. Besides lysine residues, in some rare cases, cysteine (Cadwell and Coscoy, 2005), serine, threonine (Wang et al., 2007; Wang et al., 2012), or the N-terminus (Breitschopf et al., 1998; Coulombe et al., 2004; Li et al., 2007) of a protein can also be ubiquitinated. If a protein is modified by one ubiquitin, or by multiple ubiquitins at different lysines, the modification is termed monoubiquitination or multi-monoubiquitination, respectively. Proteins can also be modified with ubiquitin polymers. This is because ubiquitin itself has amines on its seven lysine residues and on its N-terminus (Fig. 1B). All of them can be modified by another ubiquitin to form diverse polyubiquitin chains (Behrends and Harper, 2011; Miranda and Sorkin, 2007; Xu et al., 2009). The fate of the ubiquitinated proteins is mainly determined by the type of ubiquitination and polyubiquitin chains on the substrates. For example, monoubiquitination and multi-monoubiquitination can alter protein-protein interaction or change protein localization (Mukhopadhyay and Riezman, 2007). Proteins modified by K48-linked polyubiquitin chains are degraded by the 26S proteasome (Wilkinson et al., 1995), whereas K63-linked polyubiquitin chains have functions in endocytosis, DNA repair, and activation of protein kinases (Deng et al., 2000). Protein ubiquitination can also be reversed by deubiquitinating enzymes (DUBs) (Amerik and Hochstrasser, 2004). Hence, protein ubiquitination is dynamically regulated in cells.
Figure 1. Sequence alignment of ubiquitin from different organisms and the crystal structure of human ubiquitin.

(A) Ubiquitin has highly conserved amino acid sequence across different organisms. The ubiquitin sequences from Homo sapiens Mus musculus Rattus norvegicus Drosophila melanogaster Caenorhabditis elegans, and Saccharomyces cerevisiae are aligned. There are only three amino acids (red text in yellow background) that are different between human and yeast ubiquitin. In addition, ubiquitin does not have cysteine, tryptophan, and internal methionine. This can be exploited in protocols to enrich for the ubiquitinated portion of proteins.
(B) The crystal structure of human ubiquitin (PDB code: 1UBI). The ubiquitin C-terminal Gly can be conjugated to its substrates through an enzymatic cascade. The seven lysine residues in ubiquitin are colored and depicted in stick mode. Each of the seven lysines and the N-terminus can be modified by another ubiquitin molecule, forming diverse polyubiquitin chain topologies.
In order to understand the biological function of ubiquitination and its roles in diseases, it is important to identify protein ubiquitination sites. Defects in protein ubiquitination can cause many diseases, such as cancer (Hoeller and Dikic, 2009; Hoeller et al., 2006; Spataro et al., 1998), neurodegenerative diseases (Cole and Timiras, 1987; Giasson and Lee, 2003; Shimura et al., 2000; Shimura et al., 2001), cardiovascular disease (Herrmann et al., 2004; Powell et al., 2012), and inflammation (Ahmed et al., 2011; Gerlach et al., 2011; Tokunaga and Iwai, 2012). Although previous studies have advanced our understanding of the roles of protein ubiquitination in these diseases, the mechanisms and the molecular pathways involved in these diseases are still elusive. One common feature in these diseases is that they are often caused by the abnormal activities of ubiquitin ligases or DUBs (Hashizume et al., 2001; Kitada et al., 1998; Lucking et al., 2000; Pramstaller et al., 2005; Ruffner et al., 2001), which determine the substrate specificity and dynamic regulation of ubiquitination. Since these enzymes are the major players in protein ubiquitination, it is important to determine the ubiquitinated proteins and their modification sites that are regulated by these enzymes. Knowing the substrates of these enzymes can help to design approaches to study the mechanisms of ubiquitin-related diseases and hence to develop new therapeutic strategy to treat them.
It is particularly challenging to identify ubiquitination sites due to the following reasons. First, the abundance of ubiquitinated proteins is very low in cells under normal physiological conditions (Mann and Jensen, 2003). This is because many ubiquitinated proteins are either rapidly degraded by the proteasome (Wilkinson et al., 1995) or dynamically regulated in cell signaling pathways (Mori et al., 1995). Second, only one or a few lysine residues are modified in a ubiquitinated protein (Jadhav and Wooten, 2009). Third, ubiquitin has a larger size than many other PTMs, such as phosphorylation and acetylation, which increases the difficulty in their direct detection by MS. In addition, unlike phosphorylation, the ubiquitination sites do not appear to have major conserved motifs (Danielsen et al., 2011; Kim et al., 2011b; Xu et al., 2010), which prevents accurate prediction of ubiquitination sites for an unknown protein via bioinformatics tools. Therefore, in order to successfully identify protein ubiquitination sites, approaches for efficient enrichment of ubiquitinated proteins or peptides containing ubiquitination sites are required.
In this review, we discuss the approaches used to identify ubiquitination sites, focusing on the isolation of ubiquitinated proteins, and then evaluate the advantages and disadvantages of these methods. The recently developed high-throughput proteomic approach, ubiquitin remnant profiling, is described and its application is discussed in detail.
Conventional Approaches for the Identification of Protein Ubiquitination Sites
Many methods have been developed to identify ubiquitination sites in proteins of interest. However, in order to study the dynamic regulation of protein ubiquitination in a specific cell signaling pathway, or to identify protein ubiquitination events regulated by an E3 ligase or a DUB in an unbiased manner, MS-based proteomic approaches are more attractive since they provide the possibility to monitor ubiquitination at a proteome-wide level. Below, we discuss approaches for identifying protein ubiquitination sites at a single protein level and at the proteome level.
IDENTIFICATION OF UBIQUITINATION SITES FOR A SINGLE PROTEIN
Biochemical Approaches for the Identification of Protein Ubiquitination and its Modification Sites
Traditionally, the identification of ubiquitinated proteins and their corresponding modification sites was carried out through biochemical approaches. The first known ubiquitinated protein, A24 (the monoubiquitinated form of histone 2A), was observed on a two-dimensional gel electrophoresis of a nuclear protein fraction from rat liver (Goldknopf et al., 1975). This protein was further demonstrated to have a branched structure containing histone and ubiquitin (Goldknopf and Busch, 1975; Goldknopf and Busch, 1977; Hunt and Dayhoff, 1977). In addition, the ubiquitination site was mapped to its 119th residue (Goldknopf and Busch, 1977). Later on, a standard approach was developed to determine whether a protein is ubiquitinated and to identify the ubiquitination sites. In this approach, a putative ubiquitinated protein is purified by immunoprecipitation and its ubiquitination level is evaluated by anti-ubiquitin Western blotting (Shanklin et al., 1987). In order to determine the modification sites, the suspected ubiquitinated lysine residues are mutated to arginine to generate ubiquitination-resistant mutants. The tagged mutants and wild-type counterpart are then expressed, immunoprecipitated, and analyzed by an anti-ubiquitin Western blot to determine whether the mutated lysines are the modification sites based on the reduction of ubiquitination level after mutation (Baboshina and Haas, 1996; Gregori et al., 1990; Treier et al., 1994). This approach uses standard molecular biology and biochemistry techniques and is straightforward to carry out. Therefore, it remains a popular approach for the detection and validation of ubiquitination of proteins and lysine residues at a single protein level.
However, this approach has several shortcomings. First, these experiments are laborious and time-consuming, especially when multiple mutants have to be made to determine the exact modification sites, or if multiple sites are involved in the ubiquitination (Hou et al., 1994). Second, when the major ubiquitination site is mutated, other nearby lysines can become targeted as well (Bhat et al., 2010; Pickart, 2001; Treier et al., 1994). Third, extensive lysine-to-arginine mutation may disrupt protein structure (Sokalingam et al., 2012), and interfere with its normal ubiquitination pathways. Most importantly, an observed reduction in ubiquitination level after mutation may be caused by disrupting the interaction between the mutated protein and its E3 ligase so that the E3 ligase cannot ubiquitinate the mutant any more (Bustos et al., 2012). Therefore, approaches, such as MS-based approaches, which can directly detect ubiquitination sites from biological samples, are needed to confidently identify ubiquitinated proteins and their modification sites under physiological conditions.
Mass Spectrometry-Based Proteomic Approaches for the Identification of Ubiquitination Sites
(1) Principle for the identification of ubiquitination sites by MS
Ubiquitination sites can be identified by MS through the detection of peptide adducts derived from ubiquitin. The C-terminus of the mature ubiquitin has the amino acid sequence KESTLHLVLRLRGG, in which the last Gly can be conjugated to lysine residues on target proteins. The peptide bonds at the carboxyl side of Arg and Lys can be cleaved by trypsin regardless of whether they are in the target proteins, in the conjugated ubiquitin, or in the free ubiquitin, as long as these amino acids are not modified. When the conjugated ubiquitin is cleaved with trypsin, it leaves two Gly residues on the modified lysine residues, generating a new type of peptide (Fig. 2). These peptides are termed as “ubiquitin remnant-containing peptides” (Xu et al., 2010) or “ubiquitin signature peptides” (Peng et al., 2003). The monoisotopic mass of this diglycine adduct is 114.04 Da. In some very rare cases, trypsin cleaves the peptide bond between Arg and Leu but misses the one between Arg and Gly on the conjugated ubiquitin. Therefore, a four-amino acid peptide, LRGG, will be retained on the substrates, which generates an adduct with a monoisotopic mass of 383.23 Da on the modification sites (Denis et al., 2007; Warren et al., 2005). These two modifications (GG and LRGG) on the lysine residues do not generate complicated fragments in MS/MS spectra, which does not complicate the interpretation of the MS/MS spectra. Therefore, the modification sites can be readily detected. If different proteases are used to hydrolyze ubiquitinated proteins, the peptide fragment derived from the conjugated ubiquitin and retained on the modified proteins could be longer. For example, if Lys-C or Glu-C is used to digest the ubiquitinated proteins, longer peptides with sequences of ESTLHLVLRLRGG or STLHLVLRLRGG are retained on the modification sites. The unique peptides can unambiguously determine that the modification is from ubiquitin. Detection of these unique mass differences or the unique peptides on lysine residues by MS allows for the identification of both the ubiquitination sites and the corresponding ubiquitinated proteins.
Figure 2. Unique peptides derived from the ubiquitinated portion of proteins can be obtained after digestion of ubiquitinated proteins with different proteases.

In general, ubiquitin modifies lysine residues through its C-terminus. Depending on the proteases, such as trypsin, Lys-C, and Glu-C, which are used to digest the ubiquitin-protein conjugates, different amino acid sequences from the conjugated ubiquitin can be retained on the modified lysine residues. These unique modifications can be used to identify the ubiquitination sites through mass spectrometry.
(2) Identification of ubiquitination sites by MALDI-TOF MS
The ubiquitination sites can be identified through the measurement of the exact masses of the modified peptides which are derived from the ubiquitinated portion of proteins. In this approach, a protein of interest is purified by immunoprecipitation with antibodies targeting this protein or through an affinity tag, such as FLAG, HA, hexahistidine (His6), and glutathione S-transferase (GST), which is engineered at the terminus of the protein. Because ubiquitin has a molecular weight of ∼8.5 kDa, the ubiquitinated proteins have an increase in their molecular weight compared to the non-modified form and the exact molecular weight of the modified proteins depends on the number of ubiquitin on the modified proteins. Therefore, different forms of the protein can be separated by gel electrophoresis. The low mobility bands corresponding to the addition of ubiquitin molecules are extracted and the protein is digested in gel with Lys-C, which cleaves peptide bonds after lysines. If a protein is ubiquitinated, new peptides containing the remnant from the C-terminus of ubiquitin appear. Detection of the exact masses of these peptides by MALDI-TOF MS can identify the ubiquitinated peptides and ubiquitination sites. In order to confidently identify the modified peptides, a control experiment for the non-modified protein is carried out simultaneously to confirm the appearance of new peptides in the ubiquitinated protein (Fig. 3). For example, exogenously expressed HA- and FLAG-tagged macroH2A1.2 was purified through its C-terminal tags and new peptides derived from the ubiquitinated portion of proteins were identified based on the detection of the addition of the mass corresponding to the peptide of ESTLHLVLRLRGG, which was generated from the conjugated ubiquitin after Lys-C digestion. In this experiment, two ubiquitination sites on macroH2A1.2 were detected (Ogawa et al., 2005).
Figure 3. Mass spectrometric approaches to identify ubiquitination events for a protein of interest.

The target protein is expressed in cells without or with an affinity tag, such as FLAG and Myc tags. The target protein is then immunoprecipitated with the specific antibody targeting the protein of interest or with the anti-tag antibody. The purified proteins are resolved in SDS-PAGE and visualized by Coomassie brilliant blue staining or by silver staining. The bands corresponding to the ubiquitinated proteins are excised and digested. The resulting peptides are analyzed by mass spectrometry. In order to identify the modification sites by MALDI-TOF MS, peptides from the non-modified protein are also processed in parallel with peptides from the modified proteins. New peaks appearing in the modified sample correspond to the masses of the modified peptides. If LC-MS/MS is used, the peptide identification can be achieved through the MS/MS spectra.
Use of Lys-C as a protease for sample preparation has two advantages in the above case. The first one is that due to the presence of many lysine and arginine residues in macroH2A1.2, trypsin generates many short peptides which are difficult to detect and to determine its identity by MS. Instead, Lys-C can generate longer peptides than trypsin in macroH2A1.2. Many of them can be detected by MALDI-TOF MS, which results in the higher sequence coverage. The second advantage is the ubiquitin remnant generated by Lys-C is a long peptide with a unique sequence. The determination of the additional mass can unambiguously identify the modification sites. If trypsin was used for this experiment, the peptide derived from one of the ubiquitination site (GG-ε-KR) would be too short to detect. However, for many proteins, trypsin can also be used for the identification of ubiquitination sites. For example, an in vitro ubiquitination assay was carried out for the ubiquitination of GST-tagged Ubc5 and a Fourier transform-ion cyclotron resonance mass spectrometer was used to determine the exact masses of the tryptic peptides from the GST-tagged ubiquitinated Ubc5. The ubiquitination sites were identified by the detection of the additional mass on the modified lysines (Cooper et al., 2004).
Another example is the identification of the major ubiquitination site in the Mad homology 2 (MH2) domain of Smad4 using MALDI-TOF MS (Moren et al., 2003). The Smad4 MH2 domain was purified by a two-step immunoprecipitation using a FLAG tag and an HA tag and the samples were resolved by SDS-PAGE. The non-modified and modified proteins were visualized by silver stain, excised, and digested in-gel with trypsin. Based on the protein sequence, a comparison of the MS spectra between the non-modified and ubiquitinated forms of the protein suggested the presence of one ubiquitination site. This ubiquitination site is highly conserved in all Smad proteins. The mono- or oligoubiquitination of Smad4 promotes the formation of hetero-oligomerization with receptor-regulated Smads and thus maintains its normal transcriptional activity, whereas its polyubiquitination mostly occurs in cancer cells with Smad4 mutants, which causes its degradation. Since MALDI-TOF MS does not provide any information about protein sequence, proper control and validation experiments have to be carried out to draw a confident conclusion about the ubiquitination sites.
Ubiquitination sites can be unambiguously identified through the post-source decay analysis by MALDI-TOF MS. This approach can not only determine the amino acid sequence of regular tryptic peptides, but also identify the sequence of peptides derived from the ubiquitinated portion of proteins. However, the MS/MS spectra obtained from post-source decay are typically weak for small peptide fragments and the signals from b-ions may complicate the identification of the amino acid sequence. In order to simplify the MS/MS spectra obtained from post-source decay analysis, Cotter and coworkers (Wang et al., 2005) used 4-sulfophenyl isothiocyanate (SPITC) to chemically modify the N-terminus and lysine amines after trypsin digestion of ubiquitinated proteins. After sulfonation, the signals for the b-ions are significantly reduced while the y-ions maintain their signals under low-energy collision induced dissociation (CID). Therefore, the resulting MS/MS spectra only contain y-ions, and the amino acid sequence can be easily determined. In addition, the MS/MS spectra contain two portions: a sequence portion and a signature portion. In the signature portion, the sulfonated tags generate characteristic masses, which can validate the amino acid sequence between the N-terminus and the ubiquitination sites. This approach simplifies the identification of ubiquitination sites by MALDI-TOF MS. In addition, MALDI-TOF-TOF (Cotter et al., 2005) and ESI-MS/MS (Wang et al., 2006) have also been used to validate the sulfonated ubiquitin remnant-containing peptides, which contain two SPITC moieties.
Although detecting the exact masses of the conjugated peptides after proteolysis or the use of post-source decay in MALDI-TOF MS can identify ubiquitination sites, it has many shortcomings. First, highly pure samples for the non-modified protein and the ubiquitinated protein are needed. In general, one dimensional SDS-PAGE is used to separate these two forms of proteins, which may introduce contaminants and reduce peptide yield after trypsin digestion. The MALDI-TOF MS measurement has to be carried out simultaneously for two samples in order to find the peptides generated from the modified proteins containing the modification site(s). If the peptide derived from the ubiquitinated portion contains other modifications, such as phosphorylation, acetylation, methylation, deamidation, and methionine oxidation, it will be difficult to identify the modification site(s) using this approach. If the sample is contaminated during the preparation, or if multiple ubiquitination sites are present in the protein, it is not easy to identify all the modified peptides. Typically, the MALDI-TOF MS signal is relative low for the peptides derived from the ubiquitinated portion of proteins. Therefore, thorough examination of new MS peaks from the modified sample is required. In addition, the throughput of this approach is very low, which makes it difficult to rapidly identify multiple ubiquitination sites. Tandem MS is therefore needed in order to resolve these shortcomings.
(3) Identification of ubiquitination sites for a protein of interest by LC-MS/MS
LC-MS/MS can identify protein ubiquitination sites for a protein of interest. Two methods have been used to isolate the ubiquitinated forms for the identification of ubiquitination sites by LC-MS/MS. A common approach is to isolate the target protein both in the ubiquitinated and non-modified forms, which are further separated by SDS-PAGE. The gel bands corresponding to the modified form of proteins are excised and digested with trypsin. The resulting peptides are analyzed by LC-MS/MS and identified by a database search using a computational algorithm, such as Mascot, Sequest, X!Tandem, OMSSA, Phenyx, etc. The peptides from the modified portion of proteins can also be identified when they have suitable size and sufficient abundance.
One of the earliest examples in this scenario is the identification of the ubiquitination sites for the G protein α subunit (Gpa1) (Marotti et al., 2002). In this experiment, Gpa1was purified using His6-tag or anti-Gpa1 antibody and the ubiquitinated protein in SDS-PAGE was digested with trypsin, and analyzed on an ion trap mass spectrometer using CID fragmentation mode. A lysine ubiquitination site was revealed in the α helical structure of this protein. Further analysis of this protein demonstrated that the ubiquitination of this protein regulates its turnover. Similar approaches have also been used to identify multiple ubiquitination sites for cIAP2-mediated ubiquitination in tumor necrosis factor receptor-associated factor 1 (Lee et al., 2004), epidermal growth factor (EGF) receptor under EGF stimulus (Huang et al., 2006), HSP70 and HSP90 (Kundrat and Regan, 2010), inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) (Sliter et al., 2011), etc.
In order to more efficiently and unambiguously identify ubiquitination sites, a bimolecular affinity purification approach has been developed to purify ubiquitinated forms of proteins under fully denaturing conditions. In this approach, two different tags are engineered to the target protein and the ubiquitin N-terminus, respectively, to facilitate the purification of ubiquitinated form of the target protein (Maine et al., 2009). Proteins of interest are expressed with a tobacco etch virus (TEV) cleavage site (sequence: ENLYFQ↓S, ↓ representing the cleavage site) (Carrington et al., 1989) and a biotin acceptor peptide (BAP) comprising 76 amino acids derived from Propionibacterium shermanii transcarboxylase (Cronan, 1990) at the C-terminus or the N-terminus. The BAP can accept a biotin molecule on a lysine residue in cells. Therefore, the tagged protein can be isolated with streptavidin agarose. In addition, a His6-tagged ubiquitin was co-expressed with the tagged protein. Under denaturing conditions, all the ubiquitinated proteins were purified with Ni-NTA resin. Ubiquitinated proteins of interest, such as COMMD1, were further isolated with streptavidin and eluted by the TEV protease, which cleaves the peptide bond at the engineered TEV cleavage site. The purified ubiquitinated protein was resolved on SDS-PAGE. The ubiquitinated proteins were processed as described above and analyzed by LC-MS/MS. The ubiquitination sites were identified by detecting the Gly-Gly modification on the lysine residues. This approach has successfully identified the ubiquitination sites for several proteins, such as COMMD1, RelA, and HIF-1α (Maine et al., 2009).
Although these approaches have identified ubiquitination sites for many proteins, they have several disadvantages. These methods are low throughput and cannot be used to identify ubiquitination sites for a large number of proteins simultaneously. They also require time for making the expression construct. In addition, when a protein has multiple modification sites or is polyubiquitinated, the ubiquitinated proteins form a smear on the gel, which increases the size of the gel band to be excised and can therefore introduce contamination. In addition, some high molecular weight bands, which are not easily visible, may be missed and the abundance of the modified peptide underestimated. Therefore, alternative approaches are required to identify ubiquitinated proteins and the modification sites in a high-throughput manner.
IDENTIFICATION OF UBIQUITINATION SITES AT THE PROTEOME LEVEL
Due to the low abundance of ubiquitinated proteins in cells, it is necessary to enrich for the ubiquitinated proteins in order to successfully identify these proteins and their ubiquitination sites. Here, we briefly discuss the approaches that have been developed for the isolation of ubiquitinated proteins from cells and tissues for the identification of ubiquitination sites at the proteome level.
Expression of Tagged Ubiquitin for the Isolation of Ubiquitinated Proteins in Cells
Several small affinity tags have been used to label ubiquitin and to isolate ubiquitinated proteins from yeast and mammalian cells (Fig. 4A). The most common small affinity tags which have been used to label ubiquitin are His6 (Jeon et al., 2007; Kirkpatrick et al., 2005; Meierhofer et al., 2008; Peng and Cheng, 2005; Peng et al., 2003), BAP (Meierhofer et al., 2008; Tagwerker et al., 2006), and Strep (Danielsen et al., 2011) for proteomic identification of ubiquitination sites. These short peptide tags are engineered at the N-terminus of ubiquitin and the tagged ubiquitin is expressed in cells. When a protein is ubiquitinated, it is also labeled by the affinity tag on the ubiquitin. Therefore, tagged ubiquitinated proteins can be isolated using commercially available resins (Ni-NTA or TALON for His6, Streptavidin for BAP, and Strep-Tactin for Strep) and identified by tandem MS. The first such example was carried out in yeast, in which His6-tagged ubiquitin was expressed instead of wild-type ubiquitin. In this experiment, 110 ubiquitination sites in 72 proteins were identified by MS/MS (Peng et al., 2003) after the purification of ubiquitinated proteins, trypsin digestion, and peptide fractionation. The ubiquitination site was determined by the identification of the 114.04 Da Gly-Gly adduct on the modified lysine residues. In addition, more than 1000 proteins were also detected based on the identification of regular tryptic peptides but not the ubiquitin remnant-containing peptides. These proteins were called “putative ubiquitinated proteins”. This approach has also been extended to identify ubiquitinated proteins in mammalian cells (Jeon et al., 2007; Kirkpatrick et al., 2005; Meierhofer et al., 2008). In these experiments, tagged ubiquitin is expressed at a much lower level than the endogenous ubiquitin. Therefore, tagging of ubiquitin does not appear to alter protein ubiquitination pathways (Kirkpatrick et al., 2005).
Figure 4. Identification of ubiquitination sites by affinity enrichment of tagged ubiquitin.

(A) Ubiquitinated proteins from cell lysates are purified by affinity purification and digested with trypsin. The tryptic peptides are fractionated and desalted, and then subjected to LC-MS/MS analysis. The peptides are identified by computerized database search algorithms. In most cases, the identified peptides are unmodified tryptic peptides which come from the unmodified part of the target protein. However, a small number of peptides are identified with a diglycine modification on the lysine residue, which demonstrates that the protein is ubiquitinated.
(B) Ubiquitinated proteins are chemically cleaved at methionine or cysteine residues. Because ubiquitin does not have cysteines or internal methionines, ubiquitin and its tag remain intact during chemical cleavage. Therefore, protein fragments with the tagged ubiquitin can be isolated through the affinity tag. The purified ubiquitinated fragments are further digested with trypsin and identified by MS. Alternatively, when the lysine residues in ubiquitin are mutated to arginine, the ubiquitinated proteins can be digested with Lys-C while the tagged ubiquitin is kept intact. Then the ubiquitinated fragments are purified by the tag on the ubiquitin and further digested with trypsin for subsequent identification.
A short BAP peptide (GLNDIFEAQKIEWHE), which was discovered during a screen using combinatorial peptide libraries (Beckett et al., 1999), was appended to the N-terminus of ubiquitin to isolate ubiquitinated proteins. The lysine residue in this peptide can accept an exogenously supplied biotin in the presence of an Escherichia coli biotin holoenzyme synthetase, BirA. Therefore, the ubiquitinated proteins can be significantly enriched by the strong interaction between biotin and streptavidin prior to MS identification. This approach has been used to study protein ubiquitination in primary neurons (Franco et al., 2010) and resulted in the identification of 48 previously unknown ubiquitinated proteins in Drosophila.
Another small tag used for the purification of ubiquitinated proteins is Strep tag, which is an eight-amino acid peptide (WSHPQFEK) and can bind strongly to Strep-Tactin. This binding can be disrupted under mild elution conditions containing low concentrations of desthiobiotin or biotin (Schmidt and Skerra, 2007). A proteomic approach using Strep-HA-ubiquitin in human embryonic kidney 293T (HEK293T) cells to isolate ubiquitinated proteins by Strep-Tactin resin was successfully developed (Danielsen et al., 2011). In their experiments, 753 ubiquitination sites were identified among 5756 putative ubiquitinated substrates on a linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer. These results demonstrate that the use of affinity-tagged ubiquitin allows for the identification of hundreds of ubiquitination sites.
The unique nature of the amino acid sequence of ubiquitin can also be utilized to simplify peptides derived from the ubiquitinated portion of proteins. As shown in Fig. 1, ubiquitin does not contain internal methionine and cysteine residues. Chemical methods which cleave peptide bonds at these amino acids can potentially isolate protein fragments from the ubiquitinated portion of proteins (Fig. 4B). For example, cyanogen bromide (CNBr) can hydrolyze peptide bonds after methionines (Gross and Witkop, 1962) while 2-nitro-5-thiocyanobenzoic acid (NTCB) cleaves peptide bonds at cysteines (Tang and Speicher, 2004). However, ubiquitin itself cannot be cleaved by these reagents due to the lack of these amino acids except the presence of the initiator methionine. Therefore, the enrichment of ubiquitinated portion of proteins can be achieved by the affinity tag at the N-terminus of ubiquitin after the chemical cleavage of the substrates. These approaches have been applied to identify ubiquitination sites from rat heart (Jeon et al., 2007) and yeast cells (Starita et al., 2012) which expressed His8-tagged ubiquitin. In addition, when all the lysine residues in the wild-type ubiquitin are mutated to arginine, Lys-C cannot cleave the Lys-free ubiquitin mutant. Use of Lys-C to digest proteins conjugated with Lys-free ubiquitin and the affinity tag at the N-terminus of ubiquitin can substantially remove the non-ubiquitinated portion of proteins. After purification of these ubiquitin-containing fragments and digestion with trypsin, the peptides derived from the ubiquitinated portion of proteins are significantly enriched, which facilitates the identification of ubiquitination sites. This experiment led to the detection of 1392 ubiquitination sites in 794 proteins from HEK293T cells using an LTQ Orbitrap mass spectrometer (Oshikawa et al., 2012).
Although these small affinity tags improved the identification of ubiquitination sites, these approaches also have disadvantages. In order to purify His-tagged ubiquitinated protein, Ni-NTA or TALON resins are used under denaturing conditions to reduce non-specifically bound proteins. However, these resins also interact with many histidine-rich proteins in mammalian cells. In the case of 15-amino acid BAP tag, BirA has to be expressed simultaneously in cells and exogenous free biotin has to be supplied in the culture media. After purification, the BAP-tagged ubiquitinated proteins attached on the beads may not be efficiently eluted due to the strong interaction between biotin and avidin. To overcome this problem, on-bead trypsin digestion may be used instead of performing elution and digestion in solution. It should also be noted that a few endogenously biotinylated proteins will also be co-purified using avidin-based resins. During the purification of Strep-tagged ubiquitinated proteins, protein-protein interactions may not be completely disrupted, which leads to the co-isolation of non-ubiquitinated interacting partners. However, mild elution conditions can keep non-specifically bound and non-interacting proteins on the resin. Finally, in all these approaches, free tagged ubiquitin is also isolated together with ubiquitinated proteins, which may obscure the low abundance peptides from ubiquitinated proteins. The comparison between different tags that have been used in isolating ubiquitinated proteins is summarized in Table 1. The selection of the affinity tags depends on the application and the purification conditions. In general, purification under denaturing conditions works better for disrupting protein-protein interaction and to remove non-specifically bound proteins.
Table 1.
Peptide tags that have been used to label the N-terminus of ubiquitin for affinity purification of ubiquitinated proteins.
| # | Tags | Amino acid sequence |
Resins or antibodies for purification | Advantages | Disadvantages | Examples |
|---|---|---|---|---|---|---|
| 1 | Hexahistidine (His6) | HHHHHH | Ni-NTA, TALON |
|
|
(Jeon et al., 2007; Kirkpatrick et al., 2005; Meierhofer et al., 2008; Peng and Cheng, 2005; Peng et al., 2003) |
| 2 | Biotin acceptor peptide (BAP) | GLNDIFEAQ KIEWHE | Streptavidin, Neutravidin, Monomeric avidin |
|
|
(Meierhofer et al., 2008; Tagwerker et al., 2006) |
| 3 | Strep | WSHPQFEK | Strep-Tactin |
|
|
(Danielsen et al., 2011) |
| 4 | FLAG | DYKDDDDK | Anti-FLAG antibody |
|
|
(Argenzio et al., 2011; Kim et al., 2011a; Oshikawa et al., 2012) |
A common advantage is that all the tags are very small and their presence at the N-terminus of ubiquitin does not significantly affect the ubiquitination pathways.
A common disadvantage is that free ubiquitin is purified simultaneously.
The use of two tags sequentially to label ubiquitin for affinity purification of ubiquitinated proteins may overcome some of the limitations discussed above. Indeed, an approach using His6-BAP-ubiquitin to purify ubiquitinated proteins from cells under fully denaturing conditions has been described (Tagwerker et al., 2006). In addition, co-expression of BAP-ubiquitin and His6-ubiquitin in cells and purification of ubiquitinated proteins through the His6-tag and biotin-tag in BAP sequentially resulted in the specific enrichment and identification of proteins which are multi-monoubiquitinated and/or polyubiquitinated (Ota et al., 2008).
Use of Anti-Ubiquitin Antibodies to Isolate Ubiquitinated Proteins
Expressing tagged ubiquitin can facilitate the isolation of ubiquitinated proteins. However, this approach is difficult to be applied to determine ubiquitination events from non-modified cells, tissues, or patient samples. Therefore, approaches that do not rely on tagged ubiquitin are appealing.
Several types of anti-ubiquitin antibodies are available for the detection of free ubiquitin and ubiquitin conjugation. (I) Rabbit polyclonal antibodies were produced against denatured conjugated ubiquitin attached to keyhole limpet hemocyanin (KLH). These antibodies have a higher affinity for conjugated ubiquitin than for free ubiquitin (Haas and Bright, 1985) and can therefore be used for immunodetection and immunoprecipitation of ubiquitinated proteins. (II) Monoclonal antibodies recognizing free ubiquitin, such as P4D1, were raised against native ubiquitin or synthetic peptides from ubiquitin. These antibodies can be used for Western blotting, immunostaining, enzyme-linked immunosorbent assay, immunohistochemistry, flow cytometry, and immunoprecipitation. However, it cannot distinguish conjugated ubiquitin from free ubiquitin. (III) Monoclonal antibodies recognizing ubiquitin conjugates were also developed. FK2 and FK1 are two most commonly used such kind of antibodies (Fujimuro et al., 1994). The FK2 antibody can recognize both monoubiquitinated proteins and polyubiquitinated proteins while the FK1 antibody can only recognize polyubiquitinated proteins. Neither of them recognizes free ubiquitin. The FK1 and FK2 antibodies have been used for immunostaining (Takada et al., 1995), quantification (Fujimuro et al., 1997), and isolation (Takada et al., 2001) of polyubiquitin chains. (IV) Monoclonal antibodies which can detect specific ubiquitin chain linkages, such as K11-, K48-, K63-linkages, were developed through the phage display technology. These antibodies have been used for immunoblotting, immunostaining, and immunoprecipitation of polyubiquitin chains with specific chain linkages (Matsumoto et al., 2010; Newton et al., 2008). In general, monoclonal antibodies recognizing conjugated ubiquitin are more suitable for the isolation of ubiquitinated proteins.
Some of the above antibodies have been used for the isolation of ubiquitinated proteins for proteomic identification. For example, the FK2 antibody has been used to isolate endogenous ubiquitinated proteins from diseased cells (Matsumoto et al., 2005; Vasilescu et al., 2005; Vasilescu et al., 2007). Using FK2 antibodies, seventy putative ubiquitinated proteins and several ubiquitination sites were identified in a breast cancer cell line treated with MG132, a proteasome inhibitor (Vasilescu et al., 2005). This was the first proteomic analysis of endogenous ubiquitinated proteins from an untransfected diseased cell line. Using a similar approach, 21 ubiquitination sites were detected from a lung adenocarcinoma cell line (Vasilescu et al., 2007). This approach has also been improved by the incorporation of two dimensional LC-MS/MS, which led to the identification of 18 ubiquitination sites and 670 distinct putative ubiquitinated proteins in HEK293T cells (Matsumoto et al., 2005). This approach is broadly applicable to characterize ubiquitination in samples from animal tissues or patients without the need for genetic manipulation.
There are several advantages and limitations in the immunoprecipitation of untagged ubiquitinated proteins. The major advantage is that this approach can identify ubiquitination of endogenous proteins under physiological conditions, from diseased cells, or from tissues and patient samples. Another advantage is that certain types of chain-linkage specific antibodies, such as K11-, K48-, K63-linkage specific antibodies (Matsumoto et al., 2010; Newton et al., 2008), are available for the isolation of ubiquitinated proteins with specific chain linkages. The data resulting from these experiments can provide additional important information about the functions of ubiquitinated proteins with specific class of ubiquitin linkages. The primary disadvantages of this approach are the relatively high cost of antibodies and potential non-specifically bound proteins recovered during immunoprecipitation. Some of these antibodies may cross-react with NEDD8, which has a similar C-terminal amino acid sequence (LHLVLALRGG) as ubiquitin (LHLVLRLRGG), although this modification is much less abundant than ubiquitination. Here, the italic letters represent the different amino acids in the C-terminal regions of these two proteins.
Isolation of Polyubiquitinated Proteins via Ubiquitin-Binding Domains
Another approach to isolate ubiquitinated proteins for MS identification uses ubiquitin-binding domains (UBDs). Some E3s, DUBs, and ubiquitin receptors contain UBDs, which recognize ubiquitin (Haglund and Dikic, 2005) and can be utilized to enrich for polyubiquitinated proteins. Proteins containing multiple UBDs, such as ubiquitin-associating domain (UBA) and ubiquitin-interacting motif (UIM), are first covalently linked to a solid support. The ubiquitinated proteins are then isolated via the interaction between the UBDs and polyubiquitin chains. The proteasome subunit, S5a, contains two UIMs (Young et al., 1998) and selectively binds to polyubiquitin chains containing four or more ubiquitins prior to the degradation of the ubiquitinated proteins (Walters et al., 2002). Tan et al. used S5a to pull down polyubiquitinated proteins and identified 19 ubiquitination sites from liver cells (Tan et al., 2008). Multimerization of the UBAs or UIMs has also shown to be more powerful in the purification of polyubiquitinated proteins, presumably by the increase in binding affinity due to the enhanced avidity. Use of two UBAs from Arabidopsis or three UIMs from S5a to generate multiple UBD-containing proteins for the isolation of polyubiquitinated proteins has resulted in the identification of 294 proteins and 85 ubiquitination sites with the aid of the multidimensional LC-MS/MS (Maor et al., 2007). Amazingly, Shi et al. (Shi et al., 2011) generated a GST fusion protein with four UBA domains from ubiquilin-1 to isolate endogenous polyubiquitinated proteins from untransfected HEK293T cells and identified 294 ubiquitination sites on a high resolution mass spectrometer. The use of UBDs has resulted in the identification of many ubiquitinated proteins and their ubiquitination sites. These results indicate that it is a promising approach for future ubiquitome studies.
One of the advantages using UBDs is that this approach could employ diverse UBDs that recognize different ubiquitin chain topologies with high affinity. Recently, four tandem UIMs derived from S5a were used to generate a tandem ubiquitin binding entity (TUBE), which has high affinity towards polyubiquitinated proteins and has been used to selectively isolate polyubiquitinated proteins (Hjerpe et al., 2009; Hjerpe and Rodriguez, 2008). UBDs derived from other proteins, such as Rabex-5 (Lee et al., 2006; Penengo et al., 2006) and Dsk2 (Ohno et al., 2005), which have high affinity toward ubiquitin (Hurley et al., 2006), could also be used to develop high affinity ubiquitin-interacting proteins. It is possible to use multiple different UBAs or UIMs to generate versatile TUBEs and enrich for polyubiquitin chains with specific chain linkages for the identification of selected pools of ubiquitinated proteins. Indeed, fluorescent sensors using engineered proteins containing multiple optimized UIMs have been developed and used to detect the dynamic regulation of endogenous K63-linked polyubiquitin chains (Sims et al., 2012). The limitation of this approach is that the affinity of current UBDs with monoubiquitinated proteins is relative weak, which necessitates the use of other approaches to identify monoubiquitinated proteins.
The advantages and disadvantages of the above three approaches for the isolation of ubiquitinated proteins are summarized in Table 2.
Table 2.
Comparison of three different approaches in the isolation of ubiquitinated proteins for proteomic identification of ubiquitination sites.
| # | Tags | Advantages | Disadvantages | Examples |
|---|---|---|---|---|
| 1 | Enrichment with affinity-tagged ubiquitin |
|
|
See Table 1. |
| 2 | Immunoprecipitation with anti-ubiquitin antibodies |
|
|
(Matsumoto et al., 2005; Vasilescu et al., 2005; Vasilescu et al., 2007) |
| 3 | Isolation with UBDs |
|
|
(Maor et al., 2007; Shi et al., 2011; Tan et al., 2008) |
Ubiquitin Remnant Profiling for the Identification of Ubiquitination Sites at a Proteome-Wide Level
As mentioned above, ubiquitination sites can be identified by MS through the identification of ubiquitin remnant-containing peptides. A common major disadvantage of these proteomic approaches is that many of the peptides that are detected in MS do not contain modified lysine residues (Danielsen et al., 2011; Jeon et al., 2007; Matsumoto et al., 2005). This makes it difficult to tell whether proteins from which those peptides derive are ubiquitin-interacting proteins, non-specifically bound proteins, or the actual ubiquitinated proteins. Indeed, for any ubiquitinated protein, the majority of the peptides detected on MS will not contain modified lysine residues, which significantly reduces the efficiency of the mass spectrometric analysis in the identification of ubiquitination sites. In order to unambiguously determine if a protein is indeed ubiquitinated, it is necessary to identify the peptide which contains the modified lysine residue. The enrichment and detection of ubiquitin remnant-containing peptides can be achieved by antibodies which can bind lysines containing the ubiquitin remnant.
DEVELOPMENT OF ANTI-DIGLYCYL LYSINE ANTIBODIES
Epitope Selection for Ubiquitinated Proteins
Several epitopes could be chosen for the generation of antibodies recognizing peptides derived from the ubiquitinated portion of proteins. As shown in Fig. 2, Glu-C, Lys-C, and trypsin can generate different peptides from conjugated ubiquitin on the modified lysine. Glu-C and Lys-C generate unique 12 or 13 amino acid peptides while trypsin generates a diglycine modification, or in some rare case, a tretrapeptide with a sequence of Leu-Arg-Gly-Gly if one cleavage is missed. The 12 and 13 amino acid peptides have the optimal lengths for antibody production. However, the antibodies targeting these two peptides would most likely recognize free ubiquitin as well and the specificity for the conjugated peptide may not be high enough to distinguish them from free ubiquitin. In addition, these long peptides contain basic amino acids, such as two arginines and one histidine, which can generate multiple positive charges and complicate peptide fragments in mass spectrometers when it is covalently linked to a regular tryptic peptide. This will create technical challenges in the database search of MS/MS spectra for their identification. On the other hand, trypsin digestion generates a diglycine remnant on the modified lysine residues. This modification is relative small so that it was thought that antibodies targeting this epitope would not have high affinity and specificity. However, the affinity and specificity of this antibody could be increased due to the presence of the free amine at the N-terminus of the first glycine and the conjugation between the second glycine and the modified lysine residue. The size and chemical structure of this epitope (Gly-Gly-ε-Lys) is very similar to that of a tretrapeptide Gly-Gly-Gly-Gly, which has successfully been used to generate a polyclonal antibody with allotypic specificity (Escribano, 1974). Moreover, diglycine modification on the lysine residue is very stable and does not generate complicated ions during MS fragmentation. In addition, trypsin digests proteins more efficiently than either Lys-C or Glu-C. Therefore, the diglycine remnant is an appealing epitope for antibody generation.
Strategies for the Generation of Monoclonal Antibodies Targeting Diglycine-Modified Lysines
The first publication describing the use of ubiquitin remnant specific antibodies for profiling ubiquitination was from the Jaffrey group (Xu et al., 2010). These investigators generated ubiquitin remnant-specific antibodies by using t-butyloxycarbonyl (Boc)-Gly-Gly-N-hydroxysuccinimide to modify all lysine residues in a lysine-rich histone protein. They then used trifluoroacetic acid to remove the Boc protecting group. This Gly-Gly-modified histone mimics diglycine-modified lysines in diverse backbone amino acid sequences and was used as an antigen to produce monoclonal antibodies (Fig. 5) (Xu et al., 2010). A subsequent publication used an antibody generated by Cell Signaling Technology. This antibody was prepared through a related approach. In that approach, a 13 amino acid peptide library was made. The peptides in this library contain a diglycine-modified lysine residue in the middle, which was surrounded by any of 18 amino acids (except Trp and Cys) at other positions, and a Cys at the peptide N-terminus. This peptide library was then crosslinked to KLH carrier protein and used to generate the monoclonal anti-diglycyl lysine antibody from rabbits (Fig. 5) (Kim et al., 2011b). These studies provided the first ubiquitin-remnant specific antibodies.
Figure 5. Two antigens used to generate anti-diglycyl lysine antibodies.

(A) Antigen I is a lysine-rich protein, such as histone, which is chemically modified with diglycine on the lysine residues. This was used in Xu, et al. 2010.
(B) Antigen II uses KLH as a carrier protein to couple a peptide library which consists of diglycine-modified lysine in the center and random amino acids at other positions. X represents any of amino acids except Cys and Trp. This was used in Kim, et al., 2011.
CHARACTERIZATION OF ANTI-DIGLYCYL LYSINE ANTIBODIES
The specificity of the antibodies is important for their use in proteomic application. In general, peptides with a length of 8–20 amino acids are optimal for antibody recognition. It was commonly thought that small epitopes like methyl groups and diglycine could not confer the specificity and affinity needed for antibodies suitable for immunoprecipitation. A second issue is that the antibodies must recognize ubiquitin remnant-modified lysine in diverse sequence contexts. The unique strategies that were used to generate anti-diglycyl lysine antibodies used multiple epitopes in one antigen with a variety of backbone amino acid sequences, which increases the likelihood of selecting for antibodies capable of binding diverse sequences. Each of these features was measured when validating the antibodies.
The anti-diglycyl lysine antibody can recognize diglycine-modified proteins in a variety of context and immunoprecipitate peptides with diglycine-modified lysines. One of the anti-diglycine antibodies was extensively characterized prior to its use for immunoprecipitation of ubiquitin remnant-containing peptides from complex mixtures (Xu et al., 2010). To do so, a series of protein samples, including histone and rat brain lysate, with diglycine-modified lysines were generated by the same chemical reactions that were used to generate Antigen I (Fig. 5). Using these test samples, an antibody (clone GX41), which has the highest specificity among many hybridoma cell lines, was identified. The anti-diglycyl lysine Western blotting confirmed that the antibody can efficiently recognize diglycine-modified proteins and does not interact with the non-modified counterparts (Fig. 6A). In addition, the result from the rat brain lysates, which showed a smear on the Western blot, demonstrated that this antibody can recognize epitopes in a variety of contexts, regardless of the amino acid sequence in protein backbones. In order to further confirm the ability of this antibody for immunoprecipitation, a peptide mixture containing similar amount of non-modified and diglycine-modified peptides was prepared and the diglycine-modified peptides were immunopurified with the immobilized antibody. The MALDI-TOF MS measurement for the samples prior to and after the immunoprecipitation demonstrated that only modified peptides were present after immunoprecipitation (Fig. 6B). In addition, this antibody also pulled down diglycine-modified peptides from the trypsin digest of a complex mixture of diglycine-modified and non-modified bovine serum albumin. The MS/MS spectra of two diglycine-modified peptides from bovine serum albumin provided excellent b-ions and y-ions and precisely identified the modification sites (Fig. 6C). These results confirmed the ability of this antibody to immunoprecipitate ubiquitin remnant-containing peptides, and the ability of MS/MS to efficiently reveal the sequence of these peptides.
Figure 6. Characterization of the anti-diglycyl lysine antibody (clone GX41).

(A) The diglycyl lysine antibody can recognize diglycine-modified lysine-containing proteins but not the non-modified counterparts. Samples I and II are non-modified and diglycine-modified histone or rat brain lysate. The Western blot analysis demonstrates that this antibody only recognizes sample II.
(B) The anti-diglycyl lysine antibody can immunoprecipitate peptides that contain diglycine-modified lysine residues. Top panel: MALDI-TOF MS of the mixture of a five-lysine-containing peptide and its diglycine-modified forms prior to immunopurification. In this panel, the non-modified and modified peptides are present in a similar abundance. Bottom panel: MALDI-TOF MS of anti-diglycyl lysine antibody purified peptides. In this panel, only the peptides with diglycine-modified lysines are retained.
(C) The anti-diglycyl lysine antibody can be used to isolate diglycine-lysine containing peptides from a complex peptide mixture. Lysine residues in bovine serum albumin were partially modified with diglycine and digested with trypsin. The diglycine-modified peptides are purified and identified by LC-MS/MS. The peptide sequence and the diglycine modification can be determined by the MS/MS spectra as shown in the two examples above. The symbols, \, / and |, represent b-ions, y-ions, and both b-ions and y-ions, respectively.
HIGH-THROUGHPUT IDENTIFICATION OF UBIQUITINATION SITES BY UBIQUITIN REMNANT PROFILING
Immunoprecipitation of Ubiquitin Remnant-Containing Peptides via Anti-Diglycyl Lysine Antibodies
The first use of anti-diglycyl lysine antibodies to perform ubiquitin remnant profiling was described by Xu et al. (Xu et al., 2010). In this study, proteins are extracted from cell lysates and digested with trypsin. Ubiquitin remnant-containing peptides are enriched by immunoprecipitation with anti-diglycyl lysine antibodies. The identification of these peptides is achieved by detecting a 114.04 Da adduct on the modified lysine residues. This strategy is termed as “ubiquitin remnant profiling”, which can significantly improve the identification of ubiquitination sites from complex protein samples (Fig. 7). Subsequently, this approach was used by numerous other groups. These studies identified more than twenty thousand ubiquitination sites in mammalian cell lines (Emanuele et al., 2011; Kim et al., 2011b; Lee et al., 2011; Na et al., 2012; Udeshi et al., 2012; Wagner et al., 2011; Xu et al., 2010). Together, these studies demonstrated that ubiquitin remnant profiling is a powerful tool for the identification of ubiquitination events and can significantly advance the understanding of protein ubiquitination.
Figure 7. The ubiquitin remnant profiling technique for proteome-wide profiling of ubiquitination sites.

Proteins extracted from cell lysates or tissues are digested with trypsin. The ubiquitin remnant-containing peptides are immunoprecipitated using anti-diglycyl lysine antibodies and analyzed by tandem MS. The presence of the diglycine remnant on lysine residues can be identified by the 114.04 Da change in the monoisotopic mass between the modified and non-modified y-ions or b-ions.
Using High Resolution Mass Spectrometers to Reduce False Positive Identification of Ubiquitination Sites
It is well known that the assignment of ubiquitin modification sites is notoriously difficult because many other chemical moieties are isobaric to the 114 Da diglycine modification. Many modifications, such as hydoxyprolyl, isoleucyl, leucyl, norleucyl, asparagyl, ornithyl, and aspartyl, are isobaric to diglycine modification within a 2.0 Da window of mass accuracy (Table 3). Many of them cannot be distinguished by the detection of peptide masses if an ion-trap mass spectrometer is used, which has a 2.0 Da window of mass accuracy. Therefore, previously identified ubiquitination sites using ion-trap mass spectrometers with low mass accuracy have to be carefully verified for further functional studies. High resolution mass spectrometers, such as Q-TOF or Orbitrap with a mass accuracy of below 10 ppm or 1 ppm, respectively, are required for the confident identification of ubiquitination sites. These instruments can significantly reduce the false discovery rate. Using these instruments, only two isobaric modifications, Asn and dicarbamidomethyl modification, cannot be completely excluded.
Table 3.
Possible modifications isobaric to the diglycine modification.
| # | Modification | Modification Site |
Formula | Monoisotopic mass (Da) |
Structure |
|---|---|---|---|---|---|
| 1 | Acrolein112 | Lys and N-term | C6H8O2 | 112.05243 | 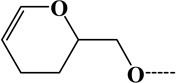 |
| 2 | Hydroxyproline | Pro | C5H7O2N | 113.04767 | 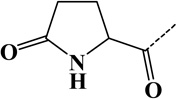 |
| 3 | Acetylhypusine | Lys | C6H11ON | 113.08406 | 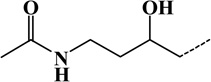 |
| 4 | Ile | - | C6H11ON | 113.08406 | 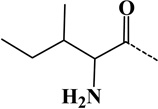 |
| 5 | Leu | - | C6H11ON | 113.08406 | 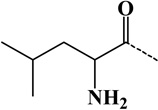 |
| 6 | norleucyl | - | C6H11ON | 113.08406 | 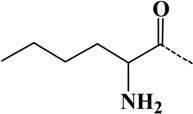 |
| 7 | GlyGly(diglycine) | Lys | C4H6O2N2 | 114.04293 | 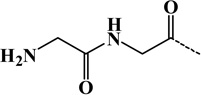 |
| 8 | Dicarbamidomethyl | Lys | C4H6O2N2 | 114.04293 | 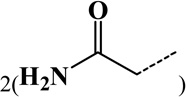 |
| 9 | Asn | - | C4H6O2N2 | 114.04293 | 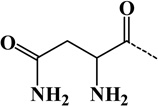 |
| 10 | t-Amyloxycarbonyl | Amine | C6H10O2 | 114.06807 | 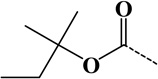 |
| 11 | Ornithyl | Amine | C5H10ON2 | 114.07930 | 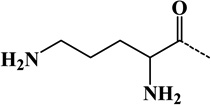 |
| 12 | Asp | - | C4H5O3N | 115.02694 | 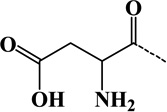 |
| 13 | Ala→Trp | Ala | C8H5N | 115.04220 | 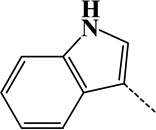 |
| 14 | 2-Succinyl | Cys | C4H4O4 | 116.01096 | 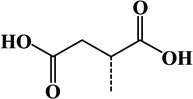 |
The dashed line at the right side of the structure is covalently linked to the modification site.
The modifications for 4, 5, 6, 9, and 12 can be at any position in the modified peptides and the amino group can be used to form a peptide bond with neighboring amino acid in the peptides.
Improvement in MS Database Search and Sample Preparation to Reduce Isobaric Modification
Two situations can cause modifications with exactly the same mass addition to diglycine modification. One is the presence of Asn in the proximity of lysine residues. Database search in proteomics usually provides multiple best matches for the MS/MS spectra. If the identified ubiquitin remnant-containing peptides have corresponding sequences which have an Asn residue close to the modified lysine residue, these sequences will also be reported and will have similar or better scores as those of peptides with diglycine modifications. Therefore, this situation can be readily identified through the careful examination of other best matches in the search results.
Another modification isobaric to diglycine modification is the dicarbamidomethyl modification on lysine residues, which can be introduced during the alkylation step prior to trypsin digestion. At high temperatures, such as 37 °C, and high concentrations, such as 50 mM, of common alkylating agent, iodoacetamide, lysines could be artificially modified by two acetamide moieties (Fig. 8). This modification has the same chemical formula and molecular weight as the diglycine modification and could be misinterpreted as a diglycine modification (Nielsen et al., 2008). Decreasing the reaction temperature to 21 °C and reducing the concentration of iodoacetamide to 10–30 mM (Bustos et al., 2012; Na et al., 2012) or replacing iodoacetamide with a more specific alkylating agent, chloroacetamide (Nielsen et al., 2008), can reduce this artificial modification to the undetected level. In addition, the anti-diglycyl lysine antibody can enrich for ubiquitin remnant-containing peptides >1000 times more than the dicarbamidomethyl modification, which can significantly reduce the presence of this artificial modification in the purified peptide mixture (Bustos et al., 2012; Na et al., 2012). Therefore, ubiquitin remnant profiling can significantly reduce the misassignment of modifications isobaric to the diglycine modification. We expect this would hold true for other modifications isobaric to the diglycine modification because they have significant differences in their structures (Table 3). In summary, the ubiquitin remnant profiling technique can significantly reduce the false positive hits stemmed from other isobaric modifications.
Figure 8. Chemical structure of ubiquitin remnant-containing peptides (A) and pseudo-ubiquitin remnant-containing peptides (B).

The ubiquitin remnant-containing peptides are mainly derived from trypsin digestion of ubiquitinated proteins. The pseudo-ubiquitin remnant-containing peptides are potentially introduced during the alkylation of thiols with iodoacetamide at high concentrations and high temperatures. Both adducts have same chemical formula but different chemical structures. The anti-diglycyl lysine antibody can recognize and immunoprecipitate peptide A but not peptide B. In addition, the formation of pseudo-ubiquitin remnant-containing peptides during sample preparation can be eliminated when the alkylation step is carried out at low concentrations of iodoacetamide and at low temperatures. Alternatively, the replacement of iodoacetamide with a more specific alkylating agent, chloroacetamide, can prevent the generation of pseudo-ubiquitin remnant-containing peptides.
Identification of Peptides with a Diglycine-Modified C-Terminal Lysine
In general, trypsin does not cleave the peptide bonds after the lysine which has been modified by ubiquitin (Peng et al., 2003). However, several situations can generate peptides with the C-terminal diglycine modification. The first one is that a protein ends with a lysine at its C-terminus and this C-terminal lysine is ubiquitinated. Indeed, Peng and coworkers detected three C-terminal diglycine-modified peptides from rat brain lysate (Na et al., 2012). In the second situation, peptides containing multiple lysine residues at the C-terminus are ubiquitinated and the diglycine modifications are assigned to the C-terminus of the peptides by database search algorithm. In this case, the database search program may have preference to assign the modified lysine to the C-terminal amino acids when multiple positive charges are present, which generate more complicated MS/MS spectra. Manual inspection of the MS/MS spectra may be required to correctly locate the true ubiquitinated lysine. Indeed, Peng and coworkers found that most of the C-terminal diglycine-modified peptides fall into this category (Na et al., 2012). New computational algorithm which can precisely locate the ubiquitination site for peptides with multiple lysine residues will improve the identification. The third possibility is that the proteins are cleaved after lysine residues by active proteases in cells and this terminal lysine is ubiquitinated as well. Therefore, a diglycine-modified C-terminal lysine is formed after trypsin digestion.
Proteomic Techniques for the Improvement in the Identification of Ubiquitination Sites
Because trypsin usually does not cleave peptide bonds after the ubiquitinated lysine residues, ubiquitin remnant-containing peptides, on average, are longer than regular tryptic peptides. In addition, these peptides have an additional positive charge which is located on the glycine amine on the ubiquitin remnant. This charge is in additional to the N-terminal amine and the amine at the C-terminus of lysine or arginine (Note: peptides derived from the C-terminus of a protein may not have this charge). In general, these peptides can bear three or more positive charges in electrospray ionization. Hence, traditional CID may not be the best fragmentation method to generate high quality MS/MS spectra for confident identification of long ubiquitin remnant-containing peptides. Instead, electron transfer dissociation (ETD) can perform better fragmentation for long peptides with multiple positive charges (Wiesner et al., 2008). Selective fragmentation of > +2 charged precursor ions may result in the identification of more ubiquitinated peptides (Wagner et al., 2011). High energy dissociation (HCD) generates peptide fragments with high resolution and high mass accuracy, which allows for their unambiguous assignment during database search. These three peptide fragmentation methods are complementary (Shen et al., 2011) and simultaneous use of them could result in the identification of more ubiquitinated peptides.
Protein databases contain limited number of naturally occurring protein variants. However, many ubiquitinated proteins are mistranslated and cannot fold properly. These proteins are ubiquitinated and degraded by the proteasome. De novo sequencing of non-matched MS/MS spectra may improve the identification of ubiquitination events whose actual amino acid sequence is not present in the databases.
A Limitation of the Ubiquitin Remnant Profiling Approach
Two ubiquitin-like modifiers, NEDD8 and ISG15, also have a C-terminal sequence of Arg-Gly-Gly and their C-termini can be conjugated to the lysine residues in target proteins. After trypsin digestion of these modified proteins, the resulting modified peptides are indistinguishable from ubiquitin remnant-containing peptides. However, ISG15 and NEDD8-mediated modifications in cells seem to be restricted to a small number of specific subsets of proteins. For instance, the expression of ISG15 is triggered by type I interferon, and the ISG15 level is very low under normal cell culture conditions (Zhang and Zhang, 2011). NEDD8 mainly modifies cullin family proteins (Hori et al., 1999; Rabut and Peter, 2008) although it has been reported that it can also covalently modify other proteins (Xirodimas, 2008; Xirodimas et al., 2004; Xirodimas et al., 2008). Therefore, these ubiquitin-like modifications only account for a very small percentage of diglycine-modified peptides (Bustos et al., 2012; Kim et al., 2011b). Alternatively, an enrichment step which isolates ubiquitinated proteins before trypsin digestion can solve this problem (Xu et al., 2010). To do this, one of the previously mentioned approaches for the isolation of ubiquitinated proteins can be applied.
In summary, the ubiquitin remnant profiling approach can significantly enrich diglycine-modified peptides from a complex tryptic digest and can reduce the false discovery rate in the identification of protein ubiquitination sites.
APPLICATIONS OF THE UBIQUITIN REMNANT PROFILING TECHNIQUE
The ubiquitin remnant profiling technique can significantly improve the efficiency of the identification of ubiquitination events in a high-throughput manner. Since its inception, this approach has been utilized to profile ubiquitination events that occur in signaling pathways or that are regulated by specific regulatory enzymes participated in the ubiquitin proteasome system (UPS). The resulting datasets from this approach have led to the finding that ubiquitination is the second most highly documented PTM after phosphorylation (Wagner et al., 2011). Here we briefly discuss the various applications of this technique.
Discovery of the Diverse Regulatory Functions of Protein Ubiquitination
It is well known that protein ubiquitination plays roles in almost every aspect of cellular processes. However, there was no high-throughput approach to quickly distinguish which ubiquitination events have regulatory functions in cell signaling. Wagner et al. (Wagner et al., 2011) and Kim et al. (Kim et al., 2011b) used the ubiquitin remnant profiling approach to study protein ubiquitination in human cell lines and identified more than 20,000 diglycine-modification sites, the majority of which are derived from ubiquitinated proteins. In these datasets, almost half of the ubiquitination sites have non-degradative functions and the ubiquitination levels of many sites are dramatically decreased after proteasome inhibition. They also discovered that a significant number of proteins are ubiquitinated on multiple lysine residues and that the ubiquitination status of each lysine residue in one protein can be independently regulated, which is consistent with a previous finding (Xu et al., 2010). These results suggested that different ubiquitination sites in the same protein may have completely different functions.
Using Gene Ontology (GO) annotation, Wagner et al. further confirmed that ubiquitinated proteins have diverse cellular functions, such as cell signaling, receptor endocytosis, DNA replication, DNA damage repair, and cell cycle progression (Wagner et al., 2011). Identification of ubiquitination of cell surface proteins would be critical to study cell signaling pathways (Hicke, 1999). Wagner et al. discovered the ubiquitination of many cell surface receptors, which are involved in a variety of signaling pathways, such as Wnt, Notch, and G-protein coupled receptor signaling. Ubiquitination was also detected on many cytoplasmic tyrosine and serine threonine kinases which are downstream regulators of cell surface receptors. These discoveries can potentially elucidate the ubiquitination signaling pathways in signal transduction.
Another remarkable finding from these experiments was that the majority of ubiquitination events occur on newly synthesized proteins (Kim et al., 2011b), which reflects “quality control” ubiquitination events. These ubiquitination events serve to remove nascent proteins that are not properly folded. The nascent misfolded proteins are ubiquitinated and degraded through the UPS. Unlike quality control ubiquitination events, regulatory ubiquitination events have roles in cellular signaling pathways. To distinguish regulatory ubiquitination events from quality control events, Kim et al. (Kim et al., 2011b) used an inhibitor to block protein synthesis. They found that more than 70% proteins had a significant drop in the ubiquitination level, which suggests that these ubiquitination events are likely to occur on newly synthesized proteins and therefore are quality control events.
Profiling Ubiquitination Events that are Regulated by Specific E3 Ligases
One important question in the ubiquitination field is to identify the substrates for specific E3 ligases. Several strategies have been developed for this purpose. One strategy uses protein microarray technology to detect the ubiquitin signal after the conjugation of ubiquitin molecules to the immobilized substrates in the presence of an E3 ligase in vitro (Andrews et al., 2009; Gupta et al., 2007; Loch et al., 2011; Lu et al., 2008). A second strategy measures the change of protein stability after the alteration of a specific E3 ligase activity in cells to identify the putative substrates of a specific E3 ligase (Burande et al., 2009; Hor et al., 2009; Yen and Elledge, 2008). The third approach is to use tagged ubiquitin to isolate ubiquitinated proteins after the alteration in the expression of E3 ligases and then to quantify their relative abundance by quantitative proteomics (Song et al., 2011). However, none of these approaches directly measures the change of ubiquitination levels for the ubiquitination sites upon the reduction of the E3 ligase activity in vivo.
The ubiquitin remnant profiling technique can directly determine the substrates of E3 ligases. Kim et al. (Kim et al., 2011b) and Emanuele et al. (Emanuele et al., 2011) both identified substrates of cullin RING ligases (CRLs) using ubiquitin remnant profiling and quantitative proteomics for the samples in the presence and absence of an inhibitor of CRLs, MLN4924. This drug blocks neddylation, which activates CRLs. These experiments identified hundreds of proteins that are candidate substrates for CRLs. It is also valuable to identify the substrates for a specific E3 ligase, which can determine the specific targets of this E3 ligase and evaluate whether it would be a potential therapeutic target. Using quantitative proteomics, Lee et al. (Lee et al., 2011) identified more than one hundred substrates and their ubiquitination sites for HRD1 (synoviolin), a RING domain-containing E3 ligase, which is linked to rheumatoid arthritis and other human diseases.
Deciphering the Ubiquitination Events Regulated by DUBs
The ubiquitin remnant profiling approach provides a convenient means to directly identify DUB targets. Udeshi et al. (Udeshi et al., 2012) applied a general DUB inhibitor, PR-619, to suppress the activities of all DUBs in human T cell leukemia cells, and used ubiquitin remnant profiling to perform the first comprehensive proteomic profiling for DUB substrates. Upon DUB inhibition, their substrates are expected to exhibit increased levels of ubiquitination. Indeed, PR-619 treatment profoundly affected the ubiquitination landscape and ubiquitin remnant profiling identified hundreds of DUB substrates. The results of this study suggested that DUBs play broad roles in regulating ubiquitination processing and that ubiquitin remnant profiling can discover specific targets of DUBs.
FUTURE APPLICATIONS OF THE UBIQUITIN REMNANT PROFILING APPROACH
Determining Ubiquitination Events with Specific Ubiquitin Chain Linkages
The function of protein ubiquitination is closely linked to the type of ubiquitination and the specific polyubiquitin chain linkage (Xu et al., 2009). Therefore, it is very valuable to determine the ubiquitination events which are modified by different types of ubiquitin chain linkages and to understand their dynamic regulation in cell signaling pathways. It is expected that with the development of new separation techniques to isolate ubiquitinated proteins with different ubiquitin chain linkages by either linkage-specific antibodies or UBDs, ubiquitination events with different ubiquitin chain linkages can be determined using ubiquitin remnant profiling.
Identifying Ubiquitination Events that are Induced by Cell Signaling Pathways
Many ubiquitination events are triggered by the activation of cell signaling pathways. Indeed, a proteomic experiment, which determined the ubiquitinated proteins without any information about the ubiquitination sites, has been carried out to study the ubiquitin network under EGF signaling (Argenzio et al., 2011). By performing ubiquitin remnant profiling in the presence or absence of a signaling molecule, ubiquitination events elicited during signaling can be readily identified. In fact, many cell surface signaling proteins were identified to be ubiquitinated by ubiquitin remnant profiling (Wagner et al., 2011).
Unraveling the Interacting Network in the UPS
Bioinformatics analyses of the human genome have discovered hundreds of ubiquitin regulatory enzymes that appear to influence cellular ubiquitination pathways. An E1 enzyme can activate and transfer ubiquitin to tens of E2 enzymes and each E2 enzyme can form different protein-protein interaction complexes with multiple E3 ligases to mediate target ubiquitination. Each E3 ligase may be able to interact with and ubiquitinate many protein substrates. Moreover, a protein could be ubiquitinated by several E3 ligases and the ubiquitination can be reversed by DUBs. Therefore, these enzymes and their substrates form a complex and dynamically regulated interacting network. Identifying ubiquitinated proteins and their ubiquitination sties for specific E3 ligases will help to determine whether a ubiquitination event is regulated by multiple E3 ligases and to understand their biological functions and their roles in disease progression and in drug development. The ubiquitin remnant profiling technique can identify these events in a high-throughput manner and can decipher the complex interacting network in the UPS. This information can help to assess which specific E3 ligase could be a potential drug target or whether targeting multiple E3 ligases would benefit disease treatment.
Elucidating the Crosstalk between Ubiquitination and other PTMs
Acetyltransferases introduce acetyl groups on lysine resides, which may therefore prevent ubiquitination on the same residue (Arif et al., 2010; Iyer et al., 2011). Among previously identified lysine acetylated sites (Choudhary et al., 2009), about 24% are also the sites of ubiquitination, which indicates the potential large-scale crosstalk between these two PTMs. In addition, many ubiquitin-like modifiers, such as SUMO, ISG15, and NEDD8, also utilize lysine residues as their modification sites. The function of these modifications is very different and each of these modifications on the same lysine residue can lead to different effects on target proteins. For example, SUMOylation of lysine 164 in proliferating cell nuclear antigen enhances its stability while ubiquitination of this residue is involved in DNA repair (Hoege et al., 2002). In addition, some PTMs, such as phosphorylation, are required for protein ubiquitination (Harper, 2002; Kong and Chock, 1992). It will be very interesting to investigate the dynamic regulation of different types of PTMs and to determine how specific PTMs regulate the extent of ubiquitination in signaling pathways.
Conclusions
Mass spectrometric analysis has become the most commonly used technique to study protein PTMs and to precisely locate the sites of modification. The identification of modification sites allows researchers to create modification-resistant mutants for further functional analysis of the modification. Ubiquitination is one of the most difficult PTMs to be identified due to the size, low abundance, and dynamic regulation of the modification. The ubiquitin remnant profiling technique initially developed in the Jaffrey laboratory has significantly improved the efficiency in the identification of ubiquitination events. By incorporating other sample preparation techniques and quantitative proteomics, the complexity of protein ubiquitination in cell signaling and disease states will become gradually elucidated, which will help to decipher the roles of this PTM in disease progression and in drug discovery.
Acknowledgement
The work was supported by the National Natural Science Foundation of China (Grant 31270874), start-up funding from Soochow University (GX), a project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions (GX), NIH-NIMH (MH086128, S.R.J.), and NIH-NCI (T32CA062948, G.X.).
Abbreviations
- PTM
post-translational modification
- DUB
deubiquitinating enzyme
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- Q-TOF
quadrupole time-of-flight
- LTQ
linear trap quadrupole
- ESI
electrospray ionization
- LC
liquid chromatography
- CID
collision induced dissociation
- ETD
electron transfer dissociation
- HCD
high energy dissociation
- His6
hexahistindine
- BAP
biotin acceptor peptide
- GST
glutathione S-transferase
- TEV
tobacco etch virus
- TUBE
tandem ubiquitin binding entity
- CNBr
cyanogen bromide
- NTCB
2-nitro-5-thiocyanobenzoic acid
- SPITC
4-sulfophenyl isothiocyanate
- UBD
ubiquitin-binding domain
- UBA
ubiquitin-associating domain
- UIM
ubiquitin-interacting motif
- KLH
keyhole limpet hemocyanin
- EGF
epidermal growth factor
- PS
ubiquitin proteasome system
- RING
really interesting new gene
- CRL
cullin RING ligase
References
- Ahmed N, Zeng M, Sinha I, et al. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nature Immunology. 2011;12:1176–1183. doi: 10.1038/ni.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochimica et Biophysica Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Andrews PS, Schneider S, Yang E, et al. Identification of substrates of SMURF1 ubiquitin ligase activity utilizing protein microarrays. Assay and Drug Development Technologies. 2009;8:471–487. doi: 10.1089/adt.2009.0264. [DOI] [PubMed] [Google Scholar]
- Argenzio E, Bange T, Oldrini B, et al. Proteomic snapshot of the EGF-induced ubiquitin network. Molecular Systems Biology. 2011;7:462. doi: 10.1038/msb.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif M, Senapati P, Shandilya J, et al. Protein lysine acetylation in cellular function and its role in cancer manifestation. Biochimica et Biophysica Acta. 2010;1799:702–716. doi: 10.1016/j.bbagrm.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Baboshina OV, Haas AL. Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2EPF and RAD6 are recognized by 26 S proteasome subunit 5. Journal of Biological Chemistry. 1996;271:2823–2831. doi: 10.1074/jbc.271.5.2823. [DOI] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Science. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nature Structural and Molecular Biology. 2011;18:520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Truax AD, Greer SF. Phosphorylation and ubiquitination of degron proximal residues are essential for class II transactivator (CIITA) transactivation and major histocompatibility class II expression. Journal of Biological Chemistry. 2010;285:25893–25903. doi: 10.1074/jbc.M110.127746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitschopf K, Bengal E, Ziv T, et al. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO Journal. 1998;17:5964–5973. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burande CF, Heuze ML, Lamsoul I, et al. A label-free quantitative proteomics strategy to identify E3 ubiquitin ligase substrates targeted to proteasome degradation. Molecular and Cellular Proteomics. 2009;8:1719–1727. doi: 10.1074/mcp.M800410-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos D, Bakalarski CE, Yang Y, et al. Characterizing ubiquitination sites by peptide based immunoaffinity enrichment. Molecular and Cellular Proteomics. 2012;11 doi: 10.1074/mcp.R112.019117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- Carrington JC, Cary SM, Parks TD, et al. A second proteinase encoded by a plant potyvirus genome. EMBO Journal. 1989;8:365–370. doi: 10.1002/j.1460-2075.1989.tb03386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Cole GM, Timiras PS. Ubiquitin-protein conjugates in Alzheimer's lesions. Neuroscience Letters. 1987;79:207–212. doi: 10.1016/0304-3940(87)90698-7. [DOI] [PubMed] [Google Scholar]
- Cooper HJ, Heath JK, Jaffray E, et al. Identification of sites of ubiquitination in proteins: a fourier transform ion cyclotron resonance mass spectrometry approach. Analytical Chemistry. 2004;76:6982–6988. doi: 10.1021/ac0401063. [DOI] [PubMed] [Google Scholar]
- Cotter RJ, Iltchenko S, Wang D, et al. Tandem time-of-flight (TOF/TOF) mass spectrometry and proteomics. Journal of the Mass Spectrometry Society of Japan. 2005;53:7–17. doi: 10.5702/massspec.53.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe P, Rodier G, Bonneil E, et al. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Molecular and Cellular Biology. 2004;24:6140–6150. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE., Jr Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. Journal of Biological Chemistry. 1990;265:10327–10333. [PubMed] [Google Scholar]
- Danielsen JM, Sylvestersen KB, Bekker-Jensen S, et al. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Molecular and Cellular Proteomics. 2011;10 doi: 10.1074/mcp.M110.003590. M110 003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, et al. Activation of the κB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Denis NJ, Vasilescu J, Lambert JP, et al. Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics. 2007;7:868–874. doi: 10.1002/pmic.200600410. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano MJ. Antibodies of restricted heterogeneity obtained in rabbits immunized with tetraglycine peptide conjugated to an autologous carrier. European Journal of Immunology. 1974;4:793–798. [Google Scholar]
- Franco M, Seyfried NT, Brand AH, et al. A novel strategy to isolate ubiquitin conjugates reveals wide role for ubiquitination during neural development. Molecular and Cellular Proteomics. 2010;10 doi: 10.1074/mcp.M110.002188. M110 002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimuro M, Sawada H, Yokosawa H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Letters. 1994;349:173–180. doi: 10.1016/0014-5793(94)00647-4. [DOI] [PubMed] [Google Scholar]
- Fujimuro M, Sawada H, Yokosawa H. Dynamics of ubiquitin conjugation during heat-shock response revealed by using a monoclonal antibody specific to multi-ubiquitin chains. European Journal of Biochemistry. 1997;249:427–433. doi: 10.1111/j.1432-1033.1997.00427.x. [DOI] [PubMed] [Google Scholar]
- Gerlach B, Cordier SM, Schmukle AC, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM. Are ubiquitination pathways central to Parkinson's disease? Cell. 2003;114:1–8. doi: 10.1016/s0092-8674(03)00509-9. [DOI] [PubMed] [Google Scholar]
- Goldknopf IL, Busch H. Remarkable similarities of peptide fingerprints of histone 2A and nonhistone chromosomal protein A24. Biochemical and Biophysical Research Communications. 1975;65:951–960. doi: 10.1016/s0006-291x(75)80478-5. [DOI] [PubMed] [Google Scholar]
- Goldknopf IL, Busch H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:864–868. doi: 10.1073/pnas.74.3.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldknopf IL, Taylor CW, Baum RM, et al. Isolation and characterization of protein A24, a "histone-like" non-histone chromosomal protein. Journal of Biological Chemistry. 1975;250:7182–7187. [PubMed] [Google Scholar]
- Gregori L, Poosch MS, Cousins G, et al. A uniform isopeptide-linked multiubiquitin chain is sufficient to target substrate for degradation in ubiquitin-mediated proteolysis. Journal of Biological Chemistry. 1990;265:8354–8357. [PubMed] [Google Scholar]
- Gross E, Witkop B. Nonenzymatic cleavage of peptide bonds: the methionine residues in bovine pancreatic ribonuclease. Journal of Biological Chemistry. 1962;237:1856–1860. [PubMed] [Google Scholar]
- Gupta R, Kus B, Fladd C, et al. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Molecular Systems Biology. 2007;3:116. doi: 10.1038/msb4100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AL, Bright PM. The immunochemical detection and quantitation of intracellular ubiquitin-protein conjugates. Journal of Biological Chemistry. 1985;260:12464–12473. [PubMed] [Google Scholar]
- Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO Journal. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW. A phosphorylation-driven ubiquitination switch for cell-cycle control. Trends in Cell Biology. 2002;12:104–107. doi: 10.1016/s0962-8924(01)02238-3. [DOI] [PubMed] [Google Scholar]
- Hashizume R, Fukuda M, Maeda I, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. Journal of Biological Chemistry. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- Herrmann J, Ciechanover A, Lerman LO, et al. The ubiquitin-proteasome system in cardiovascular diseases-a hypothesis extended. Cardiovascular Research. 2004;61:11–21. doi: 10.1016/j.cardiores.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annual Review of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hicke L. Gettin' down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends in Cell Biology. 1999;9:107–112. doi: 10.1016/s0962-8924(98)01491-3. [DOI] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, et al. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Reports. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Rodriguez MS. Efficient approaches for characterizing ubiquitinated proteins. Biochemical Society Transactions. 2008;36:823–827. doi: 10.1042/BST0360823. [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, et al. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nature Reviews Cancer. 2006;6:776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Hor S, Ziv T, Admon A, et al. Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantitation identify new substrates for the MARCH9 transmembrane E3 ligase. Molecular and Cellular Proteomics. 2009;8:1959–1971. doi: 10.1074/mcp.M900174-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Osaka F, Chiba T, et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18:6829–6834. doi: 10.1038/sj.onc.1203093. [DOI] [PubMed] [Google Scholar]
- Hou D, Cenciarelli C, Jensen JP, et al. Activation-dependent ubiquitination of a T cell antigen receptor subunit on multiple intracellular lysines. Journal of Biological Chemistry. 1994;269:14244–14247. [PubMed] [Google Scholar]
- Huang F, Kirkpatrick D, Jiang X, et al. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Molecular Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Hunt LT, Dayhoff MO. Amino-terminal sequence identity of ubiquitin and the nonhistone component of nuclear protein A24. Biochemical and Biophysical Research Communications. 1977;74:650–655. doi: 10.1016/0006-291x(77)90352-7. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochemical Journal. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, Fairlie DP, Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunology and Cell Biology. 2011;90:39–46. doi: 10.1038/icb.2011.99. [DOI] [PubMed] [Google Scholar]
- Jadhav T, Wooten MW. Defining an embedded code for protein ubiquitination. Journal of Proteomics and Bioinformatics. 2009;2:316. doi: 10.4172/jpb.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon HB, Choi ES, Yoon JH, et al. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochemical and Biophysical Research Communications. 2007;357:731–736. doi: 10.1016/j.bbrc.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Kim JY, Anderson ED, Huynh W, et al. Proteomic survey of ubiquitin-linked nuclear proteins in interferon-stimulated macrophages. Journal of Interferon and Cytokine Research. 2011a;31:619–628. doi: 10.1089/jir.2011.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011b;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Weldon SF, Tsaprailis G, et al. Proteomic identification of ubiquitinated proteins from human cells expressing His-tagged ubiquitin. Proteomics. 2005;5:2104–2111. doi: 10.1002/pmic.200401089. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kong SK, Chock PB. Protein ubiquitination is regulated by phosphorylation. An in vitro study. Journal of Biological Chemistry. 1992;267:14189–14192. [PubMed] [Google Scholar]
- Kundrat L, Regan L. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. Journal of Molecular Biology. 2010;395:587–594. doi: 10.1016/j.jmb.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Hong US, Lee TH, et al. Mass spectrometric analysis of tumor necrosis factor receptor-associated factor 1 ubiquitination mediated by cellular inhibitor of apoptosis 2. Proteomics. 2004;4:3376–3382. doi: 10.1002/pmic.200401000. [DOI] [PubMed] [Google Scholar]
- Lee KA, Hammerle LP, Andrews PS, et al. Ubiquitin ligase substrate identification through quantitative proteomics at both the protein and peptide levels. Journal of Biological Chemistry. 2011;286:41530–41538. doi: 10.1074/jbc.M111.248856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tsai YC, Mattera R, et al. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nature Structural and Molecular Biology. 2006;13:264–271. doi: 10.1038/nsmb1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RF, Shang Y, Liu D, et al. Differential ubiquitination of Smad1 mediated by CHIP: implications in the regulation of the bone morphogenetic protein signaling pathway. Journal of Molecular Biology. 2007;374:777–790. doi: 10.1016/j.jmb.2007.09.082. [DOI] [PubMed] [Google Scholar]
- Loch CM, Eddins MJ, Strickler JE. Protein microarrays for the identification of praja1 e3 ubiquitin ligase substrates. Cell Biochemistry and Biophysics. 2011;60:127–135. doi: 10.1007/s12013-011-9180-x. [DOI] [PubMed] [Google Scholar]
- Lu JY, Lin YY, Qian J, et al. Functional dissection of a HECT ubiquitin E3 ligase. Molecular and Cellular Proteomics. 2008;7:35–45. doi: 10.1074/mcp.M700353-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. New England Journal of Medicine. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Maine GN, Gluck N, Zaidi IW, et al. Bimolecular affinity purification (BAP): Tandem affinity purification using two protein baits. Cold Spring Harbor Protocols. 2009 doi: 10.1101/pdb.prot5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature Biotechnology. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- Maor R, Jones A, Nuhse TS, et al. Multidimensional protein identification technology (MudPIT) analysis of ubiquitinated proteins in plants. Molecular and Cellular Proteomics. 2007;6:601–610. doi: 10.1074/mcp.M600408-MCP200. [DOI] [PubMed] [Google Scholar]
- Marotti LA, Jr, Newitt R, Wang Y, et al. Direct identification of a G protein ubiquitination site by mass spectrometry. Biochemistry. 2002;41:5067–5074. doi: 10.1021/bi015940q. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hatakeyama S, Oyamada K, et al. Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 2005;5:4145–4151. doi: 10.1002/pmic.200401280. [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Molecular Cell. 2010;39:477–484. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Meierhofer D, Wang X, Huang L, et al. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. Journal of Proteome Research. 2008;7:4566–4576. doi: 10.1021/pr800468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Sorkin A. Regulation of receptors and transporters by ubiquitination: new insights into surprisingly similar mechanisms. Molecular Interventions. 2007;7:157–167. doi: 10.1124/mi.7.3.7. [DOI] [PubMed] [Google Scholar]
- Moren A, Hellman U, Inada Y, et al. Differential ubiquitination defines the functional status of the tumor suppressor Smad4. Journal of Biological Chemistry. 2003;278:33571–33582. doi: 10.1074/jbc.M300159200. [DOI] [PubMed] [Google Scholar]
- Mori S, Claesson-Welsh L, Okuyama Y, et al. Ligand-induced polyubiquitination of receptor tyrosine kinases. Biochemical and Biophysical Research Communications. 1995;213:32–39. doi: 10.1006/bbrc.1995.2094. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Na CH, Jones DR, Yang Y, et al. Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. Journal of Proteome Research. 2012;11:4722–4732. doi: 10.1021/pr300536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Nielsen ML, Vermeulen M, Bonaldi T, et al. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Ono T, Wakata Y, et al. Histone variant macroH2A1.2 is mono-ubiquitinated at its histone domain. Biochemical and Biophysical Research Communications. 2005;336:204–209. doi: 10.1016/j.bbrc.2005.08.046. [DOI] [PubMed] [Google Scholar]
- Ohno A, Jee J, Fujiwara K, et al. Structure of the UBA domain of Dsk2p in complex with ubiquitin molecular determinants for ubiquitin recognition. Structure. 2005;13:521–532. doi: 10.1016/j.str.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Oshikawa K, Matsumoto M, Oyamada K, et al. Proteome-wide identification of ubiquitylation sites by conjugation of engineered lysine-less ubiquitin. Journal of Proteome Research. 2012;11:796–807. doi: 10.1021/pr200668y. [DOI] [PubMed] [Google Scholar]
- Ota K, Kito K, Iemura S, et al. A parallel affinity purification method for selective isolation of polyubiquitinated proteins. Proteomics. 2008;8:3004–3007. doi: 10.1002/pmic.200800271. [DOI] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, et al. Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell. 2006;124:1183–1195. doi: 10.1016/j.cell.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Peng J, Cheng D. Proteomic analysis of ubiquitin conjugates in yeast. Methods in Enzymology. 2005;399:367–381. doi: 10.1016/S0076-6879(05)99025-3. [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nature Biotechnology. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annual Review of Biochemistry. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Powell SR, Herrmann J, Lerman A, et al. The ubiquitin-proteasome system and cardiovascular disease. Progress in Molecular Biology and Translational Science. 2012;109:295–346. doi: 10.1016/B978-0-12-397863-9.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramstaller PP, Schlossmacher MG, Jacques TS, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Annals of Neurology. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- Rabut G, Peter M. Function and regulation of protein neddylation. 'Protein modifications: beyond the usual suspects' review series. EMBO Reports. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner H, Joazeiro CA, Hemmati D, et al. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TG, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature Protocols. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Shanklin J, Jabben M, Vierstra RD. Red light-induced formation of ubiquitin-phytochrome conjugates: Identification of possible intermediates of phytochrome degradation. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:359–363. doi: 10.1073/pnas.84.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Tolic N, Xie F, et al. Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. Journal of Proteome Research. 2011;10:3929–3943. doi: 10.1021/pr200052c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chan DW, Jung SY, et al. A data set of human endogenous protein ubiquitination sites. Molecular and Cellular Proteomics. 2011;10 doi: 10.1074/mcp.M110.002089. M110 002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature Genetics. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, et al. Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, et al. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nature Methods. 2012;9:303–309. doi: 10.1038/nmeth.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Aguiar M, Gygi SP, et al. Activated inositol 1,4,5-trisphosphate receptors are modified by homogeneous Lys-48- and Lys-63-linked ubiquitin chains, but only Lys-48-linked chains are required for degradation. Journal of Biological Chemistry. 2011;286:1074–1082. doi: 10.1074/jbc.M110.188383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokalingam S, Raghunathan G, Soundrarajan N, et al. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS One. 2012;7:e40410. doi: 10.1371/journal.pone.0040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Hakala K, Weintraub ST, et al. Quantitative proteomic identification of the BRCA1 ubiquitination substrates. Journal of Proteome Research. 2011;10:5191–5198. doi: 10.1021/pr200662b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spataro V, Norbury C, Harris AL. The ubiquitin-proteasome pathway in cancer. British Journal of Cancer. 1998;77:448–455. doi: 10.1038/bjc.1998.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starita LM, Lo RS, Eng JK, et al. Sites of ubiquitin attachment in Saccharomyces cerevisiae. Proteomics. 2012;12:236–240. doi: 10.1002/pmic.201100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagwerker C, Flick K, Cui M, et al. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Molecular and Cellular Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- Takada K, Hirakawa T, Yokosawa H, et al. Isolation of ubiquitin-E2 (ubiquitin-conjugating enzyme) complexes from erythroleukaemia cells using immunoaffinity techniques. Biochemical Journal. 2001;356:199–206. doi: 10.1042/0264-6021:3560199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Nasu H, Hibi N, et al. Immunoassay for the quantification of intracellular multi-ubiquitin chains. European Journal of Biochemistry. 1995;233:42–47. doi: 10.1111/j.1432-1033.1995.042_1.x. [DOI] [PubMed] [Google Scholar]
- Tan F, Lu L, Cai Y, et al. Proteomic analysis of ubiquitinated proteins in normal hepatocyte cell line Chang liver cells. Proteomics. 2008;8:2885–2896. doi: 10.1002/pmic.200700887. [DOI] [PubMed] [Google Scholar]
- Tang HY, Speicher DW. Identification of alternative products and optimization of 2-nitro-5-thiocyanatobenzoic acid cyanylation and cleavage at cysteine residues. Analytical Biochemistry. 2004;334:48–61. doi: 10.1016/j.ab.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Iwai K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes and Infection. 2012;14:563–572. doi: 10.1016/j.micinf.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Udeshi ND, Mani DR, Eisenhaure T, et al. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Molecular and Cellular Proteomics. 2012;11:148–159. doi: 10.1074/mcp.M111.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilescu J, Smith JC, Ethier M, et al. Proteomic analysis of ubiquitinated proteins from human MCF-7 breast cancer cells by immunoaffinity purification and mass spectrometry. Journal of Proteome Research. 2005;4:2192–2200. doi: 10.1021/pr050265i. [DOI] [PubMed] [Google Scholar]
- Vasilescu J, Zweitzig DR, Denis NJ, et al. The proteomic reactor facilitates the analysis of affinity-purified proteins by mass spectrometry: application for identifying ubiquitinated proteins in human cells. Journal of Proteome Research. 2007;6:298–305. doi: 10.1021/pr060438j. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111 013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters KJ, Kleijnen MF, Goh AM, et al. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–1777. doi: 10.1021/bi011892y. [DOI] [PubMed] [Google Scholar]
- Wang D, Kalume D, Pickart C, et al. Identification of protein ubiquitylation by electrospray ionization tandem mass spectrometric analysis of sulfonated tryptic peptides. Analytical Chemistry. 2006;78:3681–3687. doi: 10.1021/ac051904b. [DOI] [PubMed] [Google Scholar]
- Wang D, Xu W, McGrath SC, et al. Direct identification of ubiquitination sites on ubiquitin-conjugated CHIP using MALDI mass spectrometry. Journal of Proteome Research. 2005;4:1554–1560. doi: 10.1021/pr050104e. [DOI] [PubMed] [Google Scholar]
- Wang X, Herr RA, Chua WJ, et al. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. Journal of Cell Biology. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herr RA, Hansen TH. Ubiquitination of substrates by esterification. Traffic. 2012;13:19–24. doi: 10.1111/j.1600-0854.2011.01269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren MR, Parker CE, Mocanu V, et al. Electrospray ionization tandem mass spectrometry of model peptides reveals diagnostic fragment ions for protein ubiquitination. Rapid Communications in Mass Spectrometry. 2005;19:429–437. doi: 10.1002/rcm.1798. [DOI] [PubMed] [Google Scholar]
- Wiesner J, Premsler T, Sickmann A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics. 2008;8:4466–4483. doi: 10.1002/pmic.200800329. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Tashayev VL, O‘Connor LB, et al. Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry. 1995;34:14535–14546. doi: 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochemical Society Transactions. 2008;36:802–806. doi: 10.1042/BST0360802. [DOI] [PubMed] [Google Scholar]
- Xirodimas DP, Saville MK, Bourdon JC, et al. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Xirodimas DP, Sundqvist A, Nakamura A, et al. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Reports. 2008;9:280–286. doi: 10.1038/embor.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nature Biotechnology. 2010;28:868–873. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Elledge SJ. Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science. 2008;322:923–929. doi: 10.1126/science.1160462. [DOI] [PubMed] [Google Scholar]
- Young P, Deveraux Q, Beal RE, et al. Characterization of two polyubiquitin binding sites in the 26S protease subunit 5a. Journal of Biological Chemistry. 1998;273:5461–5467. doi: 10.1074/jbc.273.10.5461. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. Journal of Interferon and Cytokine Research. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]